MLC1 protein: a likely link between leukodystrophies and brain channelopathies
 Maria S. Brignone†
Maria S. Brignone†  Angela Lanciotti†
Angela Lanciotti†  Serena Camerini
Serena Camerini  Chiara De Nuccio
Chiara De Nuccio  Tamara C. Petrucci
Tamara C. Petrucci  Sergio Visentin
Sergio Visentin  Elena Ambrosini*
Elena Ambrosini*- Department of Cell Biology and Neuroscience, Istituto Superiore di Sanità, Rome, Italy
Megalencephalic leukoencephalopathy with subcortical cysts (MLCs) disease is a rare inherited, autosomal recessive form of childhood-onset spongiform leukodystrophy characterized by macrocephaly, deterioration of motor functions, epileptic seizures and mental decline. Brain edema, subcortical fluid cysts, myelin and astrocyte vacuolation are the histopathological hallmarks of MLC. Mutations in either the MLC1 gene (>75% of patients) or the GlialCAM gene (<20% of patients) are responsible for the disease. Recently, the GlialCAM adhesion protein was found essential for the membrane expression and function of the chloride channel ClC-2 indicating MLC disease caused by mutation in GlialCAM as the first channelopathy among leukodystrophies. On the contrary, the function of MLC1 protein, which binds GlialCAM, its functional relationship with ClC-2 and the molecular mechanisms underlying MLC1 mutation-induced functional defects are not fully understood yet. The human MLC1 gene encodes a 377-amino acid membrane protein with eight predicted transmembrane domains which shows very low homology with voltage-dependent potassium (K+) channel subunits. The high expression of MLC1 in brain astrocytes contacting blood vessels and meninges and brain alterations observed in MLC patients have led to hypothesize a role for MLC1 in the regulation of ion and water homeostasis. Recent studies have shown that MLC1 establishes structural and/or functional interactions with several ion/water channels and transporters and ion channel accessory proteins, and that these interactions are affected by MLC1 mutations causing MLC. Here, we review data on MLC1 functional properties obtained in in vitro and in vivo models and discuss evidence linking the effects of MLC1 mutations to brain channelopathies.
Introduction
Channelopathies are a heterogeneous group of disorders of genetic or acquired origin caused by altered function of ion channel subunits or the proteins that regulate them. These include diseases of the cardiovascular, muscular, respiratory, urinary, endocrine, and immune systems. Due to the fundamental role played by ion channels in neuronal activity, channelopathies are involved in a growing number of nervous system disorders (Kim, 2014 and references therein). Channelopathies leading to dysfunction of astrocytes, the glial cells responsible for the control of ion homeostasis in the central nervous system (CNS), have also been described (Wingerchuk et al., 2007; Vaknin-Dembinsky et al., 2014). The rare leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts (MLC) has recently emerged as a potential brain channelopathy associated to astrocyte dysfunction (Van der Knaap et al., 2012; Lanciotti et al., 2013).
Leukodystrophies comprise a large number of rare genetic disorders that affect mainly the CNS with different etiology and a common target. The European Leukodystrophies Association (ELA) offers a classification including the following types: peroxisomal, lysosomal, vacuolating, hypomyelinating, atypical, undetermined. Against such a heterogeneous etiopathogenesis, the common target is myelin: “either the myelin does not form, degrades, or is too abundant” (from the official ELA site: http://ela-asso.com).
