Disparate roles of zinc in chemical hypoxia-induced neuronal death
 Sujeong Kim
Sujeong Kim Jung-Woo Seo
Jung-Woo Seo Shin Bi Oh1
Shin Bi Oh1  Joo-Yong Lee
Joo-Yong Lee- 1Asan Institute for Life Sciences, Asan Medical Center, Seoul, South Korea
- 2Department of Neurology, University of Ulsan College of Medicine, Seoul, South Korea
Accumulating evidence has provided a causative role of zinc (Zn2+) in neuronal death following ischemic brain injury. Using a hypoxia model of primary cultured cortical neurons with hypoxia-inducing chemicals, cobalt chloride (1 mM CoCl2), deferoxamine (3 mM DFX), and sodium azide (2 mM NaN3), we evaluated whether Zn2+ is involved in hypoxic neuronal death. The hypoxic chemicals rapidly elicited intracellular Zn2+ release/accumulation in viable neurons. The immediate addition of the Zn2+ chelator, CaEDTA or N,N,N’N’-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN), prevented the intracellular Zn2+ load and CoCl2-induced neuronal death, but neither 3 hour later Zn2+ chelation nor a non-Zn2+ chelator ZnEDTA (1 mM) demonstrated any effects. However, neither CaEDTA nor TPEN rescued neurons from cell death following DFX- or NaN3-induced hypoxia, whereas ZnEDTA rendered them resistant to the hypoxic injury. Instead, the immediate supplementation of Zn2+ rescued DFX- and NaN3-induced neuronal death. The iron supplementation also afforded neuroprotection against DFX-induced hypoxic injury. Thus, although intracellular Zn2+ release/accumulation is common during chemical hypoxia, Zn2+ might differently influence the subsequent fate of neurons; it appears to play a neurotoxic or neuroprotective role depending on the hypoxic chemical used. These results also suggest that different hypoxic chemicals may induce neuronal death via distinct mechanisms.
Introduction
Zinc (Zn2+) contributes to neuronal injury according to various experimental models of excitotoxic brain injury (Sensi et al., 2011). Exposing cortical cultures to high levels of Zn2+ induces extensive neuronal and glial cell death (Choi et al., 1988). The intracellular release of Zn2+ subsequent to exposure to oxidative or nitrosative agents leads to neuronal degeneration in cultured neurons (Bossy-Wetzel et al., 2004; Hwang et al., 2008). In animal models of acute brain injury, including cerebral ischemia, epilepsy, and trauma, a large accumulation of Zn2+ occurs in degenerating neurons as demonstrated by the Zn2+-specific fluorescence dyes (Frederickson et al., 1988; Tønder et al., 1990; Koh et al., 1996; Lee et al., 2000; Suh et al., 2000). Intracellular Zn2+ release/accumulation obviously precedes neuronal death in these experimental models (Koh et al., 1996) since Zn2+ chelators, such as ethylenediaminetetraacetic acid (EDTA; Koh et al., 1996; Lee et al., 2000; Frederickson et al., 2002; Suh et al., 2004) and N,N,N′,N′,-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN; Bossy-Wetzel et al., 2004; Cho et al., 2010), intercept intracellular Zn2+ load to suppress neuronal death.
Cerebral hypoxia develops when the brain suffers from oxygen shortage due to the blockage of blood flow, resulting in extensive neuronal death in selective vulnerable areas (Sharp and Bernaudin, 2004). Since the involvement of Zn2+ in neuronal death in the hippocampal CA1 area following transient global cerebral ischemia was reported (Koh et al., 1996), studies have suggested that excessive Zn2+ release/accumulation leads to neuronal injury after hypoxia/ischemia (Sensi et al., 2011). When mouse hippocampal slices are subjected to oxygen and glucose deprivation (OGD)—which is a typical experimental model of hypoxia/ischemia—intracellular Zn2+ becomes prominent in degenerating neurons, whereby the Zn2+ chelator CaEDTA attenuates both Zn2+ accumulation and neuronal death (Yin et al., 2002; Medvedeva et al., 2009). Similarly, hypobaric hypoxia causes Zn2+-mediated inflammation and apoptosis in neurons of the mouse hippocampus, which are also reversed by CaEDTA (Malairaman et al., 2014). Recent studies have provided that Zn2+ promotes hypoxic cell death by upregulating hypoxia-inducible transcription factor-1α (HIF1α) via an activation of NADPH oxidase or poly(ADP-ribose) polymerase (PARP; Pan et al., 2013; Malairaman et al., 2014).
While the precise control of oxygen level is crucial to simulate hypoxic condition in cell culture, it is difficult, so various in vitro models of neuronal hypoxia have been provided containing OGD models. Some divalent cations such as cobalt (Co2+), nickel (Ni2+), and the iron-chelator deferoxamine (DFX), have been applicable to mimic hypoxic conditions in cultured cells because they activate hypoxic signals by stabilizing the expression of HIF1α (Ho and Bunn, 1996). Sodium azide (NaN3) and potassium cyanide (KCN) are also potent inhibitors of cytochrome c oxidase (i.e., complex IV of the mitochondrial respiratory chain) to induce chemical hypoxia (Roemgens et al., 2011). However, although the hypoxic chemicals have helped us to understand the molecular events that underlie the hypoxic neuronal death, it remains unclear whether chemical hypoxia also involves Zn2+-mediated neuronal injury in cultured neurons.
In this study, we found that intracellular Zn2+ release/accumulation occurs in primary neuronal cells shortly after exposure to CoCl2, DFX, or NaN3, whereas the effects of Zn2+ chelation on neuronal fate differ depending on the hypoxia-inducing chemicals used. This study shows the disparate roles of Zn2+ in neuronal death following chemical hypoxia.
Materials and Methods
Primary Cortical Neuron Cultures
We used ICR mice in this study, in accordance with the Guidelines of the Asan Institute for Life Sciences for the Care and Use of Laboratory Animals. Cerebral cortical tissues were dissected from the brains of fetal ICR mice (Koatech, Pyeongtaek, Korea) at embryonic day E14, dissociated in Ca2+/Mg2+-free Hank’s balanced salt solution (HBSS; Invitrogen, Carlsbad, CA, USA) containing 0.25% trypsin-EDTA (Invitrogen), and filtered through 40-µm nylon cell strainer (BD Biosciences, Durham, NC, USA). Cells were washed in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and penicillin/streptomycin (Invitrogen), and resuspended in serum-free Neurobasal medium (Invitrogen) containing the B27 supplement (Invitrogen), L-glutamine (2 mM; Invitrogen) and penicillin/streptomycin. Cells were plated at a density of 5 × 105–106 cells/well on poly-L-lysine-coated well culture dishes and grown in a humidified 5% CO2 incubator at 37°C. Cultures were treated with cytosine arabinoside (Ara-C, 2 µM; Sigma, St. Louis, MO, USA) for 24 h at 3 days in vitro (DIV3) to halt the growth of non-neuronal cells, and maintained in fresh Neurobasal medium with B27 until used in experiments between DIV10–11.
