Genetic aspects of autism spectrum disorders: insights from animal models
 Swati Banerjee
Swati Banerjee Maeveen Riordan
Maeveen Riordan Manzoor A. Bhat
Manzoor A. Bhat- Department of Physiology, Center for Biomedical Neuroscience, School of Medicine, University of Texas Health Science Center, San Antonio, TX, USA
Autism spectrum disorders (ASDs) are a complex neurodevelopmental disorder that display a triad of core behavioral deficits including restricted interests, often accompanied by repetitive behavior, deficits in language and communication, and an inability to engage in reciprocal social interactions. ASD is among the most heritable disorders but is not a simple disorder with a singular pathology and has a rather complex etiology. It is interesting to note that perturbations in synaptic growth, development, and stability underlie a variety of neuropsychiatric disorders, including ASD, schizophrenia, epilepsy, and intellectual disability. Biological characterization of an increasing repertoire of synaptic mutants in various model organisms indicates synaptic dysfunction as causal in the pathophysiology of ASD. Our understanding of the genes and genetic pathways that contribute toward the formation, stabilization, and maintenance of functional synapses coupled with an in-depth phenotypic analysis of the cellular and behavioral characteristics is therefore essential to unraveling the pathogenesis of these disorders. In this review, we discuss the genetic aspects of ASD emphasizing on the well conserved set of genes and genetic pathways implicated in this disorder, many of which contribute to synapse assembly and maintenance across species. We also review how fundamental research using animal models is providing key insights into the various facets of human ASD.
Introduction
Autism spectrum disorders (ASDs) are a complex set of heterogeneous neurodevelopmental disorders categorized by a triad of key behavioral anomalies. Characteristic behavioral abnormalities consist of restricted interests accompanied by repetitive behavior, deficits in language and communication, and the inability to engage in reciprocal social interactions (Abrahams and Geschwind, 2008; Betancur et al., 2009; Levitt and Campbell, 2009; Peca et al., 2011b; Zoghbi and Bear, 2012). Autism is not a singular disease entity. The disorder encompasses a spectrum of wide ranging phenotypic manifestations which span from debilitating impairments to mild behavioral and personality traits. Therefore, autism is rightfully referred to as “autism spectrum disorders” (Persico and Bourgeron, 2006).
Autism spectrum disorder appears to be involved in early brain development. Obvious signs and symptoms show early onset within the first 3 years of life and persist into adulthood. According to the recent reports from the Center for Disease Control, an estimated 1 in 88 children has been identified with ASD. Interestingly, these disorders show a gender bias where males are affected almost five times more than females (http://www.cdc.gov/Features/CountingAutism/). ASD is among the most heritable disorders evidenced by family and twin studies with a concordance rate of 70–90% for monozygotic twins (Folstein and Rutter, 1977; Steffenburg et al., 1989; Bailey et al., 1995; Folstein and Rosen-Sheidley, 2001). Nevertheless, heritability in this case is more complex due to the differences in manifestations of its core symptoms, gradual changes over time, and differing degrees of response to interventions (Abrahams and Geschwind, 2008; Levitt and Campbell, 2009). 10–25% of ASD cases seem to have an underlying genetic disorder such as fragile X syndrome, tuberous sclerosis (TSC), and Rett syndrome (Betancur et al., 2009).
Recent studies have highlighted numerous potential risk factors that may contribute to ASD. These risk factors range from genetic, to epigenetic, to environmental factors. Detection of copy number variations (CNV), point mutations, and identification of rare variants in synaptic cell adhesion proteins and pathways are some of the ways researchers are providing insight into the pathophysiology of ASD (Sebat et al., 2007; Malhotra and Sebat, 2012; Zoghbi and Bear, 2012). It is worthwhile to note that the genes and the genetic pathways implicated in ASD, and the identification of any causal rare variants are accessible to modeling in experimental systems. Research findings both from studying human genetics and animal models of ASD suggest that disruption of synapse formation and stabilization processes is a key underlying feature in ASD etiology. Dysfunctions in the assembly or structure of transmembrane and scaffolding proteins needed for building and maintaining synapses, and disruption in cellular signaling pathways controlling synaptogenesis are major contributing factors in ASD.
A large portion of this review will emphasize the well-conserved sets of genes and genetic pathways implicated in ASD, many of which contribute to synapse assembly and maintenance across different species. Given the complexity and heterogeneity of this disorder, it has proved challenging to unravel the underlying causes of ASD from human clinical population alone. Nevertheless, numerous animal models have been utilized that have enormously contributed toward understanding specific aspects that constitute the spectrum of these disorders.
Historical Overview of Autism Spectrum Disorders
Leo Kanner, a psychiatrist, initially described autism well over half a century ago (Kanner, 1943, 1968, 1971). Studies on the relationship between autism and abnormal electroencephalogram were among the first to suggest autism as a disorder of brain function (Creak and Pampiglione, 1969). Despite these groundbreaking observations on autism, early identification of autism was marred by lack of adequate diagnostic criteria. It was not until the introduction of the concept of “autism triad” that highlighted the now well-established characteristics of impairment in social interaction, language and communication did Autism become a recognizable disorder. Since then the clinical conceptualizations of ASD have consistently evolved together with a steady rise in the number of ASD cases. Our current understanding of ASD is that of a complex neurological disorder that continues to challenge our ability to identify the underlying causal mechanisms.
Many Facets of Autism Spectrum Disorders
Genetics – Copy Number Variation
Copy number variation is among the most widespread of structural variations in the human genome, and is increasingly being implicated as a major contributor to the pathophysiology of complex neurodevelopmental disorders (Sebat et al., 2007, 2009; Christian et al., 2008; Kumar et al., 2008; Marshall et al., 2008; Weiss et al., 2008; Bucan et al., 2009; Glessner et al., 2009; Merikangas et al., 2009; Luo et al., 2012; Malhotra and Sebat, 2012). CNVs largely comprise of duplications and deletions and can be de novo or familial. De novo CNVs are more prevalent in causing sporadic genomic disorders (McCarroll et al., 2008). The duplication or deletion events disrupt gene structure, expression, and function and are a common cause of developmental delay. Several studies suggest important role of CNVs in disease etiology, susceptibility, and inheritance (Beckmann et al., 2007; Estivill and Armengol, 2007). Large-scale genome-wide association studies are credited for detection of CNVs in rare cases of ASD (Ma et al., 2009a). Duplications and microdeletions in many loci are associated with ASD. Several studies identified duplication CNVs within 15q13 (Christian et al., 2008; Miller et al., 2009) and microdeletions at many loci in 16p11.2 (Sebat et al., 2007; Marshall et al., 2008; Weiss et al., 2008; Levy et al., 2011; Sanders et al., 2011), Williams syndrome locus 7q11.23, DiGeorge syndrome locus 22q11.2, 1q21.1, and Prader–Willi and Angelman syndromes at 15q11-13 (Glessner et al., 2009; Sanders et al., 2011).
Interestingly, genes associated with CNVs in ASD are involved in regulating synaptogenesis. Some of the genes include NEUROLIGIN 4 (NLGN4; Jamain et al., 2003; Laumonnier et al., 2004), SHANK3 (Durand et al., 2007; Moessner et al., 2007; Gauthier et al., 2009), TBX1, PCDH10, and NHE9 (Morrow et al., 2008). Recent findings further reiterate a correlation between synapse formation and autism (Glessner et al., 2009; Mitne-Neto et al., 2011). In addition to these genes, some of the other genes recognized as risk factors in ASD include NEUREXIN 1 (NRXN1; Kim et al., 2008; Bucan et al., 2009), SHANK2 (Berkel et al., 2010), CNTN4 (Fernandez et al., 2004), CNTNAP2 (Bakkaloglu et al., 2008; Penagarikano et al., 2011), DPYD and DPP6 (Marshall et al., 2008); NLG1 (Glessner et al., 2009) and SYNGAP1, DLGAP2 (Pinto et al., 2010). A detailed list of genes, their potential functions and genetic pathways linked to ASD are summarized in Table 1.
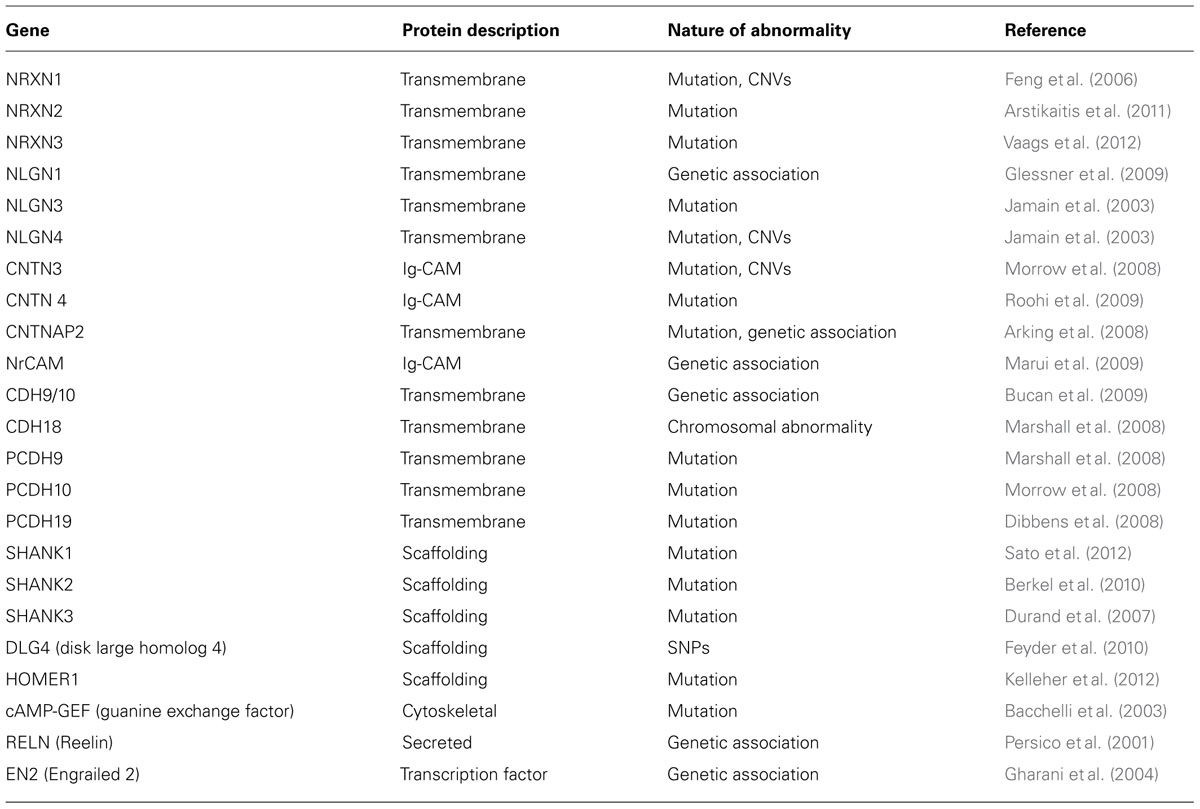
TABLE 1. Conserved genes implicated in ASD.
Epigenetics
Epigenetic mechanisms underlie several human neurodevelopmental disorders. Genomic imprinting, epimutations, DNA methylation, and histone modifications are all examples of epigenetic mechanisms linked to the development of certain disorders. These mechanisms involve modifications of nucleotides or chromosomes without altering the genetic sequence (Zoghbi, 2003; Egger et al., 2004). Thus causing modifications in gene expression that may increase the likelihood of developing a particular disease. Epigenetic mechanisms are believed to function at the interface between genetic and environmental factors (Jiang et al., 2004; Qiu, 2006). Studies linking these two factors are gaining importance for understanding the etiologies of complex disorders and could play a role in the development of ASD.
While epigenetic mechanisms are implicated in the development of many disorders, they are also an intrinsic phenomenon for normal brain development. Genomic imprinting is an example of an epigenetic mechanism that occurs normally throughout life. This is when one of the two parental alleles for an imprinted gene becomes inactive due to DNA methylation resulting in monoallelic gene expression. This phenomenon occurs quite frequently in humans but was also discovered in fungi, plants, and other animals (Martienssen and Colot, 2001; Jiang and Kohler, 2012). Using genome-wide scans, several areas on chromosomes known as, hot spots for genomic imprinting, were located on loci 7q and 15q (Reik and Walter, 2001; Luedi et al., 2007). Interestingly, these loci are highly affected in individuals with ASD (International Molecular Genetic Study of Autism Consortium, 2001; Lamb et al., 2005). Several studies have linked duplication or deletion events on the active chromosome to ASD (Cook et al., 1997; Schroer et al., 1998; Koochek et al., 2006). Individuals with Angelman syndrome (Mabb et al., 2011; Huang et al., 2012) and Prader–Willi syndrome (Miyake et al., 2012) show a defect in the active allele that leads to loss of gene expression. Such correlations provide compelling evidence for the role of genetic and epigenetic mechanisms in the etiology of ASD.