Leukodystrophies have an incidence of 1 in 7,663 live births (Bonkowsky et al., 2010) and manifest themselves mostly during childhood or adolescence. MLC disease, first described in 1995, belongs to the childhood-onset vacuolating leukodystrophy group (Van der Knaap et al., 1995a, b, 1996; Singhal et al., 1996). The incidence of MLC is unknown, although it seems to be more frequent in countries surrounding the Mediterranean basin and in the Indian Agarwal community (Topcu et al., 1998; Ben-Zeev et al., 2002; Singhal et al., 2003). Clinically, MLC is characterized by macrocephaly, deterioration of motor functions with ataxia and spasticity, epileptic seizures and mental decline; minor head trauma or common infections can lead to worsening of clinical conditions often with seizures, prolonged unconsciousness and motor deterioration (Van der Knaap et al., 1995a,b, 1996; Singhal et al., 1996; Topcu et al., 1998; Bugiani et al., 2003). Brain magnetic resonance imaging (MRI) shows diffuse white matter swelling, subcortical cysts (mainly in the temporal and fronto-parietal regions) and, in some patients, severe neurodegeneration (Mejaski-Bosnjak et al., 1997). Analysis of brain biopsies has revealed the presence of liquid vacuoles between the outer lamellae of the myelin sheaths, suggesting defects in lamellae compaction or their splitting along the intraperiod line (Van der Knaap et al., 1996; Pascual-Castroviejo et al., 2005). Reactive and stressed astrocytes with vacuoles and swelling in the end-feet contacting blood vessels have also been observed (Van der Knaap et al., 1996; López-Hernández et al., 2011b). Despite extensive white matter damage, the disease phenotype is less severe than in other leukodystrophies in which similar myelin vacuolation is described (Van der Knaap et al., 2012; Lanciotti et al., 2013).
The first gene responsible for MLC was mapped to chromosome 22qtel and identified as MLC1 (Topçu et al., 2000; Leegwater et al., 2001). The MLC1 gene consists of 12 exons with an untranslated first exon, spanning at least 24 kilobases (Steinke et al., 2003). A broad spectrum of MLC1 pathogenic mutations (>60, including missense, splice site, insertions and deletions) have been identified in about 80% of affected individuals (Boor et al., 2006). The mutations are distributed along the whole MLC1 protein sequence and show no clear correlation with the severity of the disease phenotype (Patrono et al., 2003; Montagna et al., 2006). The MLC1 gene encodes a protein that is mainly expressed in brain astrocytes, particularly at the astrocyte end-feet contacting the blood–brain barrier and the pial membrane. Recently, mutations in a second gene encoding the cell adhesion protein GlialCAM have been found in about 50% of MLC patients not carrying mutations in MLC1 (<20% of the MLC-affected population; López-Hernández et al., 2011a). GlialCAM is highly expressed in the liver and in the CNS, particularly in neurons and glial cells, where it binds MLC1 (López-Hernández et al., 2011b). The identification of GlialCAM as the molecular chaperon and functional modulator of the chloride channel ClC-2 (Jeworutzki et al., 2014) has allowed for the first time to explain the similarities between brain alterations found in ClC-2 KO mice (Blanz et al., 2007) and those characteristic of MLC patients. Notably, this finding also led to identify MLC disease caused by mutations in the GlialCAM gene as the first leukodystrophy among brain channelopathies. Although GlialCAM has been initially identified as the molecular chaperon transporting both MLC1 and ClC-2 to the astrocyte plasma membrane, the relationship between MLC1 and the ClC-2 channel is not fully understood yet. MLC1 protein structural features, molecular interactors (see below) and brain alterations observed in MLC patients (edema, fluid cysts, astrocyte, and myelin vacuolation) suggest that MLC1 can play a role in the regulation of ion and water fluxes and cell volume. However, although several recent studies support this hypothesis (Ridder et al., 2011; Lanciotti et al., 2012; Brignone et al., 2014; Hoegg-Beiler et al., 2014; Dubey et al., 2015), the exact function of MLC1 is still unknown.
In this article we shall present and discuss data on MLC1 structural and functional properties obtained in different experimental settings, suggesting a link between MLC1 mutation-induced alterations in MLC disease and brain channelopathies.
The Puzzling MLC1 Protein: Biochemical Features and Cellular Localization
Molecular Structure and Post-Translational Modifications
After the discovery of mutations in the MLC1 gene as the main cause of MLC, several research groups have investigated the physiological function of MLC1 with the aim of disclosing the pathogenic mechanisms underlying the disease. This has been a particularly challenging task. Analysis of the primary nucleotide sequence indicated that the human MLC1 gene encodes a 377-amino acid highly hydrophobic protein containing eight predicted transmembrane domains and short amino and carboxylic- cytoplasmic tails (Figure 1; Meyer et al., 2001; Teijido et al., 2004). MLC1 also contains an internal repeat structure resulting in a partial homology of the primary sequence between the two halves of the protein that is also found in several ion channels (Teijido et al., 2004). However, blast sequence analysis indicates that MLC1 has no similarities with known proteins, with the exception of a very low homology with the shaker-related voltage gated potassium (K+) channel Kv1.1 α subunit (less than 20% amino acid identity; Teijido et al., 2004).