Induction of Chemical Hypoxia
All chemicals used in this study, except CoEDTA (TCI, Tokyo, Japan), were purchased from Sigma-Aldrich or Fluka (St. Louis, MO, USA).
To induce chemical hypoxia, cells were treated with CoCl2 (1 mM) (Fang et al., 2008; Zhang et al., 2011) or DFX (3 mM) (Almli et al., 2001; Guelman et al., 2004) for 2 h, or NaN3 (2 mM) for 1 h (Garnier et al., 2003; Selvatici et al., 2009) in glucose-free MEM, and then the media was freshly replaced. To define the roles of the intracellular metals in neurons during chemical hypoxia, we added the metal chelator EDTA with various salts (CaEDTA, ZnEDTA, CoEDTA, or FeEDTA), or TPEN to the media at 10 min or 3 h after hypoxic chemical treatment.
Cell Viability Assessment
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Stanciu et al., 2000; White et al., 2001). Cortical neurons were grown on poly-L-lysine coated 24-well plates, and treated with MTT (final concentration, 0.5 mg/mL in culture media; Amresco, Solon, OH, USA) at 37°C for 2 h. After culture medium was completely removed, the insoluble formazan crystals were dissolved in dimethyl sulfoxide (DMSO; 200 µL). The reaction products (in 100 µL aliquots) were measured at 570 nm using a microplate reader (Synergy H1 Hybrid; BioTek Instruments, Winooski, VT, USA). All experiments were consisted of at least three independent repeats, and each experiment contained three parallel cultures. Duplicate measurements of MTT absorbance were performed for each sample. Resultantly, percentage of viable cells in drug-treated cultures was determined relative to vehicle-treated control cells.
In addition, neuronal cell death was visually detected by staining the nuclei with Hoechst 33342 or propidium iodide (PI). After the cells were incubated in the presence of Hoechst 33342 (10 µg/mL; Invitrogen) and PI (1 µg/mL; Sigma) for 15 min, the fluorescent phenotypes of the nuclei were examined under a fluorescence inverted microscope (Axio Observer.Z1; Carl Zeiss, Göttingen, Germany) using a DAPI filter (beam splitter, 395 nm; excitation, 365 ± 50 nm; emission, 445 ± 50 nm) and a Set20 filter (beam splitter, 560 nm; excitation, 546 ± 12 nm; emission, 575–640 nm), respectively. PI-fluorescent red nuclei-containing neurons were considered dead or dying as the dye is excluded by viable cells.
Detection and Measurement of Intracellular Zn2+
To assess the levels of intracellular Zn2+, cells were incubated with 2 µM FluoZin-3 AM (Kd for Zn2+, about 15 nM) (Molecular Probes, Eugene, OR, USA) for 30 min and washed with fresh culture medium (Gee et al., 2002). FluoZin-3 reactive cells were examined or photographed under consistent imaging conditions with an inverted fluorescence microscope (Axio Observer.Z1) using a FITC filter (beam splitter, 495 nm; excitation, 450–490 nm; emission, 500–550 nm) equipped with self-adjusting lamps and an AxioCam digital camera (Carl Zeiss).
To quantify the level of intracellular Zn2+, we took the photographs (magnification, 100×) from three spots randomly selected from each culture well, and measured the mean intensity of FluoZin-3-fluorescence in neurons using ImagePro Plus software (Media Cybernetics, Silver Spring, MD, USA). After subtracting the background intensity (which was determined by assessing areas without cells), the average intensity of FluoZin-3-fluorescence per neuron was reported as the level of intracellular Zn2+.
Statistical Analysis
Values were expressed as the mean ± standard errors of mean (SEM). Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by the post hoc Student–Newman–Keuls test using GraphPad InStat (GraphPad Software, La Jolla, CA, USA). P values < 0.05 were considered to indicate statistical significance.
Results
Since an early study implicated Zn2+ in neuronal death following transient global cerebral ischemia in rats (Koh et al., 1996), a large body of evidence has attributed excitotoxic neuronal injury to Zn2+ overload in neurons (Sensi et al., 2011). This is principally based on the proof-of-concept that intracellular Zn2+ overload occurs in degenerating neurons (correlation) before death (precedence), and that such pathological phenomena are eliminated when Zn2+ is chelated or removed (interference) (Koh et al., 1996). However, to our knowledge, there are a few study regarding that intracellular Zn2+ indeed takes part in hypoxic neuronal death, except experiments in which cerebral organ cultures (but not neuronal cells) were exclusively subjected to OGD conditions (Büchner et al., 2002; Yin et al., 2002; Miyawaki et al., 2004; Medvedeva et al., 2009). Thus, we first performed our current study to determine if Zn2+ release/accumulation occurs in association with neuronal death in primary neuronal cultures exposed to a hypoxic chemical, CoCl2, DFX, or NaN3.
Intracellular Zn2+ Release/Accumulation in Neurons Exposed to Hypoxic Chemicals
Intracellular Zn2+ was detected using the Zn2+-specific fluorescent indicator FluoZin-3 (Gee et al., 2002), which reacted with Zn2+ to emit bright green fluorescence in cortical neurons 30 min after the addition of 200 µM ZnCl2 (Figures 1B,M). Three hours later, FluoZin-3-fluorescence was significantly attenuated in the ZnCl2-treated neuron cultures (Figures 1C,M).