Additionally, DNA methylation is an important basic step in epigenetic gene control. Methyl-CpG binding proteins bind to the methylated DNA regions to control gene expression. Mutations in methyl-CpG binding protein 2 (MeCP2) cause Rett syndrome which shows characteristic autistic-like behavior in addition to seizures, ataxia, and stereotypic hand movements (Amir et al., 1999). More recently, MeCP2 was shown to regulate several genes involved in synaptic plasticity, neuronal cell proliferation and neuronal transcription factors including: brain-derived neurotrophic factor (BDNF), distal-less homeobox 5 (DlX5), and insulin-like growth factor binding protein 3 (IGF3; Chen et al., 2003; Martinowich et al., 2003; reviewed in Miyake et al., 2012). Thus, epigenetic misregulation of synaptic genes could potentially contribute to ASD (Beaudet, 2007). Yet another set of studies suggest that extrinsic factors like the environment can alter epigenetic make up leading to defective neuronal functions (Jessberger et al., 2007; Ma et al., 2009b).
Environment and other Factors
Environmental contributions and other modulating factors are emerging as potential risk factors for ASD. Heavy metals, parental age, immunological proteins, environmental pesticides and insecticides, and food contaminants are thought to act as modulators of ASD (Durkin et al., 2008). These factors could contribute toward an increase in the prevalence of ASD but may not be sufficient to cause ASD. Nonetheless, a major challenge is to identify environmental factors relevant to ASD that could influence susceptibility, severity, and intervention outcomes. Heritable genetic vulnerabilities can magnify the adverse effects triggered by environmental factors. If both genes and environment converge, a resulting dysfunction of neurotransmitters and signaling pathways could take place at key developmental time points (Pessah et al., 2008). The toxicological literature point toward several environmental chemicals of concern to human health that can either directly or indirectly affect signaling pathways impaired in ASD.
Prenatal exposure to certain pesticides and insecticides are known to inhibit acetylcholine (ACh) and γ-aminobutyric acid (GABA; Shelton et al., 2012). Studies show these neurotransmitter systems are altered in a subset of autistic individuals. Similarly, environmentally induced alterations in calcium signaling pathways caused by organic pollutants, impact a broad range of neurotransmitter systems like the cholinergic, GABAergic, and dopaminergic systems (Pessah et al., 2008; Corrales and Herbert, 2011). In addition to disruptions in important neuronal signaling pathways, pesticides can cause oxidative stress, neuroinflammation, and mitochondrial dysfunction, all contributors to neuronal cell-death and dysfunction (Herbert, 2010; Shelton et al., 2012). Furthermore, cytokine-mediated influences and immune-related proteins are also listed as modulating factors for ASD (Ashwood et al., 2011; Onore et al., 2012). Families with ASD often show clustering of autoimmune disorders (Croen et al., 2005; Currenti, 2010). Several reports indicate the presence of serum antibody reactivity against human cortical and cerebellar regions of the brain in autistic patients (Silva et al., 2004). This process is thought to begin in utero and is associated with placental transfer of maternal autoantibodies to neuronal proteins potentially leading to neuronal dysfunction.
Animal Models of Autism Spectrum Disorders
Autism spectrum disorder is a complex disorder with no singular pathology and because of this a collective and collaborative approach is the key to understanding its etiology and design of rational interventions. Studies in animals are aimed at modeling the core phenotypes associated with ASD, including communication and social impairments, restricted interests, and repetitive behaviors in an attempt to uncover the mechanisms that underscore the entire spectrum of the disorder. In this section, we will uncover the wide range of both invertebrate and vertebrate model systems utilized by researchers that have collectively made significant contributions toward understanding the mechanisms that underlie ASD (summarized in Table 3).
Non-Human Primates
One of the animal models largely believed to help bridge the gap between humans and lower vertebrate systems is the non-human primate (NHP) model. NHP model is relevant for understanding ASD due to its high degree of correspondence to human behavior and their striking homology in the anatomy of neural circuits that mediate social behavior (for review, see Ongur and Price, 2000; Watson and Platt, 2012). Some of the behavioral correlates that NHPs have with human behavioral deficits seen in ASD include repetitive behaviors (Lutz et al., 2003; Alarcon et al., 2008), social communication (Ghazanfar and Santos, 2004), and their ability to follow other’s gazes, a tendency that is compromised in Autism. For example, ablation studies in NHPs especially of the superior temporal sulcus region reveal difficulties in responding to social cues like eye gaze (Campbell et al., 1990). The lesion model involving the amygdala in NHPs is used to study alterations in socio-emotional behaviors (Amaral et al., 2003). Some papers speculate on the involvement of mirror neurons in the development of ASD (Oberman et al., 2005). In early childhood development, mirror neurons may play a key role in mimicking behaviors, actions, and language. A failure in the development or proper organization of mirror neurons might be linked to some of the behavioral phenotypes associated with ASD (Williams et al., 2001). NHP, like humans, possess mirror neurons and their use as a model system could provide some insight into the involvement of mirror neurons in ASD. NHP models are also used to investigate immunological factors in the etiology of ASD. Autoantibodies present in children with ASD have prompted investigators to analyze the affects of maternal IgG antibodies on the fetal brain during gestation. Injections of IgG antibodies from human mothers who had multiple children with ASD to pregnant rhesus monkeys resulted in abnormal stereotyped behaviors in offspring, and increased activity of offspring compared to controls (Martin et al., 2008). Although use of NHP model has the capability of contributing to some of the more behavioral aspects of ASD research, the absence of genetic knockouts in NHPs modeling ASD together with the careful considerations of ethical implications of NHP research tend to pose limitations that can be better addressed using rodents and invertebrate model systems.
Rodents
Mouse models recapitulating symptoms of ASD through selective manipulations of genes and neural circuitry is a much more amenable model system compared to NHP models. Currently, there is a sizeable number of autism mouse models available made possible due to generation of specific gene knockouts; mutations in these genes are thought to contribute to ASD together with the emergence of CNVs and high-end genome-wide sequencing studies. Mouse models of human disorders have limitations in recapitulating the entire phenotypic spectrum (Arguello and Gogos, 2006). The validity of mouse models of human disorders are based on three criteria: (i) construct validity as provided by knock outs that carry a mutation in a gene that is affected in the human disorder (Peca et al., 2011a), (ii) face validity as reflected in animals that bear many of the core and ancillary physical or behavioral resemblances to the human disorder (Crawley, 2004), and (iii) predictive validity, which by far, is the most challenging to accomplish and indicates a similar response in the mouse model to an intervention that is known to be effective in human patients with that disorder.
Some of the mouse models representing syndromic forms of ASD include mice modeling Phelan–McDermid syndrome (SHANK3; Bangash et al., 2011; Peca et al., 2011a), Rett syndrome (MeCP2; Shahbazian et al., 2002; Moretti et al., 2006), fragile X syndrome (FMR1; Ronesi et al., 2012), Timothy syndrome (TS; CACNA1C; Bader et al., 2011), and others (see also Table 2). Neuroligin 3 knock out mice serve as a model for non-syndromic autism (Baudouin et al., 2012). Other examples of mouse models to study characteristics of ASD include Purkinje-specific knock out of TSC1 (Tsai et al., 2012), chromosome-engineered mouse model for human 15q11-13 (Nakatani et al., 2009), model for 16p11.2 lesion found in autism (Horev et al., 2011), 22q11.2 mice lacking PTEN (Zhou et al., 2009), CNTNAP2 (Penagarikano et al., 2011), SHANK2 (Won et al., 2012), and SCN1A (Han et al., 2012). The impressive array of mouse models displaying behaviors that are reflective of the human behavioral and cognitive ASD symptoms is highly informative. On the other hand, the behavioral phenotypes between mouse models with ablation of the same gene show variations based on either their genetic backgrounds (Crabbe et al., 1999), or how individual laboratories conduct their behavioral assays further underscoring the impact of the genetic background or the local environment on the displayed phenotypes. In any case, a complete understanding of the similarities and differences in the behavioral phenotypes across the ASD mouse models will provide key insights into the underlying neural circuitry behind these behaviors.
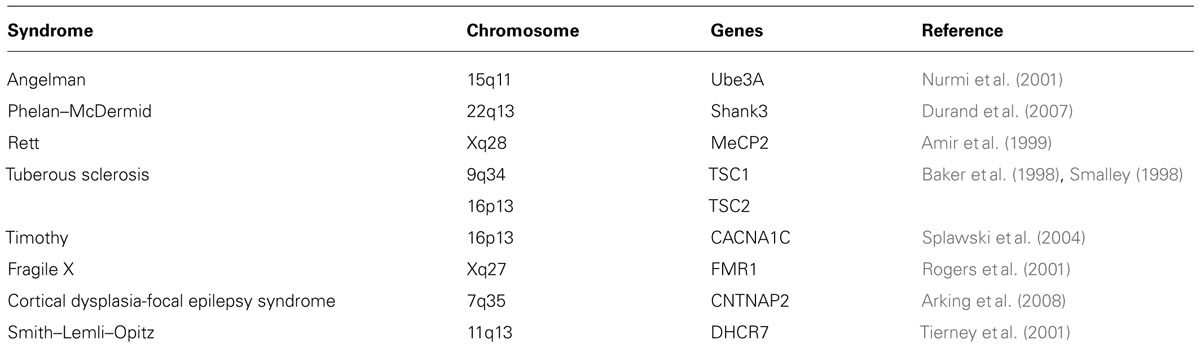
TABLE 2. Genetic syndromes with ASD-related phenotypes.
Other emerging rodent models of ASD include rat and prairie vole (McGraw and Young, 2010). Rats that are injected with valproic acid (VPA) serve as an environmentally triggered model of autism and this method has emerged as a new way to study ASD in rats (Rodier et al., 1997). VPA injected to gestational mothers before neural tube closure causes autistic-like phenotypes in offspring such as a reduction in the number of cerebellar Purkinje cells and disruption in inhibitory circuits (Gogolla et al., 2009). Furthermore, these animals show similar behavioral phenotypes associated with autism including lower sensitivity to pain and higher sensitivity to non-painful stimuli, repetitive behaviors, hyperactivity, and decreased number of social behaviors. They also show delayed mental impairments and lower body weight (Schneider and Przewlocki, 2005; Favre et al., 2013). Prairie voles also generated interest in the area of ASD because of their ability to form lasting social bonds and their nurturing behavior. Impaired social behaviors and deficits in various aspects of social cognition are some of the signature of ASD in humans. Thus, development of genetic, molecular, and genomic tools in prairie vole will likely be useful in basic and translational research that may be relevant to ASD (McGraw and Young, 2010).
Zebrafish
Zebrafish are widely used as a model for studying vertebrate development and although not as popular as the mouse model for studying ASD, zebrafish are being used to dissect the genetic basis of autism due to a multitude of genetic techniques available. These include lineage tracing using fluorescent tracers or labeling cells with lipophilic dyes, loss of function analyses using chemicals, transposable elements, and gain of function assays such as those involving microinjection of synthetic mRNA. In addition, morpholino technology is widely used as an efficient reverse genetic approach to understand gene function (Tropepe and Sive, 2003). Furthermore, fast oogenesis and embryogenesis, and high fecundity allows for rapid experimental assays in this model. Transparency and external development of embryos allows for the study of growth and development of cells and tissues in live embryos (Tropepe and Sive, 2003). Additionally, zebrafish are an excellent model for use in carrying out genetic screens to identify new genes of interest and are useful in designing genetic screens to uncover specific enhancers or suppressors of particular phenotype (Dooley and Zon, 2000). These screens identified several candidate genes, such as Reelin and MET that confers susceptibility to human ASD in zebrafish (Rice and Curran, 2001). The presence of structurally and functionally homologous regions in zebrafish brain, which are perturbed in human autistic patients, is another avenue that zebrafish researchers are taking advantage of to study brain development and neuronal connections. Although zebrafish is an excellent model to study some of the genetic aspects of ASD, the behavioral phenotypes associated with ASD are difficult to recapitulate (Tropepe and Sive, 2003). Thus, other model systems might be useful in studying some of the behavioral phenotypes associated with ASD.
Songbirds
Songbirds can be used both as a molecular and a behavioral model for understanding the etiology of ASD. Songbirds are socially sophisticated and display characteristically human traits like monogamy and cultural inheritance while demonstrating the ability to learn vocalizations (Clayton et al., 2009). Vocal learning is an important element of language. Impairments in vocal and language learning are some of the core deficits in autism. Thus, understanding vocal learning through songbirds has emerged as an important model to study some aspects of ASD.
