NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia
- Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, PA, USA
Schizophrenia is a disabling mental illness that is now recognized as a neurodevelopmental disorder. It is likely that genetic risk factors interact with environmental perturbations to affect normal brain development and that this altered trajectory results in a combination of positive, negative, and cognitive symptoms. Although the exact pathophysiology of schizophrenia is unknown, the N-methyl-D-aspartate receptor (NMDAR), a major glutamate receptor subtype, has received great attention. Proper expression and regulation of NMDARs in the brain is critical for learning and memory processes as well as cortical plasticity and maturation. Evidence from both animal models and human studies implicates a dysfunction of NMDARs both in disease progression and symptoms of schizophrenia. Furthermore, mutations in many of the known genetic risk factors for schizophrenia suggest that NMDAR hypofunction is a convergence point for schizophrenia. In this review, we discuss how disrupted NMDAR function leads to altered neurodevelopment that may contribute to the progression and development of symptoms for schizophrenia, particularly cognitive deficits. We review the shared signaling pathways among the schizophrenia susceptibility genes DISC1, neuregulin1, and dysbindin, focusing on the AKT/GSK3β pathway, and how their mutations and interactions can lead to NMDAR dysfunction during development. Additionally, we explore what open questions remain and suggest where schizophrenia research needs to move in order to provide mechanistic insight into the cause of NMDAR dysfunction, as well as generate possible new avenues for therapeutic intervention.
Introduction
Schizophrenia is a devastating psychological disorder that consists of a complex set of positive, negative, and cognitive symptoms. Although the pathophysiological mechanisms associated with this disease remain unclear, the dopamine (DA) hypothesis has dominated the theories of schizophrenia for several decades (Howes and Kapur, 2009; Abi-Dargham, 2012). It was proposed that hyperactivity in the mesolimbic DA pathway is the mediator of positive symptoms of schizophrenia, whereas hypoactivity in the mesocortical DA pathway mediates the negative and cognitive symptoms of schizophrenia. However, focusing on the DA system has led to limited progress in understanding the pathophysiological processes in schizophrenia, and subsequently has led to minimal development of novel therapeutics (Miyamoto et al., 2012). In the past two decades, hypotheses of schizophrenia have progressed beyond the DA hypothesis. In a major paradigm shift on the etiology of schizophrenia, it has been proposed that numerous genetic and environmental risk factors converge on the N-methyl-D-aspartate receptors (NMDAR)-mediated glutamatergic system and result in NMDAR hypofunction in the limbic system during neurodevelopment.
NMDARs are widely thought to be crucial in synaptic plasticity and circuit formation for pre- and early postnatal stages of brain development, otherwise known as the “critical developmental window.” Numerous studies have indicated that the maturation of brain circuitry is usually coincident with the NMDAR subunit switch (e.g., NR2B-to-NR2A and NR3A-to-NR3B) that occurs at the onset of the critical period of development (Monyer et al., 1994; Sheng et al., 1994; Quinlan et al., 1999; Wang et al., 2008; Roberts et al., 2009; Wang and Gao, 2009; Snyder et al., 2013). The NMDAR subunit shift therefore marks the transition from juvenile to “adult” neural processing (Dumas, 2005; Henson et al., 2010) and the subunit switch makes the NMDARs extremely vulnerable to genetic and environmental risk factors (Spear, 2000). Because NMDARs regulate DA neurons and DA transmission, hypofunction of NMDARs may be responsible for the abnormal DA activity associated with the symptoms of schizophrenia. Indeed, the NMDAR-mediated glutamatergic model provides an alternate approach for conceptualizing the brain abnormalities associated with schizophrenia (Harrison and Weinberger, 2005; Lewis and Moghaddam, 2006; Lisman et al., 2008). Although it remains unclear what changes induce the onset of cognitive dysfunction, NMDAR dysfunction appears to be a convergence point for progression and symptoms of schizophrenia, especially for cognitive deficits. There have been several elegant review articles; some issues on a specific topic, such as neuregulin1, circuit-level glutamatergic hypothesis and metabotropic glutamate receptors, can be found in these references (Moghaddam, 2003; Coyle, 2006; Lisman et al., 2008; Banerjee et al., 2010; Marek et al., 2010; Niswender and Conn, 2010; Geddes et al., 2011; Lin et al., 2012; Millan et al., 2012; Vinson and Conn, 2012). Below we focus on the current literature and explain how the hypothesis of NMDA hypofunction is formulated, why NMDA hypofunction could be a convergence point for the progression and symptoms of schizophrenia, what mechanisms are associated with regulation of NMDAR function, as well as possible signaling pathways related to the regulation of NMDAR function by high-risk genes for schizophrenia. It is likely that convergent mechanisms target NMDAR, which in turn contribute to negative symptoms and neurocognitive dysfunction directly (Lau and Zukin, 2007), as well as to positive symptoms via dysregulation of brain DA systems indirectly (Howes and Kapur, 2009; Abi-Dargham, 2012).
Evidence for Abnormal Glutamate Transmission and NMDAR Hypofunction in Schizophrenia
In the past two decades, the abnormalities found in human subjects with schizophrenia and the various animal models for schizophrenia all point to an important contribution of the glutamatergic system to the disease (Moghaddam and Jackson, 2003; Javitt, 2004; Millan, 2005). Accumulating studies have shown that aberrant NMDAR function, namely NMDAR hypofunction, in the limbic brain region, may underlie many aspects of molecular, cellular, and behavioral abnormalities associated with schizophrenia (Mohn et al., 1999; Olney et al., 1999; Tamminga, 1999; Dracheva et al., 2001; Krystal et al., 2002; Moghaddam and Jackson, 2003; Javitt, 2004; Coyle, 2006). First, mice with reduced NMDAR expression display behaviors related to schizophrenia (Mohn et al., 1999). Second, NMDAR antagonists, such as phencyclidine (PCP), dizocilpine (MK-801), and ketamine, produce “schizophrenia like” symptoms in healthy individuals (Javitt and Zukin, 1991; Krystal et al., 1994; Lahti et al., 1995). Compelling evidence has suggested that the NMDAR antagonist PCP and its analog compounds can produce a pattern of metabolic, neurochemical, and behavioral changes that reproduce almost exactly those seen in patients with schizophrenia, with remarkable regional specificity (Morris et al., 2005). This finding has provided considerable insight into the processes that lead to the development of the disease, emphasizing the potential importance of NMDAR hypofunction. Third, a majority of the genes that are associated with an increased risk for schizophrenia can influence the function of NMDARs or related receptor-interacting proteins and signal transduction pathways (Moghaddam, 2003; Harrison and Weinberger, 2005) (see below for detail). Fourth, dysregulated NMDAR subunits are usually seen in postmortem tissue from patients with schizophrenia (Akbarian et al., 1996; Gao et al., 2000; Kristiansen et al., 2007; Geddes et al., 2011; Weickert et al., 2012) and in animal models of NMDAR antagonism (Lisman et al., 2008; Gunduz-Bruce, 2009). Postmortem studies also show changes in glutamate receptor binding, transcription, and subunit protein expression in the prefrontal cortex (Akbarian et al., 1996; Kristiansen et al., 2006; Beneyto and Meador-Woodruff, 2008), thalamus (Ibrahim et al., 2000; Clinton and Meador-Woodruff, 2004; Clinton et al., 2006; Dracheva et al., 2008), and hippocampus (Gao et al., 2000; Beneyto et al., 2007; McCullumsmith et al., 2007) of subjects with schizophrenia (Geddes et al., 2011). These changes include decreased NR1, increased excitatory amino-acid transporter, and altered NMDA receptor-affiliated intracellular proteins such as post synaptic density protein 95 (PSD95) and synapse associated protein 102 (SAP102) in the prefrontal cortex and thalamus [see (Geddes et al., 2011) Table 1 for detail]. Fifth, glutamatergic neurons also interact with other neurons that have been strongly implicated in the pathophysiology of schizophrenia, including morphologically altered GABAergic interneurons (Lewis et al., 2005) and antipsychotic drug-targeted DA neurons (Howes and Kapur, 2009; Abi-Dargham, 2012; Grace, 2012).