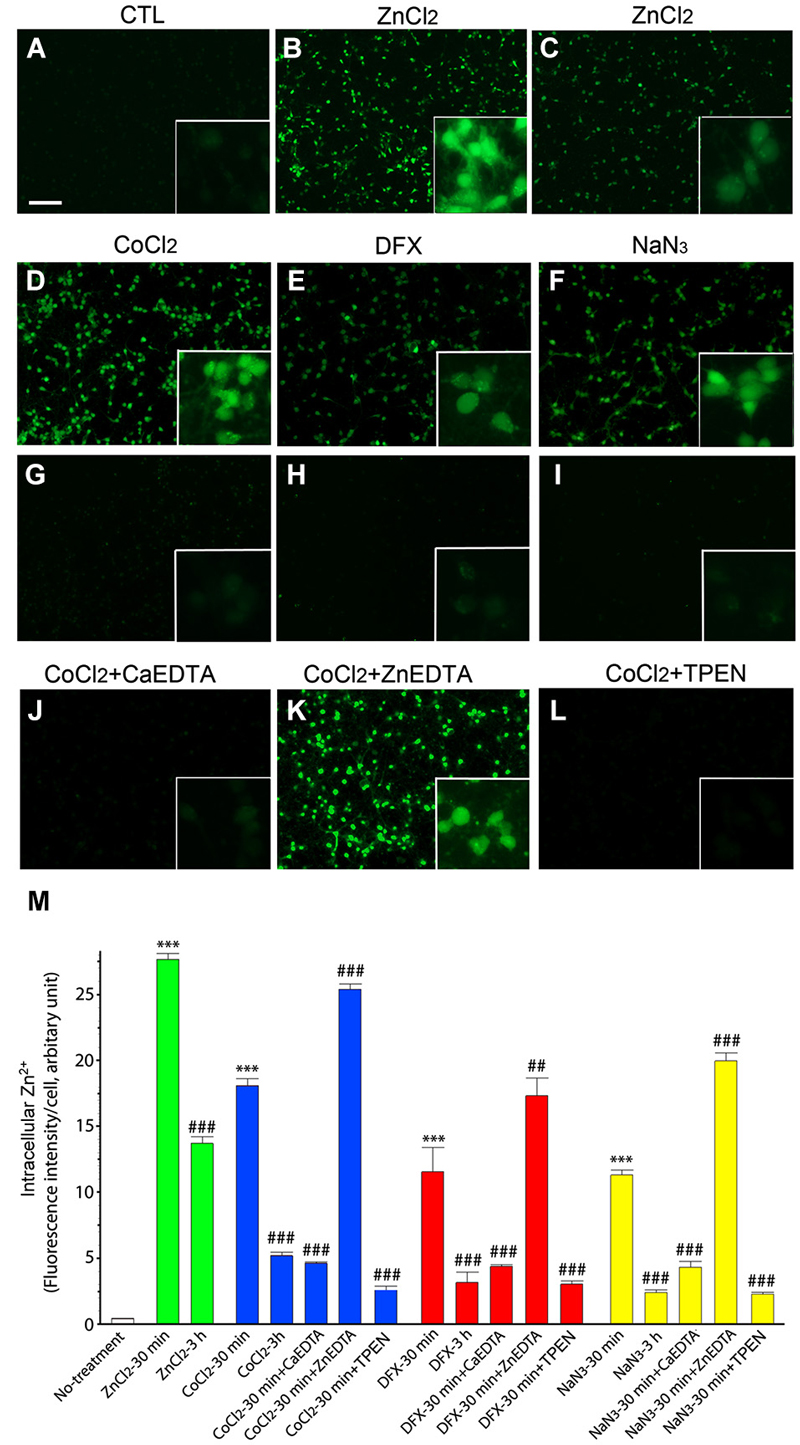
Figure 1. Intracellular Zn2+ release/accumulation in neurons exposed to hypoxia-inducing chemicals. At 30 min (A,B,D–F) or 3 h (C,G–I) after treatment with 200 µM ZnCl2 (B,C), 1 mM CoCl2 (D,G), 3 mM DFX (E,H), or 2 mM NaN3 (F,I), cultured neurons were incubated with 2 µM FluoZin-3 AM for 30 min. CoCl2-exposed neurons were also added with 1 mM CaEDTA (J), 1 mM ZnEDTA (K) or TPEN (0.5 µM; L) 10 min later. Insets show highly magnified FluoZin-3–stained neurons. Scale bar, 100 µm. The intensity of FluoZin-3 fluorescence was quantified in neuronal cultures under a variety of chemical combinations (M). Bars denote the mean intensity of the intracellular Zn2+ fluorescence per neuron after subtracting the background level taken from the cell-free area. All experiments were performed in four independent replications (n = 4), and the intensity of FluoZin-3 fluorescence was quantified by measuring 3 random spots per each independent sample and expressed as arbitrary units. Data are the mean ± SEM of quadruplicate experiments. ## < 0.01, or *** or ###p < 0.001 in comparison with the corresponding control according to one-way ANOVA followed by the Student–Newman–Keuls post hoc test.
Similarly, we noted the rapid evolution of FluoZin-3-fluorescence in neurons following 30 min-exposures to the hypoxia-inducing chemicals CoCl2 (1 mM; Figures 1D,M), DFX (3 mM; Figures 1E,M), and NaN3 (2 mM; Figures 1F,M). Three hours later, the intensity of the intracellular fluorescence was significantly reduced in the hypoxic chemical-treated neurons (Figures 1G–I,M). These results thus indicate that Zn2+ was released and accumulated in neurons shortly after exposure to hypoxic chemicals, and thereafter gradually disappears as the time progresses.
Intracellular Zn2+ Release/Accumulation Precedes Neuronal Death During Chemical Hypoxia
We assessed neuronal death for 24 h after hypoxic insult. On the basis of PI exclusion assay, we found that neurons were still intact at 30 min after ZnCl2- or hypoxic chemical treatment (Figure 2), when Zn2+ had highly accumulated in neurons (Figure 1). However, neuronal death began to appear about 3 h later (Figure 3) when intracellular Zn2+-fluorescence decreased (Figure 1), and gradually increased as time progressed, a phenomenon that was further evidenced by MTT cell viability assay (Figure 3). Hence, these data indicate that intracellular Zn2+ release/accumulation precedes neuronal death after chemical-induced hypoxia, thus providing the Zn2+-induced delayed neuronal death.
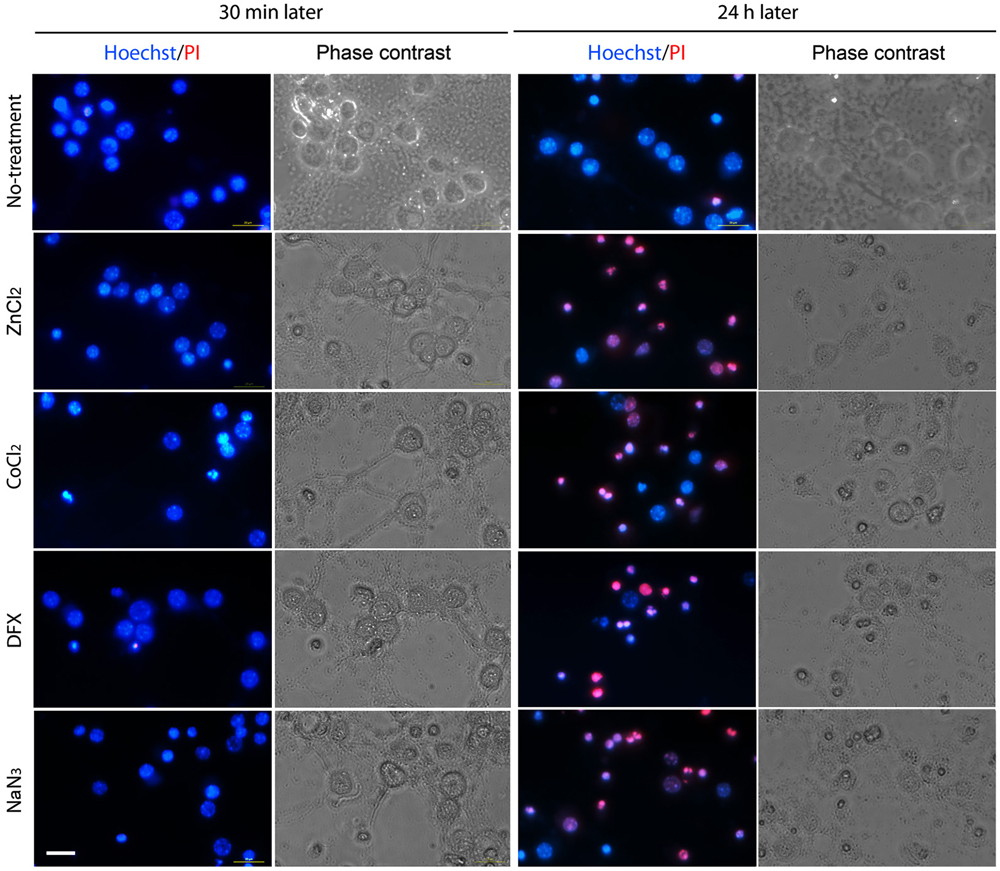
Figure 2. Morphological determination of chemical hypoxia-induced neuronal death. Neuronal cultures were photographed in the absence (2nd and 4th columns) or presence (1st and 3rd columns) of double staining with Hoechst 33342 (blue) and propidium iodide (PI; red) at 30 min (1st and 2nd columns) or 24 h (3rd and 4th columns) after non-treatment (1st row) or exposure to 1 mM CoCl2 (2nd row), 3 mM DFX (3rd row) or 2 mM NaN3 (4th row). Scale bar, 20 µm.