Speech in humans and bird songs display striking parallels in that both seem to have a critical developmental time window for learning, a homologous underlying neural circuitry involving a loop between the cerebral cortex, basal ganglia, and thalamus, and a role for social influences in the learning of vocalizations (Panaitof, 2012). Studies from songbird indicate that CNTNAP2, which is implicated in human ASD and is enriched in human language related neural circuits (Alarcon et al., 2008), might play a role in vocal communication in songbirds as well (Panaitof et al., 2010). Similar to developing human brain where CNTNAP2 shows a gradient distribution in frontal cortical areas, Cntnap2 expression is either enhanced or reduced in key song control nuclei in songbird brain. In the absence of an animal model that can address language deficits, songbird model may prove useful for further exploration of the cellular and molecular mechanisms underlying the homologous neural circuitry that underscore language development in humans. Recently, using microarray and in situ hybridization analyses, large databases have been compiled that reveal expression patterns of certain genes in specific regions of the brain (Warren et al., 2010). Expansion on the molecular aspects of these observations has further increased the validity of behavioral songbird research. While linking behavior and genetics in songbirds is a tall order, it might still provide important clues about neuronal circuitry and language acquisition in the complex and heterogeneous nature of ASD and its behavioral manifestations.
Invertebrate Models
Despite being millions of years apart on the evolutionary scale there is a surprising degree of genetic conservation between invertebrates and humans. Invertebrate models have made seminal contributions toward a basic understanding of human neurological disorders that are hard to ignore. One such classic invertebrate model for studying human neurodevelopmental disorders is the fruit fly, Drosophila. The fruit fly has proven to be an important model time and again to study various disorders where a single, causative genetic defect has been identified in Rett syndrome (Cukier et al., 2008), fragile X syndrome (Morales et al., 2002), and Angelman syndrome (Wu et al., 2008; see also Table 2). Drosophila studies have advanced our fundamental understanding of some of these human disease gene functions, which show features of ASD. Recent studies in Drosophila have started to unravel some of the key genes, such as Neurexin 1 (Li et al., 2007; Zeng et al., 2007), Neuroligin 1 (Banovic et al., 2010), and Neuroligin 2 (Chen et al., 2012), which are the fly homologs of human NRXNs and NLGNs, respectively, that are implicated in ASD (De Jaco et al., 2005; Sudhof, 2008). With the unmatched power of Drosophila genetics and the potential of carrying out large scale screens using the sophisticated genetic tools available, Drosophila will undoubtedly continue to provide key mechanistic insights to dissect the genetic basis of ASD and likely facilitate the design of therapeutics. Apart from Drosophila, other invertebrate models that are being used to study aspects of ASD are C. elegans (Calahorro and Ruiz-Rubio, 2011, 2012) and Aplysia (Choi et al., 2011; Ye and Carew, 2011).
A recent study in Aplysia showed that trans-synaptic Neurexin–Neuroligin interactions govern synaptic remodeling and regulates signaling required for the storage of long-term memory, including emotional memory, an ability that is affected in ASD patients (Choi et al., 2011). This study showed that long-term facilitation and associated pre-synaptic growth were compromised when neurexin or neuroligin was depleted from pre- and post-synaptic machineries. In addition, an introduction of R451C mutation of NLGN3 associated with human ASD blocked both intermediate- and long-term facilitation in Aplysia (Choi et al., 2011).
Another genetically tractable animal model is the nematode, C. elegans, which are utilized to understand the underlying mechanisms and abnormalities in neuronal synaptic communications in complex human neurological disorders like Alzheimer’s and ASD (Calahorro and Ruiz-Rubio, 2012). C. elegans have orthologs for ASD-related genes such as NLGNs, NRXNs, and SHANK. Recent reports highlight behavioral phenotypes in C. elegans reminiscent of ASD following removal of neuroligin homolog, nlg-1 (Hunter et al., 2010; Calahorro and Ruiz-Rubio, 2012) and subsequent functional phenotypic rescue by human NLGN1 (Calahorro and Ruiz-Rubio, 2012). Additionally, trans-synaptic NRX-1 and NLG-1 in C. elegans are found to mediate retrograde synaptic inhibition of neurotransmitter release at the neuromuscular junction (Hu et al., 2012) further underscoring the function of these molecules in synaptic modulation.
Thus determining the biological underpinnings of ASD will require a concerted effort involving studies from human clinical populations and several different animal models. This effort will provide complementary and critical insights toward understanding the complex and unknown ASD etiology. Since testing the causality and exploring the molecular and cellular mechanisms of ASD are either severely limited or off limits in human populations, research using various animal models will provide clues to the range of functional deficits that cause the disorder and may even hint at the underlying neural circuits that drive the behavioral deficits.
Genes Implicated in ASD
With the growing repertoire of synaptic genes implicated in ASD, it is becoming increasingly clear that synaptic dysfunction at multiple levels may underlie ASD. Synapses comprise of pre- and post-synaptic elements (Figure 1; see also review by Delorme et al., 2013) like synaptic cell adhesion molecules (CAMs), ion channels, neurotransmitter receptors, scaffolding and cytoskeletal proteins that work harmoniously to provide synaptic structural integrity and functionality (refer Tables 1 and 4). Perturbations in synaptic assembly or function are commonly reported in many neuropsychiatric disorders (Blanpied and Ehlers, 2004).
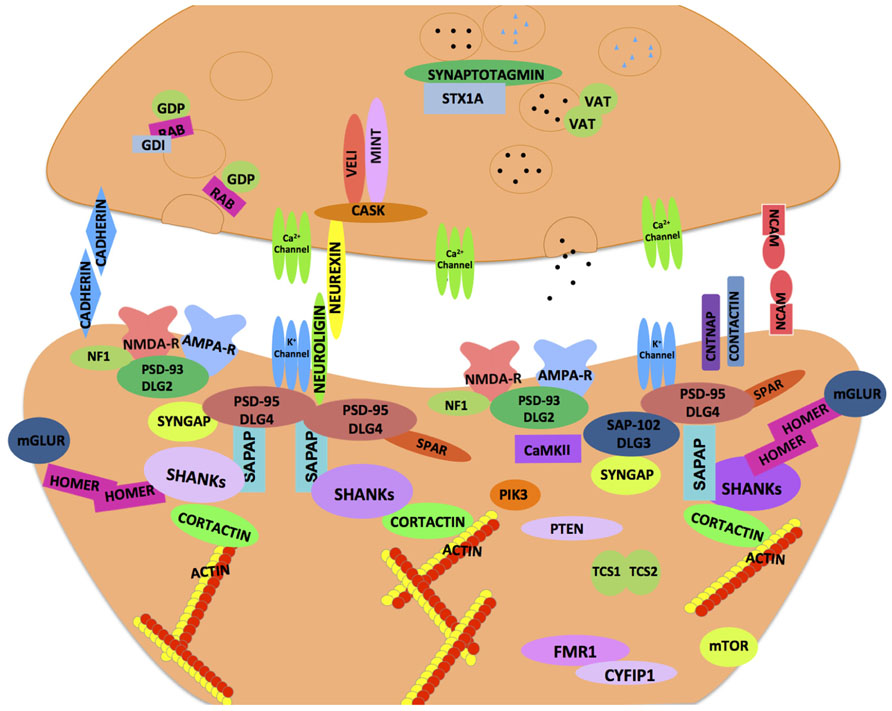
FIGURE 1. Synaptic proteins implicated in neurodevelopmental and neuropsychiatric disorders. A schematic illustration of an ensemble of pre- and post-synaptic proteins. The majority of these proteins are highly conserved across species, and thought to confer susceptibility to a host of neurodevelopmental and neuropsychiatric disorders including ASD. The cellular machinery of synapses is comprised of transmembrane heterophilic (such as Neurexin and Neuroligin) and homophilic cell-adhesion molecules (such as Cadherins and NCAM), cytoplasmic scaffolding proteins (such as PSD-95, Cask, and Shank) and cytoskeletal proteins (such as Homer and Cortactin) that link transmembrane and membrane-associated protein complexes with the underlying actin cytoskeleton. One of the emerging models in ASD is based on synaptic dysfunction in a molecular pathway that is orchestrated by trans-synaptic Neurexin–Neuroligin-dependent proteins complexes. This molecular assembly aligns the pre- and post-synaptic apparatus facilitating functional activation and modulation of ion channels that are in proximity to the neurotransmitter containing synaptic vesicles on the pre-synaptic side. These protein complexes recruit other proteins both pre- and post-synaptically and help organize functional neural networks. The cytoskeletal scaffolding protein Cask is one such notable protein which binds to the C-terminus of Neurexin. At the post-synaptic density, Neuroligin binds to PSD-95, other molecules such as PSD-93, SAP97, and the SAPAP family of proteins as well as the Shank family of scaffolding proteins help orchestrate the post-synaptic area. Homer and Shank function is thought to stabilize the post-synaptic density and serve as a platform to incorporate the post-synaptic receptors (such as NMDAR, AMPAR, and mGluR) into the machinery. The synaptic dysfunction in ASD may occur at multiple levels whereby failure to organize proper protein–protein interactions at the synapse may compromise neuronal functions (refer text for more details).

TABLE 3. Phenotypic analyses and relevant animal models of ASD.
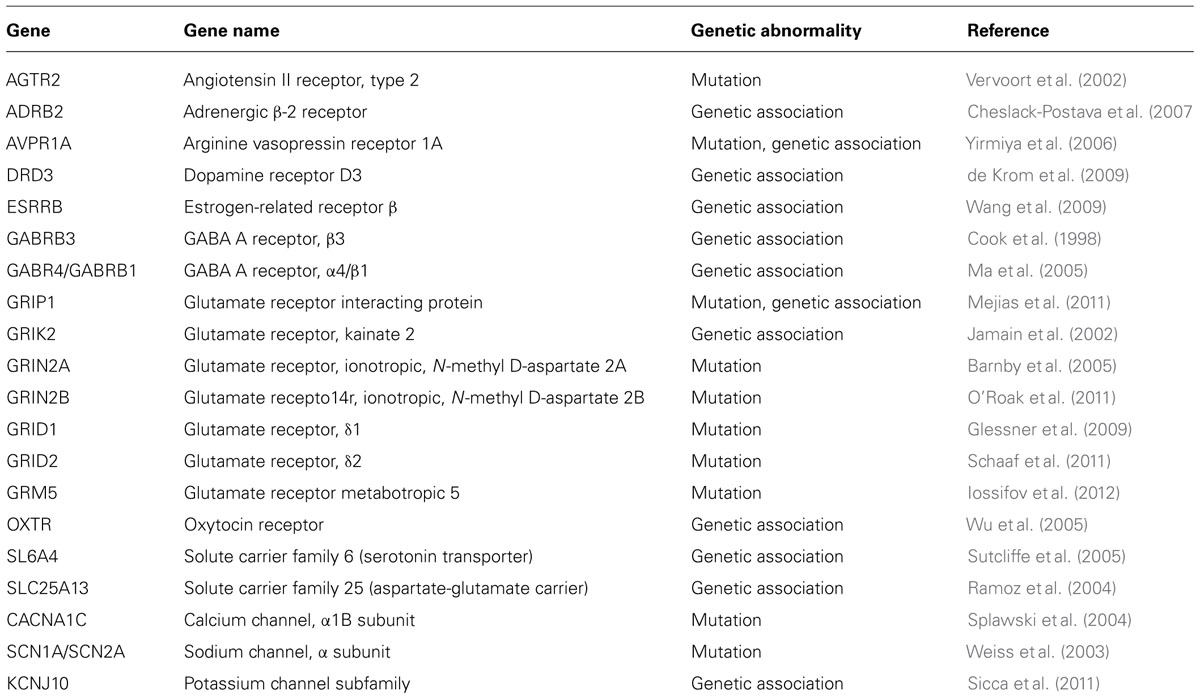
TABLE 4. Receptors, transporters, and channel proteins in ASD.
