Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease
 Ilaria Canobbio
Ilaria Canobbio Aisha Alsheikh Abubaker
Aisha Alsheikh Abubaker Caterina Visconte
Caterina Visconte Mauro Torti
Mauro Torti Giordano Pula
Giordano Pula- Department of Biology and Biotechnology, Unit of Biochemistry, University of Pavia, Pavia, Italy
Alzheimer’s disease (AD) is the most common neurodegenerative cause of dementia in the elderly. AD is accompanied by the accumulation of amyloid peptides in the brain parenchyma and in the cerebral vessels. The sporadic form of AD accounts for about 95% of all cases. It is characterized by a late onset, typically after the age of 65, with a complex and still poorly understood aetiology. Several observations point towards a central role of cerebrovascular dysfunction in the onset of sporadic AD (SAD). According to the “vascular hypothesis”, AD may be initiated by vascular dysfunctions that precede and promote the neurodegenerative process. In accordance to this, AD patients show increased hemorrhagic or ischemic stroke risks. It is now clear that multiple bidirectional connections exist between AD and cerebrovascular disease, and in this new scenario, the effect of amyloid peptides on vascular cells and blood platelets appear to be central to AD. In this review, we analyze the effect of amyloid peptides on vascular function and platelet activation and its contribution to the cerebrovascular pathology associated with AD and the progression of this disease.
Alzheimer’s Disease and Amyloid Peptides
Alzheimer’s disease (AD) is the most invalidating neurological disorder in the elderly and affects 45 million people worldwide. In parallel with population aging, AD patients are expected to rise to 115 million in 2050 worldwide (World Alzheimer Report et al., 2011).1 The social and economic burden of AD is high and has been calculated that in the US alone 600 billion dollars per year are spent for AD patients’ care (The Lancet Neurology et al., 2010). In the UK, dementia costs the economy more than cancer and heart disease combined (Alzheimer research UK site). Despite several promising advances in this field (Blennow et al., 2015), effective predictive markers of AD are still elusive and diagnosis relies on cognitive symptoms characteristic of the advanced stages of the disease. Brain scan and postmortem histological analysis of brain specimens remain the most reliable tools for AD diagnosis (Ikonomovic et al., 2008).
AD results in progressive loss of cognitive function and memory, which ultimately depends on extensive loss of cerebral tissue functionality linked to neuronal death (Gandy, 2005). The histological hallmarks of AD throughout the brain (mainly in the region of hippocampus and neocortex) are intracellular deposition of hyperphosphorylated form of the microtubule associated protein tau called neurofibrillary tangles (NTF) and extracellular accumulation of amyloid β peptides (Aβ peptides) in senile plaques (Bloom, 2014). Deposits of Aβ peptides are also observed in the cerebrovasculature, where they may develop cerebral amyloid angiopathy (CAA; Viswanathan and Greenberg, 2011).
Aβ peptides that accumulate in cerebral senile plaques and vessel walls derive from the metabolism of the larger glycoprotein called amyloid precursor protein (APP), which is a type 1 membrane glycoprotein expressed ubiquitously in the cells. APP isoform 695 is mainly expressed in neurons, whereas APP751 and APP770 that contain the Kunitz type serine protease inhibitory domain KPI are mainly expressed on peripheral cells and platelets (Van Nostrand et al., 1994). APP can be processed via two alternative pathways, amyloidogenic and non-amyloidogenic (Gandy, 2005). The amyloidogenic pathway produces Aβ peptides by the subsequent action of β- and γ-secretases. β-secretase proteolyses APP in the extracellular domain to generate soluble amyloid precursor protein β (sAPPβ) and a carboxyl terminal fragment named CTFβ (C99), which is the substrate for γ secretase to produce Aβ peptides. β secretase, or BACE, is a membrane bound aspartyl protease, for which APP is a substrate (Vassar et al., 2014). For this reason, BACE antagonists have been proposed for treatment of AD (Menting and Claassen, 2014). γ- secretase activity is contained within a molecular complex formed by the association of four essential subunits: presenilin1 or presenilin2 (either PS1 or PS2), nicastrin, presenilin-enhancer-2 (PEN-2) and anterior-pharynx-defective 1 (APH-1; Iwatsubo, 2004). The presenilins are ubiquitously expressed and represent the catalytic subunits of γ- secretase, whereas the other subunits help to stabilize the complex and to recruit the substrates to be cleaved (Smolarkiewicz et al., 2013). Alternatively, APP may be proteolysed through the non-amyloidogenic pathway, via the action of α- and γ-secretase (Buxbaum et al., 1998). α-secretase cleaves APP within the Aβ sequence, thus precluding the generation of Aβ peptides. Cleavage within Aβ sequence of APP by α-secretase generates soluble amino terminal fragments of 100–130 kDa (sAPPα) and a carboxyl terminal fragment named CTFα (C83), which is substrate for γ-secretase (Esch et al., 1990; Vingtdeux and Marambaud, 2012) to produce the non amyloidogenic peptide p3. sAPPα generated from the cleavage of APP by α-secretase shows biological functions in growth regulation and neuroprotection and, in the case of forms containing the Kunitz-type serine proteinase inhibitors KPI, in blood coagulation (Xu et al., 2007). A schematic representation of APP metabolism is presented in Figure 1.
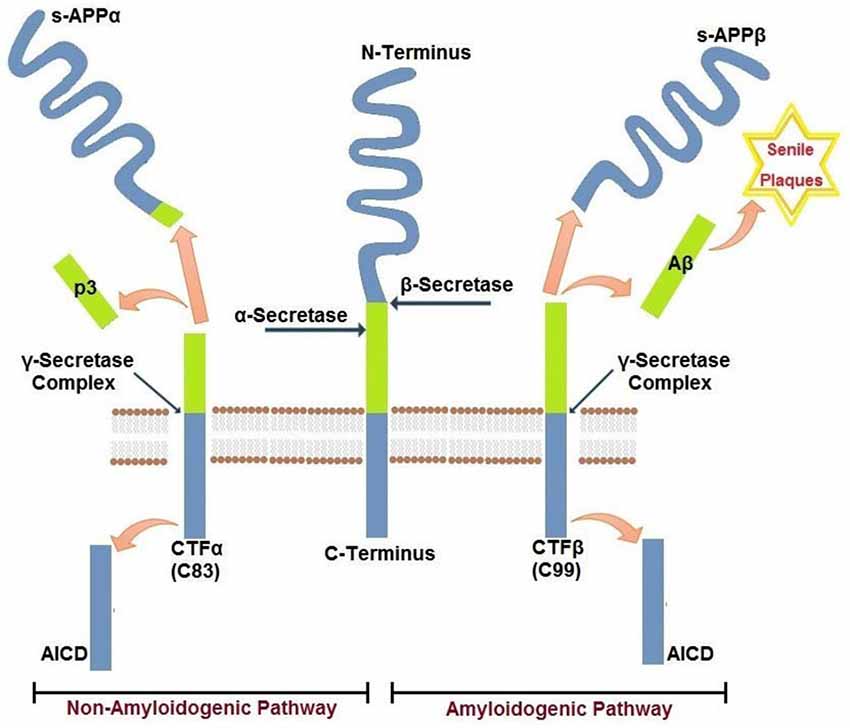
Figure 1. Amyloidogenic and non-amyloidogenic pathways of APP. Amyloid precursor protein APP is a single pass transmembrane glycoprotein. APP may be cleaved by β−γ secretases (amyloidogenic) releasing amyloid Aβ peptide(s) or by α−γ secretases (non-amyloidogenic). The two pathways are mutually exclusive and are detailed in the text.
Aβ peptides are heterogeneous short hydrophobic peptides (<5 kDa) ranging between 38 and 43 amino acids. Several different Aβ peptide species exist, but Aβ1–40 (Aβ40) is the most abundant. Aβ40 peptides contain 16 amino acid residues in the amino extracellular domain of APP, and 24 amino acids in the membrane-spanning domain. Aβ42 has two additional hydrophobic residues, Ile and Ala, and accounts for only 2–5% of Aβ peptides. Aβ42 peptides are more hydrophobic and fibrillogenic, have a higher aggregation potential and are the principal species deposited in the brain (Murphy and LeVine, 2010). Aβ peptides are normally generated in healthy cells (Seubert et al., 1992), but deficiencies in their clearance and/or their abnormal production and accumulation in the brain and in the cerebral blood vessels are correlated with the onset of AD. Aβ peptides in their monomeric forms are soluble, whereas their oligomerisation leads to the formation of intermolecular β-sheets and precipitation in senile plaques (Tjernberg et al., 1999). Aβ oligomers have the ability to permeabilize the plasma membrane of different cell types, which triggers a series of cellular event leading to cell dysfunction and ultimately death. This mechanism of action is common to different amyloidogenic peptides, including Aβ peptides, α-synuclein, polyglutamine and APP (Glabe, 2006).
Vascular Pathology in AD
Amyloid and Vascular Hypotheses
According to the “amyloid cascade hypothesis”, (Hardy and Higgins, 1992; Hardy and Selkoe, 2002) accumulation of Aβ peptides in the brain is the central event in the pathogenesis of AD. Abnormal deposition of Aβ peptides in the brain then cause plaque and tangle formation, neuronal and vascular damage, cell loss, and finally dementia. This hypothesis is supported by the finding that the familial AD (FAD) results from mutations in genes involved in APP metabolism: APP, PS1 and PS2 (Reznik-Wolf et al., 1998). These mutations cause abnormal production of Aβ40 and Aβ42 peptides, which accumulate in the brain and in the cerebral vessel walls. FAD affects less than 5% of AD cases with autosomal dominant inheritance. Symptoms develop before the age of 65 years, and the pathology is particularly aggressive and leads to death in 5–8 years.
It is noteworthy, however, that the majority of AD patients (95%) develop the pathology after the age of 65 (Kennedy et al., 2003). This form of AD is called late onset AD (LOAD) or sporadic AD (SAD) and has a very complex etiology. The most common risk factor for LOAD/SAD is aging. A correlation has been demonstrated for the presence of the ε4 allele for the cholesterol transporter apolipoproteinE (APOE ε4) and the risk and the age of onset of AD (Michaelson, 2014). Other risk factors for LOAD/SAD are hypercholesterolemia, hypertension, Down syndrome, metabolic syndrome, diabetes, smoking and obesity (DeFronzo and Ferrannini, 1991; Gorelick, 2004; Morris et al., 2014).
The etiology of LOAD/SAD remains elusive and the role of Aβ peptides is controversial (Lee et al., 2004; Sorrentino et al., 2014). Several lines of evidence point towards a central role of early vascular dysfunction in the onset of LOAD/SAD. The “vascular hypothesis” for AD was first proposed in 1993 by De La Torre, after the observation of extensive abnormalities of cerebral capillaries in AD brain that finally results in brain hypoperfusion and reduced cerebral blood flow (de la Torre and Mussivand, 1993). In this context, it is important to note that the first case reported by Dr. Alzheimer in 1906 revealed not only the presence of senile plaques but also the presence of cerebrovascular dysfunction. In addition, Glenner and Wong firstly isolated Aβ peptides from the meningeal vessels of a LOAD/SAD patient in 1984 (Glenner and Wong, 1984). This led to the coin of the term “vascular dementia”, which indicates the loss of cognitive functions due to cerebral blood vessel alteration and poor blood supply to the brain (de la Torre, 2000; Kara et al., 2012). Vascular dementia and AD are correlated and largely overlapping phenomena (Ahtiluoto et al., 2010), and vascular dysfunctions in the brain are recognized as a contributing factor to AD (Viswanathan et al., 2009). To date, many patients with AD present vascular symptoms, including altered cerebral blood flow, damaged cerebral vasculature, and abnormal hemostasis (de la Torre, 2004a; Brundel et al., 2012). Tolppanen et al. demonstrated that AD patients, especially younger patients, have higher risk of hemorrhagic strokes (adjusted hazard ratio of 1.34; Tolppanen et al., 2013). Similarly, a recent Taiwanese population based cohort study showed that clinical diagnosis of AD is associated with considerably increased risk of stroke development (odd ratio of 1.66–1.70; Chi et al., 2013). Moreover, clinical studies indicate that asymptomatic spontaneous cerebral emboli are highly correlated with AD (Purandare and Burns, 2009) and that cerebral microinfarctions occur more likely in AD patients than in healthy controls (Brundel et al., 2012).