On the basis of these observations, it has been postulated that the glutamatergic disturbances may involve hypofunctioning of NMDARs on gamma-aminobutyric acid (GABA) interneurons in the limbic circuit (Olney and Farber, 1995; Olney et al., 1999; Lindsley et al., 2006; Lisman et al., 2008). How might this be achieved? Activity in the corticolimbothalamic circuit is strongly regulated by local GABAergic interneurons, especially basket and chandelier cells. Output from the cortical pyramidal neurons is suppressed and coordinated by GABAergic interneurons. These cells are activated by recurrent collaterals from the pyramidal neurons and exert a powerful feedback inhibitory action on pyramidal cells via synapses onto the soma and axon hillock (Figure 1). Both basket and chandelier cells are particularly important for restraining excessive pyramidal neuron activity, the impairment of these cells leads to dramatic disinhibition of the pyramidal neuron efferent activity and elevated uncoordinated firing throughout the corticolimbic circuit. Considering the dysfunction of NMDAR subunits in patients with schizophrenia (Akbarian et al., 1996; Eastwood et al., 1997; Goff and Wine, 1997; Grimwood et al., 1999; Gao et al., 2000; Clinton et al., 2003; Clinton and Meador-Woodruff, 2004; Weickert et al., 2012), it has been speculated that NMDAR subunits distributed on interneurons may be responsible for NMDAR hypofunction (Nakazawa et al., 2012). The central pathological characteristics seem to be caused by NMDAR hypofunction acting on GABAergic interneurons, followed by the disinhibition of glutamatergic transmission and an overstimulation of non-NMDARs on pyramidal neurons (Figure 1) (Olney and Farber, 1995; Olney et al., 1999; Lindsley et al., 2006; Lisman et al., 2008). The postulated existence of disinhibited glutamatergic transmission and the subsequent cascade of excitotoxic events resulting from NMDAR hypofunction, degeneration of GABAergic interneurons, or a combination of both, have suggested diverse experimental therapeutic interventions for schizophrenia, such as facilitation of NMDA receptor-mediated neurotransmission and potentiation of GABAergic inhibition (Coyle and Tsai, 2004; Javitt, 2004). Recently, a heuristic model for the pathophysiology of schizophrenia that attempts to reconcile the neuropathological and neurocognitive features of the disorder has been proposed (Lisman et al., 2008).
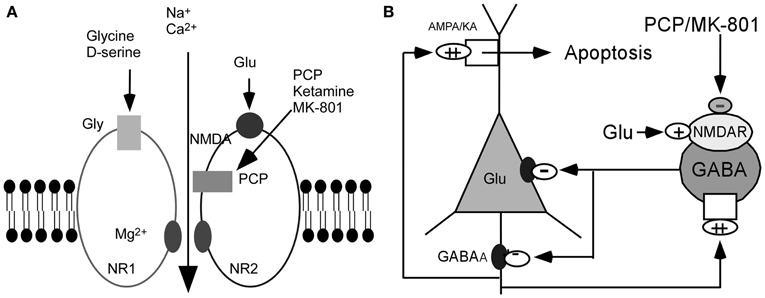
Figure 1. Hypothesis of NMDAR hypofunction. (A) Schematic diagram of NMDAR complex. (B) NMDAR hypoactivity and glutamate neurotoxicity. PCP/MK801 ⇒ NMDAR hypofunction on GABAergic neurons ⇒ disinhibition of pyramidal neurons ⇒ more glutamate release ⇒ AMPA/KA receptors excessively stimulated ⇒ excitotoxic damage [Figure 1B was modified from (Olney et al., 1999)].
When does the hypofunction of NMDAR occur and what are the mechanisms involved? Specifically, it is crucial to understand which neurons express altered glutamate receptor subtypes, whether these neurons are inhibitory or excitatory, and how the circuitries are affected. It is possible that the hypofunction of the NMDAR on GABAergic interneurons disrupts the functional integrity of the corticolimbic circuit, causing cognitive impairments and negative symptoms. Based on this hypothesis, it is reasonable to speculate that the NMDARs on frontal cortical and limbic GABAergic interneurons are most sensitive to these antagonists and therefore may be an important site of pathology resulting in NMDAR dysfunction. To address these possibilities, we have examined the developmental changes and functions of NMDARs in identified prefrontal neurons. Interestingly, we found that the development of NR2 subunits in pyramidal neurons and GABAergic interneurons of rat prefrontal cortex is cell type-specific (Wang et al., 2008; Wang and Gao, 2009). NR2B levels remain high until adulthood, without significant NR2B-to-NR2A subunit switch, in layer 5 pyramidal neurons in the prefrontal cortex (Wang et al., 2008); however, they are gradually replaced by NR2A subunits in fast-spiking interneurons (Wang and Gao, 2009). Particularly, fast-spiking interneurons in the prefrontal cortex undergo dramatic changes in glutamatergic receptors during the adolescent period (Wang and Gao, 2009, 2010) and consequently, a cell type-specific change of NMDAR subunits in parvalbumin-positive interneurons is clearly evidenced (Xi et al., 2009). These findings strongly suggested that fast-spiking or parvalbumin-positive interneurons are more sensitive to pharmacological or environmental stimulation. Indeed, we found that MK-801 induces distinct changes of AMPA and NMDARs in the fast-spiking interneurons and pyramidal cells in adolescent rat prefrontal cortex (Wang and Gao, 2012). Furthermore, when the NR1 subunit was selectively eliminated in parvalbumin-positive interneurons in forebrain cortices and hippocampus in early (neonatal) development, the rats exhibited reduced glutamic acid decarboxylase 67 (GAD67) and parvalbumin as well as distinct schizophrenia-related symptoms that emerged after adolescence; in contrast, post-adolescent deletion of NR1 did not result in such abnormalities (Belforte et al., 2010). These basic studies in NMDAR development in the prefrontal cortex have been extremely useful in the formulation of an NMDAR hypofunction hypothesis. The high vulnerability of corticolimbic fast-spiking interneurons to genetic predispositions and early environmental insults such as excitotoxicity and oxidative stress could help to better explain their significant contribution to the development of schizophrenia (Nakazawa et al., 2012). Given that both DA and GABA systems are indeed the targets of NMDAR disruption, it is plausible to propose that dysfunction of NMDARs in the DA neurons and GABAergic cells induce DA hyperactivity or GABA downregulation, which in turn results in psychosis.
Still, this does not completely explain the pathophysiology of schizophrenia, as there is evidence of NMDAR dysfunction in other key brain areas, especially during development. In addition to the prefrontal cortex, the hippocampus is a brain region that is consistently implicated in schizophrenia (Bogerts et al., 1990; Medoff et al., 2001; Harrison, 2004; Witthaus et al., 2009). In hippocampus, like other cortical regions, proper NMDAR subunit expression and function is necessary for hippocampal development, with NMDAR misregulation affecting synaptogenesis and circuit maturation (Roberts et al., 2009; Brigman et al., 2010; Gambrill and Barria, 2011; John Gray et al., 2011). Therefore, misregulation of NMDAR subunit composition and function during hippocampal development may contribute to the pathogenesis in schizophrenia. Indeed, we recently found in the MAM neurodevelopmental schizophrenia model, that NMDAR function is disrupted in CA1 pyramidal neurons early in hippocampal development (Snyder et al., 2013). Understanding when and how NMDAR function is disrupted in regards to schizophrenia progression is a key area of research.
Schizophrenia is a Neurodevelopmental Disorder with Multiple Susceptibility Genes Converging on NMDARs
It is increasingly recognized that schizophrenia is a neurodevelopmental disorder that involves disrupted alterations in brain circuits (Weinberger, 1987; Lewis and Gonzalez-Burgos, 2008; Jaaro-Peled et al., 2009). Although psychosis usually emerges in late adolescence or early adulthood, we still do not understand all of the changes in normal or abnormal development prior to and during this period. It is particularly unclear what factors alter the excitatory-inhibitory synaptic balance in the juvenile brain and what changes induce the onset of cognitive dysfunction. Current studies suggest that problems related to schizophrenia are evident much earlier than the juvenile stage of development. The emerging picture from genetic and epigenetic studies indicates that early brain development is affected. However, after many years of intensive investigations, no single gene has been found to be responsible for schizophrenia. Although recent findings have generated great interest in the copy number variations of genes in schizophrenia patients, they are rare and are unlikely to account for the majority of cases of the disorder (Allen et al., 2008; O'Donovan et al., 2008; Stefansson et al., 2008). Rather, a number of high-risk genes have been identified as increasing susceptibility for schizophrenia (Allen et al., 2008), including the catechol-o-methyltransferase gene (COMT) (Weinberger et al., 2001; Bilder et al., 2004; Cannon, 2005; Harrison and Weinberger, 2005; Savitz et al., 2006; Tunbridge et al., 2006; Tan et al., 2009), neuregulin 1 (NRG1) (Roy et al., 2007; Mei and Xiong, 2008; Kato et al., 2011), disrupted in schizophrenia-1 (DISC-1) (Lipina et al., 2010; Niwa et al., 2010), and dystrobrevin-binding protein 1 (dysbindin) (Iizuka et al., 2007; Ji et al., 2009; Papaleo and Weinberger, 2011; Papaleo et al., 2012), among others. Many of these genetic variants associated with schizophrenia are involved with neurodevelopment that is related to the glutamatergic system in the brain (Hahn et al., 2006; Allen et al., 2008; Shi et al., 2008; Papaleo et al., 2012).
Recent studies indicate that single genes may not be sufficient to cause schizophrenia. Instead, multiple “susceptibility” genes could possibly work together to trigger disease onset with each susceptibility gene coding for a subtle molecular abnormality in transmitter receptors, enzymes, protein kinases, transcription, and translation (Harrison and Weinberger, 2005). These subtle changes could disrupt neurodevelopment, intracellular signaling pathways and neurotransmission, consequently resulting in disturbed information processing in brain circuits that mediate the symptoms of schizophrenia. It is therefore not surprising that many of the susceptibility genes for schizophrenia regulate not only neuronal proliferation, neuronal migration, and synaptogenesis during early development, but also have functions linked to glutamate neurotransmission, especially the NMDA receptor, in postnatal development (Straub and Weinberger, 2006; Karam et al., 2010).