Cell Adhesion Molecules
Neuroligins
Neuroligins (NLGNs) are post-synaptic CAMs localized at glutamatergic or GABAergic synapses (Song et al., 1999; Varoqueaux et al., 2004). NLGNs have a large extracellular cholinesterase-like domain, and a small intracellular domain with PDZ-binding motif. There are five NLGNs, which include two X-linked genes (NlgN3 and NlgN4X) and one Y-linked (NlgN4Y). The identification of ASD-linked mutations in NLGN3 and NLGN4X was an important finding that implicated these genes in the etiology of ASD (Jamain et al., 2003) and linked ASD to molecules with synaptic function. In vitro studies using mutant forms of NlgN3 and NlgN4X showed retention of the mutant proteins in endoplasmic reticulum resulting in reduced cell surface binding to Neurexin (NRXN; Chih et al., 2004; Comoletti et al., 2004; Boucard et al., 2005). Some of the mutations of NLGNs seen in ASD patients were generated in mice and other model systems. For example, NlgN3 R451C knock-in mice showed challenged social interactions, enhanced inhibitory synaptic transmission, and altered spatial learning abilities (Jamain et al., 2003; Tabuchi et al., 2007; Chadman et al., 2008). This arginine to cysteine point mutation at the analogous position as the human NLGN3 R451C was recently made in Aplysia revealing abnormal synaptic facilitation (Choi et al., 2011). NLGN3 knock out mice showed reduced ultrasound vocalization together and a lack of social novelty preference (Radyushkin et al., 2009). NlgN4 knock out mice displayed reduced reciprocal social interactions and vocalizations consistent with observations in human ASD patients (Jamain et al., 2008). Studies on NlgN1 knock out mice showed impaired NMDA receptor signaling, while NlgN2 knock out mutants revealed reduced inhibitory synaptic transmission (Chubykin et al., 2007). Studies in vertebrate and invertebrates alike have now established that NLGNs are essential for proper synapse maturation, maintenance, and function as opposed to initial synapse formation (Chih et al., 2005; Varoqueaux et al., 2006; Sudhof, 2008; Banovic et al., 2010; Chen et al., 2012).
Recent studies on null mutants of Drosophila neuroligin 2 (dnlg2), which is the fly homolog of human NlgN3 showed reduced synaptic bouton numbers and synaptic transmission (Chen et al., 2012). Interestingly, dnlg2 is required both pre- and post-synaptically for proper synapse structure and function. Another study in C. elegans reported presence of Neuroligin at both pre- and post-synaptic regions (Feinberg et al., 2008). These studies highlight the exceptions to the traditional role and localization of NLGNs at the post-synaptic terminals as seen at most vertebrate synapses. It also raises interesting questions about how synaptic organization might be fine tuned, and how signaling pathways might regulate the expression of pre- and post-synaptic proteins during synaptic development and function. Thus, studies on NLGNs using various model systems will provide key insights into how these synaptic CAMs are involved in human ASD.
Neurexins
Neurexins (NRXNs) are predominantly presynaptic CAMs (Ichtchenko et al., 1995), although they were also reported to be expressed post-synaptically (Taniguchi et al., 2007). There are three Nrxn genes, Nrxn1, Nrxn2, and Nrxn3, each of which encode α- and β-isoforms. The α-Neurexin extracellular domain consists of six LNS domains interspersed by three EGF-like repeats and interacts with various proteins in the synaptic cleft. Mouse knock out mutants of individual Nrxns do not show gross abnormalities in synaptic ultrastructure or in synapse number while triple α-Nrxn knock out mice die prenatally due to respiratory complications. These mice show impaired synaptic transmission, but not synapse formation suggesting that like their Nlgn ligands, α-NRXNs are required for proper synaptic maintenance and function, and not initial synapse formation (Missler et al., 2003). Pre-synaptic calcium channel function is also disrupted in α-Nrxn knock out mice. Interestingly, compared to three NRXNs in mammals, Drosophila has a single neurexin-1 (dnrx) gene which like its vertebrate counterparts is pre-synaptic and is required for proper synaptic growth and neurotransmission (Li et al., 2007).
NRXN1 has emerged as a strong candidate in ASD since the identification of overlapping de novo deletions in Nrxn1 in individuals with ASD. Although rare, missense mutations (Feng et al., 2006; Kim et al., 2008) and deletions, and chromosomal aberrations in the NRXN1 were also found in ASD patients (Marshall et al., 2008; Zahir et al., 2008; Glessner et al., 2009). Interestingly, NRXN1 deletions also confer risk for schizophrenia pointing to an overlap between the two neurodevelopmental disorders (Betancur et al., 2009; Rujescu et al., 2009).
Contactins
The Contactins (CNTNs) are glycosyl phosphatidyl-inositol (GPI) anchored immunoglobulin (Ig) superfamily proteins with diverse functions ranging from myelination (Berglund et al., 1999; Bhat et al., 2001) to synapse formation and plasticity (Betancur et al., 2009). Disruption of CNTN4 is associated with ASD (Fernandez et al., 2004). Deletion and duplication in CNTN4 and small deletions near CNTN3 have been identified in various patients with ASD (Roohi et al., 2009). Since CNTN3 and CNTN4 expression and localization overlaps with synaptogenesis in the developing brain, it raises the possibility that mutations or genomic rearrangements in these genes seen in ASD could be attributed to altered synapse formation and function.
Contactin-associated protein like 2
Contactin-associated protein like 2 (CNTNAP2) is a member of Neurexin superfamily and is a locus that is significantly associated with susceptibility for ASD (Alarcon et al., 2008; Arking et al., 2008). CNTNAP2 encodes CASPR2, a multidomain transmembrane protein that is best known for clustering potassium channels at the juxtaparanodes in myelinated axons (Poliak et al., 2003). CNTNAP2 localizes at high levels in human fetal brain prior to myelination (Abrahams et al., 2007). It also shows a distribution gradient as frontal cortical enrichment in the developing human brain, indicative of a role in patterning circuits that underlie higher cognition and language. Thus, CNTNAP2 might play a role in the developing brain regions that are likely to be affected in ASD. A recessive frameshift mutation in CNTNAP2 was identified in individuals with cortical dysplasia focal epilepsy syndrome, a congenital disorder, where majority of individuals displayed characteristic features of ASD (Strauss et al., 2006). In addition, other studies that attribute CNTNAP2 to ASD include rare single base pair mutations and common variations in the CNTNAP2 locus identified in patients with ASD (Alarcon et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Falivelli et al., 2012). Recent phenotypic characterization of Cntnap2 mutant mice revealed deficits in the three core ASD behavioral domains with hyperactivity and epileptic seizures (Penagarikano et al., 2011). These mutant mice also showed neuronal migration abnormalities, a significant reduction in the number of interneurons, and abnormal neuronal network activity before the onset of seizures. Most importantly, treatment with the FDA-approved drug risperidone led to amelioration of the repetitive behaviors in the mutant mice further demonstrating a functional role for CNTNAP2 in neuronal development and opening of new avenues for therapeutic intervention in ASD (Penagarikano et al., 2011).
NrCAM
NrCAM is a CAM that has gene homology to NgCAM and is capable of homophilic cell adhesion as well as heterophilic interactions with other non-NrCAM molecules such as Contactin-1, Contactin-2/TAG1, and Neurofascin (Suter et al., 1995; Volkmer et al., 1996; Pavlou et al., 2002). More recently, association analysis has linked NrCAM to ASD (Sakurai et al., 2006). This study showed over transmission of particular haplotypes of NrCAM that modulate NrCAM expression in the brain, are associated with a specific subset of autism with a severe obsessive–compulsive behavior. Several single nucleotide polymorphisms (SNPs) in the NrCAM gene were also found to be associated with autism (Marui et al., 2009) further underscoring NrCAM as a strong candidate gene in ASD.
Cadherins
Cadherins (CDH) and protocadherins (PCDH) include a large family of CAMs, a number of which are required for synaptic formation and function (Weiner et al., 2005; Arikkath and Reichardt, 2008). CDHs mostly undergo homophilic cell adhesion and are involved in intracellular signaling pathways associated with neuropsychiatric disorders. Many of the CDHs have specific spatio-temporal expression patterns in the brain and loss of CDHs leads to altered functional connectivity and neuronal information processing in human brain (Redies et al., 2012). Recent studies have identified de novo translocation deleting CDH18 in ASD (Marshall et al., 2008). This study also reported CNVs associated with PCDH9 gene in ASD. Homozygous deletions in PCDH10 have also been shown in autistic children (Morrow et al., 2008). Other CDHs and PCDHs are disrupted in disorders related to mental retardation and intellectual disabilities (Weiner and Jontes, 2013).
ION Channels
Ion channels are essential for regulating axonal conduction of electrical activity and maintaining the optimum level of excitability within the nervous system. Recent studies linked neuronal excitation alterations with ASD pointing to a potential role for ion channels in the etiology of ASD. Mutations in calcium, sodium, and potassium ion channels seem to enhance neuronal excitability. ASD-linked ion channel mutations involve the SCN1A (Nav1.1), CACNA1C (Cav1.2), KCNMA1 (BK Ca2+), and KCNJ10 (Kir4.1) channels (Ji et al., 2009; Liao and Soong, 2010; Li et al., 2011; Sicca et al., 2011).
Nav1.1
SCN1A encodes the alpha subunit of the sodium channel type 1 (Nav1.1) which belongs to the voltage-gated sodium channel family necessary for axonal conduction and action potential propagation. These transmembrane proteins possess a large pore-forming alpha subunit and two auxiliary beta subunits. This organization is important for allowing sodium ions to move through the axonal membrane to initiate and propagate action potentials. Recently, SCN1A has emerged as the most important gene in epilepsy (Mulley et al., 2005). More that 70% of individuals with epileptic encephalopathy posses a mutation in the region encoding SCN1A causing severe myoclonic epilepsy in infancy, also known as Dravet syndrome (DS; Harkin et al., 2007). This disorder is often accompanied by certain behavioral abnormalities such as hyperactivity, sleep-disorder, anxiety, attention deficit, impaired social interactions, restricted interests, and severe cognitive defects (Weiss et al., 2003; Ramoz et al., 2008; O’Roak et al., 2012). Such behaviors are very similar to those observed in patients with ASD and emerging evidence has linked SCNA1 and ASD (Li et al., 2011). Researchers found that mice with a loss of function mutation for SCN1A phenocopy DS and show autistic-like behaviors (Han et al., 2012). It was suggested that the autism-related traits in DS mice might be caused by a decrease in inhibitory neurotransmission in GABAergic interneurons due to SCN1A haploinsufficiency providing further evidence that impaired GABAergic signaling may underlie ASD (Chao et al., 2010).
Cav1.2
The calcium channels, voltage-dependent, L type, alpha 1C subunit, also known as Cav1.2 encoded by the gene CACNA1C has been implicated in ASD. Cav1.2 channels are important in the activation of transcription factors and play a key role in promoting neuronal survival and dendritic arborization (Krey and Dolmetsch, 2007). A mutation in the G406R region of the CACNA1C gene is known to cause TS, a rare genetic disorder that results in malformations of multi-organ systems, neurological and developmental defects, and autism (Liao and Soong, 2010). The mutation in this region causes prolonged inward current and has dramatic effects on calcium channel inactivation (Splawski et al., 2004). The cellular and molecular consequences of this mutation are not yet known. Future studies should address the physiological relationship between calcium channel inactivation and ASD.
Kir4.1 and BKCa
Recent studies have linked potassium channel proteins Kir4.1 and BKCa to ASD. Mutational screens identified several missense mutations in the KCNJ10 region encoding the potassium ion channel Kir4.1. This ATP sensitive inward rectifier type potassium channel is characterized by having a greater tendency to allow potassium ions to flow into the cell and is suggested to be responsible for the buffering action of glial cells in the brain. Individuals who possess a mutation in the region show symptoms consistent with the DSM-IV-TR criteria for ASD along with seizure and intellectual disability (Sicca et al., 2011). BKCa is a potassium channel known for its large conductance of potassium ions across cell membranes. The gene KCNMA1 encodes BKCa. which is thought to function as a synaptic regulator of neuronal excitability which seems to be disrupted in patients with ASD (Laumonnier et al., 2006). Disruption of this gene caused a decrease in BKCa channel activity and haploinsufficiency in Autism patients further implicating excessive ion channel activity to ASD (Ji et al., 2009).
Scaffolding Proteins
Scaffolding proteins are essential molecules of the synaptic architecture. They are enriched in post-synaptic densities (PSDs) and function in synapse biogenesis by trafficking and anchoring synaptic proteins and clustering of membrane-associated proteins. Most importantly, the scaffolding proteins serve to link post-synaptic receptors with their downstream signaling components and regulate cytoskeletal dynamics (Verpelli et al., 2012).
Shank
Shank protein family is one such synaptic scaffolding family of proteins that includes Shank1, Shank2, and Shank3. They have multiple protein–protein interaction domains and are also known as proline-rich synapse-associated proteins (ProSAPs). Shank proteins are enriched in PSDs and stabilize the PSD-95/SAPAP/Shank/Homer complex (Tu et al., 1999; Sala et al., 2001). Additionally, Shank interacts with NMDA receptors/PSD-95/GKAP complex and actin regulatory protein, Cortactin (Naisbitt et al., 1999; refer Figure 1). Strong genetic and molecular evidence has linked SHANK2 and SHANK3 to the development of ASD phenotypes.
