Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis
- 1Achucarro—Basque Center for Neuroscience, Zamudio, Spain
- 2Department of Neuroscience, University of the Basque Country EHU/UPV, Leioa, Spain
- 3Ikerbasque—Basque Foundation for Science, Bilbao, Spain
- 4Neural Reconstruction Group, Institute of Reconstructive Neurobiology, University of Bonn, Bonn, Germany
Microglia are the resident brain macrophages and they have been traditionally studied as orchestrators of the brain inflammatory response during infections and disease. In addition, microglia has a more benign, less explored role as the brain professional phagocytes. Phagocytosis is a term coined from the Greek to describe the receptor-mediated engulfment and degradation of dead cells and microbes. In addition, microglia phagocytoses brain-specific cargo, such as axonal and myelin debris in spinal cord injury or multiple sclerosis, amyloid-β deposits in Alzheimer's disease, and supernumerary synapses in postnatal development. Common mechanisms of recognition, engulfment, and degradation of the different types of cargo are assumed, but very little is known about the shared and specific molecules involved in the phagocytosis of each target by microglia. More importantly, the functional consequences of microglial phagocytosis remain largely unexplored. Overall, phagocytosis is considered a beneficial phenomenon, since it eliminates dead cells and induces an anti-inflammatory response. However, phagocytosis can also activate the respiratory burst, which produces toxic reactive oxygen species (ROS). Phagocytosis has been traditionally studied in pathological conditions, leading to the assumption that microglia have to be activated in order to become efficient phagocytes. Recent data, however, has shown that unchallenged microglia phagocytose apoptotic cells during development and in adult neurogenic niches, suggesting an overlooked role in brain remodeling throughout the normal lifespan. The present review will summarize the current state of the literature regarding the role of microglial phagocytosis in maintaining tissue homeostasis in health as in disease.
Definition of Phagocytosis
Phagocytosis is a Greek-derived term which literally means the cellular process of eating. It describes the recognition, engulfment, and degradation of large (>0.5 μm), particulated organisms or structures (Mukherjee et al., 1997). Phagocytosis of invading microorganisms by immune cells was first discovered by the father of cellular immunology Ilya Metchnikoff in 1882, for which he was awarded the Nobel Prize. Most cell types, including unicellular organisms, have the capacity to phagocytose. In multicellular organisms, and more particularly in animals with a well-developed immune system, phagocytosis is mostly performed by specialized, professional phagocytes: macrophages, dendritic cells (DCs), and neutrophils. Together with inflammation, phagocytosis composes the first line of defense against multicellular organisms by the innate immune system. In jawed vertebrates, including mammals, phagocytosis also helps to initiate the more specific adaptive immune response through antigen-presentation to T lymphocytes (Litman et al., 2005). In the mammalian central nervous system (CNS), the innate immune response is orchestrated by microglia.
Microglia are the brain resident macrophages. They are of myeloid origin and share many properties with other well-studied tissue macrophages. However, they have a different origin and are a unique macrophage cell type in the adult organism. Microglia are yolk sac-derived, invade the brain during early embryonic development and then locally proliferate in the brain (Ginhoux et al., 2010; Schulz et al., 2012). In contrast to other yolk sac-derived macrophages, they are not replaced during the postnatal period and later life by liver- or bone marrow-derived macrophages (Hoeffel et al., 2012). Cell biology of phagocytosis has been mainly established on bone marrow-derived tissue macrophages. In this review we directly extrapolate basic phagocytic mechanisms from bone marrow-derived tissue macrophages to the less-known processes of microglia, but would like to point out that these assumed similarities might not fully hold true to the yolk sac-derived microglia. Particularly, yolk sac-derived macrophages including microglia may possibly have different tasks since they are confronted with different target structures, mainly apoptotic cells during developmental tissue remodeling, while bone marrow-derived macrophages have a higher chance to be confronted with pathogens, mainly during defense against invading microbes. Overall, microglial phagocytosis is a relatively unknown process in terms of the receptors involved, the mechanisms of its execution, its beneficial or detrimental consequences, and its ultimate impact in maintaining tissue homeostasis. To further complicate the issue, macrophage phagocytosis has two main targets: dead resident cells (apoptotic or necrotic) and live invading microorganisms; whereas microglial phagocytosis appears to be adapted to the brain environment for remodeling tasks such as engulfment of synapses, axonal and myelin debris, or clearance of proteins with very high turnover such as amyloid beta (Aβ) protein.
One major limitation is that in most studies, microglial phagocytosis is assessed exclusively in vitro, utilizing a model of phagocytosis in which primary or transformed microglial cells are “fed” with a variety of targets, ranging from latex beads to primary or transformed apoptotic cells, cell debris, or Aβ. These models have proved to be extremely valuable to study the molecular pathways involved in the recognition, engulfment, and degradation of the targets. However, the lack of molecular tools to specifically block these three steps has precluded our understanding of the global impact of microglial phagocytosis in vivo. In fact, many in vivo experiments rely on the use of phagocytosis “markers” such as CD68 (ED1 or macrosialin) as a proxy. One major problem is that the function of CD68 is unknown. While located in the lysosomal compartment, anti-CD68 antibodies do not block macrophage phagocytosis in vitro (Damoiseaux et al., 1994), and macrophages from mice deficient in CD68 have no defects in phagocytosis of bacteria (Song et al., 2011). In the adult hippocampus, the expression of ED1 in microglia phagocytosing apoptotic cells is similar to non-phagocytic microglia and much lower than the expression induced by inflammatory challenge (Sierra et al., 2010). However, a higher expression of ED1 seems to correlate with the capacity of postnatal microglia to engulf synapses (Schafer et al., 2012). Furthermore, few studies have attempted to quantify the extent of microglial phagocytosis in vivo (Ashwell, 1990; Dalmau et al., 2003; Parnaik et al., 2000; Sierra et al., 2010) and many rely on a qualitative observation of microglial engulfment to determine whether microglia is phagocytosing or not.
The rapid clearance time of dead cells is likely behind this qualitative rather than quantitative assessment. In vitro, live imaging has determined that the clearance time to completely eliminate an apoptotic cell by microglia is 25–95 min in a co-culture system (Parnaik et al., 2000); in vivo, the microglial clearance time of apoptotic cells in physiological conditions has been estimated to be 70–90 min (Sierra et al., 2010). These numbers are in agreement with a general 1–2 h clearance time for macrophages (Henson and Hume, 2006). Thus, the number of apoptotic cells observed in particular time point represents only a small fraction of the actual number of apoptotic cells generated (or total number of cells which disappear). For example, if 400 cells die in a 24 h period, a clearance time of 1.5 h would mean that only 25 dead cells can be observed at any time point [for more details on estimation of the clearance time, see Barres et al. (1992); Sierra et al. (2010)]. The phagocytosis of smaller particles (synapses, axons, protein aggregates) is likely to be even faster. The promptness of microglia to eliminate cell debris is therefore an important parameter to take into account when analyzing the dynamics of cell death and phagocytosis.
In addition to the clearance time, two main parameters to quantify microglial phagocytosis of apoptotic cells in vivo using immunofluorescence and confocal microscopy have been established: the phagocytic index, or proportion of apoptotic cells which are three-dimensionally engulfed by microglia over the total number of apoptotic cells; and the phagocytic capacity, or proportion of phagocytosing microglia multiplied by the number of phagocytic pouches (i.e., number of apoptotic cells engulfed) over the total number of microglia (Sierra et al., 2010). More recently, a similar parameter was described to quantify microglial phagocytosis of synaptic inputs (allegedly, presynaptic terminals) using high resolution confocal microscopy and three-dimensional rendering to estimate the volume of internalized inputs over the total volume of microglia (Schafer et al., 2012). Similarly, microglial phagocytosis of axonal or myelin debris can be quantified using confocal microscopy to determine the percentage of microglia containing neurofilament-positive axonal material (Hosmane et al., 2012) or myelin basic protein (MBP) (Nielsen et al., 2009). The utilization of these or similar parameters is a necessary starting point to obtain a quantitative picture of microglial phagocytosis across a range of physiological and pathological conditions, which will help us to address many of the open questions. Over the next sections, we will provide an overall description of the mechanical process of phagocytosis; its beneficial and detrimental consequences; and the particular details of phagocytosis of different targets: cells and cell debris, microorganisms, tumor cells, spines, and Aβ deposits.
The Mechanics of Phagocytosis
Our current understanding of the mechanical process of phagocytosis is summarized in the three-step model: find-me, eat-me, and digest-me (Savill et al., 2002), which addresses the engulfment and degradation of apoptotic cells and microorganisms by macrophages and DCs. While it is likely that microglia utilizes similar mechanisms, direct evidence of the molecular details of these events in microglia, beyond the receptors involved in the recognition of the targets, is largely missing. In this section, we will address the featured molecular mechanisms in the process of phagocytosis by microglia (Figure 1).
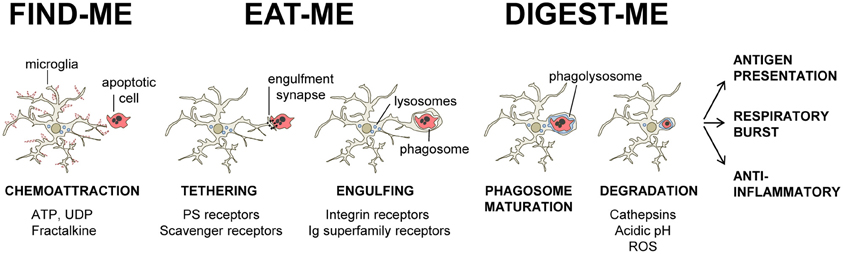
Figure 1. Three-step model of microglial phagocytosis. In physiological conditions, microglial processes are highly motile and respond to chemoattractant molecules released by damaged or apoptotic cells (“find-me” signals) such as fractalkine and extracellular nucleotides (ATP, UDP). Next, an engulfment synapse is formed between a series of microglial receptors and their ligands in the membrane of the apoptotic cell (“eat-me” signals), leading to the tethering and engulfing of the apoptotic cell in a phagosome. The phagosome becomes mature by fusing with lysosomes and other organelles, and the apoptotic cell is fully degraded in the phagolysosome in less than 2 h (see text for details).
Find-Me
The process is initiated when the phagocyte encounters a target, either randomly or triggered by signals from the target. Microglial processes are highly motile (Davalos et al., 2005; Nimmerjahn et al., 2005) and their constant surveillance of the brain parenchyma supports their capacity to fortuitously encounter targets to engulf. In addition, apoptotic cells can release signals to attract macrophages and DCs, such as the extracellular nucleotides ATP and UTP (Elliott et al., 2009). In microglia, UDP, the product of degradation of UTP by extracellular ectonucleotidases, acts on P2Y6 receptors to facilitate phagocytosis (Koizumi et al., 2007). Another important chemotactic signal released by apoptotic cells is fractalkine/CX3CL1 (Truman et al., 2008; Noda et al., 2011). Microglia express the fractalkine receptor (CX3CR1), which promotes phagocytosis of apoptotic cells (Noda et al., 2011) but shifts microglia toward a phenotype with less capacity to phagocytose Aβ (Lee et al., 2010; Liu et al., 2010). Once the phagocyte has reached the target a direct cell membrane contact is established via a receptor-ligand interaction and phagocytosis takes place.
Eat-Me
The process of recognition and engulfment of the targets is, arguably, the most important step in phagocytosis and where most research is concentrated. Phagocytes are equipped with a likely complementary array of receptors which enable them to recognize their targets (the so-called “eat-me” signals) and discriminate them from the remaining parenchyma, particularly from living cells (which express “don't-eat-me” signals) (Savill et al., 2002; Ravichandran, 2010). Some of these receptors serve to tether the phagocyte and the target together; others actually trigger internalization (Underhill and Goodridge, 2012). These receptors can be classified into two main types depending on their targets. Detection of pathogen-associated molecular patterns (PAMPS) is mediated through scavenger receptors in conjunction with Toll-like receptors (TLRs) such as the CD14/TLR4 complex, or receptors of the immunoglobulin superfamily (e.g., c-type lectins). Detection of apoptotic cells-associated cellular patterns (ACAMPs), of which exposure of phosphatidylserine (PS) in the outer leaflet of the cell membrane is the main exponent, is mediated directly by several receptors, including brain-specific angiogenesis inhibitor 1 [BAI-1; reviewed in Armstrong and Ravichandran (2011)], and by bridging molecules such as milk fat globule-epidermal growth factor (MFG-E8). Another important receptor that signals internalization is triggering receptor expressed on myeloid cells-2 (TREM2), whose loss of function prevents microglial phagocytosis (Takahashi et al., 2005; Hsieh et al., 2009). TREM2 is known to interact with the signaling adapter protein named DNAX-activation protein of 12 kD (DAP12) (Paloneva et al., 2002), however its ligand in apoptotic cells remains elusive. Several candidates have been proposed, namely, anionic oligosaccharides such as bacterial lipopolysaccharides (Daws et al., 2003; Quan et al., 2008) and heat shock protein 60 (Hsp60) (Stefano et al., 2009). Hsp60 is exposed in the surface of apoptotic cells (Goh et al., 2011) and increases the phagocytic activity of microglia (Stefano et al., 2009). In addition to the direct recognition of the targets by microglial cell membrane receptors, engulfment can also triggered by soluble opsonins that bind to receptors signaling internalization in the microglia. Antibodies such as IgG and proteins of the complement system such as C3b, bind to phagocyte Fc receptors and complement receptor 3 (CR3), respectively, and mediate phagocytosis (Underhill and Goodridge, 2012). Further examples of receptors and their targets are summarized in Table 1 and Figure 2 and will be provided in the following sections. The level of expression of the receptors involved in phagocytosis may change under different stimuli such as inflammation (Falsig et al., 2008), but it is not known whether they ultimately impact the efficiency of microglial phagocytosis.