Numerous susceptibility genes have been shown to be able to regulate various elements of NMDAR mediated signaling. Dysbindin, neuregulin, and DISC1 all function to affect NMDAR function through a variety of mechanisms. Both dysbindin and neuregulin regulate the formation and function of the postsynaptic density (PSD), a set of proteins that interacts with the postsynaptic membrane to provide structural and functional regulatory elements for neurotransmission and for NMDARs (Numakawa et al., 2004; Hahn et al., 2006). Neuregulin also activates an Erb signaling system that is co-localized with NMDARs (Hahn et al., 2006). This Erb signaling system is a member of the receptor tyrosine kinase and neurotrophin signal transduction system, interacts with PSD, and is involved in neuroplasticity mediated by NMDARs (Huang et al., 2000). Furthermore, neuregulin has been shown to alter NMDAR expression (Ozaki et al., 1997; Li et al., 2007; Mei and Xiong, 2008; Banerjee et al., 2010) [see (Geddes et al., 2011) for detail]. Preventing NRG1/ErbB4 signaling leads to loss of NMDA synaptic currents and dendritic spines (Li et al., 2007). Dysbindin also regulates the activity of the vesicular glutamate transporter, vGluT (Fanous et al., 2005), and may contribute to NMDAR dysfunction (Karlsgodt et al., 2011). Furthermore, the degree of dysbindin-induced NR1 degradation correlates with impairment in spatial working memory performance (Karlsgodt et al., 2011). This is strong evidence that dysbindin's effects on NMDAR expression could contribute to the cognitive symptoms of schizophrenia.
DISC1 affects presynaptic glutamate release from axonal terminals (Maher and LoTurco, 2012), and regulates cyclic adenosine monophosphate (cAMP) signaling, which would affect the functions of glutamate neurotransmission mediated by metabotropic glutamate receptors (mGluR) (Millar et al., 2005). DISC1 also binds to and stabilizes serine racemase (SR), the enzyme that generates D-serine, an endogenous co-agonist of the NMDA receptor. In a mouse model of selective and inducible expression of mutant DISC1 in astrocytes, the main source of D-serine in the brain, Ma et al. found that mutant DISC1 leads to SR degradation, resulting in D-serine deficiency that coincides with behavioral changes indicative of altered NMDAR neurotransmission (Ma et al., 2012). While not yet specifically tested, these changes would likely lead to reduced function of NMDARs at synapses. In addition, the DAOA gene encodes a protein that activates the enzyme D-amino acid oxidase, which degrades the co-transmitter D-serine that acts at glutamate synapses and at NMDARs. DAOA activates this enzyme, so abnormalities in this gene would be expected to alter the metabolism of D-serine, which in turn would alter glutamate neurotransmission at NMDARs (Stahl, 2007a).
Thus, there is strong evidence that the known susceptibility genes for schizophrenia converge on glutamate synapses, specifically at NMDARs. These observations support the notion that the NMDAR hypofunction hypothesis is a plausible theory for schizophrenia (Stahl, 2007a) and NMDAR dysfunction is a convergence point for schizophrenia (Kantrowitz and Javitt, 2010). Genes that code for any subtle molecular abnormalities linked to NMDAR function in specific brain circuits theoretically could create inefficient information processing at glutamate synapses that can produce the symptoms of schizophrenia, especially cognitive dysfunctions. If these genetically mediated abnormalities occur simultaneously in a permissive environment, the syndrome of schizophrenia could be induced and onset of symptoms will be triggered (Stahl, 2007b).
Molecular Mechanisms Associated with NMDAR Regulation and NMDAR Hypofunction in Schizophrenia
As discussed above, there are many risk genes associated with schizophrenia. However, changes in their expression and function are unlikely to entirely account for the pathophysiology of schizophrenia. A fundamental question is what causes the alteration of NMDAR during neurodevelopment in schizophrenia. In addition to genetic modifications, there are several possible mechanisms, including altered transcription/translation and posttranslational modifications that could contribute to NMDAR hypofunction in schizophrenia. For example, NMDAR hypofunction could result from reduced levels of mRNA and translation and in fact, there is evidence of reduced mRNA levels of some NMDAR subunits in postmortem tissue of schizophrenics (Dracheva et al., 2001; Beneyto and Meador-Woodruff, 2008; Weickert et al., 2012) but plenty of evidence also suggests an increase or no change in some subunits (Akbarian et al., 1996; Geddes et al., 2011; Weickert et al., 2012). Given the complexity of the disorder and the numerous risk genes involved, it is likely that several mechanisms work in concert. Fortunately, substantial knowledge exists as to how NMDARs are translated, trafficked to synaptic membranes, stabilized, exocytosed, and removed for recycling or degradation (Sans et al., 2003; Wenthold et al., 2003; Perez-Otano and Ehlers, 2004; Lau and Zukin, 2007). However, any disruption of this well-regulated process can lead to NMDAR hypofunction and contribute to altered development and symptomatology seen in schizophrenia. Thus, it becomes a daunting challenge to understand the pathophysiological processes involved.
An exciting avenue of research in schizophrenia and other psychiatric disorders is evaluating the epigenetic changes that occur in these illnesses. Epigenetics is a broad term that describes changes to chromatin which alter the frequency of gene transcription without changing the genetic sequence. These changes include DNA methylation and a variety of histone modifications. In general, increasing DNA methylation, particularly at CpG islands of promoter sequences, will decrease gene expression (Bird, 2002). Therefore, even if a gene is not found to be definitively altered in human schizophrenic patients by standard genome-wide association study (GWAS), it is possible that epigenetic changes are contributing to altered neurodevelopment and cognitive symptoms in schizophrenia (Borrelli et al., 2008; Day and Sweatt, 2011; Rodenas-Ruano et al., 2012). Indeed, a role for histone acetylation and methylation in cognition is increasingly being appreciated (Jeremy Day and Sweatt, 2011). Other data suggest that chromatin modifications by histone deacetylases (HDACs) may underlie cognitive dysfunctions in a variety of mental disorders (Fischer et al., 2010). Thus far, epigenetic modulation of several genes, including GAD1 and RELN, has been found to be altered in schizophrenia (Abdolmaleky et al., 2005; Ruzicka et al., 2007). Additionally, the DNA methylating enzyme, DNA-methyltransferase 1 (DNMT1), showed increased expression in cortical interneurons in postmortem tissue from schizophrenics (Veldic et al., 2005). This change in DNMT1 correlated with the alterations in GAD1 and RELN. However, it is possible that other genes and associated interacting proteins are also similarly affected. For example, animal research has shown that NMDAR subunit expression can be altered through various epigenetic changes (Stadler et al., 2005; Jiang et al., 2010; Rodenas-Ruano et al., 2012). Furthermore, DNA methylation changes have been found in the promoter sequence for NR3B in major psychosis (Mill et al., 2008). These studies suggest that epigenetic regulation of NMDARs could contribute to the pathophysiology of schizophrenia. Still, it is unclear how epigenetic factors control the expression of NMDARs, particularly mRNA expression of individual subunits. It is possible that CpG islands in the promoter region of a NMDAR subunit are regulated by chromatin modification (Rodenas-Ruano et al., 2012). Gene mutation or environmental risk factors could alter gene promoter sequences via either DNA methylation or histone modification and thus result in mis-expression of NMDARs.
Furthermore, NMDAR subunits undergo several post-translation modifications including phosphorylation, palmitoylation, and polyubiquitination. Dysregulation of any of these processes can greatly impact channel function and expression and consequently contribute to NMDAR hypofunction. The most-studied posttranslational modification of NMDARs is phosphorylation, which is a well-characterized means for regulating synaptic localization, stabilization, and channel kinetics. Therefore, changes in NMDAR phosphorylation have important implications both for synaptic plasticity and cognitive symptoms in schizophrenia (Rosenblum et al., 1996; Lu et al., 1998; Li et al., 2009). This dynamic process not only involves the direct phosphorylation of NMDARs, but also kinase activation and subsequent phosphorylation of other synaptic proteins (Lau and Zukin, 2007; Lau et al., 2010). Moreover, the NR2 subunit's large C-terminus has many putative sites for phosphorylation which can affect channel gating and stabilization at the synapse (Monyer et al., 1992; Kornau et al., 1995). NMDAR subunits are phosphorylated at serine or threonine and at tyrosine residues (Raymond et al., 1994; Wang and Salter, 1994; Kohr and Seeburg, 1996; Tingley et al., 1997). These sites are substrates for phosphorylation by a variety of kinases including the Src family of kinases (SFK), cAMP-dependent protein kinase A (PKA), protein kinase C (PKC), cyclin-dependent kinase 5 (Cdk5), casein kinase 2 (CK2), and CaMKII (Omkumar et al., 1996; Raman et al., 1996; Li et al., 2001; Chung et al., 2004). In fact, the activity and expression of many of these kinases are altered in postmortem tissue from human schizophrenic patients (Aksenova et al., 1991; Engmann et al., 2011; Funk et al., 2012). This provides strong evidence that altered kinase signaling likely plays a role in NMDAR function in schizophrenia.