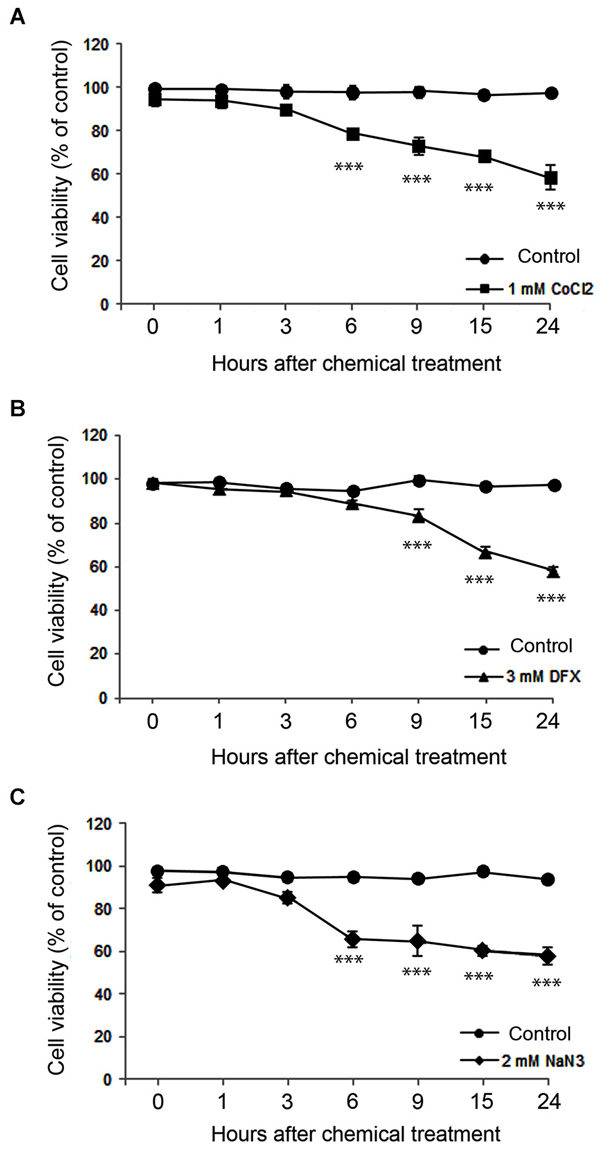
Figure 3. Time course of neuronal death assessed using the MTT cell viability assay at various time points after 1 mM CoCl2- (A), 3 mM DFX- (B), or 2 mM NaN3- (C) treatment. Data are the mean ± SEM of quadruplicate independent experiments, which contained three parallel cultures. ***p < 0.001 in comparison with the corresponding control treatment according to one-way ANOVA followed by the Student–Newman–Keuls post hoc test.
Effects of Zn2+ Chelation on Chemical Hypoxia-Induced Neuronal Death
To relieve intracellular Zn2+ overload, we added CaEDTA or ZnEDTA (each 1 mM) to the media at 10 min after exposure of the neurons to ZnCl2 or hypoxic chemicals, and examined FluoZin-3-fluorescence 30 min later. Consistent with our expectations that EDTA would fully chelate intracellular Zn2+ (Frederickson et al., 2002), CaEDTA reduced FluoZin3-fluorescence in the ZnCl2- or hypoxic chemical-exposed neurons (Figure 1) whereas ZnEDTA increased fluorescence (Figures 1K,M). Furthermore, TPEN (0.5 µM) perfectly depleted it (Figures 1L,M).
Previous studies using various experimental models of neurological disease have reported that CaEDTA (but not ZnEDTA) efficiently blocks neuronal death (Koh et al., 1996). Hence, in order to define the Zn2+-specific actions on chemical hypoxia-induced neuronal death, we added various salt forms of EDTA (0.1–1.0 mM) or TPEN (0.1–0.5 µM) to the culture media at 10 min or 3 h after exposure to ZnCl2 or a hypoxic chemical, and then determined cell viability using the MTT assay (Figures 4–7).
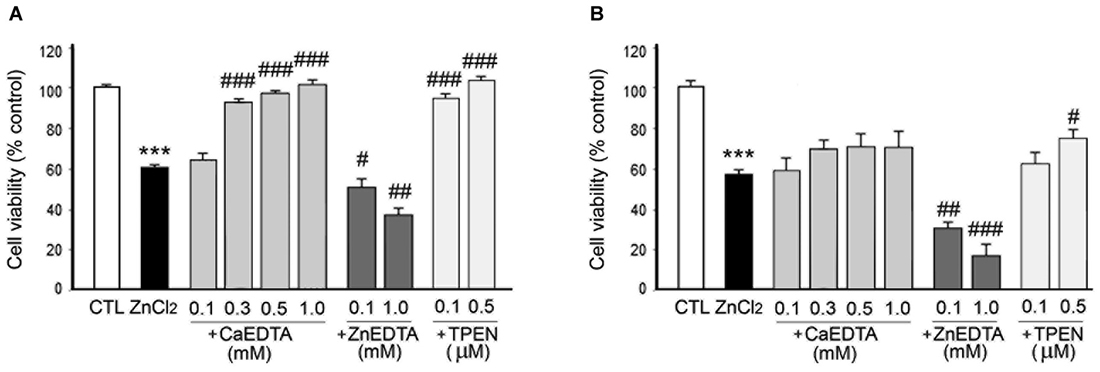
Figure 4. Effects of metal chelation on ZnCl2-induced neuron death. ZnCl2 (200 µM)-exposed neurons were followed by the immediate (A) or 3 h later (B) addition of CaEDTA (0.1–1.0 mM), ZnEDTA (0.1 or 1.0 mM), or TPEN (0.1 or 0.5 µM). Twenty-four hours after ZnCl2 application, neuronal death was assessed by the MTT cell viability assay. Bars denote the mean ± SEM of at least three independent experiments, which each consisted of three parallel cultures. Values were expressed as percentages of non-treated control cells. #p < 0.05, ##p < 0.01, or *** or ###p < 0.001 in the corresponding comparison according to one-way ANOVA followed by the Student–Newman–Keuls post hoc test.