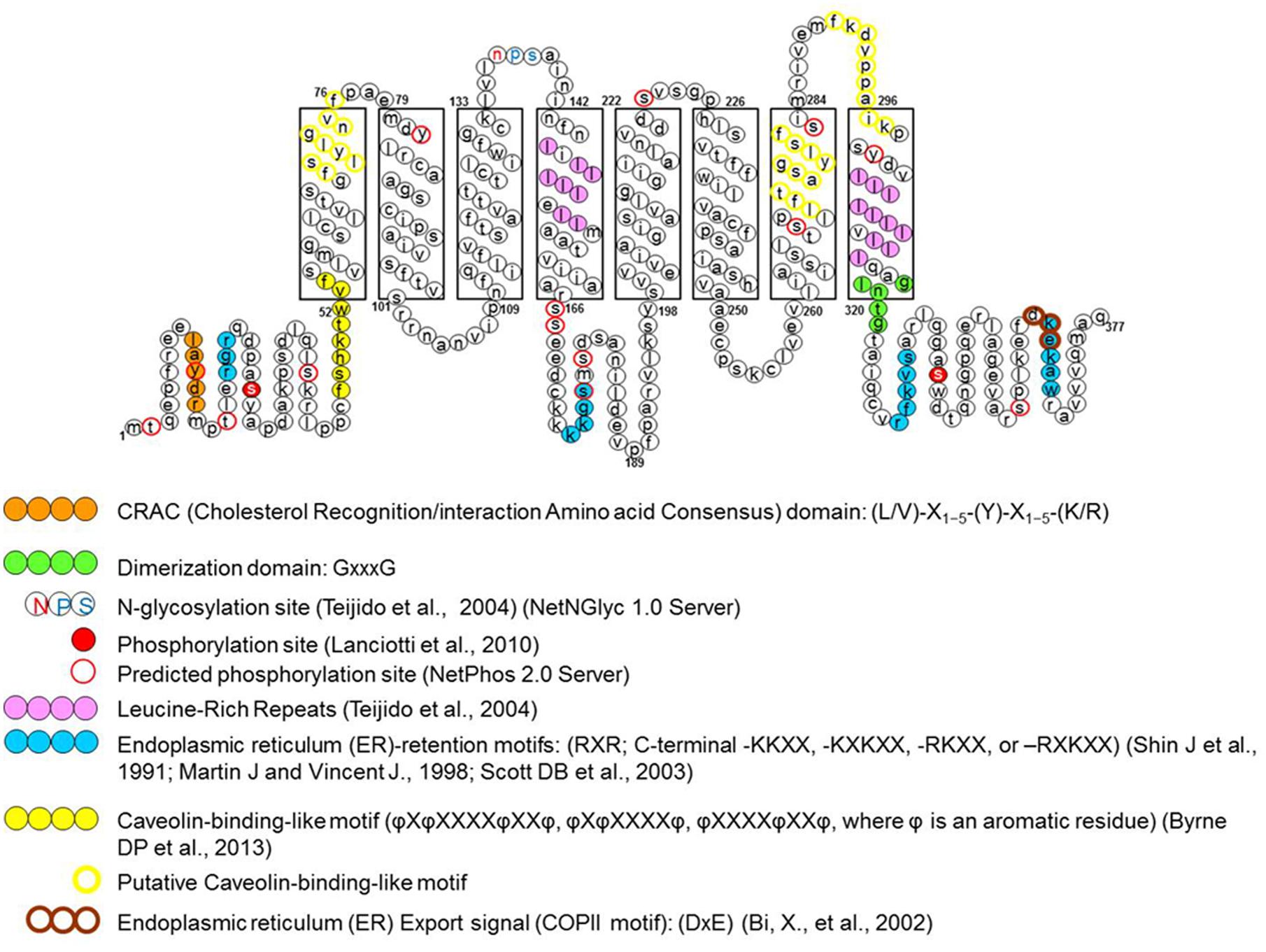
FIGURE 1. Schematic representation of specific amino acid consensus motifs found in the human megalencephalic leukoencephalopathy with subcortical cysts (MLC1) protein sequence. The picture shows consensus motifs for post-translational modifications (glycosylation and phosphorylation), protein–protein or protein–lipid interactions (protein dimerization and cav-1 and cholesterol binding sites) and protein trafficking signals (endoplasmic reticulum export and retention signals), either putative (empty circles), or functionally identified (filled circles) (modified from Lanciotti et al., 2012).