It is clear that the interaction between synaptic scaffolding proteins and the NR2 subunit C-terminal tails are critical for NMDAR synaptic targeting and thus could contribute to NMDAR hypofunction. PDZ-containing proteins can bind directly to NR2 subunits via PDZ recognition sequences in the distal portions of their C-termini, and this association is critical for targeting NDMARs to the synapse (Mori et al., 1998; Steigerwald et al., 2000; Lin et al., 2004). Further, both NR2A and NR2B are known to interact with membrane-associated guanylate kinase (MAGUK) family of proteins, including PSD-95, PSD-93, and SAP102 (Al-Hallaq et al., 2007). Interestingly, the neuregulin receptor ErbB4 also associates with similar PDZ domains, positioning NRG-Erb signaling to affect NMDAR function (Garcia et al., 2000). Furthermore, ErbB4 interacts with FYN, a member of SFKs. SFKs phosphorylate tyrosine residues on both NR2A and NR2B subunits affecting channel gating and increasing NMDAR currents (Wang and Salter, 1994; Kohr and Seeburg, 1996; Hisatsune et al., 1999; Nakazawa et al., 2001; Takasu et al., 2002). NRG1-Erb signaling can prevent Src upregulation of NMDAR-mediated currents by inhibiting NR2B phosphorylation (Li et al., 2001; Bjarnadottir et al., 2007; Pitcher et al., 2011). Additionally, NMDAR tyrosine phosphorylation is important for synaptic plasticity. NR2B tyrosine phosphorylation is increased following long-term potentiation (LTP) and inhibiting Src activation prevents LTP induction (Grant et al., 1992; Rosenblum et al., 1996; Rostas et al., 1996; Lu et al., 1998). In hippocampus, NRG-Erb signaling can suppress LTP (Kwon et al., 2005; Pitcher et al., 2008). Therefore, NRG1 could contribute to cognitive dysfunction in schizophrenia by altering NMDAR function and/or affecting synaptic plasticity (Mei and Xiong, 2008). Similarly, DISC1 is a known binding partner of PDE4B, which regulates cAMP activity and thus PKA activity (Millar et al., 2005; Clapcote et al., 2007). PKA-mediated phosphorylation of NMDARs can affect their release from the endoplasmic reticulum, and regulate expression levels of NR2B (Scott et al., 2003; Llansola et al., 2004). However, it has not been directly tested whether mutations in DISC1 affect NMDAR expression and function. Additionally, it remains an open question if disruption of dysbindin would produce similar modifications in NMDARs. If and how the schizophrenia risk genes affect NMDAR phosphorylation and thus expression and function is an area of research that needs to be further explored.
Another crucial mechanism for proper NMDAR function is the maintenance of appropriate levels of NMDARs in the synapse. This process requires a balance between NMDAR insertion and endocytosis. Specialized endocytic zones involving clathrin-coated pits have been described lateral to the PSD for glutamatergic synapses, and serve to internalize NMDARs (Blanpied et al., 2002; Petralia et al., 2003; Nong et al., 2004). Altered dysbindin expression can alter NMDAR surface expression through clathrin-dependent endocytosis (Jeans et al., 2011). Further, palmitoylation and ubiquitination can also regulate NMDAR synaptic numbers. Palmitoylation is a reversible process that involves the covalent attachment of palmitate group to proteins via thioester bonds at cysteine residues. Palmitoylation is a critical regulator of many cellular processes involved in neuronal development and synaptic plasticity (Fukata and Fukata, 2010). Therefore, dysregulation of palmitoylation could contribute to synaptic dysfunction and cognitive symptoms in schizophrenia. Furthermore, key proteins implicated in schizophrenia, including GAD65 and PSD-95 are known to be regulated dynamically through palmitoylation (El-Husseini Ael et al., 2002; Kanaani et al., 2008). More recently, it was discovered that palmitoylation can regulate NR2A and NR2B trafficking (Hayashi et al., 2009). In fact, palmitoylation can promote synaptic stabilization or sequestering of NMDARs in the Golgi apparatus to affect the level of NMDARs at synapses. Interestingly, altered protein palmitoylation was found in a mouse model of 22q11.2 deletion, a high risk factor of developing schizophrenia (Madry et al., 2008). However, it remains unknown if NMDAR palmitoylation is disrupted in schizophrenia and if or how other schizophrenia risk genes may be involved.
Equally as important as trafficking and stabilizing proteins in the synapse is the process of targeting proteins for removal and degradation. It is known that ubiquitin-based protein degradation of NMDARs is an important homeostatic regulator of NMDAR levels at synapses (Ehlers, 2003). For example, down-regulation of synaptic NR1 has been associated with polyubiquitination (Groblewski and Stafford, 2010; Bangash et al., 2011). Additionally, ubiquitination of scaffolding proteins, such as Shank3, is linked to NR2B downregulation (Mao et al., 2009a). Also, NR2B itself is ubiquitinated in a Fyn dependent manner (Jurd et al., 2008). Given NRG1-ErbB4 interactions with Fyn, it is possible that their signaling could contribute to ubiquitination of NR2B. However, this relationship has not been tested experimentally. Therefore, while there is evidence that the ubiquitin proteasome pathway is disrupted in schizophrenia (Nilsson et al., 2007), it is currently unknown how ubiquitination of NMDARs and other synaptic proteins contribute to the disease process. Exploring this relationship as well as how schizophrenia risk genes could alter these processes is an important line of research.
Given the diverse set of mechanisms that could contribute to NMDAR hypofunction, it is not surprising that multiple signaling pathways are implicated in schizophrenia. For example, both PLC/IP3R/Ca2+ and Ras/MEK/ERK (extracellular signal-regulated kinase) signaling pathways are involved in the neuregulin-induced reduction of NMDAR currents, which likely occurs through enhancing NR1 internalization via an actin-dependent mechanism (Gu et al., 2005). While the candidate genes discussed activate many signaling cascades to affect neurodevelopment and NMDAR function, the AKT (also known as protein kinase B) signaling pathway, and its downstream target glycogen synthase kinase 3β (GSK-3β) may serve as a convergence point or common pathway. AKT is a serine/threonine kinase that serves in a variety of processes including regulation of protein synthesis, neurodevelopment, and neuronal plasticity (Sanna et al., 2002; Jiang et al., 2005; Balu et al., 2012). Further, DISC1, NRG1, and dysbindin all contribute to these cellular processes, and are all known regulators of AKT and GSK3β (Lemke, 1996; Huang et al., 2000; Kamiya et al., 2005; Ghiani et al., 2010; Lee et al., 2011). DISC1 regulates the AKT-GSK3β signaling pathway to affect neurodevelopment and adult neurogenesis (Kim et al., 2009; Mao et al., 2009b). Furthermore, knockdown of DISC1 with siRNA caused a decrease in AKT phosphorylation, which would in turn increase GSK3β activity (Hashimoto et al., 2006). Interestingly, reducing GSK3β activity was able to correct behavioral deficits in DISC1 mutant mice, strongly implicating DISC1 affects GSK3β in schizophrenia pathogenesis (Lipina et al., 2011, 2012). Similarly, both NRG1 and dysbindin can regulate AKT phosphorylation (Numakawa et al., 2004; Guo et al., 2010). Additionally, AKT protein levels and phosphorylation of GSK3β are altered (Emamian et al., 2004) and NRG1-stimulated phosphorylation of AKT is reduced in schizophrenia (Keri et al., 2009). Yet, how would regulation of the AKT/GSK3β signaling pathway by DISC1, NRG1, and dysbindin affect NMDAR function? It was recently demonstrated that GSK3β activity can regulate NMDAR expression and function (Li et al., 2009; Xi et al., 2011). While this evidence provides a possible common link between schizophrenia risk genes and NMDAR hypofunction, direct experimental evidence is still needed.
Conclusion and Future Perspective
In this review, we have summarized the current literature and discussed the various mechanisms that are associated with NMDAR regulation in schizophrenia. All of the findings derived from the known genetic risk factors for schizophrenia suggest that NMDARs may serve as a convergence point for the progression and symptoms of schizophrenia. Despite such progress, there are still many questions that need to be answered to confirm this intriguing hypothesis. For example, it is unclear how gene mutations in neurons and/or astrocytes and their interaction can lead to NMDAR dysfunction during development. It is also unknown how disrupted NMDAR function leads to altered neurodevelopment, which contributes to the progression and development of this devastating disease. The vast majority of schizophrenia research has focused on changes in adulthood, leaving neurodevelopmental alterations relatively unexplored. So, while it is known that proper expression and regulation of NMDARs is critical for cortical maturation and synaptic plasticity that underlie cognitive functioning, it is unknown if there is a common signaling pathway, such as AKT/GSK-3β pathway, mediates this pathophysiological process among the schizophrenia susceptibility genes. If yes, what are the downstream target substrates of AKT and/or GSK-3β that contribute to the regulation of NMDAR functions? It is possible that AKT/GSK-3β act directly upon NMDARs as our recent research suggests (Li et al., 2009; Xi et al., 2011). However, given their diverse targets (Kockeritz et al., 2006; Peineau et al., 2008; Karam et al., 2010; Li and Gao, 2011), it is also possible they indirectly affect NMDARs by acting on other targets, such as β-catenin (Mao et al., 2009b), β-arrestin (Beaulieu et al., 2005), DISC1 and/or PDE4 interaction (Mao et al., 2009b; Lipina et al., 2012), as well as the AKT/mTOR signaling pathway. Activation of mTOR has been functionally linked with local protein synthesis in synapses, resulting in the production of proteins required for synaptic formation and maturation (Kelleher et al., 2004; Hoeffer and Klann, 2010).