Figure 5. Effects of metal chelation or Zn2+ supplementation on CoCl2-induced neuronal death. CoCl2 (1 mM)-treated neurons were followed by the immediate (A) or 3 h later (B) addition of CaEDTA (1.0 mM), ZnEDTA (1.0 mM), CoEDTA (1.0 mM), FeEDTA (1.0 mM), or TPEN (0.5 µM), or by the immediate addition of ZnCl2 (0.05–1.0 mM) (C). Twenty-four hours after CoCl2 application, neuronal death was assessed using the MTT cell viability assay. Bars denote the mean ± SEM of at least three independent experiments, which each consisted of three parallel cultures. Values were expressed as percentages of non-treated control cells. ##p < 0.01, or *** or ###p < 0.001 in the corresponding comparison by the one-way ANOVA and the Student–Newman–Keuls post hoc test.
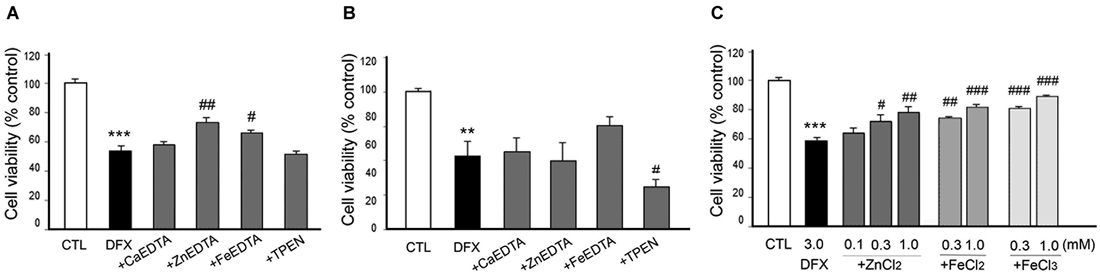
Figure 6. Effect of metal chelation or supplementation on DFX-induced neuronal death. DFX (3 mM)-exposed neurons were followed by the immediate (A) or 3 h later (B) addition of CaEDTA (1.0 mM), ZnEDTA (1.0 mM), FeEDTA (1.0 mM), or TPEN (0.5 µM), or by the immediate addition of ZnCl2 (0.1–1.0 mM), FeCl2 (0.3 or 1.0 mM) or FeCl3 (0.3 or 1.0 mM) (C). Twenty-four hours after DFX exposure, neuronal death was assessed by the MTT cell viability assay. Bars denote the mean ± SEM of at least three independent experiments, which each consisted of three parallel cultures. Values were expressed as percentages of non-treated control cells. #p < 0.05, ** or ##p < 0.01, or *** or ###p < 0.001 in the corresponding comparison by the one-way ANOVA and the Student–Newman–Keuls post hoc test.
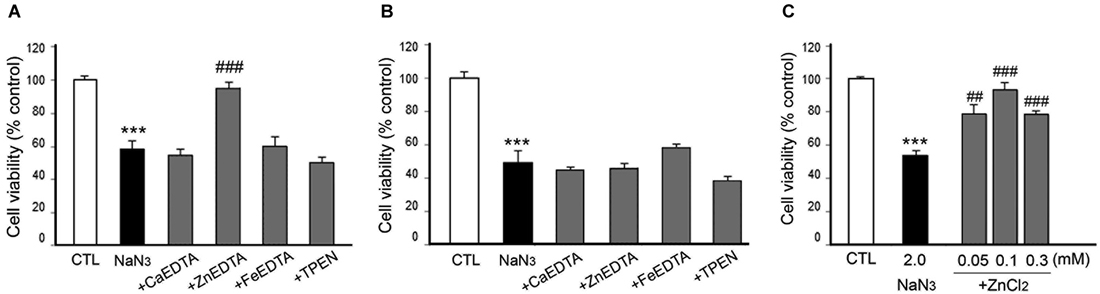
Figure 7. Effects of metal chelation or Zn2+ supplementation on NaN3-induced neuron death. NaN3 (2 mM)-exposed neurons were followed by the immediate (A) or 3 h later (B) addition of CaEDTA (1.0 mM), ZnEDTA (1.0 mM), FeEDTA (1.0 mM), or TPEN (0.5 µM), or by the immediate addition of ZnCl2 (0.05–1.0 mM) (C). Twenty-four hours after NaN3 application, neuronal death was assessed using the MTT cell viability assay. Bars denote the mean ± SEM of at least three independent experiments, which each consisted of three parallel cultures. Values were expressed as percentages of non-treated control cells. #p < 0.05, ** or ##p < 0.01, or *** or ###p < 0.001 between the corresponding comparisons by the one-way ANOVA and the Student–Newman–Keuls post hoc test.
In consistency with the previous studies (Koh et al., 1996), either CaEDTA (0.3–1.0 mM) or TPEN (0.1–0.5 µM) evidently reduced neuronal death when added at 10 min after the ZnCl2-exposure (Figure 4A), but the neuroprotective effect was less evident with the 3 h-delayed CaEDTA treatment (Figure 4B). In contrast, ZnEDTA (0.1 or 1.0 mM) aggravated the ZnCl2-induced neuronal death (Figures 4A,B). These results support the evidence for Zn2+-induced delayed neuronal death.
When either CaEDTA (1 mM) or TPEN (0.5 µM) was added to CoCl2-exposed neurons 10 min later, it significantly decreased neuronal death; however, ZnEDTA (1 mM) resulted in severe toxicity, and CoEDTA (1 mM) or FeEDTA (1 mM) had no effect (Figure 5A). Three hour post-treatment of CaEDTA or TPEN rarely affected CoCl2-induced neuronal death (Figure 5B). These findings suggest that intracellular Zn2+ can cause delayed neuronal death during CoCl2-induced hypoxia.
However, it was unexpected that ZnEDTA reduced the DFX- or NaN3-induced neuronal death when added 10 min later, but neither CaEDTA nor TPEN (Figures 6A, 7A). FeEDTA also provided some protective effects against DFX-induced hypoxic death (Figure 6A). Three hour post-treatment of EDTA or TPEN had no effect on NaN3 (Figure 6B)- or DFX (Figure 7B)-induced neuronal death.