These observations have led to the critical question: “is AD a neurodegenerative or a vascular disorder?” (de la Torre, 2004b). Cerebral blood flow is reduced and conversely many vascular defects are present in patients with AD (Bell and Zlokovic, 2009) and vascular diseases such as atherosclerosis correlate in severity with dementia and other symptoms of LOAD/SAD (Farkas and Luiten, 2001; Roher et al., 2003; Casserly and Topol, 2004; Tibolla et al., 2010). Interestingly, vascular risk factors such as aging, hypercholesterolemia, hypertension, diabetes and obesity that predispose to atherosclerosis, stroke and cardiac disease are also associated with cerebrovascular dysfunction, which might finally results in vascular dementia and the onset of AD (Morris et al., 2014). Accumulating body of evidence gathered from human studies and animal models of AD documented that cerebrovascular dysfunction precedes the development of cognitive decline and AD pathology (de la Torre, 2010; Jellinger, 2010; Kalaria, 2010). The two-hit hypothesis has been proposed regarding AD pathogenesis, where vascular dysfunction plays a primary role in causing neurological injury. Cerebrovascular abnormalities associated to AD can result in hypoperfusion, hypoxia, focal chronic inflammation and compromised function and permeability of brain blood barrier (BBB). These injuries are exacerbated by Aβ peptides deposition, which may initiate neurodegeneration and eventually cognitive decline (Zlokovic, 2005; Grammas, 2011; Kelleher and Soiza, 2013; Li et al., 2015).
To date, it has been shown that cerebrovascular dysfunction accelerates Aβ40–42 production and deposition in the cerebral vasculature (Honjo et al., 2012) and facilitates the onset of CAA (Viswanathan and Greenberg, 2011). Aβ peptides are found in plasma with the levels fluctuating widely among individuals over time both in AD and in healthy control (the amount of Aβ40 in healthy subjects varies between 16 to 659 pg/ml, the amount of Aβ42 from 4 to 149 pg/ml) (Roher et al., 2009). These data are likely to be affected by the hydrophobic nature of Aβ peptides, which makes the peptides bind to a variety of plasma and membrane proteins hindering their effective measurement in biological fluids. The source of Aβ in plasma is uncertain. Some authors suggest that plasma Aβ may derive from the central nervous system through the BBB, but most authors agree and it is now well recognized that plasma Aβ derives from endothelial and circulating cells, among them platelets (Li et al., 1998; Davies et al., 2000). Circulating blood platelets are the second source of APP protein after the brain and are the major source of Aβ peptides released in plasma (Li et al., 1998). Other peripheral sources of Aβ peptides are skeletal muscle (Kuo et al., 2000; Van Nostrand and Melchor, 2001) and endothelial vascular cells (Kitazume et al., 2012). Interestingly, Aβ peptides are able to actively pass the BBB (Deane et al., 2003, 2004), and the BBB damage typical of cerebrovascular pathologies is likely to increase the exchange of Aβ peptides between cerebral and peripheral tissues, including blood. Importantly, a recent study has demonstrated that peripheral reduction of Aβ peptides is sufficient to reduce Aβ levels in the brain, which confirms the extracerebral origin of plasma Aβ peptides (Sutcliffe et al., 2011).
AD and Vascular Inflammation
A number of morphological changes have been observed in the cerebromicrovascular; these include an overall decrease in intimal tight junctions (Stewart et al., 1992), basal membrane thickening (Thal et al., 2003; Kelleher and Soiza, 2013) (particularly found in areas of Aβ peptides deposition) and intimal atrophy (Farkas and Luiten, 2001; Truran et al., 2014). In AD brain, substantial morphological and functional cerebrovascular abnormalities were observed including, microvasculature irregularities and atrophy, basement membrane disruption and deposition of heparin sulfate proteoglycans, collagen IV and laminin, decreased cerebrovascular network density, endothelial cell alteration i.e., increased pinocytosis, decreased levels of mitochondria and detection of elevated endothelial cell markers VCAM-1 and E-selectin (Kalaria and Pax, 1995; Farkas and Luiten, 2001; Bailey et al., 2004; Christov et al., 2008; Zuliani et al., 2008). These vascular alterations are referred to as CAA, which associates with ischemic lesions, micro- and macro-hemorrhages, and impaired cerebral blood flow. Intimal inflammation and endothelial damage are also likely to contribute to platelet stimulation in AD patients (Borroni et al., 2002a,b). Vascular risk factors, frequently present in AD, preactivate endothelial cells and exacerbate vascular inflammation, which contributes to CAA (Zhang et al., 2013). The inflammatory proteins overexpressed in the AD cerebrovasculature are likely to have toxic effects on neurons, which could represent an important link between vascular inflammation and neuronal loss in AD (Tripathy et al., 2013). Failure in the clearance of Aβ peptides may therefore result in inflammation and neurotoxicity (Giri et al., 2000; Li et al., 2009) and CAA is likely to play a key part in brain damage and loss of cognitive skills typical of AD (Lee et al., 2014). In accordance to this hypothesis, human studies and animal models of AD document that cerebrovascular dysfunction precedes the development of cognitive decline and AD pathology (de la Torre, 2004b; Bell and Zlokovic, 2009; Xu et al., 2014).
Several biochemical alterations of endothelial cell physiology have been identified in response to exposure to amyloidogenic peptides (Figure 2). Amongst known inflammatory markers, cyclooxygenase 2 (COX2, a key enzyme for inflammatory reactions in platelets) expression is increased in mild cognitive impairment (MCI) and AD patients. This observation links suggestively well with several studies indicating that treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) and COX2 inhibitors may reduce the risk of AD (Szekely and Zandi, 2010). Several studies indicated elevated levels of other inflammatory mediators in AD cerebral microcirculation, where endothelial cells overexpress adhesion molecules (MCP-1, ICAM-1, CAP37), and inflammatory and stress markers such as TNFα, TGF-β, interleukins (IL-1β, IL-6, IL-8) and matrix metalloproteases (MMPs; Grammas and Ovase, 2001; Grammas et al., 2002; Thirumangalakudi et al., 2006; Yin et al., 2010; Grammas, 2011). In vitro studies on cultured endothelial cells revealed that exposure to Aβ40 increased the levels of inflammatory cytokines, including IL-6, IL-1β, MCP-1, and GRO because of JNK-AP1 activation (Vukic et al., 2009). AD senile plaques also display elevated levels of thrombin (Akiyama et al., 1992). Besides its central role in hemostasis, thrombin is a factor with complex paracrine activity that mediates a wide array of cellular processes involving inflammation, neurotoxicity and angiogenesis. Thrombin release activates endothelial cells and enhances their expression of proinflammatory proteins such as ICAM-1 and MCP-1, angiopoietin-2 release, and up-regulation of αVβ3 integrin of VEGFRs expressions (Grammas and Ovase, 2001; Tsopanoglou et al., 2002). Observations obtained from in vivo and in vitro studies, showed that thrombin can cause accelerated tau protein aggregation, neuronecrosis and can incite neurotoxic effects through multiple mechanisms (Smirnova et al., 1998; Suo et al., 2003; Mhatre et al., 2004; Park and Jin, 2008; Rao et al., 2009).
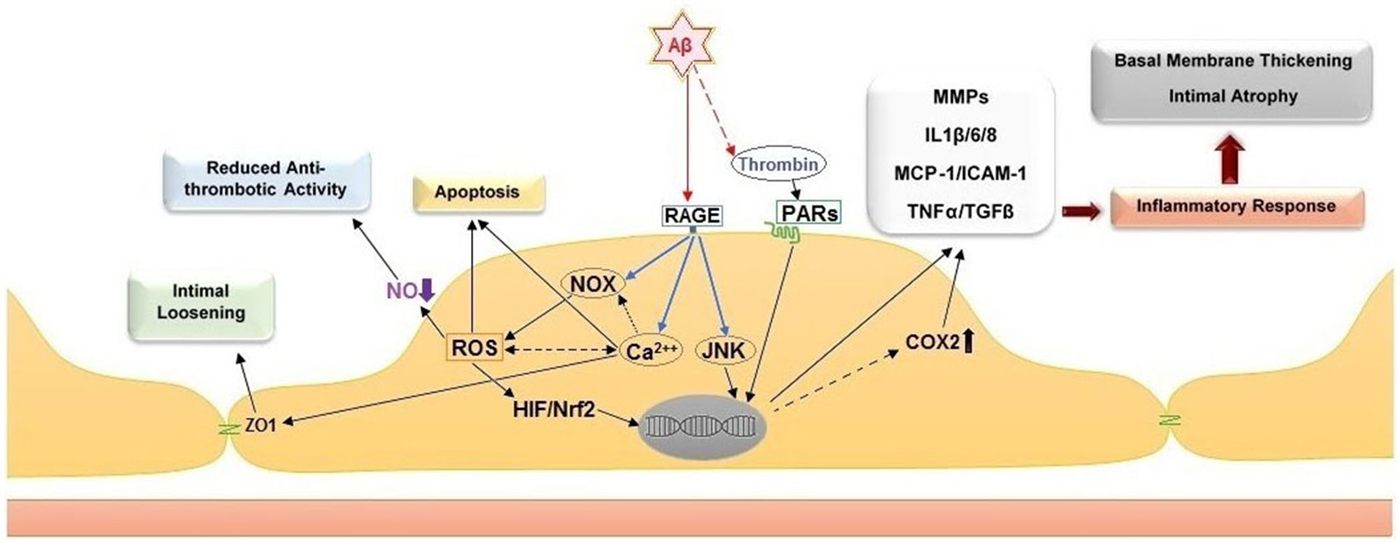
Figure 2. Cellular and histological effects of amyloid peptide β on the endothelium. Direct stimulation of the receptor for advanced glycation end products (RAGE) stimulates NADPH oxidases (NOXs), intracellular calcium increase and c-Jun N-terminal kinases (JNKs). The stimulation of NOXs and generation of reactive oxygen species (ROS) induce the activation of hypoxia-induced factor 1 (HIF) and NF-E2-related Factor 2 (Nrf2). Together with the activation of JNKs, the activity of these transcription factors leads to upregulation of pro-inflammatory genes, including cyclooxygenase 2 (COX2), metalloproteases (MMPs), interleukins 1β/6/8 (IL1β/6/8), monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), tumor necrosis factor α (TNF α) and transforming growth factor beta (TGF-β). Additional effect of ROS increase are the reduction of nitric oxide (NO) bioavailability, which leads to pro-thrombotic endothelial cell phenotype, and apoptosis. The intracellular calcium increase leads to intimal loosening via zona occludens protein 1 (ZO-1). The accumulation of thrombin in the amyloid plaques further facilitates endothelial inflammation via stimulation of the protease-activated receptors (PARs).
AD and Redox Balance in Vascular Cells
Another significant change in endothelial cells associated with AD is the overexpression of receptor for advanced glycation end products (RAGE). RAGE facilitates soluble Aβ peptides transport across BBB and across the cortical neurons intracellular space (Yan et al., 1996; Sasaki et al., 2001; Takuma et al., 2009). These findings suggest a role of RAGE in Aβ deposition in the cerebral intracellular space and thereby in the enhancement of AD-associated neuronal damage (Lue et al., 2001; Takuma et al., 2009). Moreover, Aβ interaction with RAGE at the BBB contributes to the up-regulation of endothelial CCR5 expression, in a dose- and time-dependent manner, via activating Egr-1 transcription factor through JNK, ERK and PI3K pathways (Li et al., 2009). The upregulation of CCR5 stimulates trans-endothelial migration of circulating T cells that express high levels of CXCR2 and MIP-1α. MIP-1α induces the opening of endothelial tight junctions (Li et al., 2009). It has been shown that increased RAGE expression facilitates the generation of reactive oxygen species (ROS) via NADPH oxidase (NOX) in cerebral endothelial cells and up regulates adhesion and pro-inflammatory molecules (Askarova et al., 2011). ROS are highly reactive molecules that promotes cell damage by direct modification of nucleic acids and proteins. One immediate consequence of increased ROS generation in cerebrovascular cells is the reduction of NO bioavailability through its transformation in the physiological oxidant peroxynitrite (NO3; Beckman and Koppenol, 1996). Aβ also exerts an inhibitory effect on NO signaling via binding to cell surface receptors CD36 and CD47 and inhibiting sGC/cGMP/cGK signaling pathways; thus contributing to the lack of NO signaling observed in AD (Miller et al., 2010). Because of the important antiplatelet and vasorelaxant activity of NO, its reduction in the presence of increased ROS in AD brain is likely to promote platelet activation and thromboembolic consequences. Several in vitro studies on Aβ peptides showed decreased NO production in endothelial cells in a dose dependent manner at Aβ concentrations of (10−9–10−6 M) and cerebrovascular endothelium desensitization to acetylcholine, which is an endothelium dependent vasodilator, at Aβ concentration of 10−8 M (Grammas et al., 1995; Price et al., 2001; Hayashi et al., 2009).