In addition, although psychosis manifests primarily in late adolescence or early adulthood, the emerging picture from genetic and epigenetic studies indicates that early brain development is affected, and cognitive symptoms, such as learning and memory deficits, are evident much earlier. Specifically, schizophrenia may progress from risk to prodrome in the early stage until onset of psychosis and chronic disability in the late stage (Insel, 2010). Therefore, theoretically, the key to forestall the disorder is to detect and prevent early stages of risk and prodrome with novel therapeutic targets for early treatment (Lieberman et al., 2006; Insel, 2010). However, in general, schizophrenia-related research has focused on how NMDAR function in adults contributes to psychosis and cognitive symptoms. These findings, although intriguing, are limited in that they do not reveal the changes before psychosis, specifically during neurodevelopment. In fact, there is no consensus among animal models to what changes occur pre-pubertally and how the genetic susceptibilities interact. Does the process occur simultaneously or sequentially, with various changes culminating in altered development? If it is a sequential process, when do these changes occur and is there a point of no return in terms of preventing cognitive symptoms and psychosis? It appears that adolescence is a critical period for onset of psychosis, but how and by what mechanisms? Therefore, in studying molecular mechanisms that underlie the pathophysiology of schizophrenia and related disorders, a sharp focus on the specific neurodevelopmental trajectory, especially in early development and adolescent brain maturation, is vitally important (Jaaro-Peled et al., 2009; Insel, 2010). Animal studies, particularly developmental models, will certainly help to reveal the neurodevelopmental trajectory of schizophrenia, yield disease mechanisms, and eventually offer opportunities for the development of new treatments, especially for early treatment of cognitive deficits. Utilizing multiple animal models to address similar questions will provide the greatest opportunity for determining consistent changes that most likely contribute to the progression of schizophrenia. It would also be important to definitively determine which neurons express altered glutamate receptor subtypes, whether these neurons are inhibitory or excitatory, and how the circuitries are affected by these high-risk genes.
Furthermore, it is critical to determine if there comes a point during neurodevelopment where brain circuitry is sufficiently altered that no therapeutics will halt the progression of the disease. At present, there are no approved medications for the treatment of either negative symptoms or cognitive dysfunction in schizophrenia (Ibrahim and Tamminga, 2011). However, new pharmacological and behavioral approaches aimed at potentiating glutamatergic neurotransmission, particularly at NMDARs, offer new hope for future clinical development. Although many studies support the theory of NMDAR hypofunction, they do not address the very important conceptual question of whether early treatment with mGluR agonists or other agents is able to prevent the progression or reverse the cognitive deficits and even psychosis that occur in the late stage of the disease. A failure to correct mutant phenotypes with treatment administered after symptom onset would suggest a missed critical period window and indicate that schizophrenia is a terminally differentiated phenotype of altered brain development. Earlier theories supported the notion that effective treatment for developmental disorders such as schizophrenia and autism could only occur during the critical developmental window, after which the brain would be hard wired. Indeed, recent studies demonstrated that a comprehensive phenotype correction is possible with pharmacological intervention (mGluR5 inhibitor) starting in young (3–5 postnatal weeks) animals, after development of the phenotype, in both a Fragile X syndrome model (Michalon et al., 2012) and Shank-2 knockout mice (Won et al., 2012). In addition, adolescent administration of mGluR5 PAMs not only reverse adult-onset deficits, but also prevent the emergence of cognitive impairment induced by neonatal treatment with PCP in a developmental model of schizophrenia (Clifton et al., 2013). These findings certainly offer fresh hope for schizophrenia treatment, suggesting that NMDARs could be critical targets for treatment. Currently, our experiments are under way to test this hypothesis in rats with gestational methylazoxymethanol exposure (Snyder et al., 2013) and other animal models.
Finally, if NMDAR dysfunction in schizophrenia is relative, rather than absolute, enhanced practice might be able to overcome reduced plasticity. Given the number of convergent mechanisms that may contribute to impaired NMDAR function, ideal treatment in schizophrenia may engage optimizing function within a number of convergent pathways. Combinatorial pharmacological and behavioral interventions, rather than simply targeting the point of convergence, may prove to be the most successful strategy in treating schizophrenia symptoms. Nevertheless, focusing on NMDAR hypofunction provides a wonderful opportunity for correcting cognitive impairment in schizophrenia disease progression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grant R01MH085666 to Wen-Jun Gao from the National Institutes of Health, USA and the NARSAD Young Investigator Award (2012) to Melissa A. Snyder.
Abbreviations
Akt, also known as Protein Kinase B (PKB), is a serine/threonine-specific protein kinase; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor; BDNF, brain derived neurotrophic factor; cAMP, cyclic adenosine monophosphate; CaMKII, Ca2+/calmodulin dependent protein kinase II; cdk5, cyclin-dependent kinase 5; CK2, casein kinase 2; COMT, catechol-o-methyltransferase; DA, dopamine; Dysbindin, also known as dystrobrevin-binding protein 1; DISC1, disrupted in schizophrenia-1; DAOA, D-amino acid oxidase activator; HDAC, histone deacetylase; DNMT1, DNA-methyltransferase 1; ERK, extracellular-signal-regulated kinase; GABA, gamma-aminobutyric acid; GAD65, glutamic acid decarboxylase 65; GAD67, glutamic acid decarboxylase 67; GSK-3β, glycogen synthase kinase 3β; LTP, long-term potentiation; MAGUK, membrane-associated guanylate kinase; mGluR, metabotropic glutamate receptor; MK801, dizocilpine; NMDAR, N-methyl-D-aspartate receptor; NRG1, neuregulin 1; PCP, phencyclidine; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PSD95, post synaptic density protein 95; SAP102, synapse associated protein 102; SFK, Src family of kinases; PDE4B, cAMP-specific phosphodiesterase 4B; SR, serine racemase; vGluT, vesicular glutamate transporter.
References
Abdolmaleky, H. M., Cheng, K. H., Russo, A., Smith, C. L., Faraone, S. V., Wilcox, M., et al. (2005). Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B, 60–66.
Abi-Dargham, A. (2012). The Dopa-mine Hypothesis of Schizophrenia. Schizophrenia Research Forum. Available online at: http://www.schizophreniaforum.org/for/curr/AbiDargham/default.asp
Akbarian, S., Sucher, N. J., Bradley, D., Tafazzoli, A., Trinh, D., Hetrick, W. P., et al. (1996). Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 16, 19–30.
Aksenova, M. V., Burbaeva, G. S., Kandror, K. V., Kapkov, D. V., and Stepanov, A. S. (1991). The decreased level of casein kinase 2 in brain cortex of schizophrenic and Alzheimer's disease patients. FEBS Lett. 279, 55–57.
Al-Hallaq, R. A., Conrads, T. P., Veenstra, T. D., and Wenthold, R. J. (2007). NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 27, 8334–8343.
Allen, N. C., Bagade, S., McQueen, M. B., Ioannidis, J. P., Kavvoura, F. K., Khoury, M. J., et al. (2008). Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 40, 827–834.
Balu, D. T., Carlson, G. C., Talbot, K., Kazi, H., Hill-Smith, T. E., Easton, R. M., et al. (2012). Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus 22, 230–240.
Banerjee, A., Macdonald, M. L., Borgmann-Winter, K. E., and Hahn, C. G. (2010). Neuregulin 1-erbB4 pathway in schizophrenia: from genes to an interactome. Brain Res. Bull. 83, 132–139.
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., et al. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83.
Beneyto, M., Kristiansen, L. V., Oni-Orisan, A., McCullumsmith, R. E., and Meador-Woodruff, J. H. (2007). Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32, 1888–1902.
Beneyto, M., and Meador-Woodruff, J. H. (2008). Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology 33, 2175–2186.
Bilder, R. M., Volavka, J., Lachman, H. M., and Grace, A. A. (2004). The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric pheno-types. Neuropsychopharmacology 29, 1943–1961.
Bjarnadottir, M., Misner, D. L., Haverfield-Gross, S., Bruun, S., Helgason, V. G., Stefansson, H., et al. (2007). Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/- knock-outs compared with wild-type mice. J. Neurosci. 27, 4519–4529.
Blanpied, T. A., Scott, D. B., and Ehlers, M. D. (2002). Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron 36, 435–449.
Bogerts, B., Ashtari, M., Degreef, G., Alvir, J. M., Bilder, R. M., and Lieberman, J. A. (1990). Reduced temporal limbic structure volumes on magnetic resonance images in first episode schizophrenia. Psychiatry Res. 35, 1–13.
Borrelli, E., Nestler, E. J., Allis, C. D., and Sassone-Corsi, P. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974.
Brigman, J. L., Wright, T., Talani, G., Prasad-Mulcare, S., Jinde, S., Seabold, G. K., et al. (2010). Loss of GluN2B-containing NMDA Receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 30, 4590–4600.
Cannon, T. D. (2005). The inheritance of intermediate phenotypes for schizophrenia. Curr. Opin. Psychiatry 18, 135–140.
Chung, H. J., Huang, Y. H., Lau, L. F., and Huganir, R. L. (2004). Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J. Neurosci. 24, 10248–10259.