Neuroprotective Effect of Metals in Chemical Hypoxia
Because ZnEDTA prominently reduced neuronal death following DFX- or NaN3-induced hypoxia (Figures 6A, 7A), we investigated whether Zn2+ enables neurons to survive the chemical-induced hypoxic damage (Figures 5C, 6C, 7C). As expected, ZnCl2 (0.05–1.0 mM) significantly increased the level of CoCl2–induced neuronal death (Figure 5C). However, ZnCl2 produced protective effects against neuronal death following DFX- or NaN3-induced chemical hypoxia (Figures 6C, 7C). Similar to the neuroprotection by FeEDTA against DFX-induced neuronal death (Figure 6A), supplementation with iron (0.3–1.0 mM FeCl2 or FeCl3) rendered neurons significantly more resistant to DFX-induced chemical hypoxia (Figure 6C). Therefore, apart from CoCl2-induced neuronal death that was aggravated by ZnCl2, Zn2+ is likely to protect neurons against DFX- or NaN3-induced chemical hypoxia. Plus, iron (Fe2+ or Fe3+) may also provide neuroprotective effects against DFX-induced hypoxia (Figure 6C).
Discussion
The mechanism underlying chemical hypoxia remains unclear. A line of studies have noted to the involvement of iron in stabilizing HIF1α and thereby activating hypoxic signals (Ho and Bunn, 1996). Because HIF1α is rapidly degraded by the polyubiquitination and proteasome pathway, which is manipulated by prolyl-4-hydroxylases (PHDs), it is normally present in cells at low levels (Bruick and McKnight, 2001; Epstein et al., 2001). PHDs essentially require oxygen and iron for their activity, so the depletion of iron from cells could inhibit the activity of the PHDs to stabilize HIF1α from degradation, stimulating the hypoxic responses similar to that observed due to an oxygen shortage (Bruick and McKnight, 2001; Guo et al., 2001). Transition metals (e.g., Co2+ or Ni2+) and iron chelators (e.g., DFX) could induce hypoxic responses by inhibiting PHD activity via iron replacement or depletion, respectively (Schofield and Ratcliffe, 2004; Choi et al., 2006). Although Zn2+ could be another effective replacement metal for iron in PHDs (Shibayama et al., 1986), there have been disputes regarding the roles of Zn2+ in hypoxia. Zn2+ has recently been found to elevate the intracellular expression of HIF1α through the activation of NADPH oxidase or poly(ADP ribose) polymerase (PARP; Pan et al., 2013; Malairaman et al., 2014). By contrast, Zn2+ also inhibits HIF1α activity and the activation of the hypoxia-inducible genes to block the hypoxic responses (Chun et al., 2000, 2001). Thus, while these HIF1α-modulating metal signals may suggest a mechanism of chemical hypoxia, it still remains to be defined how hypoxic chemicals induce neuron death, particularly via intracellular Zn2+ release/accumulation.
In this study, when the neuronal cultures were exposed to ZnCl2, or the hypoxic chemical CoCl2, DFX, or NaN3, we observed the intense emission of Zn2+-specific FluoZin-3-fluorescence in neurons. To confirm the intracellular Zn2+ release/accumulation, we examined that the Zn2+-chelator CaEDTA (Koh et al., 1996; Frederickson et al., 2002) evidently eliminated FluoZin-3-fluorescence from the chemical-treated cultures at the higher concentration (1 mM), despite concern that low concentration of CaEDTA perturb no response of FluoZin-3 to Zn2+ (Zhao et al., 2008). Moreover, TPEN (0.5 µM) also perfectly depleted FluoZin-3-fluorescence, but the non-Zn2+ chelator ZnEDTA (1 mM) (Koh et al., 1996) showed no attenuation of the fluorescence intensity. Therefore, these findings support that Zn2+ is robustly released and accumulated in cultured neurons shortly after the hypoxic chemical treatment. A variety of sources of releasable Zn2+ has been found in neurons, such as Zn2+-bound proteins (Aizenman et al., 2000; Lee et al., 2000, 2003) or Zn2+-containing organelles including mitochondria (Jiang et al., 2001; Sensi et al., 2002) or lysosomes (Hwang et al., 2008). In addition, since neurons survived the moment of the highest intracellular Zn2+ accumulation and then started to die along with its gradual loss, we guess that Zn2+ could cause delayed neuronal death in hypoxic chemical-treated cultures.
However, the effects of Zn2+ chelation on chemical hypoxia-induced neuronal death differed depending on the hypoxic chemical that was used. When EDTA was added immediately after CoCl2-induced hypoxia, CaEDTA evidently alleviated neuron death, but ZnEDTA potently augmented cell death. However, 3 h delayed CaEDTA rarely reduced CoCl2-induced neuronal death. CoEDTA or FeEDTA had no effects. A strong intracellular Zn2+-chelator TPEN also produced the neuroprotective effects. These results were comparable to the effect of CaEDTA or TPEN on ZnCl2-induced delayed neuronal death, where the immediate Zn2+ chelation with CaEDTA or TPEN counteracted the neuronal death but the late CaEDTA showed no protection. It appears that the late Zn2+ chelation couldn’t afford to block the death signaling process that has been already triggered by the precedent Zn2+ overload in neurons. Therefore, we believe that CoCl2-induced hypoxia rapidly triggers intracellular Zn2+ release, leading to Zn2+ overload in neurons and thereby causing their death. In contrast, there was an opposite case during DFX- or NaN3-induced hypoxia. ZnEDTA rather protected neurons from DFX- and NaN3-induced hypoxic death, but CaEDTA had no effect. Zn2+ supplementation also enabled neurons to survive DFX- or NaN3-induced hypoxic damages. These results suggest that Zn2+ may be neurotoxic or neuroprotective in neurons during chemical hypoxia; Zn2+ may directly cause hypoxic neuronal death (in CoCl2-induced hypoxia), or normally participate in neuronal survival or viability (in DFX- or NaN3-induced hypoxia). In addition, we found that iron supplementation (Fe2+ or Fe3+) can protect neurons from DFX-induced hypoxic damage, consistent with speculation that it may make up for DFX-induced iron depletion. However, it is unfortunate that there is no current explanation or information concerning how or why Zn2+ plays in the opposite roles in the chemical hypoxia-induced neuronal death.
It is well established that cytosolic calcium (Ca2+) overload triggers signal pathways to execute neuronal degeneration after hypoxic/ischemic insult (Lipton, 1999; Bano and Nicotera, 2007; Mattson, 2007; Berna-Erro et al., 2009). Our results might exclude the causative roles of Ca2+ in CoCl2-induced hypoxic neuronal death due to the evidence consistent with the earlier study (Koh et al., 1996), in which cytosolic Zn2+ overload preceded neuronal death and Zn2+-specific chelation CaEDTA (but not ZnEDTA) recovered it. Instead, as we failed to determine whether Zn2+-specific chelation inhibits neuronal death after DFX- or NaN3-inducrd hypoxia, we couldn’t rule out the possibility that Ca2+-induced excitotoxicity may contribute to hypoxic neuronal death (Koh et al., 1996; Lipton, 1999; Bano and Nicotera, 2007; Mattson, 2007; Berna-Erro et al., 2009).