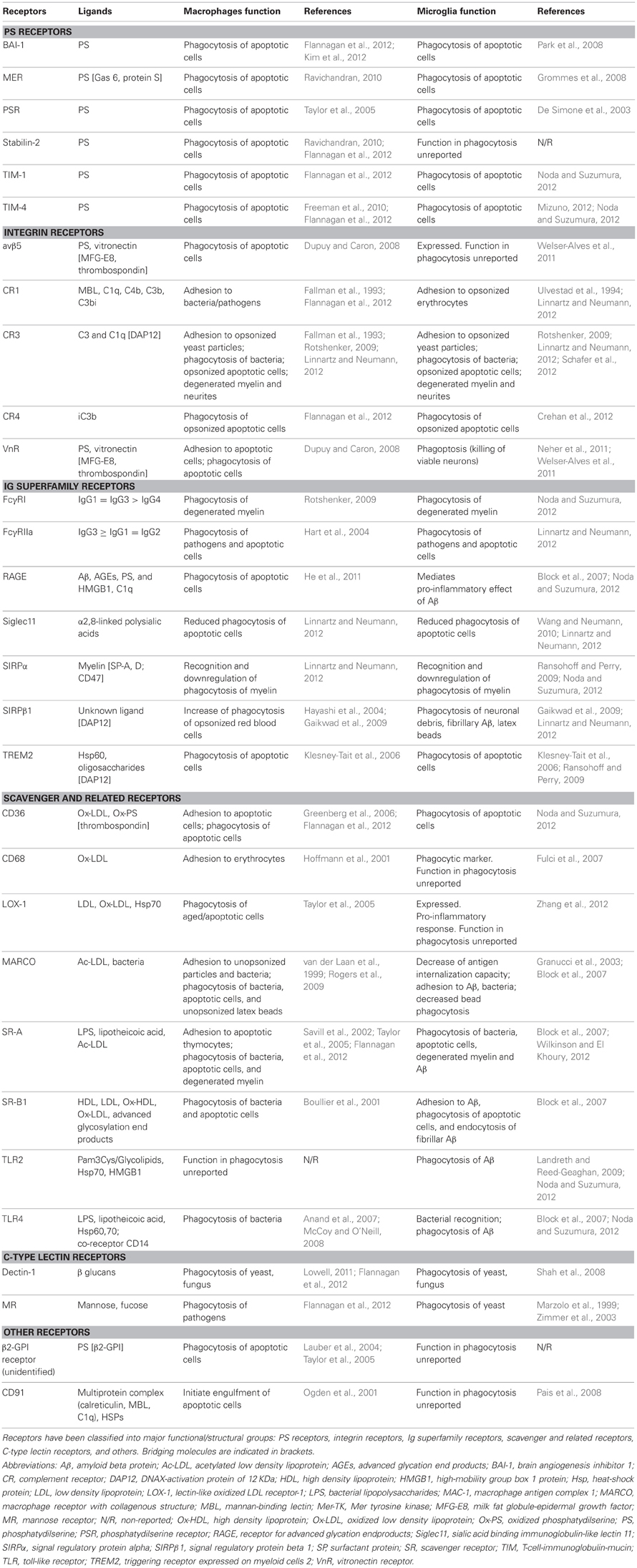
Table 1. Summary of receptors involved in macrophage and microglial phagocytosis.
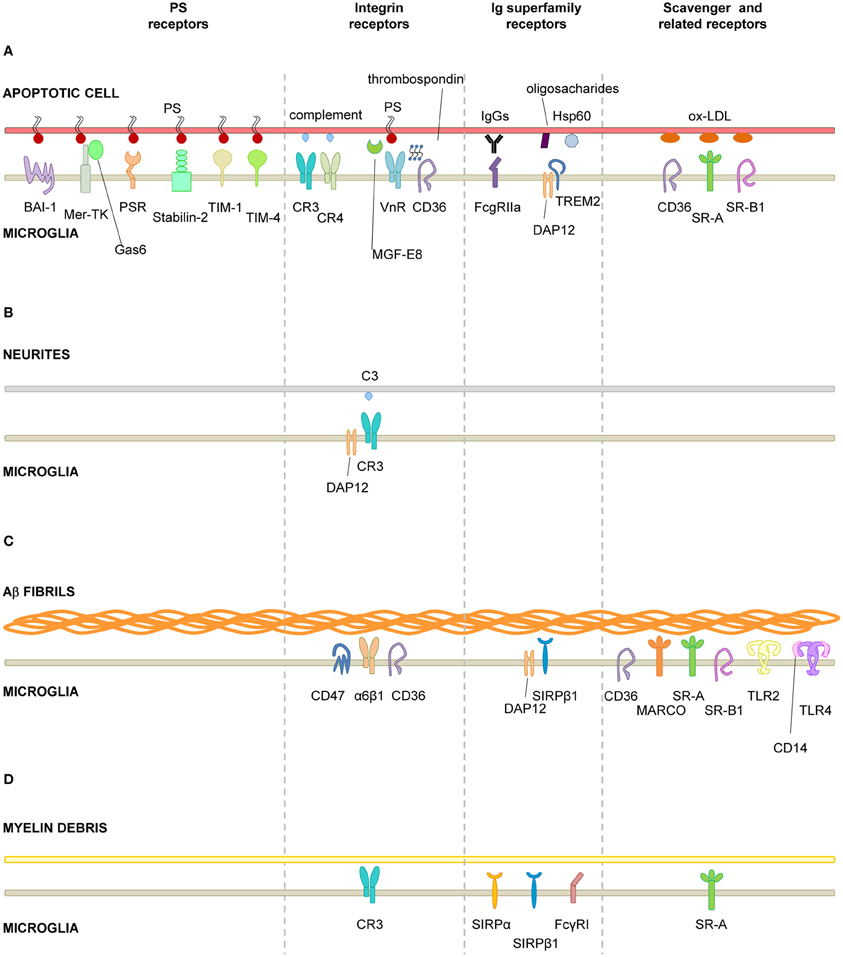
Figure 2. Receptors involved in microglial phagocytosis of endogenous structures. (A), Apoptotic cells; (B), Neurites; (C), Aβ aggregates/fibrils; (D), Myelin debris. Receptors have been classified into major functional/structural groups: phosphatidylserine (PS) receptors, integrin receptors, Ig superfamily-receptors, and scavenger and related receptors. For more details on the receptors, refer to Table 1. For receptors involved in the recognition of necrotic cells and microbes, refer to the main text. Abbreviations: Aβ, β-amyloid protein; BAI-1, brain angiogenesis inhibitor 1; CR, complement receptor; DAP12, DNAX-activation protein of 12 KDa; Hsp60, heat-shock protein of 60 KDa; MARCO, macrophage receptor with collagenous structure; Mer-TK, Mer tyrosine kinase; MFG-E8, milk fat globule-epidermal growth factor; Ox-LDL, oxidized low density lipoprotein; PS, phosphatidylserine; PSR, phosphatidylserine receptor; SIRPβ1, signal regulatory protein β1; SR-A, scavenger receptor class A; SR-B1, scavenger receptor class B1; TIM, T-cell-immunoglobulin-mucin; TLR, toll-like receptor; TREM2, triggering receptor expressed on myeloid cells 2; VnR, vitronectin receptor.
These receptors and their targets closely interact in what has been termed “engulfment synapse” or “phagocytic synapse” (Ravichandran, 2010; Dustin, 2012; Underhill and Goodridge, 2012). Similar in size (0.5 μm in diameter) and purpose (close cell–cell contact) to its immunological and neural counterparts, phagocytic synapses are specialized regions of the membrane where the apoptotic cell and the phagocyte interact through microclusters of receptors. Like the immunological synapse formed between antigen presenting cells (APCs) and T lymphocytes, and in contrast to the synapses formed between neurons, phagocytic synapses are short-lived and last only a few minutes (Dustin, 2012). Phagocytic synapses are also characterized by exclusion of phosphatases (Dustin, 2012), so that once the contact is established the signal is transduced by a variety of intracellular kinases (Syk kinase, phosphatidylinositol 3-kinase) and small GTPases (Rac, Rho) and adapter molecules such as engulfing and cell motility (ELMO) family (Gumienny et al., 2001; Lee et al., 2007). An emerging pivotal complex is that formed by ELMO-1 and dedicator of cytokinesis 180 (DOCK-180), which is activated among other receptors by BAI-1 and act as a guanine nucleotide exchange factor for Rac (Park et al., 2008; Patel et al., 2011). ELMO-1 function is regulated by phosphorylation by the hematopoietic cell kinase (HCK) (Yokoyama et al., 2005), a member of the Src family whose deficiency impairs macrophage phagocytosis (Lowell et al., 1994). These pathways lead to the remodeling of the phagocyte cytoskeleton through actin polymerization and membrane composition, triggering the formation of pseudopodia that form a phagocytic cup engulfing the target (Lee et al., 2007). Of the complex process of signal transduction and formation of the phagocytic cup, very little is known in microglia.
Digest-Me
The phagocytic cup closes and thus forms the phagosome around the target. To execute the degradation of the target, phagosomes go through a process of maturation in which they fuse sequentially with early and late endosomes, and lysosomes, to form phagolysosomes (Desjardins et al., 1994). These phagolysosomes contain hundreds of proteins, including hydrolases such as cathepsins to digest the target; and proton pumps such as vacuolar ATPases to acidify the medium (Garin et al., 2001). The acidic pH found in phagolysosomes (pH ≤ 5) is essential for the lysosomal degradation capabilities, as it is optimal for most hydrolases. Interestingly, the signaling associated with the acidic pH of lysosomes deactivates the generation of radicals from the oxidative burst (Li et al., 2010). Again, the process of phagosome formation and cargo degradation has been barely addressed in microglia. A recent study in C. elegans using live imaging showed that in this nematode microglial vATPases are required for phagosome–lysosome fusions and consequently to degrade cargo (Peri and Nusslein-Volhard, 2008). Further research is necessary to delineate the mechanisms of degradation of structurally different cargo, ranging from Aβ deposits to whole apoptotic cells, in microglia. Moreover, because phagocytosis is not only performed by ameboid, activated cells but also by terminal or en passant branches of ramified, resting microglia (Peri and Nusslein-Volhard, 2008; Sierra et al., 2010), the location where the actual degradation occur remains unexplored. Live imaging experiments show that resembling phagosomal cups are retrogradely transported to the cell soma in the mouse neocortex (Nimmerjahn et al., 2005); small puncta of apoptotic cell material are observed within branches of ramified microglia in fixed adult hippocampus, indirectly suggesting their transport (Sierra et al., 2010). Direct evidence of retrograde transport of cargo-containing phagosomes was recently found in C. elegans (Peri and Nusslein-Volhard, 2008). Collectively, these data indirectly suggest a yet unexplored role of the cytoskeleton in transporting the phagosome for the degradation of the engulfed material.
Overall, phagocytosis is considered to be essential for maintaining brain tissue homeostasis. The rapid clearance of apoptotic cells prevents their transformation into secondary necrotic cells (as it readily occurs in vitro), with the subsequent loss of permeability of the cell membranes and spillover of intracellular contents. For instance, the phagocytosis of apoptotic neutrophils and T cells during the resolution of the inflammatory response prevents the release of intracellular granules containing lysosomal enzymes and possibly acidification of the extracellular space (Magnus et al., 2001). Further, blockade of microglial phagocytosis of polymorphonuclear neutrophils, which infiltrate the brain parenchyma after focal ischemia, decreases neuronal viability in organotypic slices (Neumann et al., 2008). Phagocytosis of apoptotic neurons is also beneficial for neuronal survival. For instance, neurons can be rescued from excitotoxic death when phagocytosis is promoted by the chemokine fractalkine in neuron-microglia co-cultures, but not in neuronal cultures alone (Noda et al., 2011). In addition, myelin debris contains inhibitory molecules which inhibit axonal regeneration (Gitik et al., 2011); Aβ plaques cause physical damage of the neural tissue and lead to functional deficits if they are not removed (Wang et al., 2011); and removal of axonal debris by microglia increases axonal regeneration in vitro (Tanaka et al., 2009; Hosmane et al., 2012).
In the living brain, however, the beneficial effects of microglial phagocytosis have been harder to prove, possibly due to the lack of molecular tools to specifically interfere with microglial phagocytosis. Enhancing brain phagocytosis via intravenous delivery of TREM2-transduced bone marrow-derived precursor cells potentiates the clearance of degenerated myelin, decreases the inflammatory response and ameliorates the course of the disease in the mouse model of multiple sclerosis (MS), experimental acute encephalomyelitis (EAE) (Takahashi et al., 2007). A method recently proposed is the use of annexin V, a PS binding protein, to block phagocytosis. When administered systemically, annexin V reaches the brain and, as expected, leads to accumulation of apoptotic debris, although it has not been quantified whether it actually prevents phagocytosis (Lu et al., 2011). Treatment with annexin V impaires the generation of newborn neurons in adult neurogenic niches without affecting the proliferation of neuronal precursors, suggesting that phagocytosis is important for the differentiation and/or survival of the newborn cells (Lu et al., 2011). Direct evidence of the beneficial effects of microglial phagocytosis was shown recently in a mouse model of Rett syndrome by knocking out the methyl-CpG-binding protein Mecp2 (Derecki et al., 2012). While Rett syndrome is mostly considered a primary neuronal disease, defects in phagocytosis and inflammatory response are found in Mecp2-deficient microglia (Derecki et al., 2012). Further, bone marrow cells transplanted into irradiated Rett mice differentiate into microglia and partially arrest the pathology of the disease (Derecki et al., 2012). Interestingly, the disease is mimicked by microglial-specific depletion of Mecp2 and phagocytosis is blocked by intravenous injection of annexin V, but not by either manipulation independently. These data suggest that a failure in microglial phagocytosis does not underlie the pathology of the Rett syndrome but, nonetheless, increasing phagocytosis may be beneficial (Derecki et al., 2012).
Functional Consequences of Phagocytosis
Clearance of microbes or debris is a direct intervention of microglia to remove invading pathogens or to clean up the tissue from unwanted material. The direct consequences of this clearance function were already described in the chapter above. Furthermore, ingestion and degradation of targets has several indirect consequences for the phagocyte as well as the surrounding tissue. Phagocytosis triggers activation of several intracellular signaling pathways and remodels the cytoskeleton and cell membrane (Lee et al., 2007). In addition, microglia receive a tremendous metabolic load from the lipids, carbohydrates, proteins, and other components from the digested target (particularly in the case of apoptotic cells and microorganisms), leading to direct changes in the phagocyte's lipid and cholesterol, and possibly glucose metabolism (Han and Ravichandran, 2011). In addition, whether phagocytosis impacts proliferation, survival, or differentiation of the phagocyte remains unexplored. In contrast, three major functional consequences of microglial phagocytosis have been well-described: antigen presentation, activation of the respiratory burst, and modulation of inflammatory responses.