In addition, ROS can activate redox-sensitive transcription factors hypoxia-inducible factor 1α (HIF1 α) and nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2), which modifies gene expression of endothelial cells and can ultimately lead to the up-regulation of apoptotic/inflammatory genes (Wautier et al., 2001; Fonseca et al., 2014). Accumulative body of evidence recently shows that the main culprit behind cerebrovascular dysfunction in AD is oxidative stress and ROS production mainly by NOX induced by Aβ that eventually triggers apoptosis in association with neurovascular inflammation (Cai et al., 2003; Park et al., 2005). High levels of pro-angiogenic gene expression have also been documented in AD brains using genome-wide expression profiling and includes the expression of vascular endothelial growth factor (VEGF), HIF-1α, angiopoietin-2 and thrombin (Pogue and Lukiw, 2004; Thirumangalakudi et al., 2006). This is likely to be an adaptive yet inefficient response to poor blood flow and ischemia in AD brains. In fact, AD cerebral tissue is characterized by reduced cerebromicrovascular density (Brown and Thore, 2011). Despite the increased expression of factors that promotes angiogenesis and neovascularization, several studies demonstrated an overall decrease in vasculature density in AD brains. It has been suggested that vascular-restricted mesenchyme homeobox 2 gene (MEOX2) signaling is impaired in AD patients, which interferes with the process of angiogenesis (Jellinger, 2002; Paris et al., 2004; Wu et al., 2005).
AD and Ca2+ Homeostasis in Vascular Cells
In addition to ROS, different cell populations in AD patients and animal models of the disease documented deregulation of Ca2+ homeostasis (Berridge, 2013; Garwood et al., 2013). Aβ has been shown to form cation permeable pores in the plasma membranes (Pollard et al., 1993) and it has been demonstrated that Aβ toxicity derives from the deregulation of Ca2+ homeostasis (Demuro et al., 2005). Mitochondrial Ca2+ overload causes mitochondrial depolarization and impairs the electron transport chain, which increases ROS generation in vascular cells (Carvalho et al., 2009). Ca2+ overload can directly activate NOXs (De Bock et al., 2013) and contribute to the redox stress of AD cerebral tissue. Ca2+ modulation seems to be critical for the onset of redox stress in endothelial cells. Aβ40 induces mitochondria-mediated apoptotic endothelial cell death pathway involving ER-to-mitochondria Ca2+ transfer, decrease of mitochondrial membrane potential, and release of pro-apoptotic factors (Fonseca et al., 2013). Recent studies showed that short-term treatment with a toxic dose of Aβ40 inhibits Ca2+ retention in the endoplasmic reticulum and enhances adenosine triphosphate (ATP)-stimulated release of intracellular Ca2+ (Fonseca et al., 2014). Importantly, Aβ-RAGE interactions in cultured endothelial cells increase intracellular Ca2+ and alter tight junction proteins through the Ca2+-calcineurin pathway (Kook et al., 2012). This phenomenon is responsible for the reduction of zonula occludin-1 (ZO-1) expression and increased MMP secretion observed in cultured endothelial cells and can ultimately be the cause of enhanced MMP secretion and loss of vascular integrity in the vicinity of Aβ plaques in animal models of AD (Kook et al., 2012). The resulting Aβ-dependent alteration in the integrity of the BBB may play a significant role in the transport and accumulation of Aβ peptides from the circulation to the brain parenchyma.
Platelet Pathology in AD
APP and Aβ Peptides in Platelets
The bidirectional correlations between AD and vascular diseases points to an important role for non-neuronal cells and extraneuronal generation of Aβ peptides in the development of AD. Circulating platelets, which are anucleate cells responsible for hemostasis and thrombosis, are good candidates for the link between vascular diseases and AD. Platelets express mainly the APP isoforms 751 and 770. These isoforms contain the 56 amino acids long Kunitz type serine protease inhibitors domain KPI, which is known to inhibit serine proteases including those of the coagulation cascade (Van Nostrand et al., 1989; Xu et al., 2005, 2007). In addition, platelets express all the enzymatic machinery responsible for APP metabolism and for the generation of Aβ peptides (Evin et al., 2003; Catricalà et al., 2012). In physiological conditions, platelets metabolize APP through the non-amyloidogenic pathway: α-secretase is activated by Ca2+ and calmodulin (Canobbio et al., 2011) and releases soluble APPα. To a lesser extend platelets also release Aβ peptides. The main species of Aβ released from activated human platelets is Aβ40, while the predominant form in neuronal plaques is Aβ42. Immunoassay analysis has revealed that the inactivated platelets had an average of 84 ng/g tissue of Aβ40 and activated platelets contained an average of 57 ng/g tissue, while in both cases there were very low levels of Aβ42: 1.6 ng/g tissue and 1.7 ng/g tissue, respectively (Kokjohn et al., 2011). APP fragments and Aβ peptides are stored in platelet α-granules as revealed by immune electron microscopy (Li et al., 1994) and sucrose density gradient (Van Nostrand et al., 1990), and are released upon stimulation with physiological agonists (Li et al., 1994). Increasing evidence suggests that released Aβ peptides are able to activate platelets and promote platelet adhesion and aggregation (Shen et al., 2008a,b; Canobbio et al., 2013, 2014). This eventually results in enhanced thrombus formation in vitro and in vivo (Gowert et al., 2014; Sonkar et al., 2014). On the contrary, soluble APP fragments have been shown to regulate thrombosis and hemostasis in vivo (Xu et al., 2005, 2007).
The relatively abundant concentration of APP in platelets (9300 copies/platelet) (Rowley et al., 2011) and the predominant presence in platelets of the KPI containing isoforms suggest that APP may have a physiological role, mainly in events associated with coagulation (Van Nostrand et al., 1992; Xu et al., 2005). A minority (10%) of APP is expressed as an intact glycoprotein on the platelet surface where it may act as a receptor on the platelet surface (Kang et al., 1987; Li et al., 1994). Fibrillar Aβ but not unassembled Aβ, specifically binds to APP (Lorenzo et al., 2000; Van Nostrand et al., 2002; Sola Vigo et al., 2009). APP also can bind to sulfated proteoglycans, laminin, collagen and integrin like receptor, suggesting a role in cell-cell and cell- matrix interactions (Breen et al., 1991; Ghiso et al., 1992; Milward et al., 1992; Beher et al., 1996; Williamson et al., 1996; Verdier and Penke, 2004). Moreover several studies have reported that treatment of neuronal cell with monoclonal antibody 22C11 directed toward the N terminal domain of APP can stimulate G protein activity (Okamoto et al., 1995) and/or cause the dimerization of APP (Scheuermann et al., 2001). Notably, APP metabolism resembles that of Notch (Nakayama et al., 2011): in the canonical Notch signaling pathway, γ-secretase cleaves Notch in the plasma membrane and release an intracellular domain that shows activity in the nucleus through binding to transcription factors. Since platelets are anucleate cellular fragments this consideration deserve further investigation.
APP is proteolytically cleaved during platelet activation (Li et al., 1998; Evin et al., 2003). The released soluble APP fragment that contain KPI domain was firstly identify as Protease Nexin 2 PN-2, a chymotrypsin inhibitor and regulator of blood coagulation (Van Nostrand et al., 1989). More recently it has been shown by the same authors that PN-2 coincide with soluble APP fragment containing KPI (Van Nostrand et al., 1992). Soluble APP inhibits the activity of the blood coagulation factors IXa, XIa, and Xa, and, to a lesser extent, of factor VIIa-tissues factor complex (Smith et al., 1990; Van Nostrand et al., 1992; Schmaier et al., 1995; Scandura et al., 1997) and may therefore play a role in the coagulation cascade, thus modulating hemostasis following vascular injury by limiting thrombosis. Xu et al. generated specific transgenic mice which expressed human APP770 in platelets, under the control of rPF4 promoter. Transgenic mice express 2 fold higher APP770 amount in platelets, but not in other analyzed tissues. They investigated the APP function comparing Tg-rPF4/APP mice with wild type mice and with mice which do not express APP at all (APP KO) and demonstrated that KPI-containing forms of APP regulates cerebral thrombosis in vivo in a carotid artery thrombosis and in experimental intracerebral hemorrhage (Xu et al., 2005, 2007). In accordance to this, recombinant soluble APP has been shown to inhibit platelet aggregation and secretion induced by ADP and adrenaline in vitro (Henry et al., 1998).
Aβ Peptide-Dependent Activation of Platelets
It has been widely demonstrated by several authors that Aβ peptides are able to activate platelets (Figure 3). The first study date back to 2007 and analyzed the effects of misfolded proteins on platelet activation. Herzenik et al. showed that proteins with amyloid properties, including Aβ peptides, activate platelets. Fibrillar Aβ peptide induces platelet aggregation through the scavenger receptor CD36 and GPIbα, and activation of intracellular signaling pathways involving p38MAPK, cyclooxygenase1 (COX1) and thromboxane A2 production (Herczenik et al., 2007). More recently, the ability of amyloid peptides to activate platelets has been investigated using the synthetic peptide Aβ25–35. This undecapeptide is located in the intermembrane domain of APP and retains the toxic properties of the entire peptide representing the biologically active region of Aβ (Kang et al., 1987). Aβ25–35 toxicity resides in the ability to form oxygen radicals, nitric oxide and to disrupt calcium homeostasis (Kaminsky et al., 2010). In preliminary studies, Aβ40 and Aβ25–35 showed equal potencies in inducing platelet activation (Shen et al., 2008a). Exogenous Aβ25–35 (1–2 μM) potentiated agonist-induced platelet aggregation induced by collagen and ADP, and at higher concentrations (2–10 µM) Aβ25–35 itself directly promotes aggregation, and activation of intracellular signaling pathways through thrombin receptor PAR1 activation of p38 MAPK and cytosolic PLA2, and TxA2 formation (Shen et al., 2008a). In addition, the same authors demonstrated that Aβ25–35 stimulates PLC/PKC activation and Ca2+ mobilization (Shen et al., 2008b). Aβ25–35 also induced the activation of small GTPase RhoA and phosphorylation of myosin light chain kinase, which leads to cytoskeletal reorganization and results in shape change, granule release, integrin activation and clot retraction. An essential role for Ca2+ and ADP in Aβ -induced platelet activation and thrombus formation has been recently demonstrated by our group (Canobbio et al., 2014). We have demonstrated that the first event in Aβ-induced platelet activation is the increase of intracellular Ca2+ concentration. Our results confirmed and extended previous observations by Galeazzi et al., on Aβ25–35-induced intraplatelet Ca2+ mobilization (Galeazzi et al., 2000). Aβ-induced intracellular Ca2+ movements rely on the presence of extracellular Ca2+, this result is in line with the evidence that Aβ is able to form cation permeable pores in the plasma membranes (Pollard et al., 1993) and that Aβ toxicity derives from the deregulation of Ca2+ homeostasis (Demuro et al., 2005). Intracellular Ca2+ increase in platelets promotes acto-myosin cytoskeletal reorganization, granule secretion and ADP release. Once released ADP is able to reinforce and promote platelet activation in term of phosphorylation of selected signaling proteins, mainly the non receptor tyrosine kinase Syk, PI3K, PKC and MLC, activation of the small GTPase Rap1b, inside-out activation of integrin αIIbβ3, and finally aggregation (Canobbio et al., 2014).