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., et al. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402.
Clifton, N. E., Morisot, N., Girardon, S., Millan, M. J., and Loiseau, F. (2013). Enhancement of social novelty discrimination by positive allosteric modulators at metabotropic glutamate 5 receptors: adolescent administration prevents adult-onset deficits induced by neonatal treatment with phencyclidine. Psychopharmacology (Berl.) 225, 579–594.
Clinton, S. M., Haroutunian, V., Davis, K. L., and Meador-Woodruff, J. H. (2003). Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am. J. Psychiatry 160, 1100–1109.
Clinton, S. M., Haroutunian, V., and Meador-Woodruff, J. H. (2006). Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J. Neurochem. 98, 1114–1125.
Clinton, S. M., and Meador-Woodruff, J. H. (2004). Abnormalities of the NMDA receptor and associated intracellular molecules in the thalamus in schizophrenia and bipolar disorder. Neuropsychopharmacology 29, 1353–1362.
Coyle, J. T. (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 365–384.
Coyle, J. T., and Tsai, G. (2004). The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology 174, 32–38.
Day, J. J., and Sweatt, J. D. (2011). Epigenetic modifications in neurons are essential for formation and storage of behavioral memory. Neuropsychopharmacology 36, 357–358.
Dracheva, S., Byne, W., Chin, B., and Haroutunian, V. (2008). Ionotropic glutamate receptor mRNA expression in the human thalamus: absence of change in schizophrenia. Brain Res. 1214, 23–34.
Dracheva, S., Marras, S. A. E., Elhakem, S. L., Kramer, F. R., Davis, K. L., and Haroutunian, V. (2001). N-methyl-D-aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am. J. Psychiatry 158, 1400–1410.
Dumas, T. C. (2005). Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog. Neurobiol. 76, 189–211.
Eastwood, S. L., Kerwin, R. W., and Harrison, P. J. (1997). Immunoautoradiographic evidence for a loss of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate-preferring non-N-methyl-D-aspartate glutamate receptors within the medial temporal lobe in schizophrenia. Biol. Psychiatry 41, 636–643.
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242.
El-Husseini Ael, D., Schnell, E., Dakoji, S., Sweeney, N., Zhou, Q., Prange, O., et al. (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863.
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137.
Engmann, O., Hortobagyi, T., Pidsley, R., Troakes, C., Bernstein, H. G., Kreutz, M. R., et al. (2011). Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 134, 2408–2421.
Fanous, A. H., van den Oord, E. J., Riley, B. P., Aggen, S. H., Neale, M. C., O'Neill, F. A., et al. (2005). Relationship between a high-risk haplotype in the DTNBP1 (dysbindin) gene and clinical features of schizophrenia. Am. J. Psychiatry 162, 1824–1832.
Fischer, A., Sananbenesi, F., Mungenast, A., and Tsai, L.-H. (2010). Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci. 31, 605–617.
Fukata, Y., and Fukata, M. (2010). Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175.
Funk, A. J., McCullumsmith, R. E., Haroutunian, V., and Meador-Woodruff, J. H. (2012). Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology 37, 896–905.
Gambrill, A. C., and Barria, A. (2011). NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. U.S.A. 108, 5855–5860.
Gao, X. M., Sakai, K., Roberts, R. C., Conley, R. R., Dean, B., and Tamminga, C. A. (2000). Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am. J. Psychiatry 157, 1141–1149.
Garcia, R. A., Vasudevan, K., and Buonanno, A. (2000). The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc. Natl. Acad. Sci. U.S.A. 97, 3596–3601.
Geddes, A. E., Huang, X. F., and Newell, K. A. (2011). Reciprocal signalling between NR2 subunits of the NMDA receptor and neuregulin1 and their role in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 896–904.
Ghiani, C. A., Starcevic, M., Rodriguez-Fernandez, I. A., Nazarian, R., Cheli, V. T., Chan, L. N., et al. (2010). The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol. Psychiatry 15, 115–215.
Goff, D. C., and Wine, L. (1997). Glutamate in schizophrenia: clinical and research implications. Schizophr. Res. 27, 157–168.
Grace, A. A. (2012). Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology 62, 1342–1348.
Grant, S. G., O'Dell, T. J., Karl, K. A., Stein, P. L., Soriano, P., and Kandel, E. R. (1992). Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258, 1903–1910.
Grimwood, S., Slater, P., Deakin, J. F., and Hutson, P. H. (1999). NR2B-containing NMDA receptors are up-regulated in temporal cortex in schizophrenia. Neuroreport 10, 461–465.
Groblewski, P. A., and Stafford, J. M. (2010). When the medial prefrontal cortex fails: implications for extinction and posttraumatic stress disorder treatment. J. Neurosci. 30, 7124–7126.
Gu, Z., Jiang, Q., Fu, A. K., Ip, N. Y., and Yan, Z. (2005). Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J. Neurosci. 25, 4974–4984.
Gunduz-Bruce, H. (2009). The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res. Rev. 60, 279–286.
Guo, W. P., Fu, X. G., Jiang, S. M., and Wu, J. Z. (2010). Neuregulin-1 regulates the expression of Akt, Bcl-2, and Bad signaling after focal cerebral ischemia in rats. Biochem. Cell Biol. 88, 649–654.
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., et al. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 12, 824–828.
Harrison, P. J. (2004). The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl.) 174, 151–162.
Harrison, P. J., and Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10, 40–68.
Hashimoto, R., Numakawa, T., Ohnishi, T., Kumamaru, E., Yagasaki, Y., Ishimoto, T., et al. (2006). Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum. Mol. Genet. 15, 3024–3033.
Hayashi, T., Thomas, G. M., and Huganir, R. L. (2009). Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron 64, 213–226.
Henson, M. A., Roberts, A. C., Pérez-Otaño, I., and Philpot, B. D. (2010). Influence of the NR3A subunit on NMDA receptor functions. Prog. Neurobiol. 91, 23–37.
Hisatsune, C., Umemori, H., Mishina, M., and Yamamoto, T. (1999). Phosphorylation-dependent interaction of the N-methyl-D-aspartate receptor epsilon 2 subunit with phosphatidylinositol 3-kinase. Genes Cells 4, 657–666.
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75.
Howes, O. D., and Kapur, S. (2009). The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr. Bull. 35, 549–562.
Huang, Y. Z., Won, S., Ali, D. W., Wang, Q., Tanowitz, M., Du, Q. S., et al. (2000). Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron 26, 443–455.
Ibrahim, H. M., Hogg, A. J. Jr., Healy, D. J., Haroutunian, V., Davis, K. L., and Meador-Woodruff, J. H. (2000). Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am. J. Psychiatry 157, 1811–1823.
Ibrahim, H. M., and Tamminga, C. A. (2011). Schizophrenia: treatment targets beyond monoamine systems. Annu. Rev. Pharmacol. Toxicol. 51, 189–209.
Iizuka, Y., Sei, Y., Weinberger, D. R., and Straub, R. E. (2007). Evidence that the BLOC-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J. Neurosci. 27, 12390–12395.
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495.
Javitt, D. C. (2004). Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 9, 984–997.
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308.
Jeans, A., Malins, R., Padamsey, Z., Reinhart, M., and Emptage, N. (2011). Increased expression of dysbindin-1A leads to a selective deficit in NMDA receptor signaling in the hippocampus. Neuropharmacology 61, 1345–1353.
Ji, Y., Yang, F., Papaleo, F., Wang, H.-X., Gao, W.-J., Weinberger, D. R., et al. (2009). Role of dysbindin in dopamine receptor trafficking and cortical GABA function. Proc. Natl. Acad. Sci. U.S.A. 106, 19593–19598.
Jiang, H., Guo, W., Liang, X., and Rao, Y. (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135.
Jiang, Y., Jakovcevski, M., Bharadwaj, R., Connor, C., Schroeder, F. A., Lin, C. L., et al. (2010). Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 30, 7152–7167.
John Gray, A., Shi, Y., Usui, H., Matthew During, J., Sakimura, K., and Roger Nicoll, A. (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101.
Jurd, R., Thornton, C., Wang, J., Luong, K., Phamluong, K., Kharazia, V., et al. (2008). Mind bomb-2 is an E3 ligase that ubiquitinates the N-methyl-D-aspartate receptor NR2B subunit in a phosphorylation-dependent manner. J. Biol. Chem. 283, 301–310.
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., et al. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178.
Kanaani, J., Patterson, G., Schaufele, F., Lippincott-Schwartz, J., and Baekkeskov, S. (2008). A palmitoylation cycle dynamically regulates partitioning of the GABA-synthesizing enzyme GAD65 between ER-Golgi and post-Golgi membranes. J. Cell. Sci. 121, 437–449.
Kantrowitz, J. T., and Javitt, D. C. (2010). N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res. Bull. 83, 108–121.
Karam, C. S., Ballon, J. S., Bivens, N. M., Freyberg, Z., Girgis, R. R., Lizardi-Ortiz, J. E., et al. (2010). Signaling pathways in schizophrenia: emerging targets and therapeutic strategies. Trends Pharmacol. Sci. 31, 381–390.
Karlsgodt, K. H., Robleto, K., Trantham-Davidson, H., Jairl, C., Cannon, T. D., Lavin, A., et al. (2011). Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 69, 28–34.