In conclusion, we for the first time provide evidence that hypoxia stimulates the intracellular release/accumulation of Zn2+ in neurons, and thereby it may contribute to neuronal death or survival. The opposite roles of Zn2+ in hypoxic chemical-induced neuron death may not only indicate that different hypoxic chemicals induce neuron death via distinct mechanisms, but reflect the diverse groups of signals that essentially require Zn2+ for their functions. Otherwise, Zn2+-regulated neuronal fate may be differentially determined depending on the actual range of intracellular Zn2+ levels (Cho et al., 2010). To date, chelation study using EDTA or TPEN has focused mainly on the negative roles of Zn2+ as a main cause of neuronal death in the context of excitotoxic acute brain injury (Sensi et al., 2011). Instead, this study offers the insight into the positive aspect of Zn2+ that it could mediate neuronal survival under such neurological diseases. Further study will be warranted to elucidate the mechanism by which Zn2+ enable neurons to survive a variety of neurotoxic circumstances.
Author Contributions
Sujeong Kim and Jung-Woo Seo designed the culture experiments and performed the MTT viability analysis. Shin Bi Oh and So Hee Kim photographed the cultured neurons and performed the image-analysis. Inki Kim and Nayoung Suh managed and discussed the overall study, analyzed the data and prepared the manuscript draft. Joo-Yong Lee conceived and designed the work, approved the data analysis and interpretations, and finally completed the manuscript. All authors saw and approved the completion of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea (A092042 to Joo-Yong Lee); the Basic Science Research Program, National Research Foundation of Korea, Ministry of Education, Republic of Korea (NRF-2012R1A1A2006801 to Joo-Yong Lee); and the Asan Institute for Life Sciences, Asan Medical Center, Republic of Korea (2015-396 to Joo-Yong Lee).
References
Aizenman, E., Stout, A. K., Hartnett, K. A., Dineley, K. E., McLaughlin, B., and Reynolds, I. J. (2000). Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J. Neurochem. 75, 1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Almli, L. M., Hamrick, S. E., Koshy, A. A., Täuber, M. G., and Ferriero, D. M. (2001). Multiple pathways of neuroprotection against oxidative stress and excitotoxic injury in immature primary hippocampal neurons. Brain Res. Dev. Brain Res. 132, 121–129. doi: 10.1016/s0165-3806(01)00302-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bano, D., and Nicotera, P. (2007). Ca2+ signals and neuronal death in brain ischemia. Stroke 38, 674–676. doi: 10.1161/01.str.0000256294.46009.29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2:ra67. doi: 10.1126/scisignal.2000522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bossy-Wetzel, E., Talantova, M. V., Lee, W. D., Schölzke, M. N., Harrop, A., Mathews, E., et al. (2004). Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41, 351–365. doi: 10.1016/s0896-6273(04)00015-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruick, R. K., and McKnight, S. L. (2001). A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340. doi: 10.1126/science.1066373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Büchner, M., Huber, R., and Riepe, M. W. (2002). Trans-synaptic increase of hypoxic tolerance in hippocampus upon physical challenge with two-photon microscopy. Hippocampus 12, 765–773. doi: 10.1002/hipo.10028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, E., Hwang, J. J., Han, S. H., Chung, S. J., Koh, J. Y., and Lee, J. Y. (2010). Endogenous zinc mediates apoptotic programmed cell death in the developing brain. Neurotox. Res. 17, 156–166. doi: 10.1007/s12640-009-9085-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, S. M., Choi, K. O., Park, Y. K., Cho, H., Yang, E. G., and Park, H. (2006). Clioquinol, a Cu(II)/Zn(II) chelator, inhibits both ubiquitination and asparagine hydroxylation of hypoxia-inducible factor-1alpha, leading to expression of vascular endothelial growth factor and erythropoietin in normoxic cells. J. Biol. Chem. 281, 34056–34063. doi: 10.1074/jbc.m603913200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, D. W., Yokoyama, M., and Koh, J. (1988). Zinc neurotoxicity in cortical cell culture. Neuroscience 24, 67–79. doi: 10.1016/0306-4522(88)90312-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chun, Y. S., Choi, E., Kim, G. T., Lee, M. J., Lee, M. J., Lee, S. E., et al. (2000). Zinc induces the accumulation of hypoxia-inducible factor (HIF)-1α, but inhibits the nuclear translocation of HIF-1β, causing HIF-1 inactivation. Biochem. Biophys. Res. Commun. 268, 652–656. doi: 10.1006/bbrc.2000.2180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chun, Y. S., Choi, E., Yeo, E. J., Lee, J. H., Kim, M. S., and Park, J. W. (2001). A new HIF-1 alpha variant induced by zinc ion suppresses HIF-1-mediated hypoxic responses. J. Cell Sci. 114, 4051–4061.
Epstein, A. C., Gleadle, J. M., McNeill, L. A., Hewitson, K. S., O’Rourke, J., Mole, D. R., et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. doi: 10.1016/S0092-8674(01)00507-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fang, D., Li, Z., Zhong-Ming, Q., Mei, W. X., Ho, Y. W., Yuan, X. W., et al. (2008). Expression of bystin in reactive astrocytes induced by ischemia/reperfusion and chemical hypoxia in vitro. Biochim. Biophys. Acta 1782, 658–663. doi: 10.1016/j.bbadis.2008.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frederickson, C. J., Hernandez, M. D., Goik, S. A., Morton, J. D., and McGinty, J. F. (1988). Loss of zinc staining from hippocampal mossy fibers during kainic acid induced seizures: a histofluorescence study. Brain Res. 446, 383–386. doi: 10.1016/0006-8993(88)90899-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frederickson, C. J., Suh, S. W., Koh, J. Y., Cha, Y. K., Thompson, R. B., LaBuda, C. J., et al. (2002). Depletion of intracellular zinc from neurons by use of an extracellular chelator in vivo and in vitro. J. Histochem. Cytochem. 50, 1659–1662. doi: 10.1177/002215540205001210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garnier, P., Ying, W., and Swanson, R. A. (2003). Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J. Neurosci. 23, 7967–7973.