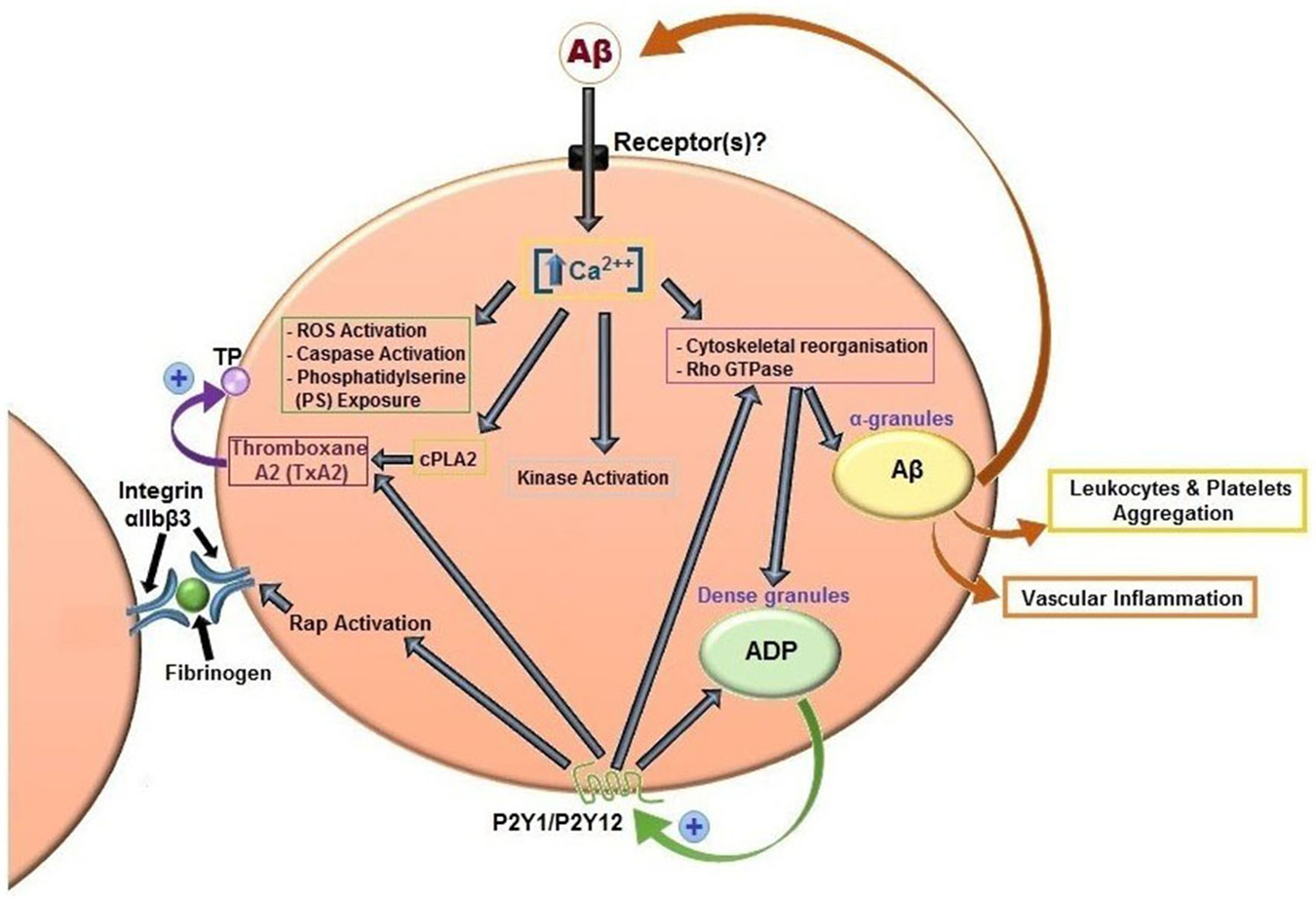
Figure 3. Aβ peptides-induced platelet activation. Aβ peptides present in plasma activate platelets inducing activation of PLC/PKC and intracellular Ca2+ movement, granule secretion, kinase activation, rap1b mediated-integrin activation and aggregation. Aβ peptides released from α-granules, ADP released by dense granules, and formation of TxA2 reinforce platelet activation. Aβ peptides also promotes ROS formation, caspase activation and membrane scrambling. Activated platelets recruit leukocytes and promotes vascular inflammation.
We have demonstrated that platelets are also able to adhere and spread over immobilized Aβ25–35 as well as Aβ40 and Aβ42, to accelerate platelet adhesion over collagen (Canobbio et al., 2013; Gowert et al., 2014), and to increase platelet spreading over fibrinogen (Sonkar et al., 2014). Interestingly, platelets adhere to Aβ peptides independently of ADP but are able to activate several intracellular signaling pathways associated with platelet stimulation dependently on the release of ADP. Aβ peptides also enhance adhesion to collagen under shear (Canobbio et al., 2014) and potentiates thrombus formation in vivo, as revealed in a mouse model of pulmonary thromboembolism (Sonkar et al., 2014). The effect of Aβ in promoting platelet activation in vivo was confirmed in injured carotid artery model (Gowert et al., 2014). More interestingly, the effect of amyloid peptides was analyzed in APP transgenic mice. APP23 and APP Dutch mice are known to develop CAA upon aging and to deposit amyloid peptides in the cerebral vessel walls. Gowert et al. analyzed brain of these transgenic mice for Aβ deposition and platelet recruitment and demonstrated that platelets adhere to vascular amyloid plaques with time and that sustained platelet recruitment may lead to full occlusion of the vessel (Gowert et al., 2014).
Aβ peptides regulate phosphatidylserine exposure, micro-particles production and caspase activation, suggesting a transition of platelets from activation to apoptosis (Gowert et al., 2014). Recently, it has been demonstrated that mitochondrial respiration is increased in Aβ-stimulated platelets (Sonkar et al., 2014). In addition, ROS generation in platelets is significantly increased by exposure to Aβ peptides (Gowert et al., 2014). Moreover, cytochrome c oxidase, an enzyme that belongs to the complex IV of the respiratory chain, has been observed to be diminished in platelets and hippocampal mitochondria of AD patients (Bosetti et al., 2002). This result was confirmed by other studies, and is correlated with increased ROS formation in AD platelets (Cardoso et al., 2004). Taken together, these studies point toward an increased oxidative stress in AD patients. There is therefore the possibility that Aβ peptides stimulate/increase platelet activation by inducing a state of redox stress, which has been shown to increase platelet responsiveness. For example, O2—has been suggested to stimulate platelet hyperactivity in anoxia/reoxygenation conditions (Leo et al., 1997) and hypercholesterolemia (Stokes et al., 2007). The dependence of platelet activation on ROS generation explains the inhibition of platelets and the anti-thrombotic effect of antioxidants (Freedman, 2008) or ROS-generating enzyme inhibitors (Vara et al., 2013).
Table 1 summarize the effects of Aβ peptides on platelet activation.
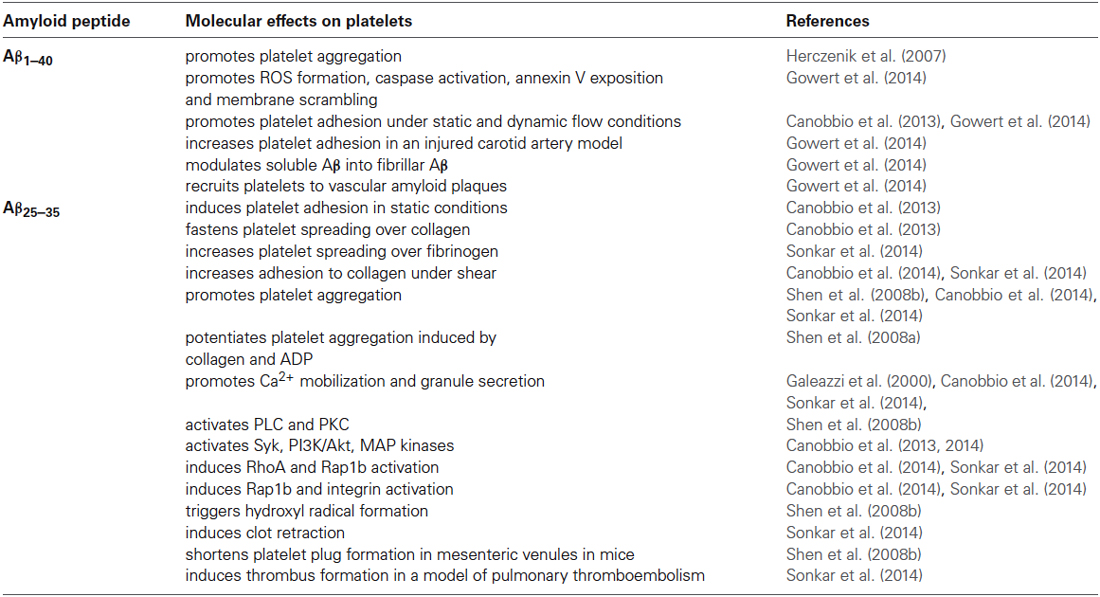
Table 1. Principal molecular effects of amyloid peptides on platelets.
Animal Models of AD and Platelet and Vascular (Dys)Functions
Since the formulation of the “amyloid cascade hypothesis” and the discovery of mutations correlated with FAD, several AD transgenic mice have been developed to study the pathophysiological role of APP and Aβ peptides. In most cases, AD transgenic mice express mutant human APP or PS1/PS2 genes (one single or more mutations correlated with AD onset in FAD). In addition, mice that do not express APP have been generated to investigate the physiological role of APP in cellular signaling. The number of murine model for AD is exponentially growing and for an extensive list of AD model mice we suggest to refer to http://www.alzforum.org/research-models. These mice have been extensively studied as a model for the onset of AD, and to investigate the mechanism underlying amyloid deposition in brain parenchyma and cerebral vessels, their correlation with cognitive impairment, and to develop new therapeutic strategy. However, only few of these murine models have been utilized for analysis of possible peripheral platelet and vascular dysfunctions. The prothrombotic phenotype observed in AD patients and related to pre-activated platelets has been demonstrated in AD transgenic mouse models. Recently, Jarre et al. analyze platelet activation in APP23 mice which carry human APP751 containing the Swedish (KM670/671NL) under the neuronal Thy promoter. These mice deposit Aβ peptides in the brain parenchyma and in the cerebral vessel walls developing CAA upon aging. In this study the authors have demonstrated that platelets of aged Alzheimer transgenic mice APP23 are in a pre-activated state and respond with enhance platelet activation upon stimulation. This finally results in a pro-thrombotic phenotype with altered hemostasis and increased thrombus formation in vivo (Jarre et al., 2014).
Not only platelets, but also cerebrovasculature is activated in AD transgenic mice. Grammas et al. analyzed two transgenic AD animal models: AD2576APPSwe and La Ferla3xTG which overexpressed Aβ in the brain and demonstrated that brain endothelial cells overexpressed Aβ, thrombin, tumor necrosis factor α, interleukin-1β and interleukin 6 and MMP9 (Grammas et al., 2014). Inflamed endothelial cells also release APP770 (Kitazume et al., 2012). Aβ and thrombin expressed from endothelial cells in turn strengthen platelet activation thus reinforcing chronic inflammation and thrombus formation.
Anti Platelet Therapies and AD
Aβ peptides released by platelets and endothelial cells result in platelet activation and vascular inflammation with dangerous consequences for the progression of AD. With this background, it seems reasonable that the use of antiplatelet agents and/or NSAIDs may slow down AD. We have previously reported in this review that treatment with COX2 inhibitors may reduce the risk of AD (Szekely and Zandi, 2010). Epidemiological studies in the past have suggested that patients taking NSAIDs or aspirin display some level of protection from AD (Henderson et al., 1997; Aisen et al., 2000; Broe et al., 2000; in ’t Veld et al., 2002; Thal et al., 2005; ADAPT Research Group et al., 2007). Aspirin is the most common anticoagulant used in prevention of stroke and myocardial infarction (Yeung and Holinstat, 2012; Zhang et al., 2013). Aspirin is an irreversible inhibitor of cyclooxygenase 1 that catalyzes the synthesis of an important platelet agonist thromboxane A2 (Loll et al., 1995). At high doses (5 g per day) aspirin inhibits prostaglandin production by white blood cells and has therefore anti-inflammatory action, whereas at low doses (75 mg) per day has mainly anti platelet activity. Aspirin is commonly used in the treatment of vascular dementia (Molnar et al., 1998). The AD 2000 collaborative group analyzed for 3 years the effect of low dose aspirin daily administration on 156 AD patients compared to 154 AD patients who did not take aspirin. The outcomes evaluated were cognition and functional ability. The results showed no benefits of aspirin on the progression of AD in the analyzed subjects. On the contrary, 8% of patients taking aspirin had serious bleed and 2% of patients in the aspirin group had fatal cerebral bleed (AD2000 Collaborative Group et al., 2008). These results advice against the use of aspirin in AD, since the risks overweigh any benefits. However, more studies on aspirin or other antithrombotic drugs are necessary to understand the potential of these drugs in the treatment of AD. It is conceivable that the administration of antithrombotic or anti-inflammatory drugs is not effective on AD when the pathology is overt, but may be important in prevention of the disease in individuals at risk.
Platelet Abnormalities in AD
Many alterations have been observed in platelets from AD patients compared to age matched control subjects, but attention must be taken when considering the data found in the literature. The most important and reliable changes observed in platelets from AD patients are reduced APP ratio, alteration of α- and β-secretase expression or activity that result in abnormal APP metabolism through the amyloidogenic pathway, and enhanced platelet activation. In addition, changes in expression and/or activity of Monoamine oxidase B (MAO-B), cytochrome c oxidase, and cicloxygenase2 have been reported in platelets isolated from AD patients. For a comprehensive and exhaustive analysis of alterations in platelets from AD patients refer to the recent paper by Veitinger et al. (2014).
Altered APP ratio
APP is present on plasma membrane as an intact glycoprotein of about 110/130 kDa and as soluble fragments of different length in platelet α-granules. Based on the electrophoretic mobility of anti-APP antibody 22C11-immunoreactive bands it is possible to reveal at least two isoforms of APP in platelets: a higher band at 130 kDa and a lower band at about 106/110 kDa. The difference in electrophoretic mobility between the 130 kDa and the 106–110 kDa bands has been attributed to the presence or absence of KPI, respectively. The APP upper to lower band ratio is lower in platelets from patients affected by MCI and SAD compared to platelet prepared from control subjects and patients affected by other kind of dementia (Di Luca et al., 2000; Borroni et al., 2005, 2010). The altered APP ratio in platelets shows a positive and specific correlation to the progression of the disease. This correlation is also present in preclinical stages of AD, suggesting that this can be as a useful biomarker for AD.