Antigen Presentation
Professional APCs (macrophages, DCs, and B lymphocytes) engulf their targets locally; next, they travel to lymph nodes, where the digested exogenous proteins (antigens) are expressed in their membrane attached to specific receptors (major histocompatibility complex class II, or MHC-II), which enable their recognition by naïve T lymphocytes, initiating the cellular arm of the adaptive immune response. Antigen presentation is a complex process beyond the scope of this review. Nonetheless, it is important to note that antigen presentation in the CNS has been a matter of hot debate in the last few years [reviewed by Ransohoff and Engelhardt (2012)]. In normal conditions, microglia do not express MHC-II and only a subpopulation expresses co-stimulatory molecules such as CD11c (Bulloch et al., 2008), and in agreement they have poor antigen presenting activity in vivo (Ford et al., 1995). Upon local inflammatory challenge, microglia show increased expression of MHC-II and CD11c in vivo and increased antigen presentation in vitro, albeit with much lower efficiency than professional APCs (Gottfried-Blackmore et al., 2009). While it is clear that they have the capacity of presenting antigens under some particular conditions, the microglial contribution to antigen presentation in vivo is thought to be mostly irrelevant (Galea et al., 2007; Ransohoff and Engelhardt, 2012). A major unresolved issue is whether microglia would have the capability of abandoning the brain parenchyma and reach the lymph nodes. In fact, antigen presentation in the CNS is thought to occur not in the parenchyma, but in the meninges and choroid plexus, which contain perivascular APCs, or by direct drainage of CNS antigens in the cerebrospinal fluid from the subarachnoidal space to channels in the cribiform plate and ultimately into deep cervical lymph nodes (Galea et al., 2007; Ransohoff and Engelhardt, 2012). Self-antigens, including CNS antigens, do not produce a response since self-reactive T cells are eliminated by clonal deletion in the thymus. In the autoimmune disease MS, however, myelin-specific T cells may escape from this tolerance mechanism and enter the brain under some circumstances, leading to demyelination (Goverman, 2011). Microglia seems to play a negative role in the disease progression of EAE, the animal model of MS, as its depletion reduces the severity of the disease (Heppner et al., 2005). Whether this potentially detrimental role of microglia in EAE is related to phagocytosis and/or antigen presentation remains unknown (Ransohoff and Engelhardt, 2012).
Respiratory Burst
During phagosome maturation the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is assembled in the phagosome. NADPH oxidase catalyzes the reaction of NADPH and oxygen to form NADP+, protons and the superoxide anion (O2−), in a process known as the respiratory burst (Minakami and Sumimotoa, 2006). In the acidic pH of the phagosome, the superoxide anion is dismutated into hydrogen peroxide, H2O2, and later transformed into other reactive oxygen species (ROS) which contribute to killing of engulfed microorganisms and degradation of other cargo (Minakami and Sumimotoa, 2006). ROS are extremely aggressive oxidants and can induce both apoptosis and necrosis (Pourova et al., 2010). Potentially toxic for the phagocyte itself and the surrounding tissue, they are thought to be released exclusively within the phagosome. In fact, NADPH assembly is a tightly controlled process in phagocytes and includes the recruitment to the phagosome of the catalytic core gp92phox and p22phox, and the phosphorylation and membrane translocation of the regulatory subunit p47phox (Quinn and Gauss, 2004). To ensure further regulation, the enzyme is rapidly deactivated, resulting in transient activity (Decoursey and Ligeti, 2005). Most of our understanding about the molecular mechanisms involved in activation or the respiratory burst comes from studies performed in neutrophils (Minakami and Sumimotoa, 2006), one of the most important components of the immune system in the defense against invading microorganisms. In microglia, the respiratory burst appears to be activated by phagocytosis through similar mechanisms (Ueyama et al., 2004) after phagocytosis of zymosan (a yeast cell wall preparation) (Newell et al., 2007) or myelin (Williams et al., 1994), although it remains to be studied whether it is also activated after phagocytosis of other types of cargo.
Recently, it has been suggested that the NADPH oxidase can be assembled in alternative locations, driving the respiratory burst in the absence of phagocytosis (Bylund et al., 2010). In microglia, the respiratory burst is triggered by hypoxia/reoxygenation (Spranger et al., 1998) and LPS (Qin et al., 2004) and results in the extracellular release of ROS. In turn, inflammation-induced respiratory burst leads to the release of glutamate by microglia, further contributing to neuronal damage (Barger et al., 2007). Interestingly, the microglial respiratory burst has been linked to induction of neuronal apoptosis in the developing cerebellum in vitro (Marin-Teva et al., 2004) and in the developing hippocampus in vivo (Wakselman et al., 2008). In both studies, ROS-trapping by free radical scavengers resulted in a significant rescue of neurons from apoptosis. However, it remains unclear whether the activation of the respiratory burst was activated as a consequence of phagocytosis or by independent mechanisms. While microglia were clearly identified as the only source of ROS, the actual intracellular location (i.e., in the phagolysosome or otherwise) was not addressed (Marin-Teva et al., 2004; Wakselman et al., 2008). The hippocampal study suggested indirectly that phagocytosis was indeed involved in the activation of the respiratory burst, because mice lacking the complement receptor CR3 or expressing mutant DAP12 showed reduced ROS production and apoptosis (Wakselman et al., 2008). In spite of the pro-phagocytic role of CR3 and DAP12, whether phagocytosis was disrupted in these transgenic mice was not addressed. Two possible scenarios remain open: in the first one, microglia would attack live (possibly healthy) neurons, phagocytose them via CR3/DAP12, and kill them intracellularly through the respiratory burst. In the second scenario, an undetermined stimulus would activate microglia to produce ROS via CR3/DAP12, which would be released extracellularly and kill neurons, subsequently phagocytosed by microglia. Clearly, more precise methods to quantify ROS (extracellular, intracellular, phagolysosomal, etc.) (Bylund et al., 2010) and to quantify phagocytosis and the nature of the cargo (Sierra et al., 2010) are necessary to unravel the role of microglial phagocytosis and respiratory burst in the developing hippocampus and cerebellum.
Modulation of Inflammatory Responses
Historically, it was believed that phagocytosis of apoptotic cells was a neutral immune event because it does not initiate an inflammatory response [reviewed by Savill et al. (2002)]. Over the past 20 years, it has been made clear that the phagocytosis of apoptotic cells is largely anti-inflammatory (Stern et al., 1996; Voll et al., 1997; Fadok et al., 1998). The inhibition of pro-inflammatory cytokines synthesis requires tethering of the phagocyte and the apoptotic cell, but not actual engulfment, followed by paracrinal release of the anti-inflammatory cytokine transforming growth factor beta (TGFβ), although the mechanism is not well-understood (Lucas et al., 2006). In addition, phagocytosis of other types of cargo such as myelin can trigger an anti-inflammatory response (Liu et al., 2006). On the contrary, phagocytosis of microbes associated with TLR stimulation is pro-inflammatory (Erdman et al., 2009). The anti-phlogistic response to phagocytosis of apoptotic cells also occurs in microglia. Microglia phagocytosing apoptotic T cells produce less tumor necrosis factor alpha (TNFα), a major pro-inflammatory cytokine, than naïve microglia after LPS challenge in vitro (Magnus et al., 2001). Similarly, phagocytosis of an apoptotic neuronal line induces an increased production of TGFβ and neural growth factor (NGF) in basal conditions; as well as a decreased production of TNFα, nitrite, and prostaglandin E2 during LPS challenge in vitro (De Simone et al., 2003). These anti-inflammatory effects are mediated by microglial PS receptor (De Simone et al., 2002), as well as by TREM2 (Takahashi et al., 2005). More recently it has been described that the immunosuppression induced by phagocytosis of apoptotic cells depends on the presence of the complement protein C1q bound to apoptotic cells, without which phagocytosis is pro-inflammatory (Fraser et al., 2010). It is important to note that complement proteins are present in the fetal bovine serum commonly used in primary cultures. Nonetheless, it remains to be determined whether the opsonin C1q is produced in the normal brain and binds to apoptotic cells phagocytosed by microglia in vivo. As a matter of fact, there is a big gap in our knowledge of the type of inflammatory response resulting from phagocytosis in vivo. The only in vivo data derives from a model of prion disease, where phagocytic microglia have been qualitatively observed not to express the pro-inflammatory cytokine interleukin 1beta (IL-1β) (Hughes et al., 2010). Because inflammation has severe effects in the brain, including neurotoxicity (Pickering et al., 2005) and epileptogenesis (Galic et al., 2012), more research is necessary to conclusively determine whether microglial phagocytosis of apoptotic cells is anti-inflammatory in vivo.
Microglial Phagocytosis in Physiological Conditions
The widely accepted theory of linear activation of microglia proposed by Gennadij Raivitch, William Streit, and collaborators (Raivich et al., 1999; Streit et al., 1999) leads to the generalized assumption that microglia have to be preactivated or primed by inflammatory challenge in order to become efficient phagocytes (Kettenmann, 2007). More current are the terms M1 and M2 to describe a spectrum of states of microglial activation, similar to the polarization of macrophages responses (Mantovani et al., 2002). The M1 phenotype or classic activation, is induced by PAMPs and pro-inflammatory cytokines, and is characterized by high expression of TLRs, TNFα, co-regulatory molecules for antigen presentation, and increased release of ROS. On the contrary, the M2 phenotype, or alternative activation, is driven by stimuli such as interleukin 4 (IL-4) or interleukin 13 (IL-13), and is characterized by the production of anti-inflammatory interleukin 10 (IL-10) and TGFβ, and higher expression of scavenger receptors. The M1 phenotype is basically neurotoxic, while the M2 phenotype is more neuroprotective (Durafourt et al., 2012). However it is not very clear what is the relationship between the M1/M2 phenotypes and phagocytosis. For instance, classic activation paradigms driving a M1 phenotype lead to increased enhanced engulfment of apoptotic cells in vitro (Chan et al., 2001; McArthur et al., 2010). In addition, expression of phagocytic receptors is triggered by many inflammatory and neurodegenerative paradigms in vivo assumedly inducing an M1 phenotype. On the other hand, a M2 phenotype driven by IL-4 and IL-13 increases the phagocytosis of myelin (Durafourt et al., 2012), whereas a M2 phenotype induced by glioma cancer stem cells reduces phagocytosis (Wu et al., 2010). Regardless of the nomenclature used to describe microglial phenotype after activation, the notion that microglia require to be preactivated to phagocytose efficiently is challenged by a number of recent papers showing that “resting” or “steady-state” microglia are efficient phagocytes of spines and apoptotic cells during physiological conditions in the adult and developing brain (Dalmau et al., 2003; Sierra et al., 2010; Schafer et al., 2012) (Figure 3).
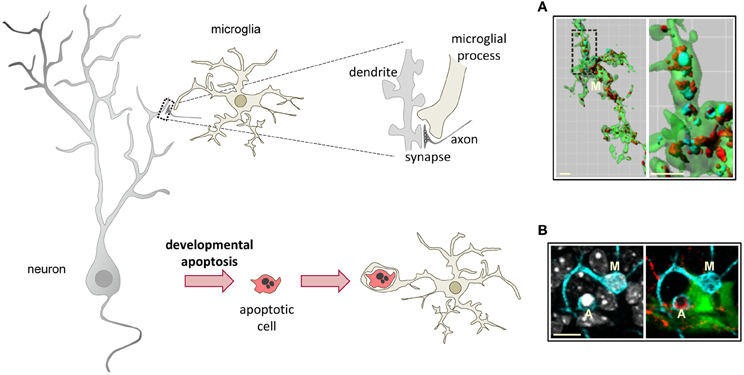
Figure 3. Microglial phagocytosis in health. Motile, ramified, unchallenged microglia phagocytose neurites (A, dendritic spines, axons) and developmentally apoptotic neurons (B) in physiological conditions in the adult and developing brain. (A) Surface-rendered CX3CR1+/EGFP microglia (M; green) from the dorsolateral geniculate nucleus engulfing ipsi- (blue, labeled with cholera-toxin (B) conjugated to Alexa 647) and contralateral (red, labeled labeled with cholera-toxin (B) conjugated to Alexa 594) inputs from retinal ganglion cells at postnatal day 5, when robust pruning occurs. The insert is shown at higher magnification on the right panel. (B) Iba-1 labeled microglia (M; cyan) branching a phagocytic pouch which engulfs a newborn apoptotic/pycnotic cell (A; labeled with the DNA dye DAPI, white), containing immunoreactivity for the neuroblast marker polysialic acid neural cell adhesion molecule (red). The cell is located in the subgranular zone of the hippocampus, where neural stem cells (visualized in nestin-GFP mice) are located and proliferate throughout adulthood. Images (A) and (B) are reprinted with permission from Elsevier. (A) is reprinted from Schafer et al. (2012). (B) is reprinted from Sierra et al. (2010).
Microglial Phagocytosis of Spines
The capacity for microglia to phagocytose synapses has just begun to be unraveled. In the juvenile visual cortex, most microglia interact with synaptic elements (spines, axon terminals, perisynaptic astrocytes, and synaptic clefts) (Tremblay et al., 2010). Furthermore, live imaging indirectly supported a role for microglia in pruning synapses, as spines contacted by microglia where more frequently eliminated than non-contacted spines (Tremblay et al., 2010). Qualitative examples of microglia containing pre- and postsynaptic material labeled respectively with SNAP25, a protein associated with presynaptic vesicles, and with PSD95, a marker of the excitatory postsynaptic density, have been shown in the developing postnatal hippocampus (Paolicelli et al., 2011). Interestingly, a transient increase in dendritic spines and immature synapses was observed in CX3CR1 knock-out mice compared to wild-type mice, possibly due to a reduction in microglial density (Paolicelli et al., 2011). Unfortunately, the authors did not carry out a quantification of microglial phagocytosis of spines in CX3CR1-KO mice and thus the role of the fractalkine receptor as an opsonin for microglial phagocytosis of spines cannot be ruled out. Direct, quantified evidence of microglia phagocytosing synapses (mostly, presynaptic elements) in the developing visual system has been recently provided using confocal and electron microscopy (Schafer et al., 2012). In the dorsal lateral geniculate nucleus (dLGN), active pruning of synaptic inputs from the retinal ganglion cells (RGCs) produces well-established ipsi- and contralateral innervations territories. This pruning is at least partially mediated by complement-mediated microglial phagocytosis, because mice deficient either for the receptor CR3 or its ligand, C3, have decreased phagocytosis of synaptic inputs, a sustained increase in synaptic density, and deficits in segregation of the eye-specific territories compared to wild-type mice (Schafer et al., 2012). Thus, unwanted synapses seem to be tagged for removal by deposition of complement proteins but—who is the decision maker? Do microglia actively instruct which synapses must be removed? In vitro, microglia produce several components of the complement cascade, including C1q and C3, which are involved in the phagocytosis of neurites via CR3 (Linnartz et al., 2012). Interestingly, removal of the monosaccharide sialic acid from the neural glycocalyx is essential for C1q binding to neurites and subsequent microglial phagocytosis in cultured neurons (Linnartz et al., 2012). It seems plausible that synapse pruning is regulated by a combination of mechanisms involving activity-dependent control of synaptic strength, alterations in the neuronal glycocalyx, and tagging of altered synapses by microglial-release of complement proteins, followed by removal of pre- and/or postsynaptic components by microglial phagocytosis. In contrast to the more established mechanism of synaptic pruning by simple retraction or degeneration of the input axon (Luo and O'Leary, 2005), microglia-dependent pruning is likely to require more time and energy, while perhaps adding more levels of control to a phenomenon which is essential to determine brain connectivity. A possible similar combination of mechanisms has been observed in the developmental pruning of dendrites in Drosophila neurons, where branch retraction and local fragmentation of dendrites are observed together with phagocytosis of caspase-labeled dendrites by blood phagocytes (Williams and Truman, 2005; Williams et al., 2006).