Based on this knowledge, the first studies aimed at identifying the biochemical and structural features of MLC1 protein. Early experiments carried out in transiently transfected heterologous cells (HeLa cells and Xenopus oocytes; Teijido et al., 2004), cultured primary rat astrocytes and rat brain tissue (Teijido et al., 2004; Ambrosini et al., 2008) showed that MLC1 is a 36–40 kDa protein that is able to form highly stable dimeric and oligomeric structures (mainly dimers but also tetramers). By performing subcellular fractionations of rat astrocytes and brain tissue it was found that the MLC1 monomeric form is present only in the cytosolic fraction (mainly organelle fraction) while the dimers are associated with the membrane compartments (plasma membrane and endoplasmic reticulum membranes; Ambrosini et al., 2008; Lanciotti et al., 2010). Database analysis of the MLC1 primary sequence revealed potential post-translational modification sites, including phosphorylation and glycosylation sites (Figure 1). However, the results of endoglycosydase treatment and site-directed mutagenesis of the putative glycosylation site (NPS in the second extracellular loop, Figure 1) ruled out that MLC1 is glycosylated when expressed in heterologous cells (Teijido et al., 2004), but did not allow to exclude that the endogenous protein is glycosylated in astrocytes. In vitro studies on recombinant peptides and endogenous protein from rat astrocytes indicated that MLC1 is phosphorylated at its NH2 and COOH terminals by both PKA and PKC and PKC alone, respectively (Lanciotti et al., 2010; Figure 1). Furthermore, it was found that treatment with substances activating PKC or PKA and phosphatase inhibitors (forskolin, okadaic acid, phorbol esters, genistein) can modify MLC1 plasma membrane expression and formation of multimeric structures (Lanciotti et al., 2010). Interestingly, analysis of the MLC1 aminoacidic sequence revealed the presence of an arginine-based ER retention motif RXR (Schutze et al., 1994; Michelsen et al., 2005) localized in the NH2 terminal near the PKC and PKA phosphorylation sites (Figure 1). Changes in the extent of phosphorylation at sites adjacent to the RXR-type ER retention motif may enable a given protein to interact with the forward secretory machinery by hiding or hampering the retention signals, therefore allowing channel/receptor trafficking to the plasma membrane (Scott et al., 2003). The ability of intracellular signaling to modulate ER export of RXR-containing protein defines a regulatory pathway that may provide dynamic control over the assembly and surface expression of a wide range of ion channels and receptors (Shikano and Li, 2003). Our data indicating that PKC and PKA activation favors MLC1 expression in the plasma membrane and the presence of an ER retention motif near the PKC/PKA phosphorylation sites suggest this possibility also for the MLC1 protein. The regulation of MLC1 membrane localization (and also activity) by PKA and PKC phosphorylation has been demonstrated for several ion channels and transporters. For example, PKA activation enhances the forward trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR; Chang et al., 2002). PKA and PKC promote the translocation to the apical membrane and activity of the transient receptor potential vanilloid-4 cation channel (TRPV4; Mamenko et al., 2013) and PKA and PKC have an additive effect in regulating the surface expression of the Na, K-ATPase pump (Kristensen et al., 2003). Protein kinases could also indirectly regulate MLC1 export toward the plasma membrane by their involvement in the formation of coat protein II (COPII)-coated vesicles (Farhan et al., 2010). The COPII protein complex forms transport vesicles from the ER and, by binding specific diacidic motifs present in the C terminal of the cargo protein, incorporates it into these vesicles (Barlowe, 2003). The presence of the canonical COPII binding motif DXE in the COOH terminal of the MLC1 protein (Figure 1) suggests that the same regulation takes place for MLC1, similarly to what observed for some ion channels and transporters (Wang et al., 2004; Sivaprasadarao et al., 2007; Zhang et al., 2011). The presence of several additional putative phosphorylation sites in the MLC1 sequence, as reported in specific databases (NetPhos 2 Server, Technical University of Denmark; Figure 1), and the identification by LC-MS analysis of several kinases and phosphatases among MLC1 interactors in primary rat astrocytes (Table 1; Ambrosini manuscript in preparation) suggest a more complex phosphorylation-mediated regulation of MLC1 localization, and probably function, in astrocytes. Experiments are in progress in our laboratory to better understand the role of kinases in MLC1 protein stability, molecular interactions, and function. This knowledge could be relevant for the therapy of MLC since several kinases are currently under investigation as drug targets to modulate the activity of ion channels involved in tumoral, inflammatory, and neurologic diseases (Son et al., 2013).