Altered secretase activation and APP metabolism
Platelets metabolize APP preferentially through non-amyloidogenic α-secretase pathway. In platelets from AD patients, the expression of α-secretase candidate ADAM10 is decreased compared to healthy subjects (Colciaghi et al., 2002; Tang et al., 2006). On the other hand, an increased β-secretase activity has been indirectly shown by a decreased ratio of its 37/56 kDa fragments (Colciaghi et al., 2004; Tang et al., 2006). The increased β-secretase activity was also demonstrated directly by enzymatic assay in the early stage of AD and MCI patients compared to control (Liu et al., 2007; Johnston et al., 2008), and by increased level of β-secretase product sAPPβ (Colciaghi et al., 2004). Taken together, this switch from non-amyloidogenic to amyloidogenic in the APP pathway is likely to be responsible for the increase in platelet-derived Aβ peptides generation.
Monoamine oxidase-B activity
MAO-B is responsible for the degradation of neurotransmitters in the nervous system (e.g., dopamine) and expressed in platelets (Paasonen et al., 1964). Several studies independently described increase in MAO-B expression and activity in brain and platelets of AD patients (Adolfsson et al., 1980; Bongioanni et al., 1997; Mészáros et al., 1998). The molecular causes of this change are currently unknown.
Platelets are preactivated in AD patients
More than ten years ago, Sevush et al analyzed platelet activation in 91 patients with probable AD and 40 age-matched control subjects. Groups were compared for percentage of circulating platelet aggregates, surface expression of P-selectin (α granule marker), formation of leukocyte-platelet complexes, and presence of circulating platelet microparticles. The results showed a significant increase in platelet aggregates, leukocyte-platelet complexes and P-selectin expression in resting platelets of AD patients demonstrating that platelets of patients with AD exhibit greater basal activation than those of controls (Sevush et al., 1998). More recently, Stellos et al. demonstrated that integrin αIIbβ3 is activated and P-selectin is expressed on plasma membrane in unstimulated platelets from AD subjects compared to control (Stellos et al., 2010; Laske, 2012). More interestingly, in one-year follow up study platelet activation parameters correlate with the rate of cognitive decline and anti-platelet therapy reduced cognitive decline in AD patients (Laske et al., 2010; Sakurai et al., 2013). As we previously reported, activated platelets have been shown to adhere to cerebral vascular deposits of Aβ in AD transgenic mice models, which are known to develop CAA upon aging. Sustain platelet recruitment to vascular amyloid plaques results in occlusion of the vessels (Gowert et al., 2014). The same authors also demonstrated that platelets are able to process soluble synthetic Aβ into fibrillar Aβ increasing its neurotoxicity in culture, which suggests an active role of platelets in promoting neuronal death in AD. Platelet localization at the site of cerebrovascular injury and in CAA lesions has been observed in several other studies, which demonstrated localization of activated platelets with deposit of Aβ peptides (Roher et al., 2009). This is likely to play an important role in the establishment of a vicious circle of platelet activation, Aβ release and neuronal cell death. Once activated, platelets can release Aβ40 in the circulation, increasing its local concentration and contributing to accumulation of amyloid peptides in the vessel of the cerebrovasculature. Amyloid accumulation also induces microvascular inflammation (Grammas and Ovase, 2001). Platelet deposition in the cerebral microvasculature may account for the hemostatic abnormalities observed in AD (Wilkerson and Sane, 2002).
Higher levels of “coated” platelets
Another important parameter of platelet activation is the presence of increased levels of coated platelets in AD patients. Coated platelets are a subset of activated platelets observed upon dual-agonist stimulation with collagen and thrombin characterized by high pro-coagulant activity (Dale, 2005). Prodan et al. demonstrated that coated platelets expressed high levels of full-length APP on their surface compared to single agonist-stimulated platelets (Prodan et al., 2006). Coated platelets are increased in early stages of AD, are elevated in patients with amnestic as compared to nonamnestic MCI, and their numbers correlate with disease progression in AD (Prodan et al., 2007, 2008). Coated platelets are also altered in patients with cerebrovascular disease and have been show to potentiate inflammation. These data suggest a role of coated platelets as a sensitive biomarker of AD (Prodan et al., 2009).
Coagulation abnormalities in AD
Besides platelet hyper activation, coagulation is also impaired in AD patients. In particular it has been observed that Aβ is able to bind fibrinogen and that fibrin clot formed in the presence of Aβ are more stable and more resistant to degradation during fibrinolysis (Ahn et al., 2010; Cortes-Canteli et al., 2010). After BBB alterations fibrinogen may deposit to brain blood vessel and accumulate in CAA and parenchyma in AD patients, and in AD mouse models (Paul et al., 2007). This could finally results in altered cerebral blood flow and worsening of the pro-thrombotic phenotype in cerebral microvasculature of AD patients.
Conclusions and Future Directions
AD is associated with platelet and vascular abnormalities. Our understanding of the mechanisms underlying thrombosis and angiopathy associated with AD is growing exponentially. Several molecular mechanisms that explain microvascular alterations associated with AD have been undercovered. Although the role of tau protein in neuronal death and cognitive ability loss is also central in AD development, the accumulation of amyloid peptides is closely related to the vascular abnormalities associated with this disease. Therefore, this was the subject of this literature review.
Future challenges in this field will be to understand fully the relevance of the microvascular symptoms of AD in dementia development and to target them in order to improve the management of this debilitating disease. This will require a truly multidisciplinary effort with basic scientists and clinicians from different areas of research (i.e., neuroscience, cardiovascular sciences and cell biology) collaborating towards the clarification of the complex etiology of AD. Biomedical research into the vascular aspects of AD provides new hopes for early disease diagnosis and the development of novel clinical strategies to reduce the rate of cognitive function loss following diagnosis.
Conflict of Interest Statement
The Guest Associate Editor Francesco Moccia declares that, despite being affiliated to the same institution as authors Ilaria Canobbio, Aisha Alsheikh Abubaker, Caterina Visconte, Mauro Torti and Giordano Pula, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by ARUK (Alzheimer’s Research UK) [grant number PPG2013B-8 (to GP and IC)] and Biotechnology and Biological Sciences Research Council [grant number BB/J002690/1 (to GP)].
Footnotes
References
AD2000 Collaborative Group, Bentham, P., Gray, R., Sellwood, E., Hills, R., Crome, P., et al. (2008). Aspirin in Alzheimer’s disease (AD2000): a randomised open-label trial. Lancet Neurol. 7, 41–49. doi: 10.1016/s1474-4422(07)70293-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
ADAPT Research Group, Lyketsos, C. G., Breitner, J. C., Green, R. C., Martin, B. K., Meinert, C., et al. (2007). Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808. doi: 10.1212/01.wnl.0000260269.93245.d2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adolfsson, R., Gottfries, C. G., Oreland, L., Wiberg, A., and Winblad, B. (1980). Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 27, 1029–1034. doi: 10.1016/0024-3205(80)90025-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahn, H. J., Zamolodchikov, D., Cortes-Canteli, M., Norris, E. H., Glickman, J. F., and Strickland, S. (2010). Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U S A 107, 21812–21817. doi: 10.1073/pnas.1010373107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahtiluoto, S., Polvikoski, T., Peltonen, M., Solomon, A., Tuomilehto, J., and Winblad, B. (2010). Diabetes, Alzheimer disease and vascular dementia: a population-based neuropathologic study. Neurology 75, 1195–1202. doi: 10.1212/WNL.0b013e3181f4d7f8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aisen, P. S., Davis, K. L., Berg, J. D., Schafer, K., Campbell, K., and Thomas, R. G. (2000). A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s disease cooperative study. Neurology 54, 588–593. doi: 10.1212/WNL.54.3.588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Akiyama, H., Ikeda, K., Kondo, H., and McGeer, P. L. (1992). Thrombin accumulation in brains of patients with Alzheimer’s disease. Neurosci. Lett. 146, 152–154. doi: 10.1016/0304-3940(92)90065-f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Askarova, S., Yang, X., Sheng, W., Sun, G. Y., and Lee, J. C. (2011). Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A2 activation in astrocytes and cerebral endothelial cells. Neuroscience 199, 375–385. doi: 10.1016/j.neuroscience.2011.09.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, T. L., Rivara, C. B., Rocher, A. B., and Hof, P. R. (2004). The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol. Res. 26, 573–578. doi: 10.1179/016164104225016272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beckman, J. S., and Koppenol, W. H. (1996). Nitric oxide, superoxide and peroxynitrite: the good, the bad and ugly. Am. J. Physiol. 271, C1424–C1437.
Beher, D., Hesse, L., Masters, C. L., and Multhaup, G. (1996). Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J. Biol. Chem. 271, 1613–1620. doi: 10.1074/jbc.271.3.1613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berridge, M. J. (2013). Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 7, 2–13. doi: 10.4161/pri.21767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blennow, K., Dubois, B., Fagan, A. M., Lewczuk, P., de Leon, M. J., and Hampel, H. (2015). Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement. 11, 58–69. doi: 10.1016/j.jalz.2014.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bongioanni, P., Gemignani, F., Boccardi, B., Borgna, M., and Rossi, B. (1997). Platelet monoamine oxidase molecular activity in demented patients. Ital. J. Neurol. Sci. 18, 151–156. doi: 10.1007/bf02048483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Agosti, C., Marcello, E., Di Luca, M., and Padovani, A. (2010). Blood cell markers in Alzheimer disease: amyloid precursor protein form ratio in platelets. Exp. Gerontol. 45, 53–56. doi: 10.1016/j.exger.2009.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Akkawi, N., Martini, G., Colciaghi, F., Prometti, P., Rozzini, L., et al. (2002a). Microvascular damage and platelet abnormalities in early Alzheimer’s disease. J. Neurol. Sci. 203–204, 189–193. doi: 10.1016/s0022-510x(02)00289-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Perani, D., Broli, M., Colciaghi, F., Garibotto, V., Paghera, B. C., et al. (2005). Pre-clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J. Neurol. 252, 1359–1362. doi: 10.1007/s00415-005-0867-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Volpi, R., Martini, G., Del Bono, R., Archetti, S., Colciaghi, F., et al. (2002b). Peripheral blood abnormalities in Alzheimer disease: evidence for early endothelial dysfunction. Alzheimer Dis. Assoc. Disord. 16, 150–155. doi: 10.1097/00002093-200207000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., et al. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 23, 371–376. doi: 10.1016/s0197-4580(01)00314-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breen, K. C., Bruce, M., and Anderton, B. H. (1991). Beta amyloid precursor protein mediates neuronal cell-cell and cell-surface adhesion. J. Neurosci. Res. 28, 90–100. doi: 10.1002/jnr.490280109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Broe, G. A., Grayson, D. A., Creasey, H. M., Waite, L. M., Casey, B. J., Bennett, H. P., et al. (2000). Anti-inflammatory drugs protect against Alzheimer disease at low doses. Arch. Neurol. 57, 1586–1591. doi: 10.1001/archneur.57.11.1586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, W. R., and Thore, C. R. (2011). Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 37, 56–74. doi: 10.1111/j.1365-2990.2010.01139.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brundel, M., Heringa, S. M., de Bresser, J., Koek, H. L., Zwanenburg, J. J., Jaap Kappelle, L., et al. (2012). High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer’s disease. J. Alzheimers Dis. 31, 259–263. doi: 10.3233/JAD-2012-120364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buxbaum, J. D., Thinakaran, G., Koliatsos, V., O’Callahan, J., Slunt, H. H., Price, D. L., et al. (1998). Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J. Neurosci. 18, 9629–9637.
Cai, H., Griendling, K. K., and Harrison, D. G. (2003). The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 24, 471–478. doi: 10.1016/s0165-6147(03)00233-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canobbio, I., Catricalà, S., Balduini, C., and Torti, M. (2011). Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim. Biophys. Acta 1813, 500–506. doi: 10.1016/j.bbamcr.2010.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canobbio, I., Catricalà, S., Di Pasqua, L. G., Guidetti, G., Consonni, A., Manganaro, D., et al. (2013). Immobilized amyloid Aβ peptides support platelet adhesion and activation. FEBS Lett. 587, 2606–2611. doi: 10.1016/j.febslet.2013.06.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canobbio, I., Guidetti, G. F., Oliviero, B., Manganaro, D., Vara, D., Torti, M., et al. (2014). Amyloid β-peptide-dependent activation of human platelets: essential role for Ca2+ and ADP in aggregation and thrombus formation. Biochem. J. 462, 513–523. doi: 10.1042/BJ20140307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cardoso, S. M., Proença, M. T., Santos, S., Santana, I., and Oliveira, C. R. (2004). Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 25, 105–110. doi: 10.1016/s0197-4580(03)00033-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carvalho, C., Correia, S. C., Santos, R. X., Cardoso, S., Moreira, P. I., Clark, T. A., et al. (2009). Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J. Bioenerg. Biomembr. 41, 433–440. doi: 10.1007/s10863-009-9247-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casserly, I. P., and Topol, E. J. (2004). Convergence of atherosclerosis and alzheimer’s disease: cholesterol, inflammation and misfolded proteins. Discov. Med. 4, 149–156.