Kato, T., Abe, Y., Sotoyama, H., Kakita, A., Kominami, R., Hirokawa, S., et al. (2011). Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: implication in neurodevelopmental hypothesis for schizophrenia. Mol. Psychiatry 16, 307–320.
Kelleher, R. J. 3rd., Govindarajan, A., and Tonegawa, S. (2004). Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 59–73.
Keri, S., Seres, I., Kelemen, O., and Benedek, G. (2009). Neuregulin 1-stimulated phosphorylation of AKT in psychotic disorders and its relationship with neurocognitive functions. Neurochem. Int. 55, 606–609.
Kim, J. Y., Duan, X., Liu, C. Y., Jang, M. H., Guo, J. U., Pow-anpongkul, N., et al. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773.
Kockeritz, L., Doble, B., Patel, S., and Woodgett, J. R. (2006). Glycogen synthase kinase-3–an overview of an over-achieving protein kinase. Curr. Drug Targets 7, 1377–1388.
Kohr, G., and Seeburg, P. H. (1996). Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J. Physiol. 492 (Pt 2), 445–452.
Kornau, H. C., Schenker, L. T., Kennedy, M. B., and Seeburg, P. H. (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269, 1737–1740.
Kristiansen, L. V., Beneyto, M., Haroutunian, V., and Meador-Woodruff, J. H. (2006). Altered NMDA receptor expression in schizophrenia. Mol. Psychiatry 11, 705.
Kristiansen, L. V., Huerta, I., Beneyto, M., and Meador-Woodruff, J. H. (2007). NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 7, 48–55.
Krystal, J. H., Anand, A., and Moghaddam, B. (2002). Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch. Gen. Psychiatry 59, 663–664.
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., et al. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cog-nitive, and neuroendocrine responses. Arch Gen. Psychiatry 51, 199–214.
Kwon, O. B., Longart, M., Vullhorst, D., Hoffman, D. A., and Buonanno, A. (2005). Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 25, 9378–9383.
Lahti, A. C., Koffel, B., LaPorte, D., and Tamminga, C. A. (1995). Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13, 9–19.
Lau, C. G., Takayasu, Y., Rodenas-Ruano, A., Paternain, A. V., Lerma, J., Bennett, M. V. L., et al. (2010). SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 30, 242–254.
Lau, C. G., and Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426.
Lee, F. H., Fadel, M. P., Preston-Maher, K., Cordes, S. P., Clapcote, S. J., Price, D. J., et al. (2011). Disc1 point mutations in mice affect development of the cerebral cortex. J. Neurosci. 31, 3197–3206.
Lewis, D. A., and Gonzalez-Burgos, G. (2008). Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33, 141–165.
Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324.
Lewis, D. A., and Moghaddam, B. (2006). Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 63, 1372–1376.
Li, B., Devidze, N., Barengolts, D., Prostak, N., Sphicas, E., Apicella, A. J., et al. (2009). NMDA receptor phosphorylation at a site affected in schizophrenia controls synaptic and behavioral plasticity. J. Neurosci. 29, 11965–11972.
Li, B., Woo, R.-S., Mei, L., and Malinow, R. (2007). The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 54, 583–597.
Li, B. S., Sun, M. K., Zhang, L., Takahashi, S., Ma, W., Vinade, L., et al. (2001). Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc. Natl. Acad. Sci. U.S.A. 98, 12742–12747.
Li, Y. C., and Gao, W. J. (2011). GSK-3beta activity and hyperdopamine-dependent behaviors. Neurosci. Biobehav. Rev. 35, 645–654.
Li, Y. C., Xi, D., Roman, J., Huang, Y. Q., and Gao, W. J. (2009). Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563.
Lieberman, J. A., Jarskog, L. F., and Malaspina, D. (2006). Preventing clinical deterioration in the course of schizophrenia: the potential for neuroprotection. J. Clin. Psychiatry 67, 983–990.
Lin, C. H., Lane, H. Y., and Tsai, G. E. (2012). Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol. Biochem. Behav. 100, 665–677.
Lin, Y., Skeberdis, V. A., Francesconi, A., Bennett, M. V., and Zukin, R. S. (2004). Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J. Neurosci. 24, 10138–10148.
Lindsley, C. W., Shipe, W. D., Wolkenberg, S. E., Theberge, C. R., Williams, D. L. Jr., Sur, C., et al. (2006). Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr. Top. Med. Chem. 6, 771–785.
Lipina, T. V., Kaidanovich-Beilin, O., Patel, S., Wang, M., Clapcote, S. J., Liu, F., et al. (2011). Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse 65, 234–248.
Lipina, T. V., Niwa, M., Jaaro-Peled, H., Fletcher, P. J., Seeman, P., Sawa, A., et al. (2010). Enhanced dopamine function in DISC1-L100P mutant mice: implications for schizophrenia. Genes Brain Behav. 9, 777–789.
Lipina, T. V., Wang, M., Liu, F., and Roder, J. C. (2012). Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology 62, 1252–1262.
Lisman, J. E., Coyle, J. T., Green, R. W., Javitt, D. C., Benes, F. M., Heckers, S., et al. (2008). Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 31, 234–242.
Llansola, M., Sanchez-Perez, A. M., Montoliu, C., and Felipo, V. (2004). Modulation of NMDA receptor function by cyclic AMP in cerebellar neurones in culture. J. Neurochem. 91, 591–599.
Lu, Y. M., Roder, J. C., Davidow, J., and Salter, M. W. (1998). Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science 279, 1363–1367.
Ma, T. M., Abazyan, S., Abazyan, B., Nomura, J., Yang, C., Seshadri, S., et al. (2012). Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol. Psychiatry. doi: 10.1038/mp.2012.97. [Epub ahead of print].
Madry, C., Betz, H., Geiger, J. R. P., and Laube, B. (2008). Supralinear potentiation of NR1/NR3A excitatory glycine receptors by Zn2+ and NR1 antagonist. Proc. Natl. Acad. Sci. U.S.A. 105, 12563–12568.
Maher, B. J., and LoTurco, J. J. (2012). Disrupted-in-schizophrenia (DISC1) functions presynaptically at glutamatergic synapses. PLoS ONE 7:e34053. doi: 10.1371/journal.pone.0034053
Mao, L.-M., Wang, W., Chu, X.-P., Zhang, G.-C., Liu, X.-Y., Yang, Y.-J., et al. (2009a). Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat. Neurosci. 12, 602–610.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., et al. (2009b). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Marek, G. J., Behl, B., Bespalov, A. Y., Gross, G., Lee, Y., and Schoemaker, H. (2010). Glutamatergic (N-methyl-D-aspartate receptor) hypofrontality in schizophrenia: too little juice or a miswired brain? Mol. Pharmacol. 77, 317–326.
McCullumsmith, R. E., Kristiansen, L. V., Beneyto, M., Scarr, E., Dean, B., and Meador-Woodruff, J. H. (2007). Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res. 1127, 108–118.
Medoff, D. R., Holcomb, H. H., Lahti, A. C., and Tamminga, C. A. (2001). Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus 11, 543–550.
Mei, L., and Xiong, W.-C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452.
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., Wettstein, J. G., et al. (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56.
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., Bouchard, L., et al. (2008). Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 82, 696–711.
Millan, M. J. (2005). N-Methyl-D-aspartate receptors as a target for improved antipsychotic agents, novel insights and clinical perspectives. Psychopharmacol (Berl.) 179, 30–53.
Millan, M. J., Agid, Y., Brüne, M., Bullmore, E. T., Carter, C. S., Clayton, N. S., et al. (2012). Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat. Rev. Drug Discov. 11, 141–168.
Millar, J. K., Pickard, B. S., Mackie, S., James, R., Christie, S., Buchanan, S. R., et al. (2005). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191.
Miyamoto, S., Miyake, N., Jarskog, L. F., Fleischhacker, W. W., and Lieberman, J. A. (2012). Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 17, 1206–1227.
Moghaddam, B., and Jackson, M. E. (2003). Glutamatergic animal models of schizophrenia. Ann. N.Y. Acad. Sci. 1003, 131–137.
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436.
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540.
Monyer, H., Sprengel, R., Schoepfer, R., Herb, A., Higuchi, M., Lomeli, H., et al. (1992). NMDA receptors: molecular and functional distinction of subtypes. Science 256, 1217–1221.
Mori, H., Manabe, T., Watanabe, M., Satoh, Y., Suzuki, N., Toki, S., et al. (1998). Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron 21, 571–580.
Morris, B. J., Cochran, S. M., and Pratt, J. A. (2005). PCP: from pharmacology to modelling schizophrenia. Curr. Opin. Pharmacol. 5, 101–106.
Nakazawa, K., Zsiros, V., Jiang, Z., Nakao, K., Kolata, S., Zhang, S., et al. (2012). GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583.
Nakazawa, T., Komai, S., Tezuka, T., Hisatsune, C., Umemori, H., Semba, K., et al. (2001). Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 276, 693–699.
Nilsson, A., Duan, J., Mo-Boquist, L. L., Benedikz, E., and Sundström, E. (2007). Characterisation of the human NMDA receptor subunit NR3A glycine binding site. Neuropharmacology 52, 1151–1159.
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322.
Niwa, M., Kamiya, A., Murai, R., Kubo, K., Gruber, A. J., Tomita, K., et al. (2010). Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65, 480–489.