SHANK2
Mutations in ProSAP1/Shank2 gene result in an upregulation of glutamate receptors in certain brain regions, an increase in Shank3 at the synapse, and a decrease in dendritic spine morphology and synaptic transmission (Schmeisser et al., 2012). ProSAP/Shank2 mutants also display behavioral phenotypes that are consistent with those seen in ASD. Shank2 mutant mice are hyperactive, exhibit repetitive grooming, and have impairments in social and vocal behaviors (Schmeisser et al., 2012). Such phenotypic manifestations are linked to the reduction of NMDAR function that results from the absence of the Shank2 protein (Won et al., 2012). Human studies using microarray analyses have identified several variants in SHANK2 that are associated with ASD and mental retardation (Berkel et al., 2010).
SHANK3
Shank3 is an important member of Shank family of proteins and interacts with NLGN (Gerrow et al., 2006) to play a key role in spine morphogenesis and synaptic plasticity (Sala et al., 2001). Recent studies on Shank3 using knockout mice suggest its involvement in the regulation of glutamatergic synapse size, shape, and structure (Jiang and Ehlers, 2013). In Shank3 knockout mice, synaptic ultrastructure is compromised. Overall, shank3 loss leads to a reduction in spine volume, decreased PSD thickness, and loss of dendritic spines (Bozdagi et al., 2010; Peca et al., 2011a; Wang et al., 2011; Jiang and Ehlers, 2013). Furthermore, Shank3 knockout mice show abnormal social behaviors, communication patterns, repetitive behaviors, and impairments in learning and memory (Bozdagi et al., 2010; Peca et al., 2011a; Wang et al., 2011).
There is growing evidence of the involvement of Shank3 in ASD. Molecular characterization of individuals with 22q13.3 deletion syndrome that display autism behavior identified a deletion disrupting Shank3 among other genes (Wilson et al., 2003). Haploinsufficiency of Shank3 has been confirmed to account for 22q13 deletion phenotype of developmental and speech delays (Durand et al., 2007). Other studies that attributed a role of Shank3 in ASD include identification of de novo splice site mutation in ASD (Gauthier et al., 2009). More recently, Shank3 mutations identified in patients with ASD show a modification in dendritic spine induction and morphology as well as actin accumulation in spines affecting growth cone motility (Durand et al., 2007). Furthermore, a microdeletion in Shank1 locus has been discovered using microarray analysis in individuals with ASD (Sato et al., 2012). Recent studies have further uncovered the functional role of Shank3 as Shank3 duplication in mice leads to hyperactivity and spontaneous seizures much like human subjects who have small duplications in the SHANK3 locus. These recent studies further underscore the function of Shank3 in neuronal function and possibly in the maintenance of a balance between the excitatory and inhibitory (E/I) synaptic mechanisms (Han et al., 2013).
SynGAP1
SynGAP1 encodes the RAS GTPase-activating protein (GAPs) which is a critical component of the PSD. At the PSD, SynGAP1 regulates synapse development and maintenance of proper synaptic function. It is known to interact with PSD-95 and colocalizes with excitatory NMDA receptor complexes (Chen et al., 1998). SynGAP is shown to play a critical role in the PSD during early postnatal development as SYNGAP1 knockout mice for die during early development (Kim et al., 2003). Furthermore, mice heterozygous for SynGAP1 show impairments in learning and memory consistent with its involvement in NMDA receptor complexes (Komiyama et al., 2002). In humans, sequencing of the SYNGAP1 locus revealed mutations linked to non-syndromic mental retardation evidencing the importance of SynGAP in synaptic plasticity and learning (Hamdan et al., 2009). SynGap1 was recently implicated ASD because many of its key interacting partners including PSD-95/DLG4, SAP-102/DLG3, PSD-93/DLG2, Neurexins, and Neuroligins have previously been associated with ASD (van de Lagemaat and Grant, 2010). Recent evidence suggests that SynGAP may play a crucial role in controlling the E/I balance in cortical neurons through the regulation on ERK signaling pathways (Wang et al., 2013). This is interesting because the E/I balance seems to be altered in individuals with ASD (Eichler and Meier, 2008; Won et al., 2013). Further characterization of SynGAP as a regulator of synaptic function will provide additional insight into its involvement in ASD.
Cytoskeletal Proteins
A set of cytoskeletal proteins is also mutated in individuals with ASD. These include factors regulating dynamics of actin cytoskeleton, such as GAPs and guanosine exchange factors (GEFs; Newey et al., 2005). Rare non-synonymous variants in cAMP-GEFII are among candidate genes for autism in chromosome 2q (Bacchelli et al., 2003). Mutations in tumor suppressor genes TSC1 and TSC2 are also linked to ASD as the mutant proteins are thought to perturb cytoskeletal dynamics and dendritic spine structure in mutant animals (Folstein and Rosen-Sheidley, 2001). More recently, microtubule associated protein, KATNAL2, has emerged as a credible risk factor for ASD (Neale et al., 2012).
Apart from CAMs, scaffolding and cytoskeletal proteins, a host of other receptors, transporters and channel proteins are known to contribute toward the etiology of ASD (summarized in Table 4). Discovery of more and more genes and genetic pathways are expanding the genetic landscape of ASD. It is interesting to note that these genes include chromatin modifiers, DNA binding proteins, ion channels, and neurotransmitter receptors (State and Sestan, 2012). Recently, CNVs in new candidate genes within GABAergic signaling and neural development pathways associated with ASD were identified using genome wide SNP array (Griswold et al., 2012). These genes include an allosteric binder of GABA receptor, DB1, the GABA receptor-associated protein, GABARAPL1 and a post-synaptic GABA transporter, SLC6A11. Other genes contributing to the genetics of ASD include glutamate receptors, such as GRID1, GRIK2, and GRIK4, synaptic regulators, such as SLC6A8 and SYN3, and transcription factors, such as Engrailed 2 (EN2; Griswold et al., 2012).
Signaling Pathways
Signaling pathways are a complex system of communication within cells that function to organize cellular activities. Signaling pathways and cascades have long been implicated in many disease models. Understanding signaling pathways may play a central role in developing pharmacological or other agents to better treat disease. Currently, disruptions in signaling pathways are being linked to the development of ASD phenotypes. One such pathway connected to ASD is the mammalian target of rapamycin (mTOR) pathway.
mTOR Pathway
Mammalian target of rapamycin is a serine/threonine protein kinase involved in the regulation of cell proliferation, cell growth, cell survival, protein synthesis, and transcription. The mTOR signaling cascade plays a very important role in synapse protein synthesis and several studies have linked this pathway to ASD (Hay and Sonenberg, 2004; Hoeffer and Klann, 2010). Many of the signaling components of the mTOR cascade are located at the synapses where they have been shown to regulate dendritic spine morphology and synaptogenesis (Kumar et al., 2005). Mutations in the proteins known to inhibit mTOR signaling including NF1, PTEN, TSC1, and TSC2 are all linked to neurological disease and autistic-like behavioral phenotypes (Williams and Hersh, 1998; Butler et al., 2005; Won et al., 2013). Furthermore, mutations in the downstream targets of the mTOR signaling cascade have been identified in patients with ASD. The mTOR signaling cascade works by the phosphorylation of 4E-BP by mTOR. This causes 4E-BP to dissociate from the eIF4E initiation factor resulting in cap-dependent translation and elongation of mRNA (Laplante and Sabatini, 2009; Wang and Doering, 2013). Genomic sequence analyses of the eIF4E promoter region identified a SNP in autism patients that enhanced the promoter activity of eIF4E (Neves-Pereira et al., 2009). Additionally, 4E-BP knockout mice as well as mice with an overexpression of eIF4E show autistic like behaviors, enhanced translational of Neuroligins, and disruptions in the E/I balance (Gkogkas et al., 2013; Won et al., 2013). Analyses of monogenetic sources of ASD found that approximately 8–10% of all ASD are involved in regulation of the mTOR pathway (Moldin et al., 2006; Kelleher and Bear, 2008; Hoeffer and Klann, 2010). Of those, 1–2% of ASD cases result due to a mutation in the gene encoding PTEN, an upstream member of the mTOR pathway (Hoeffer and Klann, 2010). The other upstream members, TSC1 and TSC2, form a heterodimer complex. Mutations in the genes encoding this complex cause TSC which is defined clinically by the appearance and growth of benign hamartomas throughout the body and brain (Smalley, 1998). TSC patients suffer from mental retardation and epilepsy. Recent studies show that 25–50% of patients with TSC show behaviors that are consistent with ASD behavioral phenotypes (Hoeffer and Klann, 2010). Pharmacological manipulations to identify therapeutic targets that may be enhancers and suppressors of mTOR signaling cascades and mRNA translation are currently being explored to combat some of the phenotypic manifestations associated with ASD (Carson et al., 2012). Further studies into the downstream and upstream targets of the mTOR signaling cascade will provide additional insights into the functional relationship between the mTOR pathway and ASD.
Concluding Remarks
Current efforts to identify the constellation of genes that confer the characteristic phenotypic manifestations within the autism spectrum have improved our understanding of this complex disorder. While modeling mutations in experimental animal model systems will highlight the underlying disruptions in conserved signaling pathways, the daunting task will still be to establish ASD-specific phenotypes at the molecular, cellular and neural circuit levels. The staggering number of genes already discovered in ASD holds the promise to translate the knowledge into designing new therapeutic interventions. The very interesting and equally challenging observation from the recent genetic studies has been a high degree of overlap of risk factors for various neurodevelopmental disorders, such as ASD, epilepsy, and schizophrenia. This pattern of overlap provides the feasibility to address which genes and genetic pathways intersect and specify the spatio-temporal sequence of events that occur within the developing human brain. The recent advent of comprehensive maps of spatio-temporal gene expression in the human brain (Kang et al., 2011) will greatly help toward providing a powerful developmentally informed approach to studying disorders such as ASD.
Although concerted efforts from studies of human clinical ASD populations and various ASD-related animal models have provided a better understanding of the genetic, molecular, and circuit level aberrations in ASD, several intriguing, yet significant questions still remain. For instance, how can the compound effects of genetics, epigenetics, and environment be consolidated in understanding ASD pathogenesis? What other events play a role in determining the appearance and trajectory of ASD symptoms? How do the majority of the genetic susceptibility loci in ASD affect synapse assembly, maintenance and functional modulation? Finally, how would the future treatments and interventions be designed and organized to accommodate the ever-changing genetic landscape of ASD?
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in our laboratory has been generously supported by the grants from the Simons Foundation Autism Research Initiative (SFARI-177037), the National Institutes of Health (GM063074, NS050356), and the University of Texas Health Science Center, San Antonio, TX, USA. We sincerely regret that the work of many authors related to the topic could not be cited here due to space limitations.