Catricalà, S., Torti, M., and Ricevuti, G. (2012). Alzheimer disease and platelets: how’s that relevant. Immun. Ageing 9:20. doi: 10.1186/1742-4933-9-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, N. F., Chien, L. N., Ku, H. L., Hu, C. J., and Chiou, H. Y. (2013). Alzheimer disease and risk of stroke: a population-based cohort study. Neurology 80, 705–711. doi: 10.1212/WNL.0b013e31828250af
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christov, A., Ottman, J., Hamdheydari, L., and Grammas, P. (2008). Structural changes in Alzheimer’s disease brain microvessels. Curr. Alzheimer Res. 5, 392–395. doi: 10.2174/156720508785132334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colciaghi, F., Borroni, B., Pastorino, L., Marcello, E., Zimmermann, M., Cattabeni, F., et al. (2002). [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 8, 67–74.
Colciaghi, F., Marcello, E., Borroni, B., Zimmermann, M., Caltagirone, C., Cattabeni, F., et al. (2004). Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology 62, 498–501. doi: 10.1212/01.wnl.0000106953.49802.9c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortes-Canteli, M., Paul, J., Norris, E. H., Bronstein, R., Ahn, H. J., Zamolodchikov, D., et al. (2010). Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron 66, 695–709. doi: 10.1016/j.neuron.2010.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dale, G. L. (2005). Coated-platelets: an emerging component of the procoagulant response. J. Thromb. Haemost. 3, 2185–2192. doi: 10.1111/j.1538-7836.2005.01274.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davies, T. A., Long, H. J., Eisenhauer, P. B., Hastey, R., Cribbs, D. H., Fine, R. E., et al. (2000). Beta amyloid fragments derived from activated platelets deposit in cerebrovascular endothelium: usage of a novel blood brain barrier endothelial cell model system. Amyloid 7, 153–165. doi: 10.3109/13506120009146830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deane, R., Wu, Z., and Zlokovic, B. V. (2004). RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35, 2628–2631. doi: 10.1161/01.str.0000143452.85382.d1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Bock, M., Wang, N., Decrock, E., Bol, M., Gadicherla, A. K., Culot, M., et al. (2013). Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 108, 1–20. doi: 10.1016/j.pneurobio.2013.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeFronzo, R. A., and Ferrannini, E. (1991). Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care 14, 173–194. doi: 10.2337/diacare.14.3.173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Torre, J. C. (2000). Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol. Aging 21, 331–342. doi: 10.1016/s0197-4580(00)00111-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Torre, J. C. (2004a). Alzheimer’s disease is a vasocognopathy: a new term to describe its nature. Neurol. Res. 26, 517–524. doi: 10.1179/016164104225016254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Torre, J. C. (2004b). Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 3, 184–190. doi: 10.1016/S1474-4422(04)00683-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Torre, J. C. (2010). Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res. Rev. 9, 218–225. doi: 10.1016/j.arr.2010.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de la Torre, J. C., and Mussivand, T. (1993). Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 15, 146–153.
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300. doi: 10.1074/jbc.m500997200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Luca, M., Colciaghi, F., Pastorino, L., Borroni, B., Padovani, A., and Cattabeni, F. (2000). Platelets as a peripheral district where to study pathogenetic mechanisms of alzheimer disease: the case of amyloid precursor protein. Eur. J. Pharmacol. 405, 277–283. doi: 10.1016/s0014-2999(00)00559-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esch, F. S., Keim, P. S., Beattie, E. C., Blacher, R. W., Culwell, A. R., Oltersdorf, T., et al. (1990). Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248, 1122–1124. doi: 10.1126/science.2111583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evin, G., Zhu, A., Holsinger, R. M., Masters, C. L., and Li, Q. X. (2003). Proteolytic processing of the Alzheimer’s disease amyloid precursor protein in brain and platelets. J. Neurosci. Res. 74, 386–392. doi: 10.1002/jnr.10745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 64, 575–611. doi: 10.1016/s0301-0082(00)00068-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fonseca, A. C., Ferreiro, E., Oliveira, C. R., Cardoso, S. M., and Pereira, C. F. (2013). Activation of the endoplasmic reticulum stress response by the amyloid-beta 1–40 peptide in brain endothelial cells. Biochim. Biophys. Acta 1832, 2191–2203. doi: 10.1016/j.bbadis.2013.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fonseca, A. C., Moreira, P. I., Oliveira, C. R., Cardoso, S. M., Pinton, P., and Pereira, C. F. (2014). Amyloid-Beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol. doi: 10.1007/s12035-014-8740-7. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freedman, J. E. (2008). Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 28, s11–s16. doi: 10.1161/ATVBAHA.107.159178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galeazzi, L., Casoli, T., Giunta, S., Fattoretti, P., Gracciotti, N., Caselli, U., et al. (2000). β-amyloid fragment 25–35 induces changes in cytosolic free calcium in human platelets. Ann. N Y Acad. Sci. 903, 451–456. doi: 10.1111/j.1749-6632.2000.tb06398.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gandy, S. (2005). The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J. Clin. Invest. 115, 1121–1129. doi: 10.1172/jci200525100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garwood, C., Faizullabhoy, A., Wharton, S. B., Ince, P. G., Heath, P. R., Shaw, P. J., et al. (2013). Calcium dysregulation in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 39, 788–799. doi: 10.1111/nan.12033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghiso, J., Rostagno, A., Gardella, J. E., Liem, L., Gorevic, P. D., and Frangione, B. (1992). A 109-amino-acid C-terminal fragment of Alzheimer’s-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem. J. 288, 1053–1059.
Giri, R., Shen, Y., Stins, M., Du Yan, S., Schmidt, A. M., Stern, D., et al. (2000). Beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am. J. Physiol. Cell Physiol. 279, C1772–C1781.
Glabe, C. G. (2006). Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575. doi: 10.1016/j.neurobiolaging.2005.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. doi: 10.1016/s0006-291x(84)80190-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorelick, P. B. (2004). Risk factors for vascular dementia and Alzheimer disease. Stroke 35, 2620–2622. doi: 10.1161/01.str.0000143318.70292.47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gowert, N. S., Donner, L., Chatterjee, M., Eisele, Y. S., Towhid, S. T., Münzer, P., et al. (2014). Blood platelets in the progression of Alzheimer’s disease. PLoS One 9:e90523. doi: 10.1371/journal.pone.0090523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grammas, P. (2011). Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflammation 8:26. doi: 10.1186/1742-2094-8-26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grammas, P., Botchlet, T., Fugate, R., Ball, M. J., and Roher, A. E. (1995). Alzheimer disease amyloid proteins inhibit brain endothelial cell proliferation in vitro. Dementia 6, 126–130. doi: 10.1159/000106934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grammas, P., Martinez, J., Sanchez, A., Yin, X., Riley, J., Gay, D., et al. (2014). A new paradigm for the treatment of Alzheimer’s disease: targeting vascular activation. J. Alzheimers Dis. 40, 619–630. doi: 10.3233/JAD-2014-132057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grammas, P., and Ovase, R. (2001). Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 22, 837–842. doi: 10.1016/s0197-4580(01)00276-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grammas, P., Yamada, M., and Zlokovic, B. (2002). The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 4, 217–223.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, S., Sato, N., Yamamoto, A., Ikegame, Y., Nakashima, S., Ogihara, T., et al. (2009). Alzheimer disease-associated peptide, amyloid beta40, inhibits vascular regeneration with induction of endothelial autophagy. Arterioscler. Thromb. Vasc. Biol. 29, 1909–1915. doi: 10.1161/ATVBAHA.109.188516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henderson, A. S., Jorm, A. F., Christensen, H., Jacomb, P. A., and Korten, A. E. (1997). Aspirin, anti-inflammatory drugs and risk of dementia. Int. J. Geriatr. Psychiatry 12, 926–930. doi: 10.1002/(sici)1099-1166(199709)12:9<926::aid-gps665>3.3.co;2-p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henry, A., Li, Q. X., Galatis, D., Hesse, L., Multhaup, G., Beyreuther, K., et al. (1998). Inhibition of platelet activation by the Alzheimer’s disease amyloid precursor protein. Br. J. Haematol. 103, 402–415. doi: 10.1046/j.1365-2141.1998.01005.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herczenik, E., Bouma, B., Korporaal, S. J., Strangi, R., Zeng, Q., Gros, P., et al. (2007). Activation of human platelets by misfolded proteins. Arterioscler. Thromb. Vasc. Biol. 27, 1657–1665. doi: 10.1161/atvbaha.107.143479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Honjo, K., Black, S. E., and Verhoeff, N. P. (2012). Alzheimer’s disease, cerebrovascular disease and the β-amyloid cascade. Can. J. Neurol. Sci. 39, 712–728. doi: 10.1017/s0317167100015547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikonomovic, M. D., Klunk, W. E., Abrahamson, E. E., Mathis, C. A., Price, J. C., Tsopelas, N. D., et al. (2008). Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131, 1630–1645. doi: 10.1093/brain/awn016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
in ’t Veld, B. A., Launer, L. J., Breteler, M. M., Hofman, A., and Stricker, B. H. (2002). Pharmacologic agents associated with a preventive effect on Alzheimer’s disease: a review of the epidemiologic evidence. Epidemiol. Rev. 24, 248–268. doi: 10.1093/epirev/mxf001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwatsubo, T. (2004). The gamma-secretase complex: machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 14, 379–383. doi: 10.1016/s0959-4388(04)00077-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jarre, A., Gowert, N. S., Donner, L., Münzer, P., Klier, M., Borst, O., et al. (2014). Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer’s disease. Cell. Signal. 26, 2040–2050. doi: 10.1016/j.cellsig.2014.05.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm. 109, 813–836. doi: 10.1007/s007020200068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jellinger, K. A. (2010). Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neurodegener. Dis. 7, 112–115. doi: 10.1159/000285518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnston, J. A., Liu, W. W., Coulson, D. T., Todd, S., Murphy, S., Brennan, S., et al. (2008). Platelet beta-secretase activity is increased in Alzheimer’s disease. Neurobiol. Aging 29, 661–668. doi: 10.1016/j.neurobiolaging.2006.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalaria, R. N. (2010). Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr. Rev. 68(Suppl. 2), S74–S87. doi: 10.1111/j.1753-4887.2010.00352.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalaria, R. N., and Pax, A. B. (1995). Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res. 705, 349–352. doi: 10.1016/0006-8993(95)01250-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaminsky, Y. G., Marlatt, M. W., Smith, M. A., and Kosenko, E. A. (2010). Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Aβ(25–35). Exp. Neurol. 221, 26–37. doi: 10.1016/j.expneurol.2009.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kara, F., Dongen, E. S., Schliebs, R., Buchem, M. A., Groot, H. J., and Alia, A. (2012). Monitoring blood flow alterations in the Tg2576 mouse model of Alzheimer’s disease by in vivo magnetic resonance angiography at 17.6 T. Neuroimage 60, 958–966. doi: 10.1016/j.neuroimage.2011.12.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelleher, R. J., and Soiza, R. L. (2013). Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 3, 197–226.
Kennedy, J. L., Farrer, L. A., Andreasen, N. C., Mayeux, R., and St George-Hyslop, P. (2003). The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science 302, 822–826. doi: 10.1126/science.1092132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitazume, S., Yoshihisa, A., Yamaki, T., Oikawa, M., Tachida, Y., Ogawa, K., et al. (2012). Soluble amyloid precursor protein 770 is released from inflamed endothelial cells and activated platelets: a novel biomarker for acute coronary syndrome. J. Biol. Chem. 287, 40817–40825. doi: 10.1074/jbc.M112.398578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kokjohn, T. A., Van Vickle, G. D., Maarouf, C. L., Kalback, W. M., Hunter, J. M., Daugs, I. D., et al. (2011). Chemical characterization of pro-inflammatory amyloid-beta peptides in human atherosclerotic lesions and platelets. Biochim. Biophys. Acta 1812, 1508–1514. doi: 10.1016/j.bbadis.2011.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kook, S. Y., Hong, H. S., Moon, M., Ha, C. M., Chang, S., and Mook-Jung, I. (2012). Aβ1–42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. J. Neurosci. 32, 8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuo, Y. M., Kokjohn, T. A., Watson, M. D., Woods, A. S., Cotter, R. J., Sue, L. I., et al. (2000). Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am. J. Pathol. 156, 797–805. doi: 10.1016/s0002-9440(10)64947-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laske, C. (2012). Alzheimer disease: blood-based biomarkers in AD–a silver lining on the horizon. Nat. Rev. Neurol. 8, 541–542. doi: 10.1038/nrneurol.2012.173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laske, C., Sopova, K., Gkotsis, C., Eschweiler, G. W., Straten, G., Gawaz, M., et al. (2010). Amyloid-β peptides in plasma and cognitive decline after 1 year follow-up in Alzheimer’s disease patients. J. Alzheimers Dis. 21, 1263–1269.