Overall, these data support a novel role for microglia in monitoring synapses in the healthy developing brain, with potentially profound consequences for diseases in which neural connectivity and microglial activation concur. One such disease is transient ischemia, in which the turnover of synapses (Zhang et al., 2005) as well as microglial activation and inflammation increase (Lambertsen et al., 2012). In the ischemic cerebral cortex microglial contacts with synaptic boutons last-longer (1 h) than in the control cortex (5 min) (Wake et al., 2009). While it is tempting to speculate that extended contacts are related to synapse pruning, only a minority of boutons disappeared after being contacted by microglia and phagocytosis was not observed (Wake et al., 2009). In autism spectrum disorders (ASD), a complex set of diseases with unknown pathophysiology, both neural hyperconnectivity (Testa-Silva et al., 2012) and inflammation (Vargas et al., 2005) develop. In MECP2-deficient mice, a model of Rett syndrome, microglia has defective phagocytosis in vitro, and preventing phagocytosis in the mice exacerbates the symptoms (Derecki et al., 2012). The potential defects in spine and/or apoptotic cell phagocytosis by microglia in Rett syndrome and other ASDs remain to be elucidated.
Microglial Phagocytosis of Apoptotic Cells During Brain Development and in Neurogenic Niches
A detailed map estimating microglial phagocytosis during development showed an overall high efficiency of microglia-removing apoptotic cells (Dalmau et al., 2003). At time points where apoptosis is maximal, microglia phagocytose 97% of apoptotic cells in the fimbria (embryonic day E18), 88 and 100% in the CA region of the hippocampus (E16 and postnatal day 0, respectively), 18 and 86% in the cerebral cortex (E16 and P0, respectively), and 93% in the dentate gyrus (P0). These estimates match well with the phagocytic efficiency of microglia in the dentate gyrus throughout adulthood, where new neurons are continuously produced. From 1 to 12 months of age in mice, microglia phagocytose over 90% of the cells undergoing apoptosis (Sierra et al., 2010). Nonetheless, the phagocytic capacity is region-specific, as microglia only phagocytose 50% of the apoptotic cells at the maximum period of cell death (P3) in the developing cerebellum (Ashwell, 1990; Marin-Teva et al., 2004; Wakselman et al., 2008).
But, what happens to the remaining apoptotic cells? A possible interpretation is that they will be engulfed by microglia at a later time point, so that the percentage of non-phagocytosed apoptotic cells is an indirect representation of the “find-me” time. Alternatively, these cells may be disposed of through other mechanisms, such as phagocytosis by other cell types. Microglia are considered the professional brain phagocytes. Non-professional phagocytes such as astrocytes delay phagocytosis for several hours (Parnaik et al., 2000) and engulf with much lower capacity (Magnus et al., 2002), at least in vitro. Nonetheless, other cell types mediate phagocytosis of apoptotic cells in vivo during development. For instance, in the developing cerebellum (P7) phagocytosis of apoptotic oligodendrocytes is carried out by specialized astrocytes, the Bergmann glia, albeit at a relative low efficiency (55% of apoptotic cells engulfed) (Parnaik et al., 2000). In the developing retina, phagocytosis is executed by microglia and Müller cells (Egensperger et al., 1996), and throughout lifetime the excess of photoreceptors membrane shredded is phagocytosed by retinal pigment epithelium (RPE) cells (Kevany and Palczewski, 2010). While considered non-professional phagocytes, RPE cells have an enormous phagocytic capacity, as they engulf up to 10% of the photoreceptors volume on a daily basis (Kevany and Palczewski, 2010). In the adult hippocampal neurogenic cascade, only microglia have been observed to phagocytose apoptotic newborn cells (Sierra et al., 2010). Nonetheless, neural-committed neuroprogenitors (neuroblasts, labeled with doublecortin) have some phagocytosing capabilities which contribute to the maintenance of the neurogenic cascade, although what they actually take in remains to be determined (Lu et al., 2011). In peripheral ganglia, the apoptotic neurons are phagocytosed by satellite glial cell precursors (Wu et al., 2009). It has been speculated that low microglial density in some regions may be related to the recruitment of non-professional phagocytes (Parnaik et al., 2000) but the reasons behind this promiscuous phagocytosis during brain development are not known. Assumedly, microglial phagocytosis contributes to the maintenance of tissue homeostasis during development and in adult neurogenic niches by rapidly removing cellular debris. It can also be speculated that microglial phagocytosis takes a more active role in regulating neurogenesis. For instance, after phagocytosis, cultured microglia produce higher levels of TGFβ and NGF (De Simone et al., 2003), which are negative and positive regulators of hippocampal neurogenesis, respectively (Buckwalter et al., 2006; Frielingsdorf et al., 2007). After stroke, microglia in the subventricular zone (SVZ) show a pro-neurogenic phenotype which includes the expression of insulin-like growth factor 1 (IGF-1) (Thored et al., 2009), another well-known inducer of neurogenesis (O'Kusky et al., 2000). Stroke-responding microglia were labeled with ED1 but unfortunately phagocytosis was not quantified. Future research will delineate the contribution of microglia to neurogenesis.
The high efficiency of phagocytosis, that is, the high coupling between apoptosis and phagocytosis, has also suggested the idea that phagocytosis executes the final stages of apoptotic cell death. Apoptosis can be initiated by two major pathways: the extrinsic pathway, driven by activation of membrane receptors such as CD95, or the TNFα receptor; and the intrinsic pathway, initiated by cellular stress (DNA mutations, deprivation of survival factors, Ca2+ overload, etc.) (Reubold and Eschenburg, 2012). While the signaling cascades are complex and varied, a common mechanism of execution of most forms of apoptosis is the formation of the apoptosome, a macromolecular complex which activates the effector caspase 3, a cystein protease responsible for DNA fragmentation, membrane blebbing, cytoskeleton degradation, and the other major hallmarks of apoptosis (Blank and Shiloh, 2007). In addition to the cell-autonomous degradation mediated by caspases, phagocytes may also contribute to carry out death. For instance, mutations in C. elegans engulfment genes, such as ced-1, permit the survival of cells that would normally die (Reddien et al., 2001). In addition, DNA fragmentation is not only mediated by cell-autonomous caspase-activated DNase (CAD) but can also be partly attributed to postengulfment degradation by lysosomal enzymes (McIlroy et al., 2000). The lysosomal DNase II of macrophages contributes to thymocyte DNA degradation during thymus development (Kawane et al., 2003). Whether this mechanism is universal to macrophages and microglia is unknown, because DNase II deficient mice embryos show severe defects in the thymus and kidney, but not in the brain (Kawane et al., 2003). Nevertheless, these evidences suggest that, since there are no stop points in apoptosis analog to the check points found in mitosis, phagocytosis is the de facto mechanism to discriminate between moribund and dead cells.
In some circumstances this effective coupling may have detrimental consequences. It has been suggested that if phagocytosis is too effective, it may lead to the removal of cells which will otherwise have time to repair themselves (Kao et al., 2011). For example, the macrophages of mice and worms deficient in the secreted glycoprotein progranulin have an enhanced phagocytic efficiency, which is perhaps related to the neurodegeneration found in human patients of familial frontotemporal lobar degeneration, mostly caused by mutations in progranulin (Kao et al., 2011). Going further, microglial phagocytosis may be the primary cause of cell death under some circumstances. Macrophages are known to interact with live cells but disengage quickly because of the “don't eat-me” signals (Ravichandran, 2010). Under some circumstances, however, phagocytes kill live cells. For instance, macrophages induce apoptosis of normal vascular endothelial cells of the hyaloid vascular system as well as papillary cells in the developing mouse eye (Lang and Bishop, 1993; Diez-Roux and Lang, 1997). Similarly, microglial phagocytosis has been reported to induce the death of viable, motile non-apoptotic polymorphonuclear neutrophils in organotypic hippocampal cultures in which ischemia was induced by oxygen and glucose deprivation (Neumann et al., 2008). Further, in inflammatory conditions driven by activation of TLR2 or TLR4, microglial phagocytosis induces cerebellar granule cell death by a complex mechanism involving the microglial release of peroxynitrite which leads to a transient exposure of PS in the neurons and opsonization with MFG-E8, followed by recognition through the vitronectin receptor and phagocytosis by microglia in vitro (Neher et al., 2011) and in the striatum in vivo (Fricker et al., 2012). However, high concentrations of peroxynitrite lead to increased intracellular calcium, exposure of PS, activation of caspases and, ultimately, apoptotic cell death (Leist et al., 1997). Neurons dying from primary phagocytosis do not express features of apoptosis or necrosis, and the actual mechanism executing death is an open area of research (Brown and Neher, 2012). Thus, phagocytosis represents a wide range of responses, from the mere passive clearing of apoptotic cells and the active execution of final stages of apoptosis during development to the aberrant killing of live, healthy neurons during inflammation.
Microglial Phagocytosis in Pathological Conditions
Overall, phagocytosis is considered a beneficial phenomenon and its alteration has been linked to autoimmune diseases (Nagata et al., 2010). In addition, the best-known case of a phagocytic system disease is the relatively rare but lethal Nasu–Hakola disease or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), due to loss-off-function mutations of TREM2 and/or DAP12 (Paloneva et al., 2002). PLOSL is characterized by defects in bone resorption by osteoclasts leading to the formation of cysts, as well as dementia (Bianchin et al., 2004). The contribution of microglia to the pathology of PLOSL remains obscure because DAP12-deficient mice do not reflect the human neurodegenerative pathology. Initial reports showed altered synaptogenesis and hypomyelination as well as behavioral impairments in mice deficient or expressing loss-off-function mutations of DAP12 (Kaifu et al., 2003; Roumier et al., 2004). In the mouse brain, DAP12 is exclusively expressed in microglia, suggesting an interesting link between microglia and synaptic plasticity through a yet unknown mechanism (Roumier et al., 2004). More recent studies have shown a strong demyelination in the brain of DAP12-deficient human patients; however this demyelination did not seem to correlate with changes in microglial density or activation (Satoh et al., 2011). Therefore, the exact pathophysiology of microglia in PLOSL patients remains unclear. Nonetheless, there are many other pathological conditions related to the microglial phagocytosis of apoptotic cells, viral and bacterial pathogens, tumor cells, Aβ, myelin, and axonal debris (Figure 4).
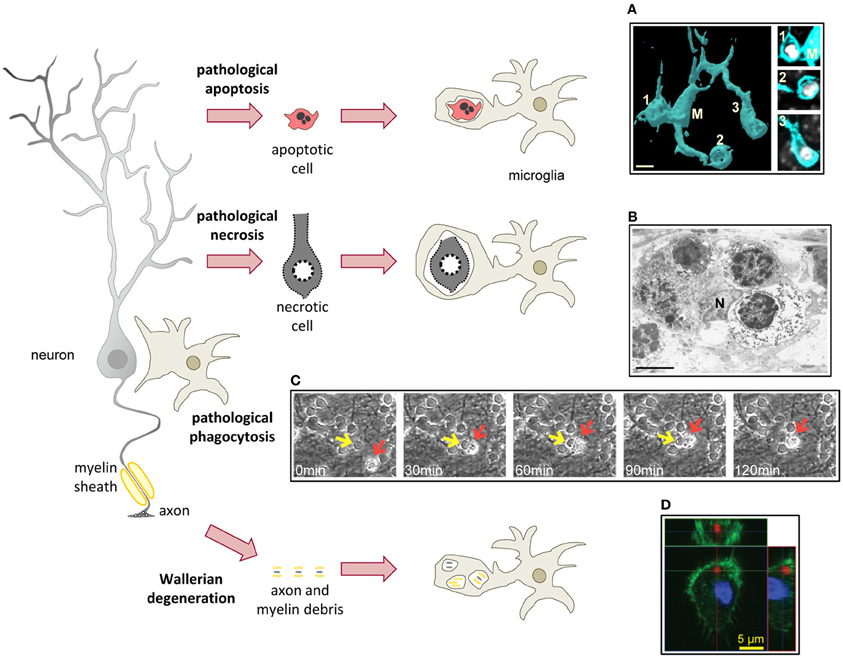
Figure 4. Microglial phagocytosis in disease. In pathological conditions microglia is challenged and usually assumes a hypertrophic morphology. Challenged microglia phagocytose apoptotic and necrotic cells (A,B). In some inflammatory conditions, microglial phagocytosis can kill healthy neurons (C). Microglia also phagocytose axonal and myelin debris resulting from Wallerian degeneration of severed axons (D). (A) Surface-rendered fms-EGFP microglia (M; cyan) from the adult hippocampus phagocytosing three apoptotic/pycnotic cells (labeled with DAPI, white), in a mouse systemically challenged with bacterial lipopolysaccharides (5 mg/kg). (B) Electron microscopy microphotograph of embryonic (E20) microglia (N, microglial nucleus) from the rat cingulate cortex following maternal hypoxia (1 day) containing four engulfed dead cells. In the developing brain, hypoxia induces a whole range of death mechanisms, including apoptosis, pathological apoptosis, and necrosis (Blomgren et al., 2007). (C) Time-lapse imaging of microglia (red arrow) engulfing a live neuron (yellow arrow) in a co-culture challenged with the TLR2 agonist lipoteichoich acid (50 μg/ml). (D) Orthogonal projection of a confocal z-stack showing the engulfment of degenerated axons (labeled with tdTomato, red) by microglia (labeled with Iba1, green; nucleus labeled with DAPI, blue) in a co-culture model. Scale bars, 5 μm. Image (A) is reprinted with permission from Elsevier from Sierra et al. (2010). Image (B) is reprinted with permission from Elsevier from Li et al. (1998). Image (C) is reprinted with permission from Neher et al. (2011), Copyright 2011. The American Association of Immunologists., Inc. Image (D) is reprinted with permission from Hosmane et al. (2012).