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Abrahams, B. S., Tentler, D., Perederiy, J. V., Oldham, M. C., Coppola, G., and Geschwind, D. H. (2007). Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc. Natl. Acad. Sci. U.S.A. 104, 17849–17854. doi: 10.1073/pnas.0706128104
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Amaral, D. G., Bauman, M. D., and Schumann, C. M. (2003). The amygdala and autism: implications from non-human primate studies. Genes Brain Behav. 2, 295–302. doi: 10.1034/j.1601-183X.2003.00043.x
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Arguello, P. A., and Gogos, J. A. (2006). Modeling madness in mice: one piece at a time. Neuron 52, 179–196. doi: 10.1016/j.neuron.2006.09.023
Arikkath, J., and Reichardt, L. F. (2008). Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 31, 487–494. doi: 10.1016/j.tins.2008.07.001
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Arstikaitis, P., Gauthier-Campbell, C., Huang, K., El-Husseini, A., and Murphy, T. H. (2011). Proteins that promote filopodia stability, but not number, lead to more axonal-dendritic contacts. PLoS ONE 6:e16998. doi: 10.1371/journal.pone.0016998
Ashwood, P., Krakowiak, P., Hertz-Picciotto, I., Hansen, R., Pessah, I., and Van De Water, J. (2011). Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 25, 40–45. doi: 10.1016/j.bbi.2010.08.003
Bacchelli, E., Blasi, F., Biondolillo, M., Lamb, J. A., Bonora, E., Barnby, G., et al. (2003). Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol. Psychiatry 8, 916–924. doi: 10.1038/sj.mp.4001340
Bader, P. L., Faizi, M., Kim, L. H., Owen, S. F., Tadross, M. R., Alfa, R. W., et al. (2011). Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc. Natl. Acad. Sci. U.S.A. 108, 15432–15437. doi: 10.1073/pnas.1112667108
Bailey, A., Le Couteur, A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 25, 63–77. doi: 10.1017/S0033291700028099
Baker, P., Piven, J., and Sato, Y. (1998). Autism and tuberous sclerosis complex: prevalence and clinical features. J. Autism Dev. Disord. 28, 279–285. doi: 10.1023/A:1026004501631
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772. doi: 10.1016/j.cell.2011.03.052
Banovic, D., Khorramshahi, O., Owald, D., Wichmann, C., Riedt, T., Fouquet, W., et al. (2010). Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron 66, 724–738. doi: 10.1016/j.neuron.2010.05.020
Barnby, G., Abbott, A., Sykes, N., Morris, A., Weeks, D. E., Mott, R., et al. (2005). Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966. doi: 10.1086/430454
Baudouin, S. J., Gaudias, J., Gerharz, S., Hatstatt, L., Zhou, K., Punnakkal, P., et al. (2012). Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132. doi: 10.1126/science.1224159
Beaudet, A. L. (2007). Autism: highly heritable but not inherited. Nat. Med. 13, 534–536. doi: 10.1038/nm0507-534
Beckmann, J. S., Estivill, X., and Antonarakis, S. E. (2007). Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat. Rev. Genet. 8, 639–646. doi: 10.1038/nrg2149
Berglund, E. O., Murai, K. K., Fredette, B., Sekerkova, G., Marturano, B., Weber, L., et al. (1999). Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 24, 739–750. doi: 10.1016/S0896-6273(00)81126-5
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Bhat, M. A., Rios, J. C., Lu, Y., Garcia-Fresco, G. P., Ching, W., St Martin, M., et al. (2001). Axon–glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30, 369–383. doi: 10.1016/S0896-6273(01)00294-X
Blanpied, T. A., and Ehlers, M. D. (2004). Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol. Psychiatry 55, 1121–1127. doi: 10.1016/j.biopsych.2003.10.006
Boucard, A. A., Chubykin, A. A., Comoletti, D., Taylor, P., and Sudhof, T. C. (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236. doi: 10.1016/j.neuron.2005.08.026
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1, 15. doi: 10.1186/2040-2392-1-15
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5:e1000536. doi: 10.1371/journal.pgen.1000536
Butler, M. G., Dasouki, M. J., Zhou, X. P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Calahorro, F., and Ruiz-Rubio, M. (2011). Caenorhabditis elegans as an experimental tool for the study of complex neurological diseases: Parkinson’s disease, Alzheimer’s disease and autism spectrum disorder. Invert. Neurosci. 11, 73–83. doi: 10.1007/s10158-011-0126-1
Calahorro, F., and Ruiz-Rubio, M. (2012). Functional phenotypic rescue of Caenorhabditis elegans neuroligin-deficient mutants by the human and rat NLGN1 genes. PLoS ONE 7:e39277. doi: 10.1371/journal.pone.0039277
Campbell, R., Heywood, C. A., Cowey, A., Regard, M., and Landis, T. (1990). Sensitivity to eye gaze in prosopagnosic patients and monkeys with superior temporal sulcus ablation. Neuropsychologia 28, 1123–1142. doi: 10.1016/0028-3932(90)90050-X
Carson, R. P., Van Nielen, D. L., Winzenburger, P. A., and Ess, K. C. (2012). Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 45, 369–380. doi: 10.1016/j.nbd.2011.08.024
Chadman, K. K., Gong, S., Scattoni, M. L., Boltuck, S. E., Gandhy, S. U., Heintz, N., et al. (2008). Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 1, 147–158. doi: 10.1002/aur.22
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chen, H. J., Rojas-Soto, M., Oguni, A., and Kennedy, M. B. (1998). A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904. doi: 10.1016/S0896-6273(00)80471-7
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Chen, Y. C., Lin, Y. Q., Banerjee, S., Venken, K., Li, J., Ismat, A., et al. (2012). Drosophila neuroligin 2 is required presynaptically and postsynaptically for proper synaptic differentiation and synaptic transmission. J. Neurosci. 32, 16018–16030. doi: 10.1523/JNEUROSCI.1685-12.2012
Cheslack-Postava, K., Fallin, M. D., Avramopoulos, D., Connors, S. L., Zimmerman, A. W., Eberhart, C. G., et al. (2007). beta2-Adrenergic receptor gene variants and risk for autism in the AGRE cohort. Mol. Psychiatry 12, 283–291. doi: 10.1038/sj.mp.4001940
Chih, B., Afridi, S. K., Clark, L., and Scheiffele, P. (2004). Disorder-associated mutations lead to functional inactivation of neuroligins. Hum. Mol. Genet. 13, 1471–1477. doi: 10.1093/hmg/ddh158ddh158
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. doi: 10.1126/science.1107470
Choi, Y. B., Li, H. L., Kassabov, S. R., Jin, I., Puthanveettil, S. V., Karl, K. A., et al. (2011). Neurexin–neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in aplysia. Neuron 70, 468–481. doi: 10.1016/j.neuron.2011.03.020
Christian, S. L., Brune, C. W., Sudi, J., Kumar, R. A., Liu, S., Karamohamed, S., et al. (2008). Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry 63, 1111–1117. doi: 10.1016/j.biopsych.2008.01.009
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Clayton, D. F., Balakrishnan, C. N., and London, S. E. (2009). Integrating genomes, brain and behavior in the study of songbirds. Curr. Biol. 19, R865–R873. doi: 10.1016/j.cub.2009.07.006
Comoletti, D., De Jaco, A., Jennings, L. L., Flynn, R. E., Gaietta, G., Tsigelny, I., et al. (2004). The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 24, 4889–4893. doi: 10.1523/JNEUROSCI.0468-04.200424/20/4889
Cook, E. H. Jr., Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., Guter, S. J., et al. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am. J. Hum. Genet. 62, 1077–1083. doi: 10.1086/301832
Cook, E. H. Jr., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 60, 928–934.
Corrales, M., and Herbert, M. (2011). “Autism and environmental genomics: synergistic systems approaches to autism complexity,” in Autism Spectrum Disorders, eds D. Amaral, G. Dawson, and D. H. Geschwind (New York: Oxford University Press), 875–892. doi: 10.1093/med/9780195371826.003.0056
Crabbe, J. C., Wahlsten, D., and Dudek, B. C. (1999). Genetics of mouse behavior: interactions with laboratory environment. Science 284, 1670–1672. doi: 10.1126/science.284.5420.1670
Crawley, J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment. Retard. Dev. Disabil. Res. Rev. 10, 248–258. doi: 10.1002/mrdd.20039
Creak, M., and Pampiglione, G. (1969). Clinical and EEG studies on a group of 35 psychotic children. Dev. Med. Child Neurol. 11, 218–227. doi: 10.1111/j.1469-8749.1969.tb01420.x
Croen, L. A., Grether, J. K., Yoshida, C. K., Odouli, R., and Van De Water, J. (2005). Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case–control study. Arch. Pediatr. Adolesc. Med. 159, 151–157. doi: 10.1001/archpedi.159.2.151
Cukier, H. N., Perez, A. M., Collins, A. L., Zhou, Z., Zoghbi, H. Y., and Botas, J. (2008). Genetic modifiers of MeCP2 function in Drosophila. PLoS Genet. 4:e1000179. doi: 10.1371/journal.pgen.1000179
Currenti, S. A. (2010). Understanding and determining the etiology of autism. Cell. Mol. Neurobiol. 30, 161–171. doi: 10.1007/s10571-009-9453-8
De Jaco, A., Kovarik, Z., Comoletti, D., Jennings, L. L., Gaietta, G., Ellisman, M. H., et al. (2005). A single mutation near the C-terminus in alpha/beta hydrolase fold protein family causes a defect in protein processing. Chem. Biol. Interact. 157–158, 371–372. doi: 10.1016/j.cbi.2005.10.057
de Krom, M., Staal, W. G., Ophoff, R. A., Hendriks, J., Buitelaar, J., Franke, B., et al. (2009). A common variant in DRD3 receptor is associated with autism spectrum disorder. Biol. Psychiatry 65, 625–630. doi: 10.1016/j.biopsych.2008.09.035
Delorme, R., Ey, E., Toro, R., Leboyer, M., Gillberg, C., and Bourgeron, T. (2013). Progress toward treatments for synaptic defects in autism. Nat. Med. 19, 685–694. doi: 10.1038/nm.3193
Dibbens, L. M., Tarpey, P. S., Hynes, K., Bayly, M. A., Scheffer, I. E., Smith, R., et al. (2008). X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 40, 776–781. doi: 10.1038/ng.149
Dooley, K., and Zon, L. I. (2000). Zebrafish: a model system for the study of human disease. Curr. Opin. Genet. Dev. 10, 252–256. doi: 10.1016/S0959-437X(00)00074-5
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Durkin, M. S., Maenner, M. J., Newschaffer, C. J., Lee, L. C., Cunniff, C. M., Daniels, J. L., et al. (2008). Advanced parental age and the risk of autism spectrum disorder. Am. J. Epidemiol. 168, 1268–1276. doi: 10.1093/aje/kwn250
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463. doi: 10.1038/nature02625
Eichler, S. A., and Meier, J. C. (2008). E-I balance and human diseases – from molecules to networking. Front. Mol. Neurosci. 1:2. doi: 10.3389/neuro.02.002.2008
Estivill, X., and Armengol, L. (2007). Copy number variants and common disorders: filling the gaps and exploring complexity in genome-wide association studies. PLoS Genet. 3:1787–1799. doi: 10.1371/journal.pgen.0030190
Falivelli, G., De Jaco, A., Favaloro, F. L., Kim, H., Wilson, J., Dubi, N., et al. (2012). Inherited genetic variants in autism-related CNTNAP2 show perturbed trafficking and ATF6 activation. Hum. Mol. Genet. doi: 10.1093/hmg/dds320
Favre, M. R., Barkat, T. R., Lamendola, D., Khazen, G., Markram, H., and Markram, K. (2013). General developmental health in the VPA-rat model of autism. Front. Behav. Neurosci. 7:88. doi: 10.3389/fnbeh.2013.00088
Feinberg, E. H., Vanhoven, M. K., Bendesky, A., Wang, G., Fetter, R. D., Shen, K., et al. (2008). GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353–363. doi: 10.1016/j.neuron.2007.11.030
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., et al. (2006). High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13. doi: 10.1016/j.neulet.2006.08.017
Fernandez, T., Morgan, T., Davis, N., Klin, A., Morris, A., Farhi, A., et al. (2004). Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 74, 1286–1293. doi: 10.1086/421474AJHG40899
Feyder, M., Karlsson, R. M., Mathur, P., Lyman, M., Bock, R., Momenan, R., et al. (2010). Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 167, 1508–1517. doi: 10.1176/appi.ajp.2010.10040484
Folstein, S., and Rutter, M. (1977). Genetic influences and infantile autism. Nature 265, 726–728. doi: 10.1038/265726a0
Folstein, S. E., and Rosen-Sheidley, B. (2001). Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2, 943–955. doi: 10.1038/35103559
Gauthier, J., Spiegelman, D., Piton, A., Lafreniere, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 421–424. doi: 10.1002/ajmg.b.30822
Gerrow, K., Romorini, S., Nabi, S. M., Colicos, M. A., Sala, C., and El-Husseini, A. (2006). A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron 49, 547–562. doi: 10.1016/j.neuron.2006.01.015
Gharani, N., Benayed, R., Mancuso, V., Brzustowicz, L. M., and Millonig, J. H. (2004). Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol. Psychiatry 9, 474–484. doi: 10.1038/sj.mp.4001498
Ghazanfar, A. A., and Santos, L. R. (2004). Primate brains in the wild: the sensory bases for social interactions. Nat. Rev. Neurosci. 5, 603–616. doi: 10.1038/nrn1473
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Gogolla, N., Leblanc, J. J., Quast, K. B., Sudhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Griswold, A. J., Ma, D., Cukier, H. N., Nations, L. D., Schmidt, M. A., Chung, R. H., et al. (2012). Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 21, 3513–3523. doi: 10.1093/hmg/dds164
Hamdan, F. F., Gauthier, J., Spiegelman, D., Noreau, A., Yang, Y., Pellerin, S., et al. (2009). Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 360, 599–605. doi: 10.1056/NEJMoa0805392
Han, K., Holder, J. L. Jr., Schaaf, C. P., Lu, H., Chen, H., Kang, H., et al. (2013). SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503, 72–77. doi: 10.1038/nature12630
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Harkin, L. A., Mcmahon, J. M., Iona, X., Dibbens, L., Pelekanos, J. T., Zuberi, S. M., et al. (2007). The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 130, 843–852. doi: 10.1093/brain/awm002
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Herbert, M. R. (2010). Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 23, 103–110. doi: 10.1097/WCO.0b013e328336a01f
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. doi: 10.1016/j.tins.2009.11.003
Horev, G., Ellegood, J., Lerch, J. P., Son, Y. E., Muthuswamy, L., Vogel, H., et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. U.S.A. 108, 17076–17081. doi: 10.1073/pnas.1114042108
Hu, Z., Hom, S., Kudze, T., Tong, X. J., Choi, S., Aramuni, G., et al. (2012). Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science 337, 980–984. doi: 10.1126/science.1224896
Huang, H. S., Allen, J. A., Mabb, A. M., King, I. F., Miriyala, J., Taylor-Blake, B., et al. (2012). Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481, 185–189. doi: 10.1038/nature10726
Hunter, J. W., Mullen, G. P., Mcmanus, J. R., Heatherly, J. M., Duke, A., and Rand, J. B. (2010). Neuroligin-deficient mutants of C. elegans have sensory processing deficits and are hypersensitive to oxidative stress and mercury toxicity. Dis. Model. Mech. 3, 366–376. doi: 10.1242/dmm.003442
Ichtchenko, K., Hata, Y., Nguyen, T., Ullrich, B., Missler, M., Moomaw, C., et al. (1995). Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81, 435–443. doi: 10.1016/0092-8674(95)90396-8
International Molecular Genetic Study of Autism Consortium. (2001). Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum. Mol. Genet. 10, 973–982. doi: 10.1093/hmg/10.9.973
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Jamain, S., Betancur, C., Quach, H., Philippe, A., Fellous, M., Giros, B., et al. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 7, 302–310. doi: 10.1038/sj.mp.4000979
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136ng1136
Jamain, S., Radyushkin, K., Hammerschmidt, K., Granon, S., Boretius, S., Varoqueaux, F., et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. U.S.A. 105, 1710–1715. doi: 10.1073/pnas.0711555105
Jessberger, S., Nakashima, K., Clemenson, G. D. Jr., Mejia, E., Mathews, E., Ure, k., et al. (2007). Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 27, 5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007
Ji, L., Chauhan, A., Brown, W. T., and Chauhan, V. (2009). Increased activities of Na+/K+-ATPase and Ca2+/Mg2+-ATPase in the frontal cortex and cerebellum of autistic individuals. Life Sci. 85, 788–793. doi: 10.1016/j.lfs.2009.10.008
Jiang, H., and Kohler, C. (2012). Evolution, function, and regulation of genomic imprinting in plant seed development. J. Exp. Bot. 63, 4713–4722. doi: 10.1093/jxb/ers145
Jiang, Y. H., Bressler, J., and Beaudet, A. L. (2004). Epigenetics and human disease. Annu. Rev. Genomics Hum. Genet. 5, 479–510. doi: 10.1146/annurev.genom.5.061903.180014
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. doi: 10.1038/nature10523
Kanner, L. (1971). Follow-up study of eleven autistic children originally reported in 1943. J. Autism Child. Schizophr. 1, 119–145. doi: 10.1007/BF01537953
Kelleher, R. J. III, and Bear, M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401–406. doi: 10.1016/j.cell.2008.10.017
Kelleher, R. J. III, Geigenmuller, U., Hovhannisyan, H., Trautman, E., Pinard, R., Rathmell, B., et al. (2012). High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS ONE 7:e35003. doi: 10.1371/journal.pone.0035003
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Kim, J. H., Lee, H. K., Takamiya, K., and Huganir, R. L. (2003). The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J. Neurosci. 23, 1119–1124.