Lee, H. G., Casadesus, G., Zhu, X., Joseph, J. A., Perry, G., and Smith, M. A. (2004). Perspectives on the amyloid-beta cascade hypothesis. J. Alzheimers Dis. 6, 137–145.
Lee, M. J., Seo, S. W., Na, D. L., Kim, C., Park, J. H., Kim, G. H., et al. (2014). Synergistic effects of ischemia and β-amyloid burden on cognitive decline in patients with subcortical vascular mild cognitive impairment. JAMA Psychiatry 71, 412–422. doi: 10.1001/jamapsychiatry.2013.4506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leo, R., Praticò, D., Iuliano, L., Pulcinelli, F. M., Ghiselli, A., Pignatelli, P., et al. (1997). Platelet activation by superoxide anion and hydroxyl radicals intrinsically generated by platelets that had undergone anoxia and then reoxygenated. Circulation 95, 885–891. doi: 10.1161/01.cir.95.4.885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Q. X., Berndt, M. C., Bush, A. I., Rumble, B., Mackenzie, I., and Friedhuber, A. (1994). Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer’s disease in human platelet and brain: surface expression on the activated human platelet. Blood 84, 133–142.
Li, J. C., Han, L., Wen, Y. X., Yang, Y. X., Li, S., Li, X. S., et al. (2015). Increased permeability of the blood-brain barrier and Alzheimer’s disease-like alterations in slit-2 transgenic mice. J. Alzheimers Dis. 43, 535–548. doi: 10.3233/JAD-141215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., Shang, D. S., Zhao, W. D., Tian, L., Li, B., Fang, W. G., et al. (2009). Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J. Immunol. 182, 5778–5788. doi: 10.4049/jimmunol.0803013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Q. X., Whyte, S., Tanner, J. E., Evin, G., Beyreuther, K., and Masters, C. L. (1998). Secretion of Alzheimer’s disease Abeta amyloid peptide by activated human platelets. Lab. Invest. 78, 461–469.
Liu, W. W., Todd, S., Craig, D., Passmore, A. P., Coulson, D. T., Murphy, S., et al. (2007). Elevated platelet β-secretase activity in mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 24, 464–468. doi: 10.1159/000110739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loll, P. J., Picot, D., and Garavito, R. M. (1995). The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat. Struct. Biol. 2, 637–643. doi: 10.1038/nsb0895-637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lorenzo, A., Yuan, M., Zhang, Z., Paganetti, P. A., Sturchler-Pierrat, C., Staufenbiel, M., et al. (2000). Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer’s disease. Nat. Neurosci. 3, 460–464. doi: 10.1038/74833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lue, L. F., Walker, D. G., Brachova, L., Beach, T. G., Rogers, J., Schmidt, A. M., et al. (2001). Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp. Neurol. 171, 29–45. doi: 10.1006/exnr.2001.7732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Menting, K. W., and Claassen, J. A. (2014). β-secretase inhibitor; a promising novel therapeutic drug in Alzheimer’s disease. Front. Aging Neurosci. 6:165. doi: 10.3389/fnagi.2014.00165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mészáros, Z., Borcsiczky, D., Máté, M., Tarcali, J., Szombathy, T., Tekes, K., et al. (1998). Platelet MAO-B activity and serotonin content in patients with dementia: effect of age, medication and disease. Neurochem. Res. 23, 863–868. doi: 10.1023/A:1022458928442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mhatre, M., Nguyen, A., Kashani, S., Pham, T., Adesina, A., and Grammas, P. (2004). Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol. Aging 25, 783–793. doi: 10.1016/s0197-4580(03)00192-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michaelson, D. M. (2014). APOE ε4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 10, 861–868. doi: 10.1016/j.jalz.2014.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, T. W., Isenberg, J. S., Shih, H. B., Wang, Y., and Roberts, D. D. (2010). Amyloid-β inhibits No-cGMP signaling in a CD36- and CD47-dependent manner. PLoS One 5:e15686. doi: 10.1371/journal.pone.0015686
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milward, E. A., Papadopoulos, R., Fuller, S. J., Moir, R. D., Small, D., Beyreuther, K., et al. (1992). The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron 9, 129–137. doi: 10.1016/0896-6273(92)90228-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molnar, F. J., Man-Son-Hing, M., St John, P., Brymer, C., Rockwood, K., and Hachinski, V. (1998). Subcortical vascular dementia: survey of treatment patterns and research considerations. Can. J. Neurol. Sci. 25, 320–324.
Morris, J. K., Honea, R. A., Vidoni, E. D., Swerdlow, R. H., and Burns, J. M. (2014). Is Alzheimer’s disease a systemic disease?. Biochim. Biophys. Acta 1842, 1340–1349. doi: 10.1016/j.bbadis.2014.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, M. P., and LeVine, H. 3rd (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakayama, K., Nagase, H., Koh, C. S., and Ohkawara, T. (2011). γ-Secretase-regulated mechanisms similar to notch signaling may play a role in signaling events, including APP signaling, which leads to Alzheimer’s disease. Cell. Mol. Neurobiol. 31, 887–900. doi: 10.1007/s10571-011-9688-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Okamoto, T., Takeda, S., Murayama, Y., Ogata, E., and Nishimoto, I. (1995). Ligand-dependent G protein coupling function of amyloid transmembrane precursor. J. Biol. Chem. 270, 4205–4208. doi: 10.1074/jbc.270.9.4205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paasonen, M. K., Solatunturi, E., and Kivalo, E. (1964). Monoamine oxidase activity of blood platelets and their ability to store 5-hydroxytryptamine in some mental deficiencies. Psychopharmacologia 6, 120–124. doi: 10.1007/bf00413224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paris, D., Patel, N., DelleDonne, A., Quadros, A., Smeed, R., and Mullan, M. (2004). Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci. Lett. 366, 80–85. doi: 10.1016/j.neulet.2004.05.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, L., Anrather, J., Zhou, P., Frys, K., Pitstick, R., Younkin, S., et al. (2005). NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. 25, 1769–1777. doi: 10.1523/jneurosci.5207-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, K. W., and Jin, B. K. (2008). Thrombin-induced oxidative stress contributes to the death of hippocampal neurons: role of neuronal NADPH oxidase. J. Neurosci. Res. 86, 1053–1063. doi: 10.1002/jnr.21571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paul, J., Strickland, S., and Melchor, J. P. (2007). Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med. 204, 1999–2008. doi: 10.1084/jem.20070304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pogue, A. I., and Lukiw, W. J. (2004). Angiogenic signaling in Alzheimer’s disease. Neuroreport 15, 1507–1510. doi: 10.1097/01.wnr.0000130539.39937.1d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pollard, H. B., Rojas, E., and Arispe, N. (1993). A new hypothesis for the mechanism of amyloid toxicity, based on the calcium channel activity of amyloid beta protein (AβP) in phospholipid bilayer membranes. Ann. N Y Acad. Sci. 695, 165–168. doi: 10.1111/j.1749-6632.1993.tb23046.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Price, J. M., Chi, X., Hellermann, G., and Sutton, E. T. (2001). Physiological levels of beta-amyloid induce cerebral vessel dysfunction and reduce endothelial nitric oxide production. Neurol. Res. 23, 506–512. doi: 10.1179/016164101101198758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prodan, C. I., Ross, E. D., Vincent, A. S., and Dale, G. L. (2007). Coated-platelets are higher in amnestic versus nonamnestic patients with mild cognitive impairment. Alzheimer Dis. Assoc. Disord. 21, 259–261. doi: 10.1097/wad.0b013e31811ec11f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prodan, C. I., Ross, E. D., Vincent, A. S., and Dale, G. L. (2008). Rate of progression in Alzheimer’s disease correlates with coated-platelet levels–a longitudinal study. Transl. Res. 152, 99–102. doi: 10.1016/j.trsl.2008.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prodan, C. I., Szasz, R., Vincent, A. S., Ross, E. D., and Dale, G. L. (2006). Coated-platelets retain amyloid precursor protein on their surface. Platelets 17, 56–60. doi: 10.1080/09537100500181913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prodan, C. I., Vincent, A. S., Padmanabhan, R., and Dale, G. L. (2009). Coated-platelet levels are low in patients with spontaneous intracerebral hemorrhage. Stroke 40, 2578–2580. doi: 10.1161/STROKEAHA.109.549014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purandare, N., and Burns, A. (2009). Cerebral emboli in the genesis of dementia. J. Neurol. Sci. 283, 17–20. doi: 10.1016/j.jns.2009.02.306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, H. V., Thirumangalakudi, L., and Grammas, P. (2009). Cyclin C and cyclin dependent kinases 1, 2 and 3 in thrombin-induced neuronal cell cycle progression and apoptosis. Neurosci. Lett. 450, 347–350. doi: 10.1016/j.neulet.2008.12.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reznik-Wolf, H., Machado, J., Haroutunian, V., DeMarco, L., Walter, G. F., and Goldman, B. (1998). Somatic mutation analysis of the APP and Presenilin 1 and 2 genes in Alzheimer’s disease brains. J. Neurogenet. 12, 55–65. doi: 10.3109/01677069809108555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roher, A. E., Esh, C. L., Kokjohn, T. A., Castaño, E. M., Van Vickle, G. D., Kalback, W. M., et al. (2009). Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 5, 18–29. doi: 10.1016/j.jalz.2008.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roher, A. E., Esh, C., Kokjohn, T. A., Kalback, W., Luehrs, D. C., Seward, J. D., et al. (2003). Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 23, 2055–2062. doi: 10.1161/01.atv.0000095973.42032.44
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rowley, J. W., Oler, A. J., Tolley, N. D., Hunter, B. N., Low, E. N., Nix, D. A., et al. (2011). Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 118, e101–e111. doi: 10.1182/blood-2011-03-339705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakurai, H., Hanyu, H., Sato, T., Kume, K., Hirao, K., Kanetaka, H., et al. (2013). Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: a pilot study. Geriatr. Gerontol. Int. 13, 90–97. doi: 10.1111/j.1447-0594.2012.00866.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, N., Toki, S., Chowei, H., Saito, T., Nakano, N., Hayashi, Y., et al. (2001). Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 888, 256–262. doi: 10.1016/s0006-8993(00)03075-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scandura, J. M., Zhang, Y., Van Nostrand, W. E., and Walsh, P. N. (1997). Progress curve analysis of the kinetics with which blood coagulation factor XIa is inhibited by protease nexin-2. Biochemistry 36, 412–420. doi: 10.1021/bi9612576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheuermann, S., Hambsch, B., Hesse, L., Stumm, J., Schmidt, C., Beher, D., et al. (2001). Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J. Biol. Chem. 276, 33923–33929. doi: 10.1074/jbc.m105410200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmaier, A. H., Dahl, L. D., Hasan, A. A., Cines, D. B., Bauer, K. A., and Van Nostrand, W. E. (1995). Factor IXa inhibition by protease nexin-2/amyloid beta-protein precursor on phospholipid vesicles and cell membranes. Biochemistry 34, 1171–1178. doi: 10.1021/bi00004a010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., et al. (1992). Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 359, 325–327. doi: 10.1038/359325a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sevush, S., Jy, W., Horstman, L. L., Mao, W. W., Kolodny, L., and Ahn, Y. S. (1998). Platelet activation in Alzheimer disease. Arch. Neurol. 55, 530–536. doi: 10.1001/archneur.55.4.530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, M. Y., Hsiao, G., Fong, T. H., Chen, H. M., Chou, D. S., Lin, C. H., et al. (2008a). Amyloid beta peptide-activated signal pathways in human platelets. Eur. J. Pharmacol. 588, 259–266. doi: 10.1016/j.ejphar.2008.04.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, M. Y., Hsiao, G., Fong, T. H., Chou, D. S., and Sheu, J. R. (2008b). Expression of amyloid beta peptide in human platelets: pivotal role of the phospholipase Cgamma2-protein kinase C pathway in platelet activation. Pharmacol. Res. 57, 151–158. doi: 10.1016/j.phrs.2008.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smirnova, I. V., Zhang, S. X., Citron, B. A., Arnold, P. M., and Festoff, B. W. (1998). Thrombin is an extracellular signal that activates intracellular death protease pathways inducing apoptosis in model motor neurons. J. Neurobiol. 36, 64–80. doi: 10.1002/(sici)1097-4695(199807)36:1<64::aid-neu6>3.3.co;2-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, R. P., Higuchi, D. A., and Broze, G. J. Jr. (1990). Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science 248, 1126–1128. doi: 10.1126/science.2111585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smolarkiewicz, M., Skrzypczak, T., and Wojtaszek, P. (2013). The very many faces of presenilins and the γ-secretase complex. Protoplasma 250, 997–1011. doi: 10.1007/s00709-013-0494-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sola Vigo, F., Kedikian, G., Heredia, L., Heredia, F., Añel, A. D., Rosa, A. L., et al. (2009). Amyloid-beta precursor protein mediates neuronal toxicity of amyloid beta through Go protein activation. Neurobiol. Aging 30, 1379–1392. doi: 10.1016/j.neurobiolaging.2007.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sonkar, V. K., Kulkarni, P. P., and Dash, D. (2014). Amyloid β peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. FASEB J. 28, 1819–1829. doi: 10.1096/fj.13-243691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sorrentino, P., Iuliano, A., Polverino, A., Jacini, F., and Sorrentino, G. (2014). The dark sides of amyloid in Alzheimer’s disease pathogenesis. FEBS Lett. 588, 641–652. doi: 10.1016/j.febslet.2013.12.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stellos, K., Panagiota, V., Kögel, A., Leyhe, T., Gawaz, M., and Laske, C. (2010). Predictive value of platelet activation for the rate of cognitive decline in Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 30, 1817–1820. doi: 10.1038/jcbfm.2010.140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stewart, P. A., Hayakawa, K., Akers, M. A., and Vinters, H. V. (1992). A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab. Invest. 67, 734–742.