Microglial Phagocytosis of Dead Cells in Pathological Conditions
Neurodegeneration by apoptosis is a major part of several brain diseases such as stroke, epilepsy, prion disease, Alzheimer's disease (AD), and Parkinson's disease (PD). Microglial phagocytosis of apoptotic cells in pathological conditions is assumed to be highly efficient, possibly because of an extrapolation of the physiological phagocytosis during brain development, however this may not be always the case. For instance, there is qualitative evidence that apoptotic neurons in a mouse model of neonatal stroke by medial cerebral artery occlusion (MCAO) are poorly phagocytosed by activated microglia compared to the contralateral healthy hemisphere, where less apoptosis occurs (Faustino et al., 2011). The reasons behind this low phagocytosis in stroke are unknown, but it has been speculated that high levels of pro-inflammatory cytokines produced by astrocytes and vessels may be the culprit (Faustino et al., 2011). However, microglial phagocytosis of apoptotic cells in vitro is increased by the pro-inflammatory interferon gamma (IFNγ), but it remains unaltered by TNFβ or TGFβ (Chan et al., 2001). Prion disease is another condition where microglial phagocytosis is impaired. In vitro, the pathogenic form of prion protein (PrPSc), reduces the phagocytosis of latex beads (Ciesielski-Treska et al., 2004); however in vivo (ME7 model of prion disease) microglia show increased phagocytosis of beads, and enhanced expression of the phagocytic machinery genes, including scavenger receptors, cathepsins, and proteins of the respiratory burst. In addition, qualitative phagocytosis of neuronal debris by microglia suggests that the microglial phagocytic machinery in prion disease is not disrupted (Hughes et al., 2010), but microglia do fail to phagocytose prion protein (PrPSc), which accumulates in the brain and leads to neurodegeneration (Hughes et al., 2010). PD is one more example in which the phagocytosis behavior of microglia is altered. Here the phagocytosis of latex beads is prevented by the aggregated form, and enhanced by the monomeric form of alpha synuclein, which are found in the parenchyma and the CSF of parkinsonian patients, respectively (Park et al., 2008). Moreover, peripheral blood monocytes from PD patients, compared to age-matched controls, exhibit decreased phagocytosis of latex beads ex vivo (Salman et al., 1999), however defects in microglial phagocytosis have not been reported in vivo so far. Again, the lack of a standardized approach to quantify microglial phagocytosis hampers our understanding of its role in brain disorders where it is expected to play a significant role in removing cellular debris.
Death by necrosis is also an important component of many brain diseases. Contrary to apoptosis, necrosis is characterized by bursting of the cell membrane and spillover of cellular contents (Savill et al., 2002). Necrosis remains an obscure process, and molecular details of its execution are not well-known. Two main forms have been recognized: accidental necrosis, or cell lysis, by exposure of toxins, physical damage, freezing, etc.; and necrosis-like programmed cell death (PCD), which involves specialized caspase-independent signaling pathways such as the apoptosis-inducing factor (AIF) or cathepsins, and evolves without chromatin condensation (Leist and Jaattela, 2001). While accidental necrosis is unavoidable unless the stimulus is removed, cells can be rescued from necrotic PCD. The mechanisms of necrotic cells recognition by macrophages and microglia are not fully understood but may involve similar signaling pathways to the recognition of apoptotic cells. For instance, in some forms of necrotic PCD, exposure of PS by calcium-independent signals triggers recognition of necrotic cells by macrophages and microglia (Hirt et al., 2000; Hirt and Leist, 2003). One alternative mechanism could be the passive release of high mobility group 1 protein (HMGB1) by necrotic cells, which binds to the receptor for advanced glycation end products (RAGE) in phagocytes and triggers inflammation (Scaffidi et al., 2002). It is worth noting that HMGB1 is not released by apoptotic cells even when they fail to be phagocytosed and transform into secondary necrotic cells (Scaffidi et al., 2002). HMGB1 is also released during ischemia (assumedly, from the necrotic core) in stroke patients and in mice subjected to MCAO, launched an inflammatory response, which is detrimental for neuronal survival (Kim et al., 2006; Muhammad et al., 2008). Initial reports suggested that microglia does not mount an inflammatory response when co-cultured with necrotic PC12 neurons (De Simone et al., 2003). Others have shown that the phagocytosis of necrotic PCD Jurkat cells reduces the release of TNFα by microglia (Hirt and Leist, 2003). More recently, it has been shown that when co-cultured with necrotic HT22 neurons microglia do indeed express higher levels of pro-inflammatory cytokines (TNFα, IL-6), pro-inflammatory enzymes (nitric oxide synthase, 2-cyclooxygenase), MHC-II, and the integrin CD11b through a mechanism involving MYD88, a TLR adapter protein (Pais et al., 2008). In summary, very little is known about the efficiency and consequences of microglial phagocytosis of necrotic cells.
Death of living cells by phagocytosis, recently termed “phagoptosis,” might contribute to the pathophysiology of some diseases where inflammation, the main trigger for phagoptosis, occurs. For instance AD, which characterized at the histological level by plaques of Aβ as well as neurofibrillary tangles, has a strong inflammatory component (Johnston et al., 2011). In vitro, low concentrations of Aβ (1–42) (nanomolar range) lead to activation of microglia and an inflammatory response, which is partly responsible for neuronal damage (Maezawa et al., 2011). In this situation, either depleting microglia, blocking vitronectin receptors, inhibiting cytoskeleton polymerization, or preventing recognition of PS with annexin V, all prevent neuronal death induced by nanomolar Aβ (Neniskyte et al., 2011). These data suggest that phagoptosis might be partly responsible for neuronal death induced by Aβ, perhaps providing an explanation for the low numbers of dead neurons found in AD (Neniskyte et al., 2011). Nonetheless, death by apoptosis has been documented in AD patients and in animal models of AD, although it is assumed to occur at low levels over a long period of time (Shimohama, 2000). We argue that the clearance time for phagoptotic neurons is likely to be longer than for phagocytosed apoptotic neurons, because phagoptotic cells fail to activate caspases and other mechanisms of self-destruction and rely exclusively on microglia for degradation (Brown and Neher, 2012). Time-lapse imaging experiments suggest that engulfing a live neuron may take microglia under 2 h (Neher et al., 2011) (Figure 4), however the time to fully degrade it has not been estimated. As a longer clearance time implies a higher probability of phagocytosis detection, it should then be possible to visualize and quantify microglial engulfment of live cells in AD or in other inflammatory diseases to ultimately determine the contribution of phagoptosis to brain pathologies in vivo.
Microglial Phagocytosis of Invading Microorganisms
In the adult and developing brain, microglia are capable of phagocytosing many pathogens, including bacteria (Escherichia coli, Streptococcus pneumonia, Staphylococcus aureus, Enterococcus faecalis), yeast (Saccharomyces cerevisiae), and fungus (Candida albicans) (Kaur et al., 2004; Falsig et al., 2008; Shah et al., 2008; Hadas et al., 2010; Ribes et al., 2010; Peppoloni et al., 2011; Kochan et al., 2012). Different PAMPs are recognized by different receptors: for instance, recognition of S. aureus peptidoglycan is mediated by TLR2 (Kielian et al., 2005); recognition of E. coli LPS is mediated by TLR4 (Sivagnanam et al., 2010); and recognition of S. cerevisiae β-glucans (such as zymosan) is mediated by Dectin1, CR3, and the mannose receptor (MR, CD206) (Hadas et al., 2010). TLR2 and 4 are constitutively expressed by microglia (Bsibsi et al., 2002) and recognition of their ligands enhances phagocytosis (Ribes et al., 2010; Kochan et al., 2012), supporting the capacity of microglia to efficiently engulf and degrade infecting microorganisms. For instance, E. coli bacteria injected into the corpus callosum of early postnatal rats are engulfed in large numbers 1–3 h after injection, and are completely eliminated in 24 h (Kaur et al., 2004). In contrast, phagosomes were found within microglial cells up to 7 days after (Kaur et al., 2004), suggesting that degradation and killing of living microorganisms is a time-consuming process. Phagocytosis of bacteria activates the inflammatory cascade in microglia, inducing the expression of pro-inflammatory cytokines (TNFα), TLRs (TLR4, TLR9), complement receptors (CR3), scavenger receptors (SRA), and MHC-II (Sivagnanam et al., 2010). Although the data on microglial phagocytosis of microorganisms is not abundant, it seems to suggest a high capacity to fight against brain infections such as meningitis.
Microglial Phagocytosis of Tumor Cells
Brain tumors are very aggressive and have extremely poor prognosis possibly due to a failure of the innate and adaptive immune response to efficiently eliminate them. Tumor-associated microglia and macrophages (TAMs) are found in large numbers in human glioblastoma and have a complex and bidirectional relationship with glioma cells which has been reviewed in detail elsewhere (Watters et al., 2005). On one hand, glioma-initiating cells contribute to the recruitment of TAMs into the tumor mass (Yi et al., 2011) and polarize them toward a M2 phenotype (Wu et al., 2010). On the other hand, TAMs contribute to glioma progression by enhancing tumor migration and proliferation through growth factors, angiogenic molecules, and enzymes degrading the extracellular matrix (Watters et al., 2005). Unfortunately, glioma cells are rarely phagocytosed by TAMs in mouse models of glioma (Galarneau et al., 2007). Microglia from human glioblastoma patients do have the capability of phagocytosing latex microbeads ex vivo (Hussain et al., 2006), however in vitro studies have shown that co-culture with tumor cells decreases bead phagocytosis after a transient increase (Voisin et al., 2010). Further, it seems that tumor cells evade phagocytosis by lacking in their membrane the appropriate “eat-me” signals. In fact, microglia do phagocytose tumor cells which have been previously induced apoptosis with the cytotoxic agent etoposide (Chang et al., 2000) or UV light (Kulprathipanja and Kruse, 2004), although many tumor cells decrease their sensitivity to apoptosis inducers during oncogenic transformation (Maher et al., 2001). This data has suggested the use of alternative therapies to promote tumor cell apoptosis, such as oncolytic viruses, which selectively replicate in tumor cells, leading to their destruction (Zeyaullah et al., 2012). This therapy, while safe, has been proven ineffectual because TAMs clear the viruses away from the tumor (Fulci et al., 2007). While the literature confirms that TAM phagocytosis of tumor cells is not very effective, others have suggested that some tumor cells, particularly in highly invasive tumors such as glioblastoma, phagocytose neighboring cells perhaps as a way to fuel their constant growing [reviewed by Huysentruyt and Seyfried (2010)].
Microglial Phagocytosis of Aβ Deposits
Aβ is a small peptide produced by proteolytic cleavage from amyloid precursor protein (APP) by β- and γ-secretases. The most pathogenic form is Aβ (1–42), which forms fibrils, insoluble aggregates found in amyloid plaques in the brains of AD patients, as well as human immunodeficiency virus (HIV)-associated neurocognitive disorders (Xu and Ikezu, 2009). A major effort has been put into developing therapies to lower the amyloid burden. Aβ reduction can occur either by decreasing its synthesis rate (e.g., with secretase inhibitors); or by increasing its elimination rate. The particular location of microglia surrounding plaques in human patients and mouse models of AD lead to the early suggestion that they could be responsible for releasing Aβ and forming the plaques (Lai and McLaurin, 2012). While microglia can synthesize Aβ in vitro (Banati et al., 1993), APP mRNA is not found in microglia of human AD brains (Scott et al., 1993). It was alternatively proposed that microglia could be responsible for phagocytosing Aβ and contribute to its clearance (Paresce et al., 1996). In vitro, microglia recognizes and engulfs fluorescently labeled fibrillary Aβ through a variety of receptors, including scavenger receptors (Paresce et al., 1996), TLR2 (Liu et al., 2012), and the TLR4-interacting molecule, CD14 (Liu et al., 2005). Further, Aβ induces a positive chemotaxis of microglia via TGFβ (Huang et al., 2010), possibly explaining their location around plaques. Recent live imaging experiments have shown that microglia is rapidly attracted to already formed plaques in a mouse model of AD with mutated APP and presenilin 1 (a protein of the γ-secretase complex) (Meyer-Luehmann et al., 2008), strongly suggesting that microglia does not participate in the initial stages of plaque formation and confirming the chemotactic nature of Aβ. Importantly, the size of the plaques remained constant and no evidence of phagocytosis or plaque clearance was obtained (Meyer-Luehmann et al., 2008). In agreement, a detailed 3D reconstruction of microglia and amyloid fibrils in APP mutated mice showed that microglial processes and amyloid fibrils were interlaced forming a network, but Aβ was not found within microglia, further suggesting that microglia does not phagocytose Aβ in vivo (Stalder et al., 2001). The failure of microglia to clear Aβ plaques of AD patients is unclear. Cultured microglia engulf and partially degrade Aβ in vitro over the first 3 days, but no additional degradation and a slow release of intact Aβ is found afterwards (Chung et al., 1999). Furthermore, microglia from old mutant APP/presenilin 1 mice have a decreased expression of phagocytic genes (SRA, CD36, RAGE, etc.) and increased expression of pro-inflammatory cytokines (TNFα, IL-1β), compared with age-matched wild type mice (Hickman et al., 2008), but both findings cannot explain the failure of microglia to sufficiently clear Aβ plaque in vivo. The role of inflammation in Aβ clearance is also not resolved. In vitro, inflammatory challenge by LPS, TNFα, IL-1β, or IFNγ inhibits fibrillary Aβ phagocytosis, whereas anti-inflammatory cytokines had no effect (Koenigsknecht-Talboo and Landreth, 2005). Aβ phagocytosis is blocked by the anti-inflammatory celecoxib (Persaud-Sawin et al., 2009), but not by minocyline (Familian et al., 2007). In vivo, LPS administered locally either transiently reduced (Herber et al., 2004), or enhanced (Qiao et al., 2001) Aβ load in mouse models of AD. The fractalkine receptor, a regulator of phagocytosis, has been found to contribute to the Aβ burden by some (Lee et al., 2010; Liu et al., 2010) but not others (Fuhrmann et al., 2010). Importantly, common treatments for HIV and AD may be detrimental for Aβ clearance, at least in vitro. HIV protease inhibitors either block Aβ degradation or enhance secretion of non-degraded Aβ by macrophages (Lan et al., 2012). Similarly, γ-secretase inhibitors prevent microglial Aβ engulfment (Farfara et al., 2011). Another point of controversy is the participation of invading macrophages. The blood-brain barrier (BBB) is partially disturbed in AD patients, facilitating the extravasation of circulating monocytes (Lai and McLaurin, 2012). Macrophages have higher Aβ capacity intake than microglia in vitro (Lai and McLaurin, 2012), and ablation experiments have suggested that the Aβ burden is cleared by blood-borne macrophages, but not resident microglia (Simard et al., 2006). The issue is further complicated by the fact that experimental interventions such as irradiation of mice lead to invasion of blood-monocytes into the brain parenchyma and that macrophages and microglia are phenotypically indistinguishable by surface markers.
Microglial Phagocytosis of Myelin and Axonal Debris
The only available data on microglial phagocytosis of axonal debris comes from in vitro models. In cortical explants where growing neurites were sectioned, the debris was cleared by added microglia (Tanaka et al., 2009). In a compartmentalized co-culture model where neurons were grown in a chamber and their axons extended in bundles through microchannels, axotomy, or nitric oxide treatment-induced axonal degeneration and microglia rapidly cleared the axonal debris (Hosmane et al., 2012). Interestingly, the mechanisms of recognition of axonal debris seem to be different from those of apoptotic cells (Tanaka et al., 2009). A candidate receptor is TIR domain-containing adapter inducing interferon beta (TRIF). The clearance of axonal debris is prevented by blocking TRIF signaling in vitro, and TRIF-deficient mice have a smaller percentage of microglia containing neurofilament-positive axonal material after dorsal root axotomy (Hosmane et al., 2012). In both set ups, axonal debris (lacking myelin) had a detrimental effect on axon regrown, which was prevented by microglial phagocytosis (Tanaka et al., 2009; Hosmane et al., 2012), suggesting that enhancing microglial phagocytosis is a novel therapeutical tool in traumatic brain injuries.