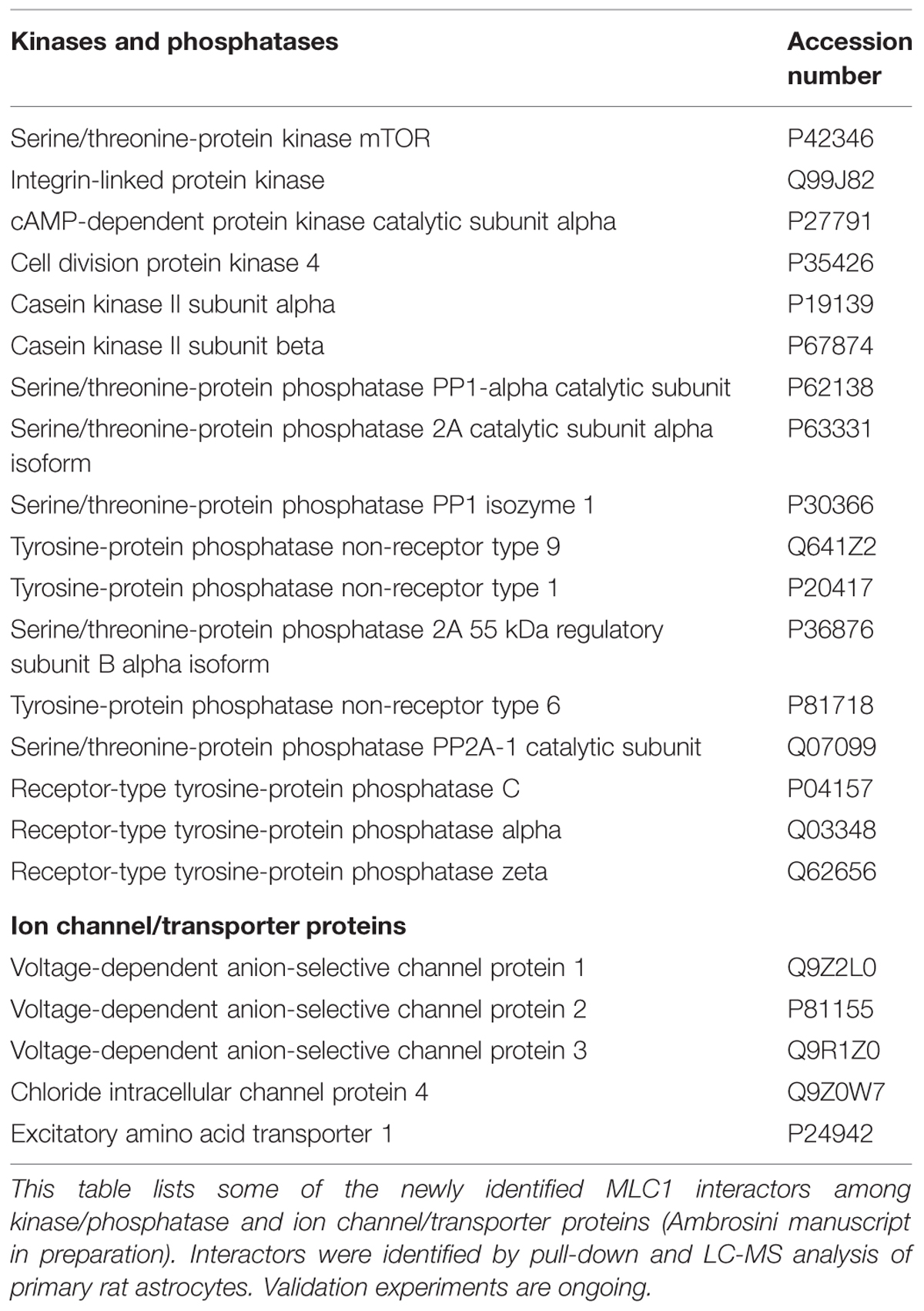
TABLE 1. Newly identified megalencephalic leukoencephalopathy with subcortical cyst (MLC1) molecular interactors.