Komiyama, N. H., Watabe, A. M., Carlisle, H. J., Porter, K., Charlesworth, P., Monti, J., et al. (2002). SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J. Neurosci. 22, 9721–9732.
Koochek, M., Harvard, C., Hildebrand, M. J., Van Allen, M., Wingert, H., Mickelson, E., et al. (2006). 15q duplication associated with autism in a multiplex family with a familial cryptic translocation t(14; 15)(q11.2; q13.3) detected using array-CGH. Clin. Genet. 69, 124–134. doi: 10.1111/j.1399-0004.2005.00560.x
Krey, J. F., and Dolmetsch, R. E. (2007). Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr. Opin. Neurobiol. 17, 112–119. doi: 10.1016/j.conb.2007.01.010
Kumar, R. A., Karamohamed, S., Sudi, J., Conrad, D. F., Brune, C., Badner, J. A., et al. (2008). Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638. doi: 10.1093/hmg/ddm376
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras–PI3K–Akt–mTOR and Ras–MAPK signaling pathways. J. Neurosci. 25, 11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005
Lamb, J. A., Barnby, G., Bonora, E., Sykes, N., Bacchelli, E., Blasi, F., et al. (2005). Analysis of IMGSAC autism susceptibility loci: evidence for sex limited and parent of origin specific effects. J. Med. Genet. 42, 132–137. doi: 10.1136/jmg.2004.025668
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594. doi: 10.1242/jcs.051011
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137AJHG40657
Laumonnier, F., Roger, S., Guerin, P., Molinari, F., M’Rad, R., Cahard, D., et al. (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry 163, 1622–1629. doi: 10.1176/appi.ajp.163.9.1622
Levitt, P., and Campbell, D. B. (2009). The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J. Clin. Invest. 119, 747–754. doi: 10.1172/JCI37934
Levy, D., Ronemus, M., Yamrom, B., Lee, Y. H., Leotta, A., Kendall, J., et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. doi: 10.1016/j.neuron.2011.05.015
Li, B. M., Liu, X. R., Yi, Y. H., Deng, Y. H., Su, T., Zou, X., et al. (2011). Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 21, 291–295. doi: 10.1016/j.yebeh.2011.04.060
Li, J., Ashley, J., Budnik, V., and Bhat, M. A. (2007). Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron 55, 741–755. doi: 10.1016/j.neuron.2007.08.002
Liao, P., and Soong, T. W. (2010). CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflugers Arch. 460, 353–359. doi: 10.1007/s00424-009-0753-0
Luedi, P. P., Dietrich, F. S., Weidman, J. R., Bosko, J. M., Jirtle, R. L., and Hartemink, A. J. (2007). Computational and experimental identification of novel human imprinted genes. Genome Res. 17, 1723–1730. doi: 10.1101/gr.6584707
Luo, R., Sanders, S. J., Tian, Y., Voineagu, I., Huang, N., Chu, S. H., et al. (2012). Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet. 91, 38–55. doi: 10.1016/j.ajhg.2012.05.011
Lutz, C., Well, A., and Novak, M. (2003). Stereotypic and self-injurious behavior in rhesus macaques: a survey and retrospective analysis of environment and early experience. Am. J. Primatol. 60, 1–15. doi: 10.1002/ajp.10075
Ma, D., Salyakina, D., Jaworski, J. M., Konidari, I., Whitehead, P. L., Andersen, A. N., et al. (2009a). A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet. 73, 263–273. doi: 10.1111/j.1469-1809.2009.00523.x
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., et al. (2009b). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077. doi: 10.1126/science.1166859
Ma, D. Q., Whitehead, P. L., Menold, M. M., Martin, E. R., Ashley-Koch, A. E., Mei, H., et al. (2005). Identification of significant association and gene–gene interaction of GABA receptor subunit genes in autism. Am. J. Hum. Genet. 77, 377–388. doi: 10.1086/433195
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303. doi: 10.1016/j.tins.2011.04.001
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241. doi: 10.1016/j.cell.2012.02.039
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
Martienssen, R. A., and Colot, V. (2001). DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293, 1070–1074. doi: 10.1126/science.293.5532.1070
Martin, L. A., Ashwood, P., Braunschweig, D., Cabanlit, M., Van De Water, J., and Amaral, D. G. (2008). Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav. Immun. 22, 806–816. doi: 10.1016/j.bbi.2007.12.007
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Marui, T., Funatogawa, I., Koishi, S., Yamamoto, K., Matsumoto, H., Hashimoto, O., et al. (2009). Association of the neuronal cell adhesion molecule (NRCAM) gene variants with autism. Int. J. Neuropsychopharmacol. 12, 1–10. doi: 10.1017/S1461145708009127
McCarroll, S. A., Kuruvilla, F. G., Korn, J. M., Cawley, S., Nemesh, J., Wysoker, A., et al. (2008). Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 40, 1166–1174. doi: 10.1038/ng.238
McGraw, L. A., and Young, L. J. (2010). The prairie vole: an emerging model organism for understanding the social brain. Trends Neurosci. 33, 103–109. doi: 10.1016/j.tins.2009.11.006
Mejias, R., Adamczyk, A., Anggono, V., Niranjan, T., Thomas, G. M., Sharma, K., et al. (2011). Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc. Natl. Acad. Sci. U.S.A. 108, 4920–4925. doi: 10.1073/pnas.1102233108
Merikangas, A. K., Corvin, A. P., and Gallagher, L. (2009). Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet. 25, 536–544. doi: 10.1016/j.tig.2009.10.006
Miller, D. T., Shen, Y., Weiss, L. A., Korn, J., Anselm, I., Bridgemohan, C., et al. (2009). Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J. Med. Genet. 46, 242–248. doi: 10.1136/jmg.2008.059907
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K., et al. (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948. doi: 10.1038/nature01755
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 20, 3642–3652. doi: 10.1093/hmg/ddr284
Miyake, K., Hirasawa, T., Koide, T., and Kubota, T. (2012). Epigenetics in autism and other neurodevelopmental diseases. Adv. Exp. Med. Biol. 724, 91–98. doi: 10.1007/978-1-4614-0653-2_7
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Moldin, S. O., Rubenstein, J. L., and Hyman, S. E. (2006). Can autism speak to neuroscience? J. Neurosci. 26, 6893–6896. doi: 10.1523/JNEUROSCI.1944-06.2006
Morales, J., Hiesinger, P. R., Schroeder, A. J., Kume, K., Verstreken, P., Jackson, F. R., et al. (2002). Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972. doi: 10.1016/S0896-6273(02)00731-6
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Mulley, J. C., Scheffer, I. E., Petrou, S., Dibbens, L. M., Berkovic, S. F., and Harkin, L. A. (2005). SCN1A mutations and epilepsy. Hum. Mutat. 25, 535–542. doi: 10.1002/humu.20178
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/S0896-6273(00)80809-0
Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K., et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. doi: 10.1016/j.cell.2009.04.024
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Neves-Pereira, M., Muller, B., Massie, D., Williams, J. H., O’Brien, P. C., Hughes, A., et al. (2009). Deregulation of EIF4E: a novel mechanism for autism. J. Med. Genet. 46, 759–765. doi: 10.1136/jmg.2009.066852
Newey, S. E., Velamoor, V., Govek, E. E., and Van Aelst, L. (2005). Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 64, 58–74. doi: 10.1002/neu.20153
Nurmi, E. L., Bradford, Y., Chen, Y., Hall, J., Arnone, B., Gardiner, M. B., et al. (2001). Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77, 105–113. doi: 10.1006/geno.2001.6617
Oberman, L. M., Hubbard, E. M., Mccleery, J. P., Altschuler, E. L., Ramachandran, V. S., and Pineda, J. A. (2005). EEG evidence for mirror neuron dysfunction in autism spectrum disorders. Brain Res. Cogn. Brain Res. 24, 190–198. doi: 10.1016/j.cogbrainres.2005.01.014
Ongur, D., and Price, J. L. (2000). The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb. Cortex 10, 206–219. doi: 10.1093/cercor/10.3.206
Onore, C., Careaga, M., and Ashwood, P. (2012). The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 26, 383–392. doi: 10.1016/j.bbi.2011.08.007
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Panaitof, S. C. (2012). A songbird animal model for dissecting the genetic bases of autism spectrum disorder. Dis. Markers 33, 241–249. doi: 10.3233/DMA-2012–2918
Panaitof, S. C., Abrahams, B. S., Dong, H., Geschwind, D. H., and White, S. A. (2010). Language-related Cntnap2 gene is differentially expressed in sexually dimorphic song nuclei essential for vocal learning in songbirds. J. Comp. Neurol. 518, 1995–2018. doi: 10.1002/cne.22318
Pavlou, O., Theodorakis, K., Falk, J., Kutsche, M., Schachner, M., Faivre-Sarrailh, C., et al. (2002). Analysis of interactions of the adhesion molecule TAG-1 and its domains with other immunoglobulin superfamily members. Mol. Cell. Neurosci. 20, 367–381. doi: 10.1006/mcne.2002.1105
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011a). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peca, J., Ting, J., and Feng, G. (2011b). SnapShot: autism and the synapse. Cell 147, 706–706.e1. doi: 10.1016/j.cell.2011.10.015
Penagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Persico, A. M., and Bourgeron, T. (2006). Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 29, 349–358. doi: 10.1016/j.tins.2006.05.010
Persico, A. M., D’Agruma, L., Maiorano, N., Totaro, A., Militerni, R., Bravaccio, C., et al. (2001). Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 6, 150–159. doi: 10.1038/sj.mp.4000850
Pessah, I. N., Seegal, R. F., Lein, P. J., Lasalle, J., Yee, B. K., Van De Water, J., et al. (2008). Immunologic and neurodevelopmental susceptibilities of autism. Neurotoxicology 29, 532–545. doi: 10.1016/j.neuro.2008.02.006
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Poliak, S., Salomon, D., Elhanany, H., Sabanay, H., Kiernan, B., Pevny, L., et al. (2003). Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 162, 1149–1160. doi: 10.1083/jcb.200305018jcb.200305018
Radyushkin, K., Hammerschmidt, K., Boretius, S., Varoqueaux, F., El-Kordi, A., Ronnenberg, A., et al. (2009). Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 8, 416–425. doi: 10.1111/j.1601-183X.2009.00487.x
Ramoz, N., Cai, G., Reichert, J. G., Silverman, J. M., and Buxbaum, J. D. (2008). An analysis of candidate autism loci on chromosome 2q24-q33: evidence for association to the STK39 gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1152–1158. doi: 10.1002/ajmg.b.30739
Ramoz, N., Reichert, J. G., Smith, C. J., Silverman, J. M., Bespalova, I. N., Davis, K. L., et al. (2004). Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am. J. Psychiatry 161, 662–669. doi: 10.1176/appi.ajp.161.4.662
Redies, C., Hertel, N., and Hubner, C. A. (2012). Cadherins and neuropsychiatric disorders. Brain Res. 1470, 130–144. doi: 10.1016/j.brainres.2012.06.020
Reik, W., and Walter, J. (2001). Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2, 21–32. doi: 10.1038/35047554