More attention has been put into the mechanisms of myelin debris clearance, particularly during MS and in spinal cord and nerve injuries. In spinal cord injury, the degeneration of the severed axons through anterograde or Wallerian degeneration (WD) is followed by degradation of myelin and apoptosis of myelinating cells, oligodendrocytes (Crowe et al., 1997). The etiology of MS remains to be fully elucidated and several hypotheses have been proposed to explain the demyelination found in patients, from an autoimmune attack to myelin followed by axonal degeneration, to the developmental, environmental, or virus-induced degeneration of axons by WD followed by myelin degradation; in both cases, myelin debris accumulates (Stys et al., 2012). Myelin proteins such as Nogo are well-known to interfere with axonal regeneration and repair, therefore an efficient myelin clearance is an absolute requirement for recovery (Wang et al., 2002). Furthermore, myelin inhibits its own phagocytosis through myelin CD47 binding to microglia and macrophages signal regulatory protein α (SIRPα) (Gitik et al., 2011). Contrary to the peripheral nervous system (PNS), myelin clearance after WD is very inefficient in the CNS [reviewed in Gaudet et al. (2011)]. In PNS lesions, the myelinating Schwann cells are the major phagocytic population in the first few days, followed by invading macrophages (Hirata and Kawabuchi, 2002). Importantly, macrophages express Nogo receptors NgR1 and NgR2, which facilitate their migration out of the healing nerve, thus resulting in the resolution of the inflammatory response (Fry et al., 2007). In culture, myelin-phagocytosing macrophages inhibit T cell proliferation, further containing the immune response (Bogie et al., 2011). The rapid clearance of myelin and resolution of the immune response is greatly responsible for regeneration after nerve injury.
In contrast, CNS regeneration after trauma does not occur, and microglial poor phagocytosing capabilities together with poor or slow recruitment of macrophages are partly to blame (Gaudet et al., 2011). In vitro, microglia recognize and phagocytose myelin through CR3, SRA, and FcRs among other receptors (Smith, 2001). Microglia do phagocytose myelin to some extent in mouse models of MS, a phenomenon which is stimulated by the presence of MBP-reactive T cells (Nielsen et al., 2009). However, myelin debris is still observed in the human spinal cord years after the injury (Buss et al., 2004). Together with insufficient activation of microglia, the absence of autoreactive antibodies, and the subsequent lack of activation of FcRs in traumatic brain injury have been suggested to explain why microglia phagocytose myelin in MS but not in spinal cord injury (Rotshenker, 2003). Microglial phagocytosis of myelin after hemisection of the ascending sensory tract is increased after treating the mice with LPS, resulting in decreased myelin debris, but, unfortunately, no axonal regeneration (Vallieres et al., 2006), possibly because of the pro-inflammatory phenotype induced by LPS. More recently, a microglial phenotype supportive of remyelination has been described (Olah et al., 2012). During the remyelination phase after cuprizone-induced demyelination in the corpus callosum, a mouse model of MS, myelin-phagocytosing microglia express genes involved not only in phagocytosis but also in the activation, migration, proliferation, and differentiation of oligodendrocytes precursor cells (Olah et al., 2012). While it remains to be directly assessed whether myelin phagocytosis triggers this remyelination-supportive phenotype, this data suggests that the beneficial consequences of enhancing microglial phagocytosis of myelin may be two-fold: clearing myelin and facilitating remyelination.
Conclusion
In conclusion, microglial phagocytosis is a pivotal mechanism of clearance of cellular debris in health and disease. Like Janus, the roman god of war and peace, microglial phagocytosis has beneficial (e.g., anti-inflammatory) and detrimental (e.g., respiratory burst) consequences for tissue homeostasis which remain largely unexplored in vivo. In particular, the establishment of standardized methods to quantify microglial phagocytosis of different types of cargo, as well as the development of novel tools to specifically block recognition, engulfment, and degradation of cargo, will undoubtedly delineate the ultimate impact of microglial phagocytosis in vivo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research of Anahita Shahraz and Harald Neumann was supported by the Hertie-Foundation and the Deutsche Forschungsgemeinschaft (FOR1336, SFB704, KFO177). Research of Oihane Abiega and Amanda Sierra was supported by Ikerbasque, the Basque Foundation of Science.
Abbreviations
ACAMPs, apoptotic cells-associated cellular patterns; AD, Alzheimer's disease; AIF, apoptosis-inducing factor; APCs, antigen presenting cells; APP, amyloid precursor protein; ASD, autism spectrum disorders; ATP, adenosine trinucleotide phosphate; Aβ, amyloid beta; BAI-1, brain-specific angiogenesis inhibitor 1; CAD, caspase-activated DNAse; CNS, central nervous system; CR1, complement receptor 1; DAP12, DNAX-activation protein X; DCs, dendritic cells; DOCK-180, dedicator of cytokinesis 180; EAE, experimental acute encephalomyelitis; ELMO, engulfing and cell motility protein; HMGB1, high mobility group 1 protein; Hsp60, heat shock protein 60; IFNγ, interferon gamma; IGF-1, insulin-like growth factor 1; IL-10, interleukin 10; IL-1β, interleukin 1beta; IL-4, interleukin 4; LPS, bacterial lipopolysaccharides; MBP, myelin basic protein; MCAO, medial cerebral artery occlusion; MFG-E8, milk fat globule-epidermal growth factor; MHC-II, major histocompatibility complex class II; MS, multiple sclerosis; NADPH, nicotinamide adenine dinucleotide phosphate; NGF, neural growth factor; PAMPS, pathogen-associated molecular patterns; PCD, programmed cell death; PD, Parkinson's disease; PLOSL, polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy; PNS, peripheral nervous system; PS, phosphatidylserine; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species; RPE, retinal pigment epithelium; SIRPα, signal regulatory protein alpha; SVZ, subventricular zone; TAMs, tumor-associated macrophages; TGFβ, transforming growth factor beta; TLRs, Toll-like receptors; TNFα, tumor necrosis factor alpha; TREM2, triggering receptor expressed on myeloid cells-2; TRIF, TIR domain-containing adapter inducing interferon beta; UDP, urydil dinucleotide phosphate; UTP, urydil trinucleotide phosphate; WD, Wallerian degeneration.
References
Anand, R. J., Kohler, J. W., Cavallo, J. A., Li, J., Dubowski, T., and Hackam, D. J. (2007). Toll-like receptor 4 plays a role in macrophage phagocytosis during peritoneal sepsis. J. Pediatr. Surg. 42, 927–932. discussion: 933.
Armstrong, A., and Ravichandran, K. S. (2011). Phosphatidylserine receptors: what is the new RAGE? EMBO Rep. 12, 287–288.
Ashwell, K. (1990). Microglia and cell death in the developing mouse cerebellum. Brain Res. Dev. Brain Res. 55, 219–230.
Banati, R. B., Gehrmann, J., Czech, C., Monning, U., Jones, L. L., Konig, G., et al. (1993). Early and rapid de novo synthesis of Alzheimer beta A4-amyloid precursor protein (APP) in activated microglia. Glia 9, 199–210.
Barger, S. W., Goodwin, M. E., Porter, M. M., and Beggs, M. L. (2007). Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem. 101, 1205–1213.
Barres, B. A., Hart, I. K., Coles, H. S., Burne, J. F., Voyvodic, J. T., Richardson, W. D., et al. (1992). Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70, 31–46.
Bianchin, M. M., Capella, H. M., Chaves, D. L., Steindel, M., Grisard, E. C., Ganev, G. G., et al. (2004). Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy–PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell. Mol. Neurobiol. 24, 1–24.
Blank, M., and Shiloh, Y. (2007). Programs for cell death: apoptosis is only one way to go. Cell Cycle 6, 686–695.
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69.
Blomgren, K., Leist, M., and Groc, L. (2007). Pathological apoptosis in the developing brain. Apoptosis 12, 993–1010.
Bogie, J. F., Stinissen, P., Hellings, N., and Hendriks, J. J. (2011). Myelin-phagocytosing macrophages modulate autoreactive T cell proliferation. J. Neuroinflammation 8:85. doi: 10.1186/1742-2094-8-85
Boullier, A., Bird, D. A., Chang, M. K., Dennis, E. A., Friedman, P., Gillotre-Taylor, K., et al. (2001). Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N.Y. Acad. Sci. 947, 214–222. discussion: 222–223.
Brown, G. C., and Neher, J. J. (2012). Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem. Sci. 37, 325–332.
Bsibsi, M., Ravid, R., Gveric, D., and van Noort, J. M. (2002). Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021.
Buckwalter, M. S., Yamane, M., Coleman, B. S., Ormerod, B. K., Chin, J. T., Palmer, T., et al. (2006). Chronically increased transforming growth factor-beta1 strongly inhibits hippocampal neurogenesis in aged mice. Am. J. Pathol. 169, 154–164.
Bulloch, K., Miller, M. M., Gal-Toth, J., Milner, T. A., Gottfried-Blackmore, A., Waters, E. M., et al. (2008). CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J. Comp. Neurol. 508, 687–710.
Buss, A., Brook, G. A., Kakulas, B., Martin, D., Franzen, R., Schoenen, J., et al. (2004). Gradual loss of myelin and formation of an astrocytic scar during Wallerian degeneration in the human spinal cord. Brain 127, 34–44.
Bylund, J., Brown, K. L., Movitz, C., Dahlgren, C., and Karlsson, A. (2010). Intracellular generation of superoxide by the phagocyte NADPH oxidase: how, where, and what for? Free Radic. Biol. Med. 49, 1834–1845.
Ciesielski-Treska, J., Grant, N. J., Ulrich, G., Corrotte, M., Bailly, Y., Haeberle, A. M., et al. (2004). Fibrillar prion peptide (106-126) and scrapie prion protein hamper phagocytosis in microglia. Glia 46, 101–115.
Crehan, H., Hardy, J., and Pocock, J. (2012). Microglia, Alzheimer's disease, and complement. Int. J. Alzheimers Dis. 2012:983640. doi: 10.1155/2012/983640
Crowe, M. J., Bresnahan, J. C., Shuman, S. L., Masters, J. N., and Beattie, M. S. (1997). Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 3, 73–76.
Chan, A., Magnus, T., and Gold, R. (2001). Phagocytosis of apoptotic inflammatory cells by microglia and modulation by different cytokines: mechanism for removal of apoptotic cells in the inflamed nervous system. Glia 33, 87–95.
Chang, G. H., Barbaro, N. M., and Pieper, R. O. (2000). Phosphatidylserine-dependent phagocytosis of apoptotic glioma cells by normal human microglia, astrocytes, and glioma cells. Neuro Oncol. 2, 174–183.
Chung, H., Brazil, M. I., Soe, T. T., and Maxfield, F. R. (1999). Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J. Biol. Chem. 274, 32301–32308.
Dalmau, I., Vela, J. M., Gonzalez, B., Finsen, B., and Castellano, B. (2003). Dynamics of microglia in the developing rat brain. J. Comp. Neurol. 458, 144–157.
Damoiseaux, J. G., Dopp, E. A., Calame, W., Chao, D., MacPherson, G. G., and Dijkstra, C. D. (1994). Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 83, 140–147.
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758.
Daws, M. R., Sullam, P. M., Niemi, E. C., Chen, T. T., Tchao, N. K., and Seaman, W. E. (2003). Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 171, 594–599.
Decoursey, T. E., and Ligeti, E. (2005). Regulation and termination of NADPH oxidase activity. Cell. Mol. Life Sci. 62, 2173–2193.
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109.
De Simone, R., Ajmone-Cat, M. A., Nicolini, A., and Minghetti, L. (2002). Expression of phosphatidylserine receptor and down-regulation of pro-inflammatory molecule production by its natural ligand in rat microglial cultures. J. Neuropathol. Exp. Neurol. 61, 237–244.
De Simone, R., Ajmone-Cat, M. A., Tirassa, P., and Minghetti, L. (2003). Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J. Neuropathol. Exp. Neurol. 62, 208–216.
Desjardins, M., Huber, L. A., Parton, R. G., and Griffiths, G. (1994). Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124, 677–688.
Diez-Roux, G., and Lang, R. A. (1997). Macrophages induce apoptosis in normal cells in vivo. Development 124, 3633–3638.
Dupuy, A. G., and Caron, E. (2008). Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J. Cell Sci. 121, 1773–1783.
Durafourt, B. A., Moore, C. S., Zammit, D. A., Johnson, T. A., Zaguia, F., Guiot, M. C., et al. (2012). Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 60, 717–727.
Egensperger, R., Maslim, J., Bisti, S., Hollander, H., and Stone, J. (1996). Fate of DNA from retinal cells dying during development: uptake by microglia and macroglia (Muller cells). Brain Res. Dev. Brain Res. 97, 1–8.
Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F., et al. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286.
Erdman, L. K., Cosio, G., Helmers, A. J., Gowda, D. C., Grinstein, S., and Kain, K. C. (2009). CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 183, 6452–6459.
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898.
Fallman, M., Andersson, R., and Andersson, T. (1993). Signaling properties of CR3 (CD11b/CD18) and CR1 (CD35) in relation to phagocytosis of complement-opsonized particles. J. Immunol. 151, 330–338.
Falsig, J., van Beek, J., Hermann, C., and Leist, M. (2008). Molecular basis for detection of invading pathogens in the brain. J. Neurosci. Res. 86, 1434–1447.
Familian, A., Eikelenboom, P., and Veerhuis, R. (2007). Minocycline does not affect amyloid beta phagocytosis by human microglial cells. Neurosci. Lett. 416, 87–91.
Farfara, D., Trudler, D., Segev-Amzaleg, N., Galron, R., Stein, R., and Frenkel, D. (2011). gamma-Secretase component presenilin is important for microglia beta-amyloid clearance. Ann. Neurol. 69, 170–180.
Faustino, J. V., Wang, X., Johnson, C. E., Klibanov, A., Derugin, N., Wendland, M. F., et al. (2011). Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 31, 12992–13001.
Flannagan, R. S., Jaumouille, V., and Grinstein, S. (2012). The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98.
Ford, A. L., Goodsall, A. L., Hickey, W. F., and Sedgwick, J. D. (1995). Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J. Immunol. 154, 4309–4321.
Fraser, D. A., Pisalyaput, K., and Tenner, A. J. (2010). C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J. Neurochem. 112, 733–743.
Freeman, G. J., Casasnovas, J. M., Umetsu, D. T., and DeKruyff, R. H. (2010). TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 235, 172–189.