Stokes, K. Y., Russell, J. M., Jennings, M. H., Alexander, J. S., and Granger, D. N. (2007). Platelet-associated NAD(P)H oxidase contributes to the thrombogenic phenotype induced by hypercholesterolemia. Free Radic. Biol. Med. 43, 22–30. doi: 10.1016/j.freeradbiomed.2007.02.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suo, Z., Wu, M., Citron, B. A., Palazzo, R. E., and Festoff, B. W. (2003). Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J. Biol. Chem. 278, 37681–37689. doi: 10.1074/jbc.m301406200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sutcliffe, J. G., Hedlund, P. B., Thomas, E. A., Bloom, F. E., and Hilbush, B. S. (2011). Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid: implications for Alzheimer’s disease. J. Neurosci. Res. 89, 808–814. doi: 10.1002/jnr.22603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szekely, C. A., and Zandi, P. P. (2010). Non-steroidal anti-inflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol. Disord. Drug Targets. 9, 132–139. doi: 10.2174/187152710791012026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takuma, K., Fang, F., Zhang, W., Yan, S., Fukuzaki, E., Du, H., et al. (2009). RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. U S A 106, 20021–20026. doi: 10.1073/pnas.0905686106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, K., Hynan, L. S., Baskin, F., and Rosenberg, R. N. (2006). Platelet amyloid precursor protein processing: a bio-marker for Alzheimer’s disease. J. Neurol. Sci. 240, 53–58. doi: 10.1016/j.jns.2005.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thal, L. J., Ferris, S. H., Kirby, L., Block, G. A., Lines, C. R., Yuen, E., et al. (2005). A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30, 1204–1215. doi: 10.1038/sj.npp.1300690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thal, D. R., Ghebremedhin, E., Orantes, M., and Wiestler, O. D. (2003). Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropathol. Exp. Neurol. 62, 1287–1301.
The Lancet Neurology. (2010). How much is dementia care worth? Lancet Neurol. 9:1037. doi: 10.1016/S1474-4422(10)70257-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thirumangalakudi, L., Samany, P. G., Owoso, A., Wiskar, B., and Grammas, P. (2006). Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimers Dis. 10, 111–118.
Tibolla, G., Norata, G. D., Meda, C., Arnaboldi, L., Uboldi, P., Piazza, F., et al. (2010). Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis 210, 78–87. doi: 10.1016/j.atherosclerosis.2009.10.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tjernberg, L. O., Callaway, D. J., Tjernberg, A., Hahne, S., Lilliehöök, C., Terenius, L., et al. (1999). A molecular model of Alzheimer amyloid beta-peptide fibril formation. J. Biol. Chem. 274, 12619–12625. doi: 10.1074/jbc.274.18.12619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tolppanen, A. M., Lavikainen, P., Solomon, A., Kivipelto, M., Soininen, H., and Hartikainen, S. (2013). Incidence of stroke in people with Alzheimer disease: a national register-based approach. Neurology 80, 353–358. doi: 10.1212/WNL.0b013e31827f08c5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tripathy, D., Sanchez, A., Yin, X., Luo, J., Martinez, J., and Grammas, P. (2013). Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 5:19. doi: 10.3389/fnagi.2013.00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Truran, S., Franco, D. A., Roher, A. E., Beach, T. G., Burciu, C., Serrano, G., et al. (2014). Adipose and leptomeningeal arteriole endothelial dysfunction induced by β-amyloid peptide: a practical human model to study Alzheimer’s disease vasculopathy. J. Neurosci. Methods 235, 123–129. doi: 10.1016/j.jneumeth.2014.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsopanoglou, N. E., Andriopoulou, P., and Maragoudakis, M. E. (2002). On the mechanism of thrombin-induced angiogenesis: involvement of alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 283, C1501–C1510. doi: 10.1152/ajpcell.00162.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., and Melchor, J. P. (2001). Disruption of pathologic amyloid beta-protein fibril assembly on the surface of cultured human cerebrovascular smooth muscle cells. Amyloid 8(Suppl. 1), 20–27.
Van Nostrand, W. E., Melchor, J. P., Keane, D. M., Saporito-Irwin, S. M., Romanov, G., Davis, J., et al. (2002). Localization of a fibrillar amyloid beta-protein binding domain on its precursor. J. Biol. Chem. 277, 36392–36398. doi: 10.1074/jbc.m204676200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., Schmaier, A. H., Farrow, J. S., and Cunningham, D. D. (1990). Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science 248, 745–748. doi: 10.1126/science.2110384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., Schmaier, A. H., Neiditch, B. R., Siegel, R. S., Raschke, W. C., Sisodia, S. S., et al. (1994). Expression, purification and characterization of the Kunitz-type proteinase inhibitor domain of the amyloid beta-protein precursor-like protein-2. Biochim. Biophys. Acta 1209, 165–170. doi: 10.1016/0167-4838(94)90180-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., Schmaier, A. H., and Wagner, S. L. (1992). Potential role of protease nexin-2/amyloid beta-protein precursor as a cerebral anticoagulant. Ann. N Y Acad. Sci. 674, 243–252. doi: 10.1111/j.1749-6632.1992.tb27493.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Nostrand, W. E., Wagner, S. L., Suzuki, M., Choi, B. H., Farrow, J. S., and Geddes, J. W. (1989). Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid beta-protein precursor. Nature 341, 546–549. doi: 10.1038/341546a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vara, D., Campanella, M., and Pula, G. (2013). The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharmacol. 168, 212–224. doi: 10.1111/j.1476-5381.2012.02130.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vassar, R., Kuhn, P. H., Haass, C., Kennedy, M. E., Rajendran, L., Wong, P. C., et al. (2014). Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 130, 4–28. doi: 10.1111/jnc.12715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veitinger, M., Varga, B., Guterres, S. B., and Zellner, M. (2014). Platelets, a reliable source for peripheral Alzheimer’s disease biomarkers? Acta Neuropathol. Commun. 2:65. doi: 10.1186/2051-5960-2-65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verdier, Y., and Penke, B. (2004). Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr. Protein Pept. Sci. 5, 19–31. doi: 10.2174/1389203043486937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vingtdeux, V., and Marambaud, P. (2012). Identification and biology of α-secretase. J. Neurochem. 120(Suppl. 1), 34–45. doi: 10.1111/j.1471-4159.2011.07477.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Viswanathan, A., and Greenberg, S. M. (2011). Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 70, 871–880. doi: 10.1002/ana.22516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Viswanathan, A., Rocca, W. A., and Tzourio, C. (2009). Vascular risk factors and dementia: how to move forward? Neurology 72, 368–374. doi: 10.1212/01.wnl.0000341271.90478.8e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vukic, V., Callaghan, D., Walker, D., Lue, L. F., Liu, Q. Y., Couraud, P. O., et al. (2009). Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 34, 95–106. doi: 10.1016/j.nbd.2008.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wautier, M. P., Chappey, O., Corda, S., Stern, D. M., Schmidt, A. M., and Wautier, J. L. (2001). Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 280, E685–E694.
Wilkerson, W. R., and Sane, D. C. (2002). Aging and thrombosis. Semin. Thromb. Hemost. 28, 555–568. doi: 10.1055/s-2002-36700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williamson, T. G., Mok, S. S., Henry, A., Cappai, R., Lander, A. D., Nurcombe, V., et al. (1996). Secreted glypican binds to the amyloid precursor protein of Alzheimer’s disease (APP) and inhibits APP-induced neurite outgrowth. J. Biol. Chem. 271, 31215–31221. doi: 10.1074/jbc.271.49.31215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
World Alzheimer Report. (2011). The Benefits of Early Diagnosis and Intervention. London, UK: Alzheimer’s disease International. Available online at: http://www.alz.co.uk/research/world-report-2011
Wu, Z., Guo, H., Chow, N., Sallstrom, J., Bell, R. D., Deane, R., et al. (2005). Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med. 11, 959–965.
Xu, F., Davis, J., Miao, J., Previti, M. L., Romanov, G., Ziegler, K., et al. (2005). Protease nexin-2/amyloid beta-protein precursor limits cerebral thrombosis. Proc. Natl. Acad. Sci. U S A 102, 18135–18140. doi: 10.1073/pnas.0507798102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, F., Kotarba, A. E., Ou-Yang, M. H., Fu, Z., Davis, J., Smith, S. O., et al. (2014). Early-onset formation of parenchymal plaque amyloid abrogates cerebral microvascular amyloid accumulation in transgenic mice. J. Biol. Chem. 289, 17895–17908. doi: 10.1074/jbc.M113.536565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, F., Previti, M. L., and Van Nostrand, W. E. (2007). Increased severity of hemorrhage in transgenic mice expressing cerebral protease nexin-2/amyloid beta-protein precursor. Stroke 38, 2598–2601. doi: 10.1161/strokeaha.106.480103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. doi: 10.1038/382685a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeung, J., and Holinstat, M. (2012). Newer agents in antiplatelet therapy: a review. J. Blood Med. 3, 33–42. doi: 10.2147/JBM.S25421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, X., Wright, J., Wall, T., and Grammas, P. (2010). Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am. J. Pathol. 176, 1600–1606. doi: 10.2353/ajpath.2010.090406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, W., Huang, W., and Jing, F. (2013). Contribution of blood platelets to vascular pathology in Alzheimer’s disease. J. Blood Med. 4, 141–147. doi: 10.2147/JBM.S45071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zlokovic, B. V. (2005). Neurovascular mechanisms of Alzheimer’s neuro-degeneration. Trends Neurosci. 28, 202–208. doi: 10.1016/j.tins.2005.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuliani, G., Cavalieri, M., Galvani, M., Passaro, A., Munari, M. R., Bosi, C., et al. (2008). Markers of endothelial dysfunction in older subjects with late onset alzheimer’s disease or vascular dementia. J. Neurol. Sci. 272, 164–170. doi: 10.1016/j.jns.2008.05.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: amyloid peptides, platelets, vascular cells, cerebrovascular disease, Alzheimer’s disease
Citation: Canobbio I, Abubaker AA, Visconte C, Torti M and Pula G (2015) Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front. Cell. Neurosci. 9:65. doi: 10.3389/fncel.2015.00065
Received: 14 January 2015; Accepted: 11 February 2015;
Published online: 03 March 2015.
Edited by:
Francesco Moccia, University of Pavia, ItalyReviewed by:
Dmitry Lim, Università del Piemonte Orientale “Amedeo Avogadro”, ItalyTiziana Casoli, Istituto Nazionale di Riposo e Cura per Anziani, Italy
Copyright © 2015 Canobbio, Abubaker, Visconte, Torti and Pula. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilaria Canobbio, Department of Biology and Biotechnology, Unit of Biochemistry, University of Pavia, Via Bassi 21, 27100 Pavia, Italy e-mail: ilaria.canobbio@unipv.it
†Present address: Aisha Alsheikh Abubaker and Giordano Pula, Department of Pharmacy and Pharmacology, Centre for Regenerative Medicine, University of Bath, Bath, UK