Rice, D. S., and Curran, T. (2001). Role of the reelin signaling pathway in central nervous system development. Annu. Rev. Neurosci. 24, 1005–1039. doi: 10.1146/annurev.neuro.24.1.1005
Rodier, P. M., Ingram, J. L., Tisdale, B., and Croog, V. J. (1997). Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol. 11, 417–422. doi: 10.1016/S0890-6238(97)80001-U
Rogers, S. J., Wehner, D. E., and Hagerman, R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 22, 409–417. doi: 10.1097/00004703-200112000-00008
Ronesi, J. A., Collins, K. A., Hays, S. A., Tsai, N. P., Guo, W., Birnbaum, S. G., et al. (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 15, 431–440, S431. doi: 10.1038/nn.3033
Roohi, J., Montagna, C., Tegay, D. H., Palmer, L. E., Devincent, C., Pomeroy, J. C., et al. (2009). Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 46, 176–182. doi: 10.1136/jmg.2008.057505
Rujescu, D., Ingason, A., Cichon, S., Pietilainen, O. P., Barnes, M. R., Toulopoulou, T., et al. (2009). Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 18, 988–996. doi: 10.1093/hmg/ddn351
Sakurai, T., Ramoz, N., Reichert, J. G., Corwin, T. E., Kryzak, L., Smith, C. J., et al. (2006). Association analysis of the NrCAM gene in autism and in subsets of families with severe obsessive–compulsive or self-stimulatory behaviors. Psychiatr. Genet. 16, 251–257. doi: 10.1097/01.ypg.0000242196.81891.c9
Sala, C., Piech, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/S0896-6273(01)00339-7
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Schaaf, C. P., Sabo, A., Sakai, Y., Crosby, J., Muzny, D., Hawes, A., et al. (2011). Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 20, 3366–3375. doi: 10.1093/hmg/ddr243
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Schneider, T., and Przewlocki, R. (2005). Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30, 80–89. doi: 10.1038/sj.npp.1300518
Schroer, R. J., Phelan, M. C., Michaelis, R. C., Crawford, E. C., Skinner, S. A., Cuccaro, M., et al. (1998). Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 76, 327–336. doi: 10.1002/(SICI)1096-8628(19980401)76:4<327::AID-AJMG8>3.0.CO;2-M
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi: 10.1126/science.1138659
Sebat, J., Levy, D. L., and Mccarthy, S. E. (2009). Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 25, 528–535. doi: 10.1016/j.tig.2009.10.004
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Shelton, J. F., Hertz-Picciotto, I., and Pessah, I. N. (2012). Tipping the balance of autism risk: potential mechanisms linking pesticides and autism. Environ. Health Perspect. 120, 944–951. doi: 10.1289/ehp.1104553
Sicca, F., Imbrici, P., D’Adamo, M. C., Moro, F., Bonatti, F., Brovedani, P., et al. (2011). Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol. Dis. 43, 239–247. doi: 10.1016/j.nbd.2011.03.016
Silva, S. C., Correia, C., Fesel, C., Barreto, M., Coutinho, A. M., Marques, C., et al. (2004). Autoantibody repertoires to brain tissue in autism nuclear families. J. Neuroimmunol. 152, 176–182. doi: 10.1016/j.jneuroim.2004.03.015
Smalley, S. L. (1998). Autism and tuberous sclerosis. J. Autism Dev. Disord. 28, 407–414. doi: 10.1023/A:1026052421693
Song, J. Y., Ichtchenko, K., Sudhof, T. C., and Brose, N. (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. U.S.A. 96, 1100–1105. doi: 10.1073/pnas.96.3.1100
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
State, M. W., and Sestan, N. (2012). Neuroscience. The emerging biology of autism spectrum disorders. Science 337, 1301–1303. doi: 10.1126/science.1224989
Steffenburg, S., Gillberg, C., Hellgren, L., Andersson, L., Gillberg, I. C., Jakobsson, G., et al. (1989). A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry 30, 405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 354, 1370–1377. doi: 10.1056/NEJMoa052773
Sudhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Sutcliffe, J. S., Delahanty, R. J., Prasad, H. C., Mccauley, J. L., Han, Q., Jiang, L., et al. (2005). Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am. J. Hum. Genet. 77, 265–279. doi: 10.1086/432648
Suter, D. M., Pollerberg, G. E., Buchstaller, A., Giger, R. J., Dreyer, W. J., and Sonderegger, P. (1995). Binding between the neural cell adhesion molecules axonin-1 and Nr-CAM/Bravo is involved in neuron-glia interaction. J. Cell Biol. 131, 1067–1081. doi: 10.1083/jcb.131.4.1067
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Taniguchi, H., Gollan, L., Scholl, F. G., Mahadomrongkul, V., Dobler, E., Limthong, N., et al. (2007). Silencing of neuroligin function by postsynaptic neurexins. J. Neurosci. 27, 2815–2824. doi: 10.1523/JNEUROSCI.0032-07.2007
Tierney, E., Nwokoro, N. A., Porter, F. D., Freund, L. S., Ghuman, J. K., and Kelley, R. I. (2001). Behavior phenotype in the RSH/Smith–Lemli–Opitz syndrome. Am. J. Med. Genet. 98, 191–200. doi: 10.1002/1096-8628(20010115)98:2<191::AID-AJMG1030>3.0.CO;2-M
Tropepe, V., and Sive, H. L. (2003). Can zebrafish be used as a model to study the neurodevelopmental causes of autism? Genes Brain Behav. 2, 268–281. doi: 10.1034/j.1601-183X.2003.00038.x
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/S0896-6273(00)80810-7
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi: 10.1016/j.ajhg.2011.11.025
van de Lagemaat, L. N., and Grant, S. G. (2010). Genome variation and complexity in the autism spectrum. Neuron 67, 8–10. doi: 10.1016/j.neuron.2010.06.026
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., et al. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. doi: 10.1016/j.neuron.2006.09.003
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456. doi: 10.1078/0171-9335-00410
Verpelli, C., Schmeisser, M. J., Sala, C., and Boeckers, T. M. (2012). Scaffold proteins at the postsynaptic density. Adv. Exp. Med. Biol. 970, 29–61. doi: 10.1007/978-3-7091-0932-8_2
Vervoort, V. S., Beachem, M. A., Edwards, P. S., Ladd, S., Miller, K. E., De Mollerat, X., et al. (2002). AGTR2 mutations in X-linked mental retardation. Science 296, 2401–2403. doi: 10.1126/science.1072191
Volkmer, H., Leuschner, R., Zacharias, U., and Rathjen, F. G. (1996). Neurofascin induces neurites by heterophilic interactions with axonal NrCAM while NrCAM requires F11 on the axonal surface to extend neurites. J. Cell Biol. 135, 1059–1069. doi: 10.1083/jcb.135.4.1059
Wang, C. C., Held, R. G., and Hall, B. J. (2013). SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS ONE 8:e83941. doi: 10.1371/journal.pone.0083941
Wang, H., and Doering, L. C. (2013). Reversing autism by targeting downstream mTOR signaling. Front. Cell. Neurosci. 7:28. doi: 10.3389/fncel.2013.00028
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Wang, X., Mccoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Warren, W. C., Clayton, D. F., Ellegren, H., Arnold, A. P., Hillier, L. W., Kunstner, A., et al. (2010). The genome of a songbird. Nature 464, 757–762. doi: 10.1038/nature08819
Watson, K. K., and Platt, M. L. (2012). Of mice and monkeys: using non-human primate models to bridge mouse- and human-based investigations of autism spectrum disorders. J. Neurodev. Disord. 4, 21. doi: 10.1186/1866-1955-4-21
Weiner, J. A., and Jontes, J. D. (2013). Protocadherins, not prototypical: a complex tale of their interactions, expression, and functions. Front. Mol. Neurosci. 6:4. doi: 10.3389/fnmol.2013.00004
Weiner, J. A., Wang, X., Tapia, J. C., and Sanes, J. R. (2005). Gamma protocadherins are required for synaptic development in the spinal cord. Proc. Natl. Acad. Sci. U.S.A. 102, 8–14. doi: 10.1073/pnas.0407931101
Weiss, L. A., Escayg, A., Kearney, J. A., Trudeau, M., Macdonald, B. T., Mori, M., et al. (2003). Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 8, 186–194. doi: 10.1038/sj.mp.40012414001241
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Williams, J. H., Whiten, A., Suddendorf, T., and Perrett, D. I. (2001). Imitation, mirror neurons and autism. Neurosci. Biobehav. Rev. 25, 287–295. doi: 10.1016/S0149-7634(01)00014-8
Williams, P. G., and Hersh, J. H. (1998). Brief report: the association of neurofibromatosis type 1 and autism. J. Autism Dev. Disord. 28, 567–571. doi: 10.1023/A:1026012414193
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Won, H., Mah, W., and Kim, E. (2013). Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front. Mol. Neurosci. 6:19. doi: 10.3389/fnmol.2013.00019
Wu, S., Jia, M., Ruan, Y., Liu, J., Guo, Y., Shuang, M., et al. (2005). Positive association of the oxytocin receptor gene (OXTR) with autism in the Chinese Han population. Biol. Psychiatry 58, 74–77. doi: 10.1016/j.biopsych.2005.03.013
Wu, Y., Bolduc, F. V., Bell, K., Tully, T., Fang, Y., Sehgal, A., et al. (2008). A Drosophila model for Angelman syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 12399–12404. doi: 10.1073/pnas.0805291105
Ye, X., and Carew, T. J. (2011). Transsynaptic coordination of presynaptic and postsynaptic modifications underlying enduring synaptic plasticity. Neuron 70, 379–381. doi: 10.1016/j.neuron.2011.04.016
Yirmiya, N., Rosenberg, C., Levi, S., Salomon, S., Shulman, C., Nemanov, L., et al. (2006). Association between the arginine vasopressin 1a receptor (AVPR1a) gene and autism in a family-based study: mediation by socialization skills. Mol. Psychiatry 11, 488–494. doi: 10.1038/sj.mp.4001812
Zahir, F. R., Baross, A., Delaney, A. D., Eydoux, P., Fernandes, N. D., Pugh, T., et al. (2008). A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1alpha. J. Med. Genet. 45, 239–243. doi: 10.1136/jmg.2007.054437
Zeng, X., Sun, M., Liu, L., Chen, F., Wei, L., and Xie, W. (2007). Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 581, 2509–2516. doi: 10.1016/j.febslet.2007.04.068
Zhou, J., Blundell, J., Ogawa, S., Kwon, C. H., Zhang, W., Sinton, C., et al. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009
Zoghbi, H. Y. (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302, 826–830. doi: 10.1126/science.1089071302/5646/826
Keywords: autism spectrum disorder, synapse, animal models, genetics, epigenetics, environment, cell adhesion molecules, scaffolding proteins
Citation: Banerjee S, Riordan M and Bhat MA (2014) Genetic aspects of autism spectrum disorders: insights from animal models. Front. Cell. Neurosci. 8:58. doi: 10.3389/fncel.2014.00058
Received: 02 January 2014; Accepted: 07 February 2014;
Published online: 24 February 2014.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Eunjoon Kim, Korea Advanced Institute of Science and Technology, South KoreaJohn Jay Gargus, University of California Irvine, USA
Copyright © 2014 Banerjee, Riordan and Bhat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swati Banerjee and Manzoor A. Bhat, Department of Physiology, Center for Biomedical Neuroscience, School of Medicine, University of Texas Health Science Center, 7703 Floyd Curl Drive, San Antonio, TX 78229-3900, USA e-mail: banerjees@uthscsa.edu; bhatm@uthscsa.edu