Fricker, M., Neher, J. J., Zhao, J. W., Thery, C., Tolkovsky, A. M., and Brown, G. C. (2012). MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 32, 2657–2666.
Frielingsdorf, H., Simpson, D. R., Thal, L. J., and Pizzo, D. P. (2007). Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol. Dis. 26, 47–55.
Fry, E. J., Ho, C., and David, S. (2007). A role for Nogo receptor in macrophage clearance from injured peripheral nerve. Neuron 53, 649–662.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat. Neurosci. 13, 411–413.
Fulci, G., Dmitrieva, N., Gianni, D., Fontana, E. J., Pan, X., Lu, Y., et al. (2007). Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 67, 9398–9406.
Gaikwad, S., Larionov, S., Wang, Y., Dannenberg, H., Matozaki, T., Monsonego, A., et al. (2009). Signal regulatory protein-beta1: a microglial modulator of phagocytosis in Alzheimer's disease. Am. J. Pathol. 175, 2528–2539.
Galarneau, H., Villeneuve, J., Gowing, G., Julien, J. P., and Vallieres, L. (2007). Increased glioma growth in mice depleted of macrophages. Cancer Res. 67, 8874–8881.
Galea, I., Bechmann, I., and Perry, V. H. (2007). What is immune privilege (not)? Trends Immunol. 28, 12–18.
Galic, M. A., Riazi, K., and Pittman, Q. J. (2012). Cytokines and brain excitability. Front. Neuroendocrinol. 33:116–125. doi: 10.1016/j.yfrne.2011.12.002
Garin, J., Diez, R., Kieffer, S., Dermine, J. F., Duclos, S., Gagnon, E., et al. (2001). The phagosome proteome: insight into phagosome functions. J. Cell Biol. 152, 165–180.
Gaudet, A. D., Popovich, P. G., and Ramer, M. S. (2011). Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 8:110. doi: 10.1186/1742-2094-8-110
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845.
Gitik, M., Liraz-Zaltsman, S., Oldenborg, P. A., Reichert, F., and Rotshenker, S. (2011). Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPalpha (signal regulatory protein-alpha) on phagocytes. J. Neuroinflammation 8:24. doi: 10.1186/1742-2094-8-24
Goh, Y. C., Yap, C. T., Huang, B. H., Cronshaw, A. D., Leung, B. P., Lai, P. B., et al. (2011). Heat-shock protein 60 translocates to the surface of apoptotic cells and differentiated megakaryocytes and stimulates phagocytosis. Cell. Mol. Life Sci. 68, 1581–1592.
Gottfried-Blackmore, A., Kaunzner, U. W., Idoyaga, J., Felger, J. C., McEwen, B. S., and Bulloch, K. (2009). Acute in vivo exposure to interferon-gamma enables resident brain dendritic cells to become effective antigen presenting cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20918–20923.
Granucci, F., Petralia, F., Urbano, M., Citterio, S., Di Tota, F., Santambrogio, L., et al. (2003). The scavenger receptor MARCO mediates cytoskeleton rearrangements in dendritic cells and microglia. Blood 102, 2940–2947.
Greenberg, M. E., Sun, M., Zhang, R., Febbraio, M., Silverstein, R., and Hazen, S. L. (2006). Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203, 2613–2625.
Grommes, C., Lee, C. Y., Wilkinson, B. L., Jiang, Q., Koenigsknecht-Talboo, J. L., Varnum, B., et al. (2008). Regulation of microglial phagocytosis and inflammatory gene expression by Gas6 acting on the Axl/Mer family of tyrosine kinases. J. Neuroimmune Pharmacol. 3, 130–140.
Gumienny, T. L., Brugnera, E., Tosello-Trampont, A. C., Kinchen, J. M., Haney, L. B., Nishiwaki, K., et al. (2001). CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41.
Hadas, S., Reichert, F., and Rotshenker, S. (2010). Dissimilar and similar functional properties of complement receptor-3 in microglia and macrophages in combating yeast pathogens by phagocytosis. Glia 58, 823–830.
Han, C. Z., and Ravichandran, K. S. (2011). Metabolic connections during apoptotic cell engulfment. Cell 147, 1442–1445.
Hart, S. P., Alexander, K. M., and Dransfield, I. (2004). Immune complexes bind preferentially to Fc gamma RIIA (CD32) on apoptotic neutrophils, leading to augmented phagocytosis by macrophages and release of proinflammatory cytokines. J. Immunol. 172, 1882–1887.
Hayashi, A., Ohnishi, H., Okazawa, H., Nakazawa, S., Ikeda, H., Motegi, S., et al. (2004). Positive regulation of phagocytosis by SIRPbeta and its signaling mechanism in macrophages. J. Biol. Chem. 279, 29450–29460.
He, M., Kubo, H., Morimoto, K., Fujino, N., Suzuki, T., Takahasi, T., et al. (2011). Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12, 358–364.
Henson, P. M., and Hume, D. A. (2006). Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 27, 244–250.
Heppner, F. L., Greter, M., Marino, D., Falsig, J., Raivich, G., Hovelmeyer, N., et al. (2005). Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 11, 146–152.
Herber, D. L., Roth, L. M., Wilson, D., Wilson, N., Mason, J. E., Morgan, D., et al. (2004). Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp. Neurol. 190, 245–253.
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360.
Hirata, K., and Kawabuchi, M. (2002). Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc. Res. Tech. 57, 541–547.
Hirt, U. A., Gantner, F., and Leist, M. (2000). Phagocytosis of nonapoptotic cells dying by caspase-independent mechanisms. J. Immunol. 164, 6520–6529.
Hirt, U. A., and Leist, M. (2003). Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 10, 1156–1164.
Hoeffel, G., Wang, Y., Greter, M., See, P., Teo, P., Malleret, B., et al. (2012). Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 209, 1167–1181.
Hoffmann, P. R., deCathelineau, A. M., Ogden, C. A., Leverrier, Y., Bratton, D. L., Daleke, D. L., et al. (2001). Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155, 649–659.
Hosmane, S., Tegenge, M. A., Rajbhandari, L., Uapinyoying, P., Kumar, N. G., Thakor, N., et al. (2012). Toll/interleukin-1 receptor domain-containing adapter inducing interferon-beta mediates microglial phagocytosis of degenerating axons. J. Neurosci. 32, 7745–7757.
Hsieh, C. L., Koike, M., Spusta, S. C., Niemi, E. C., Yenari, M., Nakamura, M. C., et al. (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156.
Huang, W. C., Yen, F. C., Shie, F. S., Pan, C. M., Shiao, Y. J., Yang, C. N., et al. (2010). TGF-beta1 blockade of microglial chemotaxis toward Abeta aggregates involves SMAD signaling and down-regulation of CCL5. J. Neuroinflammation 7:28. doi: 10.1186/1742-2094-7-28
Hughes, M. M., Field, R. H., Perry, V. H., Murray, C. L., and Cunningham, C. (2010). Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 58, 2017–2030.
Hussain, S. F., Yang, D., Suki, D., Aldape, K., Grimm, E., and Heimberger, A. B. (2006). The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 8, 261–279.
Huysentruyt, L. C., and Seyfried, T. N. (2010). Perspectives on the mesenchymal origin of metastatic cancer. Cancer Metastasis Rev. 29, 695–707.
Johnston, H., Boutin, H., and Allan, S. M. (2011). Assessing the contribution of inflammation in models of Alzheimer's disease. Biochem. Soc. Trans. 39, 886–890.
Kaifu, T., Nakahara, J., Inui, M., Mishima, K., Momiyama, T., Kaji, M., et al. (2003). Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Invest. 111, 323–332.
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4441–4446.
Kaur, C., Too, H. F., and Ling, E. A. (2004). Phagocytosis of Escherichia coli by amoeboid microglial cells in the developing brain. Acta Neuropathol. 107, 204–208.
Kawane, K., Fukuyama, H., Yoshida, H., Nagase, H., Ohsawa, Y., Uchiyama, Y., et al. (2003). Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat. Immunol. 4, 138–144.
Kevany, B. M., and Palczewski, K. (2010). Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 25, 8–15.
Kielian, T., Esen, N., and Bearden, E. D. (2005). Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia 49, 567–576.
Kim, J. B., Sig Choi, J., Yu, Y. M., Nam, K., Piao, C. S., Kim, S. W., et al. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421.
Kim, S., Park, S. Y., Kim, S. Y., Bae, D. J., Pyo, J. H., Hong, M., et al. (2012). Cross talk between engulfment receptors stabilin-2 and integrin alphavbeta5 orchestrates engulfment of phosphatidylserine-exposed erythrocytes. Mol. Cell. Biol. 32, 2698–2708.
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273.
Kochan, T., Singla, A., Tosi, J., and Kumar, A. (2012). Toll-like receptor 2 ligand pretreatment attenuates retinal microglial inflammatory response but enhances phagocytic activity toward Staphylococcus aureus. Infect. Immun. 80, 2076–2088.
Koenigsknecht-Talboo, J., and Landreth, G. E. (2005). Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 25, 8240–8249.
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095.
Kulprathipanja, N. V., and Kruse, C. A. (2004). Microglia phagocytose alloreactive CTL-damaged 9L gliosarcoma cells. J. Neuroimmunol. 153, 76–82.
Lai, A. Y., and McLaurin, J. (2012). Clearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. 7, 165–176.
Lambertsen, K. L., Biber, K., and Finsen, B. (2012). Inflammatory cytokines in experimental and human stroke. J. Cereb. Blood Flow Metab. 32, 1677–1698.
Lan, X., Kiyota, T., Hanamsagar, R., Huang, Y., Andrews, S., Peng, H., et al. (2012). The effect of HIV protease inhibitors on amyloid-beta peptide degradation and synthesis in human cells and Alzheimer's disease animal model. J. Neuroimmune Pharmacol. 7, 412–423.
Landreth, G. E., and Reed-Geaghan, E. G. (2009). Toll-like receptors in Alzheimer's disease. Curr. Top. Microbiol. Immunol. 336, 137–153.
Lang, R. A., and Bishop, J. M. (1993). Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell 74, 453–462.
Lauber, K., Blumenthal, S. G., Waibel, M., and Wesselborg, S. (2004). Clearance of apoptotic cells: getting rid of the corpses. Mol. Cell 14, 277–287.
Lee, S., Varvel, N. H., Konerth, M. E., Xu, G., Cardona, A. E., Ransohoff, R. M., et al. (2010). CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am. J. Pathol. 177, 2549–2562.
Lee, W. L., Mason, D., Schreiber, A. D., and Grinstein, S. (2007). Quantitative analysis of membrane remodeling at the phagocytic cup. Mol. Biol. Cell 18, 2883–2892.
Leist, M., and Jaattela, M. (2001). Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2, 589–598.
Leist, M., Volbracht, C., Kuhnle, S., Fava, E., Ferrando-May, E., and Nicotera, P. (1997). Caspase-mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide. Mol. Med. 3, 750–764.
Li, Q., Jagannath, C., Rao, P. K., Singh, C. R., and Lostumbo, G. (2010). Analysis of phagosomal proteomes: from latex-bead to bacterial phagosomes. Proteomics 10, 4098–4116.
Li, Y. B., Kaur, C., and Ling, E. A. (1998). Neuronal degeneration and microglial reaction in the fetal and postnatal rat brain after transient maternal hypoxia. Neurosci. Res. 32, 137–148.
Linnartz, B., Kopatz, J., Tenner, A. J., and Neumann, H. (2012). Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 32, 946–952.
Linnartz, B., and Neumann, H. (2012). Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia 61, 37–46.
Litman, G. W., Cannon, J. P., and Dishaw, L. J. (2005). Reconstructing immune phylogeny: new perspectives. Nat. Rev. Immunol. 5, 866–879.
Liu, S., Liu, Y., Hao, W., Wolf, L., Kiliaan, A. J., Penke, B., et al. (2012). TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107.
Liu, Y., Hao, W., Letiembre, M., Walter, S., Kulanga, M., Neumann, H., et al. (2006). Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J. Neurosci. 26, 12904–12913.
Liu, Y., Walter, S., Stagi, M., Cherny, D., Letiembre, M., Schulz-Schaeffer, W., et al. (2005). LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain 128, 1778–1789.
Liu, Z., Condello, C., Schain, A., Harb, R., and Grutzendler, J. (2010). CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J. Neurosci. 30, 17091–17101.
Lowell, C. A. (2011). Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb. Perspect. Biol. 3:pii: a002352. doi: 10.1101/cshperspect.a002352
Lowell, C. A., Soriano, P., and Varmus, H. E. (1994). Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 8, 387–398.
Lu, Z., Elliott, M. R., Chen, Y., Walsh, J. T., Klibanov, A. L., Ravichandran, K. S., et al. (2011). Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 13, 1076–1083.
Lucas, M., Stuart, L. M., Zhang, A., Hodivala-Dilke, K., Febbraio, M., Silverstein, R., et al. (2006). Requirements for apoptotic cell contact in regulation of macrophage responses. J. Immunol. 177, 4047–4054.
Luo, L., and O'Leary, D. D. (2005). Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 28, 127–156.
Maezawa, I., Zimin, P. I., Wulff, H., and Jin, L. W. (2011). Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 286, 3693–3706.
Magnus, T., Chan, A., Grauer, O., Toyka, K. V., and Gold, R. (2001). Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation. J. Immunol. 167, 5004–5010.
Magnus, T., Chan, A., Linker, R. A., Toyka, K. V., and Gold, R. (2002). Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: implications for the role of glial cells in the inflamed central nervous system. J. Neuropathol. Exp. Neurol. 61, 760–766.
Maher, E. A., Furnari, F. B., Bachoo, R. M., Rowitch, D. H., Louis, D. N., Cavenee, W. K., et al. (2001). Malignant glioma: genetics and biology of a grave matter. Genes Dev. 15, 1311–1333.
Mantovani, A., Sozzani, S., Locati, M., Allavena, P., and Sica, A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555.
Marin-Teva, J. L., Dusart, I., Colin, C., Gervais, A., van Rooijen, N., and Mallat, M. (2004). Microglia promote the death of developing Purkinje cells. Neuron 41, 535–547.
Marzolo, M. P., von Bernhardi, R., and Inestrosa, N. C. (1999). Mannose receptor is present in a functional state in rat microglial cells. J. Neurosci. Res. 58, 387–395.
McArthur, S., Cristante, E., Paterno, M., Christian, H., Roncaroli, F., Gillies, G. E., et al. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 185, 6317–6328.
McCoy, C. E., and O'Neill, L. A. (2008). The role of toll-like receptors in macrophages. Front. Biosci. 13, 62–70.
McIlroy, D., Tanaka, M., Sakahira, H., Fukuyama, H., Suzuki, M., Yamamura, K., et al. (2000). An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 14, 549–558.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., et al. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 451, 720–724.
Minakami, R., and Sumimotoa, H. (2006). Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int. J. Hematol. 84, 193–198.