Nong, Y., Huang, Y. Q., and Salter, M. W. (2004). NMDA receptors are movin' in. Curr. Opin. Neurobiol. 14, 353–361.
Numakawa, T., Yagasaki, Y., Ishimoto, T., Okada, T., Suzuki, T., Iwata, N., et al. (2004). Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum. Mol. Genet. 13, 2699–2708.
O'Donovan, M. C., Craddock, N., Norton, N., Williams, H., Peirce, T., Moskvina, V., et al. (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 40, 1053–1055.
Olney, J. W., and Farber, N. B. (1995). Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 52, 998–1007.
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiat. Res. 33, 523–533.
Omkumar, R. V., Kiely, M. J., Rosenstein, A. J., Min, K. T., and Kennedy, M. B. (1996). Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 271, 31670–31678.
Ozaki, M., Sasner, M., Yano, R., Lu, H. S., and Buonanno, A. (1997). Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature 390, 691–694.
Papaleo, F., and Weinberger, D. R. (2011). Dysbindin and Schizophrenia: it's dopamine and glutamate all over again. Biol. Psychiatry 69, 2–4.
Papaleo, F., Lipska, B. K., and Weinberger, D. R. (2012). Mouse models of genetic effects on cognition: relevance to schizophrenia. Neuropharmacology 62, 1204–1220.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., et al. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437.
Perez-Otano, I., and Ehlers, M. D. (2004). Learning from NMDA receptor trafficking: clues to the development and maturation of glutamatergic synapses. Neurosignals 13, 175–189.
Petralia, R. S., Wang, Y. X., and Wenthold, R. J. (2003). Internalization at glutamatergic synapses during development. Eur. J. Neurosci. 18, 3207–3217.
Pitcher, G. M., Beggs, S., Woo, R. S., Mei, L., and Salter, M. W. (2008). ErbB4 is a suppressor of long-term potentiation in the adult hippocampus. Neuroreport 19, 139–143.
Pitcher, G. M., Kalia, L. V., Ng, D., Goodfellow, N. M., Yee, K. T., Lambe, E. K., et al. (2011). Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat. Med. 17, 470–478.
Quinlan, E. M., Olstein, D. H., and Bear, M. F. (1999). Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc. Natl. Acad. Sci. U.S.A. 96, 12876–12880.
Raman, I. M., Tong, G., and Jahr, C. E. (1996). Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron 16, 415–421.
Raymond, L. A., Tingley, W. G., Blackstone, C. D., Roche, K. W., and Huganir, R. L. (1994). Glutamate receptor modulation by protein phosphorylation. J. Physiol. Paris 88, 181–192.
Roberts, A. C., Díez-García, J., Rodriguiz, R. M., López, I. P., Luján, R., Martínez-Turrillas, R., et al. (2009). Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63, 342–356.
Rodenas-Ruano, A., Chavez, A. E., Cossio, M. J., Castillo, P. E., and Zukin, R. S. (2012). REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390.
Rosenblum, K., Dudai, Y., and Richter-Levin, G. (1996). Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc. Natl. Acad. Sci. U.S.A. 93, 10457–10460.
Rostas, J. A., Brent, V. A., Voss, K., Errington, M. L., Bliss, T. V., and Gurd, J. W. (1996). Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-D-aspartate receptor in long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 93, 10452–10456.
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. U.S.A. 104, 8131–8136.
Ruzicka, W. B., Zhubi, A., Veldic, M., Grayson, D. R., Costa, E., and Guidotti, A. (2007). Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 12, 385–397.
Sanna, P. P., Cammalleri, M., Berton, F., Simpson, C., Lutjens, R., Bloom, F. E., et al. (2002). Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 22, 3359–3365.
Sans, N., Prybylowski, K., Petralia, R. S., Chang, K., Wang, Y. X., Racca, C., et al. (2003). NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat. Cell Biol. 5, 520–530.
Savitz, J., Solms, M., and Ramesar, R. (2006). The molecular genetics of cognition: dopamine, COMT and BDNF. Genes Brain Behav. 5, 311–328.
Scott, D. B., Blanpied, T. A., and Ehlers, M. D. (2003). Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45, 755–767.
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147.
Shi, J., Gershon, E. S., and Liu, C. (2008). Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr. Res. 104, 96–107.
Snyder, M. A., Adelman, A. E., and Gao, W.-J. (2013). Gestational methylazoxymethanol exposure leads to NMDAR dysfunction in hippocampus during early development and lasting deficits in learning. Neuropsychopharmacology 38, 328–340.
Spear, L. P. (2000). The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 24, 417–463.
Stadler, F., Kolb, G., Rubusch, L., Baker, S. P., Jones, E. G., and Akbarian, S. (2005). Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J. Neurochem. 94, 324–336.
Stahl, S. M. (2007a). Beyond the dopamine hypothesis to the NMDA glutamate receptor hypofunction hypothesis of schizophrenia. CNS Spectr. 12, 265–268.
Stahl, S. M. (2007b). Novel therapeutics for schizophrenia: targeting glycine modulation of NMDA glutamate receptors. CNS Spectr. 12, 423–427.
Stefansson, H., Rujescu, D., Cichon, S., Pietilainen, O. P., Ingason, A., Steinberg, S., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236.
Steigerwald, F., Schulz, T. W., Schenker, L. T., Kennedy, M. B., Seeburg, P. H., and Kohr, G. (2000). C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J. Neurosci. 20, 4573–4581.
Straub, R. E., and Weinberger, D. R. (2006). Schizophrenia genes – famine to feast. Biol. Psychiatry 60, 81–83.
Takasu, M. A., Dalva, M. B., Zigmond, R. E., and Greenberg, M. E. (2002). Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 295, 491–495.
Tan, H. Y., Callicott, J. H., and Weinberger, D. R. (2009). Prefrontal cognitive systems in schizophrenia: towards human genetic brain mechanisms. Cogn. Neuropsychiatry 14, 277–298.
Tingley, W. G., Ehlers, M. D., Kameyama, K., Doherty, C., Ptak, J. B., Riley, C. T., et al. (1997). Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 272, 5157–5166.
Tunbridge, E. M., Harrison, P. J., and Weinberger, D. R. (2006). Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 60, 141–151.
Veldic, M., Guidotti, A., Maloku, E., Davis, J. M., and Costa, E. (2005). In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc. Natl. Acad. Sci. U.S.A. 102, 2152–2157.
Vinson, P. N., and Conn, P. J. (2012). Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology 62, 1461–1472.
Wang, H. X., and Gao, W. J. (2009). Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology 34, 2028–2040.
Wang, H. X., and Gao, W. J. (2010). Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J. Physiol. 588, 2823–2838.
Wang, H. X., and Gao, W. J. (2012). Prolonged exposure to NMDAR antagonist induces cell-type specific changes of glutamatergic receptors in rat prefrontal cortex. Neuropharmacology 62, 1808–1822.
Wang, H. X., Stradtman, G. G. 3rd., Wang, X. J., and Gao, W. J. (2008). A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc. Nat. Acad. Sci. U. S. A. 105, 16791–16796.
Wang, Y. T., and Salter, M. W. (1994). Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369, 233–235.
Weickert, C. S., Fung, S. J., Catts, V. S., Schofield, P. R., Allen, K. M., Moore, L. T., et al. (2012). Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry. doi: 10.1038/mp.2012.137. [Epub ahead of print].
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669.
Weinberger, D. R., Egan, M. F., Bertolino, A., Callicott, J. H., Mattay, V. S., Lipska, B. K., et al. (2001). Prefrontal neurons and the genetics of schizophrenia. Biol. Psychiatry 50, 825–844.
Wenthold, R. J., Prybylowski, K., Standley, S., Sans, N., and Petralia, R. S. (2003). Trafficking of NMDA receptors. Annu. Rev. Pharmacol. Toxicol. 43, 335–358.
Witthaus, H., Kaufmann, C., Bohner, G., Ozgurdal, S., Gudlowski, Y., Gallinat, J., et al. (2009). Gray matter abnormalities in subjects at ultra-high risk for schizophrenia and first-episode schizophrenic patients compared to healthy controls. Psychiatry Res. 173, 163–169.
Won, H., Lee, H.-R., Gee, H. Y., Mah, W., Kim, J.-I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265.
Xi, D., Keeler, B., Zhang, W., Houle, J. D., and Gao, W. J. (2009). NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser micro dissection technique. J. Neurosci. Methods 176, 172–181.
Keywords: gene, NMDA receptors, psychiatric disorders, neurodevelopment, schizophrenia
Citation: Snyder MA and Gao W-J (2013) NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Front. Cell. Neurosci. 7:31. doi: 10.3389/fncel.2013.00031
Received: 03 December 2012; Accepted: 11 March 2013;
Published online: 27 March 2013.
Edited by:
Chao Deng, University of Wollongong, AustraliaReviewed by:
Kazu Nakazawa, National Institute of Mental Health, USAKelly A. Newell, University of Wollongong, Australia
Copyright © 2013 Snyder and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Wen-Jun Gao, Department of Neurobiology and Anatomy, Drexel University College of Medicine, 2900 Queen Lane, Room 243, Philadelphia, PA 19129, USA. e-mail: wgao@drexelmed.edu