MLC1 Expression in Mouse and Human Brain
One of the few certainties on MLC1 protein is its predominant expression in brain astrocytes, particularly in astrocytic end-feet contacting cerebral blood vessels. Due to this finding, MLC1 is emerging as a promising marker for perivascular astrocytes (Masaki et al., 2012). The first evidence of MLC1 expression in glial cells came from an in situ hybridization study in rat brain showing that MLC1 mRNA is mainly expressed in multipotent neural precursor cells during the pre- and peri-natal period, and in astrocytes, Bergmann glia, and ependymal cells, but not oligodendrocytes and neurons, in the adult brain (Schmitt et al., 2003). These researchers also found that MLC1 is developmentally regulated in the mouse brain, being maximally expressed at day 5 after birth and then decreasing to a lower and stable level from post-natal day 7 to adulthood. More recently, the temporal correspondence between the expression of MLC1 in human and mouse brain has been demonstrated (Dubey et al., 2015). Using specific antibodies, MLC1 protein was found predominantly in astrocyte compartments contacting the brain barriers, such as the astrocyte end-feet contacting blood vessels, the meninges (pial membrane) and ependymal cells lining the ventricles, and in Bergmann glia in the cerebellum, but not in neurons, oligodendrocytes, microglia, and endothelial cells (Teijido et al., 2004; Boor et al., 2005, 2007; Ambrosini et al., 2008). Outside the CNS, MLC1 is expressed in blood cells, like lymphocytes, monocytes, and macrophages differentiating in vitro (Boor et al., 2006; Petrini et al., 2013). Accordingly, defects in hyposmosis-induced Ca2+ influx (see below) were recorded in monocyte-derived macrophages from MLC patients, though the impact of such alterations is unknown since MLC patients do not show overt deficits in blood or immune system functions.
Intracellular Distribution and Trafficking of MLC1 Protein in Astrocytes
Biochemical studies in cultured astrocytes and rat brain tissue showed that MLC1 is present in specialized areas of the plasma membrane called lipid rafts (Lanciotti et al., 2010). Lipid rafts are microdomains of the plasma membrane formed by a tightly ordered lipid phase that is enriched in sterols (including cholesterol), sphingolipids (including gangliosides) and phospholipids with saturated hydrocarbon chains (Wang and Paller, 2006). Lipid rafts are defined by resistance to extraction with non-ionic detergents and low density in sucrose gradients. These microdomains are highly dynamic and possess considerable lateral mobility within the loosely ordered membrane. Caveolar rafts containing caveolae, stable flask-shaped invaginations of the plasma membrane, are a subtype of lipid rafts highly enriched in cholesterol and coated by caveolin-1 (cav-1). Cav-1, a hairpin-like palmitoylated structural protein, is thought to stabilize the invaginated caveolar structure. Caveolae have been described in several cell types, including astrocytes and astrocytoma/glioma cells (Zschocke et al., 2005; González et al., 2007), and are involved in protein trafficking and formation of cell signaling complexes (Simons and Ikonen, 1997; Brown, 2006). By performing lipid raft separation by sucrose gradient, we found that only the MLC1 dimers are distributed in the membrane caveolar raft fraction, both in cultured astrocytes and in rat brain tissue (Ambrosini et al., 2008; Lanciotti et al., 2010; Figure 2). Presence of MLC1 protein in caveolar rafts is supported by binding of cav-1 to the NH2 intracellular terminal of the MLC1 protein containing the putative caveolin/cholesterol binding site (Lanciotti et al., 2010; Figure 1).