Mizuno, T. (2012). The biphasic role of microglia in Alzheimer's disease. Int. J. Alzheimers Dis. 2012:737846. doi: 10.1155/2012/737846
Muhammad, S., Barakat, W., Stoyanov, S., Murikinati, S., Yang, H., Tracey, K. J., et al. (2008). The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 28, 12023–12031.
Nagata, S., Hanayama, R., and Kawane, K. (2010). Autoimmunity and the clearance of dead cells. Cell 140, 619–630.
Neher, J. J., Neniskyte, U., Zhao, J. W., Bal-Price, A., Tolkovsky, A. M., and Brown, G. C. (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 186, 4973–4983.
Neniskyte, U., Neher, J. J., and Brown, G. C. (2011). Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913.
Neumann, J., Sauerzweig, S., Ronicke, R., Gunzer, F., Dinkel, K., Ullrich, O., et al. (2008). Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J. Neurosci. 28, 5965–5975.
Newell, E. W., Stanley, E. F., and Schlichter, L. C. (2007). Reversed Na+/Ca2+ exchange contributes to Ca2+ influx and respiratory burst in microglia. Channels (Austin) 1, 366–376.
Nielsen, H. H., Ladeby, R., Fenger, C., Toft-Hansen, H., Babcock, A. A., Owens, T., et al. (2009). Enhanced microglial clearance of myelin debris in T cell-infiltrated central nervous system. J. Neuropathol. Exp. Neurol. 68, 845–856.
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318.
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319.
Noda, M., and Suzumura, A. (2012). Sweepers in the CNS: microglial migration and phagocytosis in the alzheimer disease pathogenesis. Int. J. Alzheimers Dis. 2012:891087. doi: 10.1155/2012/891087
Ogden, C. A., deCathelineau, A., Hoffmann, P. R., Bratton, D., Ghebrehiwet, B., Fadok, V. A., et al. (2001). C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194, 781–795.
O'Kusky, J. R., Ye, P., and D'Ercole, A. J. (2000). Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 20, 8435–8442.
Olah, M., Amor, S., Brouwer, N., Vinet, J., Eggen, B., Biber, K., et al. (2012). Identification of a microglia phenotype supportive of remyelination. Glia 60, 306–321.
Pais, T. F., Figueiredo, C., Peixoto, R., Braz, M. H., and Chatterjee, S. (2008). Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J. Neuroinflammation 5:43. doi: 10.1186/1742-2094-5-43
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662.
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458.
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553–565.
Park, J. Y., Paik, S. R., Jou, I., and Park, S. M. (2008). Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson's disease. Glia 56, 1215–1223.
Parnaik, R., Raff, M. C., and Scholes, J. (2000). Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr. Biol. 10, 857–860.
Patel, M., Pelletier, A., and Cote, J. F. (2011). Opening up on ELMO regulation: new insights into the control of Rac signaling by the DOCK180/ELMO complex. Small GTPases 2, 268–275.
Peppoloni, S., Posteraro, B., Colombari, B., Manca, L., Hartke, A., Giard, J. C., et al. (2011). Role of the (Mn)superoxide dismutase of Enterococcus faecalis in the in vitro interaction with microglia. Microbiology 157, 1816–1822.
Peri, F., and Nusslein-Volhard, C. (2008). Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916–927.
Persaud-Sawin, D. A., Banach, L., and Harry, G. J. (2009). Raft aggregation with specific receptor recruitment is required for microglial phagocytosis of Abeta42. Glia 57, 320–335.
Pickering, M., Cumiskey, D., and O'Connor, J. J. (2005). Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 90, 663–670.
Pourova, J., Kottova, M., Voprsalova, M., and Pour, M. (2010). Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiol. (Oxf.) 198, 15–35.
Qiao, X., Cummins, D. J., and Paul, S. M. (2001). Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur. J. Neurosci. 14, 474–482.
Qin, L., Liu, Y., Wang, T., Wei, S. J., Block, M. L., Wilson, B., et al. (2004). NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 279, 1415–1421.
Quan, D. N., Cooper, M. D., Potter, J. L., Roberts, M. H., Cheng, H., and Jarvis, G. A. (2008). TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal Immunol. 1, 229–238.
Quinn, M. T., and Gauss, K. A. (2004). Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J. Leukoc. Biol. 76, 760–781.
Raivich, G., Bohatschek, M., Kloss, C. U., Werner, A., Jones, L. L., and Kreutzberg, G. W. (1999). Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res. Brain Res. Rev. 30, 77–105.
Ransohoff, R. M., and Engelhardt, B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635.
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145.
Ravichandran, K. S. (2010). Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 207, 1807–1817.
Reddien, P. W., Cameron, S., and Horvitz, H. R. (2001). Phagocytosis promotes programmed cell death in C. elegans. Nature 412, 198–202.
Reubold, T. F., and Eschenburg, S. (2012). A molecular view on signal transduction by the apoptosome. Cell. Signal. 24, 1420–1425.
Ribes, S., Ebert, S., Regen, T., Agarwal, A., Tauber, S. C., Czesnik, D., et al. (2010). Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect. Immun. 78, 865–871.
Rogers, N. J., Lees, M. J., Gabriel, L., Maniati, E., Rose, S. J., Potter, P. K., et al. (2009). A defect in Marco expression contributes to systemic lupus erythematosus development via failure to clear apoptotic cells. J. Immunol. 182, 1982–1990.
Rotshenker, S. (2003). Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. J. Mol. Neurosci. 21, 65–72.
Rotshenker, S. (2009). The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci. 39, 99–103.
Roumier, A., Bechade, C., Poncer, J. C., Smalla, K. H., Tomasello, E., Vivier, E., et al. (2004). Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J. Neurosci. 24, 11421–11428.
Salman, H., Bergman, M., Djaldetti, R., Bessler, H., and Djaldetti, M. (1999). Decreased phagocytic function in patients with Parkinson's disease. Biomed. Pharmacother. 53, 146–148.
Satoh, J., Tabunoki, H., Ishida, T., Yagishita, S., Jinnai, K., Futamura, N., et al. (2011). Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology 31, 363–375.
Savill, J., Dransfield, I., Gregory, C., and Haslett, C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975.
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195.
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705.
Schulz, C., Gomez Perdiguero, E., Chorro, L., Szabo-Rogers, H., Cagnard, N., Kierdorf, K., et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90.
Scott, S. A., Johnson, S. A., Zarow, C., and Perlmutter, L. S. (1993). Inability to detect beta-amyloid protein precursor mRNA in Alzheimer plaque-associated microglia. Exp. Neurol. 121, 113–118.
Shah, V. B., Huang, Y., Keshwara, R., Ozment-Skelton, T., Williams, D. L., and Keshvara, L. (2008). Beta-glucan activates microglia without inducing cytokine production in Dectin-1-dependent manner. J. Immunol. 180, 2777–2785.
Sierra, A., Encinas, J. M., Deudero, J. J., Chancey, J. H., Enikolopov, G., Overstreet-Wadiche, L. S., et al. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495.
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502.
Sivagnanam, V., Zhu, X., and Schlichter, L. C. (2010). Dominance of E. coli phagocytosis over LPS in the inflammatory response of microglia. J. Neuroimmunol. 227, 111–119.
Smith, M. E. (2001). Phagocytic properties of microglia in vitro: implications for a role in multiple sclerosis and EAE. Microsc. Res. Tech. 54, 81–94.
Song, L., Lee, C., and Schindler, C. (2011). Deletion of the murine scavenger receptor CD68. J. Lipid Res. 52, 1542–1550.
Spranger, M., Kiprianova, I., Krempien, S., and Schwab, S. (1998). Reoxygenation increases the release of reactive oxygen intermediates in murine microglia. J. Cereb. Blood Flow Metab. 18, 670–674.
Stalder, M., Deller, T., Staufenbiel, M., and Jucker, M. (2001). 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol. Aging 22, 427–434.
Stefano, L., Racchetti, G., Bianco, F., Passini, N., Gupta, R. S., Panina Bordignon, P., et al. (2009). The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J. Neurochem. 110, 284–294.
Stern, M., Savill, J., and Haslett, C. (1996). Human monocyte-derived macrophage phagocytosis of senescent eosinophils undergoing apoptosis. Mediation by alpha v beta 3/CD36/thrombospondin recognition mechanism and lack of phlogistic response. Am. J. Pathol. 149, 911–921.
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581.
Stys, P. K., Zamponi, G. W., van Minnen, J., and Geurts, J. J. (2012). Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 13, 507–514.
Takahashi, K., Prinz, M., Stagi, M., Chechneva, O., and Neumann, H. (2007). TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4:e124. doi: 10.1371/journal.pmed.0040124
Takahashi, K., Rochford, C. D., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657.
Tanaka, T., Ueno, M., and Yamashita, T. (2009). Engulfment of axon debris by microglia requires p38 MAPK activity. J. Biol. Chem. 284, 21626–21636.
Taylor, P. R., Martinez-Pomares, L., Stacey, M., Lin, H. H., Brown, G. D., and Gordon, S. (2005). Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23, 901–944.
Testa-Silva, G., Loebel, A., Giugliano, M., de Kock, C. P., Mansvelder, H. D., and Meredith, R. M. (2012). Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb. Cortex 22, 1333–1342.
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., et al. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849.
Tremblay, M. E., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Truman, L. A., Ford, C. A., Pasikowska, M., Pound, J. D., Wilkinson, S. J., Dumitriu, I. E., et al. (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036.
Ueyama, T., Lennartz, M. R., Noda, Y., Kobayashi, T., Shirai, Y., Rikitake, K., et al. (2004). Superoxide production at phagosomal cup/phagosome through beta I protein kinase C during Fc gamma R-mediated phagocytosis in microglia. J. Immunol. 173, 4582–4589.
Ulvestad, E., Williams, K., Bjerkvig, R., Tiekotter, K., Antel, J., and Matre, R. (1994). Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J. Leukoc. Biol. 56, 732–740.
Underhill, D. M., and Goodridge, H. S. (2012). Information processing during phagocytosis. Nat. Rev. Immunol. 12, 492–502.
Vallieres, N., Berard, J. L., David, S., and Lacroix, S. (2006). Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia 53, 103–113.
van der Laan, L. J., Dopp, E. A., Haworth, R., Pikkarainen, T., Kangas, M., Elomaa, O., et al. (1999). Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J. Immunol. 162, 939–947.
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81.
Voisin, P., Bouchaud, V., Merle, M., Diolez, P., Duffy, L., Flint, K., et al. (2010). Microglia in close vicinity of glioma cells: correlation between phenotype and metabolic alterations. Front. Neuroenergetics 2:131. doi: 10.3389/fnene.2010.00131
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351.
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980.
Wakselman, S., Bechade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143.
Wang, A., Das, P., Switzer, R. C. 3rd., Golde, T. E., and Jankowsky, J. L. (2011). Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J. Neurosci. 31, 4124–4136.
Wang, K. C., Koprivica, V., Kim, J. A., Sivasankaran, R., Guo, Y., Neve, R. L., et al. (2002). Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417, 941–944.
Wang, Y., and Neumann, H. (2010). Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 30, 3482–3488.
Watters, J. J., Schartner, J. M., and Badie, B. (2005). Microglia function in brain tumors. J. Neurosci. Res. 81, 447–455.
Welser-Alves, J. V., Boroujerdi, A., Tigges, U., and Milner, R. (2011). Microglia use multiple mechanisms to mediate interactions with vitronectin; non-essential roles for the highly-expressed alphavbeta3 and alphavbeta5 integrins. J. Neuroinflammation 8:157. doi: 10.1186/1742-2094-8-157
Wilkinson, K., and El Khoury, J. (2012). Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int. J. Alzheimers Dis. 2012:489456. doi: 10.1155/2012/489456
Williams, D. W., Kondo, S., Krzyzanowska, A., Hiromi, Y., and Truman, J. W. (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236.
Williams, D. W., and Truman, J. W. (2005). Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development 132, 3631–3642.
Williams, K., Ulvestad, E., Waage, A., Antel, J. P., and McLaurin, J. (1994). Activation of adult human derived microglia by myelin phagocytosis in vitro. J. Neurosci. Res. 38, 433–443.
Wu, A., Wei, J., Kong, L. Y., Wang, Y., Priebe, W., Qiao, W., et al. (2010). Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 12, 1113–1125.
Wu, H. H., Bellmunt, E., Scheib, J. L., Venegas, V., Burkert, C., Reichardt, L. F., et al. (2009). Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat. Neurosci. 12, 1534–1541.
Xu, J., and Ikezu, T. (2009). The comorbidity of HIV-associated neurocognitive disorders and Alzheimer's disease: a foreseeable medical challenge in post-HAART era. J. Neuroimmune Pharmacol. 4, 200–212.
Yi, L., Xiao, H., Xu, M., Ye, X., Hu, J., Li, F., et al. (2011). Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 232, 75–82.
Yokoyama, N., deBakker, C. D., Zappacosta, F., Huddleston, M. J., Annan, R. S., Ravichandran, K. S., et al. (2005). Identification of tyrosine residues on ELMO1 that are phosphorylated by the Src-family kinase Hck. Biochemistry 44, 8841–8849.
Zeyaullah, M., Patro, M., Ahmad, I., Ibraheem, K., Sultan, P., Nehal, M., et al. (2012). Oncolytic viruses in the treatment of cancer: a review of current strategies. Pathol. Oncol. Res. 18, 771–781.
Zhang, D., Sun, L., Zhu, H., Wang, L., Wu, W., Xie, J., et al. (2012). Microglial LOX-1 reacts with extracellular HSP60 to bridge neuroinflammation and neurotoxicity. Neurochem. Int. 61, 1021–1035.
Zhang, S., Boyd, J., Delaney, K., and Murphy, T. H. (2005). Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J. Neurosci. 25, 5333–5338.
Keywords: microglia, phagocytosis, apoptosis, synapses, debris, myelin, amyloid, inflammation
Citation: Sierra A, Abiega O, Shahraz A and Neumann H (2013) Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 7:6. doi: 10.3389/fncel.2013.00006
Received: 10 October 2012; Paper pending published: 11 November 2012;
Accepted: 09 January 2013; Published online: 30 January 2013.
Edited by:
Marie-Eve Tremblay, Université Laval, CanadaReviewed by:
Marcel Leist, University of Konstanz, GermanyRafael Linden, Federal University of Rio de Janeiro, Brazil
Copyright © 2013 Sierra, Abiega, Shahraz and Neumann. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Amanda Sierra, Achucarro—Basque Center for Neuroscience, Laida Bidea, Building 205, Bizkaia Technological Park, 48170 Zamudio, Spain. e-mail: a.sierra@ikerbasque.org
Harald Neumann, Neural Reconstruction Group, Institute of Reconstructive Neurobiology, University Bonn, Sigmund-Freud-Str. 25, 53127 Bonn, Germany. e-mail: hneuman1@uni-bonn.de