Gee, K. R., Zhou, Z. L., Ton-That, D., Sensi, S. L., and Weiss, J. H. (2002). Measuring zinc in living cells. A new generation of sensitive and selective fluorescent probes. Cell Calcium 31, 245–251. doi: 10.1016/S0143-4160(02)00053-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guelman, L. R., Pagotto, R. M., Di Toro, C. G., and Zieher, L. M. (2004). Deferoxamine antioxidant activity on cerebellar granule cells gamma-irradiated in vitro. Neurotoxicol. Teratol. 26, 477–483. doi: 10.1016/j.ntt.2004.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, K., Searfoss, G., Krolikowski, D., Pagnoni, M., Franks, C., Clark, K., et al. (2001). Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 8, 367–376. doi: 10.1038/sj.cdd.4400810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ho, V. T., and Bunn, H. F. (1996). Effects of transition metals on the expression of the erythropoietin gene: further evidence that the oxygen sensor is a heme protein. Biochem. Biophys. Res. Commun. 223, 175–180. doi: 10.1006/bbrc.1996.0865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hwang, J. J., Lee, S. J., Kim, T. Y., Cho, J. H., and Koh, J. Y. (2008). Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J. Neurosci. 28, 3114–3122. doi: 10.1523/JNEUROSCI.0199-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, D., Sullivan, P. G., Sensi, S. L., Steward, O., and Weiss, J. H. (2001). Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 276, 47524–47529. doi: 10.1074/jbc.m108834200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koh, J. Y., Suh, S. W., Gwag, B. J., He, Y. Y., Hsu, C. Y., and Choi, D. W. (1996). The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272, 1013–1016. doi: 10.1126/science.272.5264.1013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J. Y., Cole, T. B., Palmiter, R. D., and Koh, J. Y. (2000). Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J. Neurosci. 20:RC79.
Lee, J. Y., Kim, J. H., Palmiter, R. D., and Koh, J. Y. (2003). Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp. Neurol. 184, 337–347. doi: 10.1016/s0014-4886(03)00382-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malairaman, U., Dandapani, K., and Katyal, A. (2014). Effect of Ca2EDTA on zinc mediated inflammation and neuronal apoptosis in hippocampus of an in vivo mouse model of hypobaric hypoxia. PLoS One 9:e110253. doi: 10.1371/journal.pone.0110253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P. (2007). Calcium and neurodegeneration. Aging Cell 6, 337–350. doi: 10.1111/j.1474-9726.2007.00275.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Medvedeva, Y. V., Lin, B., Shuttleworth, C. W., and Weiss, J. H. (2009). Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J. Neurosci. 29, 1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyawaki, T., Yokota, H., Oguro, K., Kato, K., and Shimazaki, K. (2004). Ischemic preconditioning decreases intracellular zinc accumulation induced by oxygen-glucose deprivation in gerbil hippocampal CA1 neurons. Neurosci. Lett. 362, 216–219. doi: 10.1016/s0304-3940(04)00326-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pan, R., Chen, C., Liu, W. L., and Liu, K. J. (2013). Zinc promotes the death of hypoxic astrocytes by upregulating hypoxia-induced hypoxia-inducible factor-1alpha expression via poly(ADP-ribose) polymerase-1. CNS Neurosci. Ther. 19, 511–520. doi: 10.1111/cns.12098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roemgens, A., Singh, S., Beyer, C., and Arnold, S. (2011). Inducers of chemical hypoxia act in a gender- and brain region-specific manner on primary astrocyte viability and cytochrome C oxidase. Neurotox. Res. 20, 1–14. doi: 10.1007/s12640-010-9213-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schofield, C. J., and Ratcliffe, P. J. (2004). Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354. doi: 10.1038/nrm1366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selvatici, R., Previati, M., Marino, S., Marani, L., Falzarano, S., Lanzoni, I., et al. (2009). Sodium azide induced neuronal damage in vitro: evidence for non-apoptotic cell death. Neurochem. Res. 34, 909–916. doi: 10.1007/s11064-008-9852-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sensi, S. L., Paoletti, P., Koh, J. Y., Aizenman, E., Bush, A. I., and Hershfinkel, M. (2011). The neurophysiology and pathology of brain zinc. J. Neurosci. 31, 16076–16085. doi: 10.1523/JNEUROSCI.3454-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sensi, S. L., Ton-That, D., and Weiss, J. H. (2002). Mitochondrial sequestration and Ca2+-dependent release of cytosolic Zn2+ loads in cortical neurons. Neurobiol. Dis. 10, 100–108. doi: 10.1006/nbdi.2002.0493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharp, F. R., and Bernaudin, M. (2004). HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 5, 437–448. doi: 10.1038/nrn1408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shibayama, N., Morimoto, H., and Miyazaki, G. (1986). Oxygen equilibrium study and light absorption spectra of Ni(II)-Fe(II) hybrid hemoglobins. J. Mol. Biol. 192, 323–329. doi: 10.1016/0022-2836(86)90367-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanciu, M., Wang, Y., Kentor, R., Burke, N., Watkins, S., Kress, G., et al. (2000). Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200–12206. doi: 10.1074/jbc.275.16.12200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suh, S. W., Chen, J. W., Motamedi, M., Bell, B., Listiak, K., Pons, N. F., et al. (2000). Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 852, 268–273. doi: 10.1016/s0006-8993(99)02095-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suh, S. W., Garnier, P., Aoyama, K., Chen, Y., and Swanson, R. A. (2004). Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol. Dis. 16, 538–545. doi: 10.1016/j.nbd.2004.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tønder, N., Johansen, F. F., Frederickson, C. J., Zimmer, J., and Diemer, N. H. (1990). Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci. Lett. 109, 247–252. doi: 10.1016/0304-3940(90)90002-q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
White, A. R., Huang, X., Jobling, M. F., Barrow, C. J., Beyreuther, K., Masters, C. L., et al. (2001). Homocysteine potentiates copper- and amyloid beta peptide-mediated toxicity in primary neuronal cultures: possible risk factors in the Alzheimer’s-type neurodegenerative pathways. J. Neurochem. 76, 1509–1520. doi: 10.1046/j.1471-4159.2001.00178.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, H. Z., Sensi, S. L., Ogoshi, F., and Weiss, J. H. (2002). Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J. Neurosci. 22, 1273–1279.
Zhang, S., Chen, X., Yang, Y., Zhou, X., Liu, J., and Ding, F. (2011). Neuroprotection against cobalt chloride-induced cell apoptosis of primary cultured cortical neurons by salidroside. Mol. Cell. Biochem. 354, 161–170. doi: 10.1007/s11010-011-0815-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, J., Bertoglio, B. A., Gee, K. R., and Kay, A. R. (2008). The zinc indicator FluoZin-3 is not perturbed significantly by physiological levels of calcium or magnesium. Cell Calcium 44, 422–426. doi: 10.1016/j.ceca.2008.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: metal chelation, iron, delayed neuronal death, neuroprotection, brain injury
Citation: Kim S, Seo J-W, Oh SB, Kim SH, Kim I, Suh N and Lee J-Y (2015) Disparate roles of zinc in chemical hypoxia-induced neuronal death. Front. Cell. Neurosci. 9:1. doi: 10.3389/fncel.2015.00001
Received: 05 November 2014; Accepted: 03 January 2015;
Published online: 23 January 2015.
Edited by:
Qi Yuan, Memorial University, CanadaReviewed by:
Hongyu Sun, University of Pennsylvania, USAXin Wang, Stanford University and Howard Hughes Medical Institute, USA
Copyright © 2015 Kim, Seo, Oh, Kim, Kim, Suh and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo-Yong Lee, Asan Institute for Life Sciences, Asan Medical Center, Seoul 138-736, South Korea e-mail: jlee@amc.seoul.kr
† These authors have contributed equally to this work.