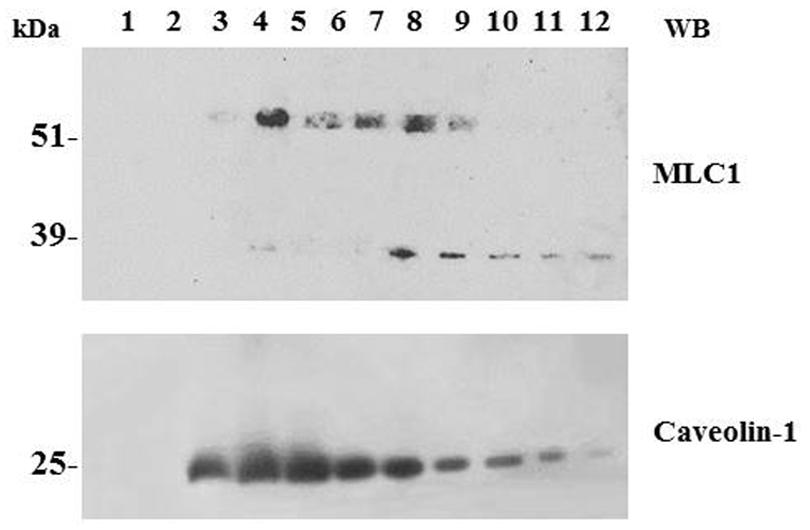
FIGURE 2. Association of MLC1 with detergent resistant membrane (DRM)/lipid raft cell compartments. WB analysis of cholesterol-rich (triton insoluble fractions: 3–6) and cholesterol-poor membrane fractions (triton soluble fractions: 10–12) of WT MLC1 expressing U251 cells. The 60–64 kDa MLC1 component is associated with the DRM fractions, whereas the 30 kDa band is detected only in the soluble fractions together with a portion of the 60–64 kDa component. The same membrane was blotted with anti-caveolin-1 antibody to track the position of caveolae-enriched membranes in the DRM fractions.
Because MLC1 function is still unknown, we do not understand at present the functional consequences of MLC1 localization in caveolar rafts. However, we have found that cholesterol and agents modulating caveolae-dependent endocytosis influence MLC1 transport from the plasma membrane to the intracellular compartments (Lanciotti et al., 2010). Our studies demonstrated that in astrocytes exposed to hyposmotic stress MLC1 is translocated to the plasma membrane and then internalized by caveolar endocytosis to be sorted to recycling or degradation pathways (Figure 3). By cell fractionation, immunofluorescence and electron microscopy analysis of rat primary astrocytes and human astrocytoma cells, MLC1 protein was detected in early endosomes (EEA1+ and Rab5+; Figures 3 and 4A,B; Lanciotti et al., 2010; Brignone et al., 2014) and in Lamp-1+ organelles and multivesicular bodies (Figure 4C; Lanciotti et al., 2010; Brignone et al., 2014), but not, or at very low levels in Golgi apparatus and clathrin vesicles (Lanciotti et al., 2010). More recently, we found that in unstimulated astrocytes MLC1 is abundantly expressed in the Rab11+ perinuclear storage/recycling compartment from which it is recruited back to the plasma membrane when cells are exposed to hyposmotic stress (Figure 3; Brignone et al., 2014). Cytoskeletal elements are involved in MLC1 intracellular trafficking since nocodazole treatment, which perturbs microtubule organization, hampers MLC1 accumulation in the Rab11+ endosomal recycling compartment in rat primary astrocytes (Lanciotti et al., 2010). It has been shown that stabilization of MLC1 in the plasma membrane of rat astrocytes cultured for 3 weeks in the presence of the antimitotic cytosine arabinoside, which leads to astrocyte maturation/differentiation, depends on intact actin filaments, but not microtubules or intermediate filaments (Duarri et al., 2011). In this context, post-translational modifications can exert an important role in MLC1 forward trafficking and stabilization in the plasma membrane (see above). Overall, these results provide evidence that the membrane expression level of MLC1 is spatially and temporally regulated by agents modulating intracellular trafficking, cytoskeletal organization and lipid composition of the plasma membrane, as observed for many ion channels and transporters (Dart, 2010; Curran and Mohler, 2014).
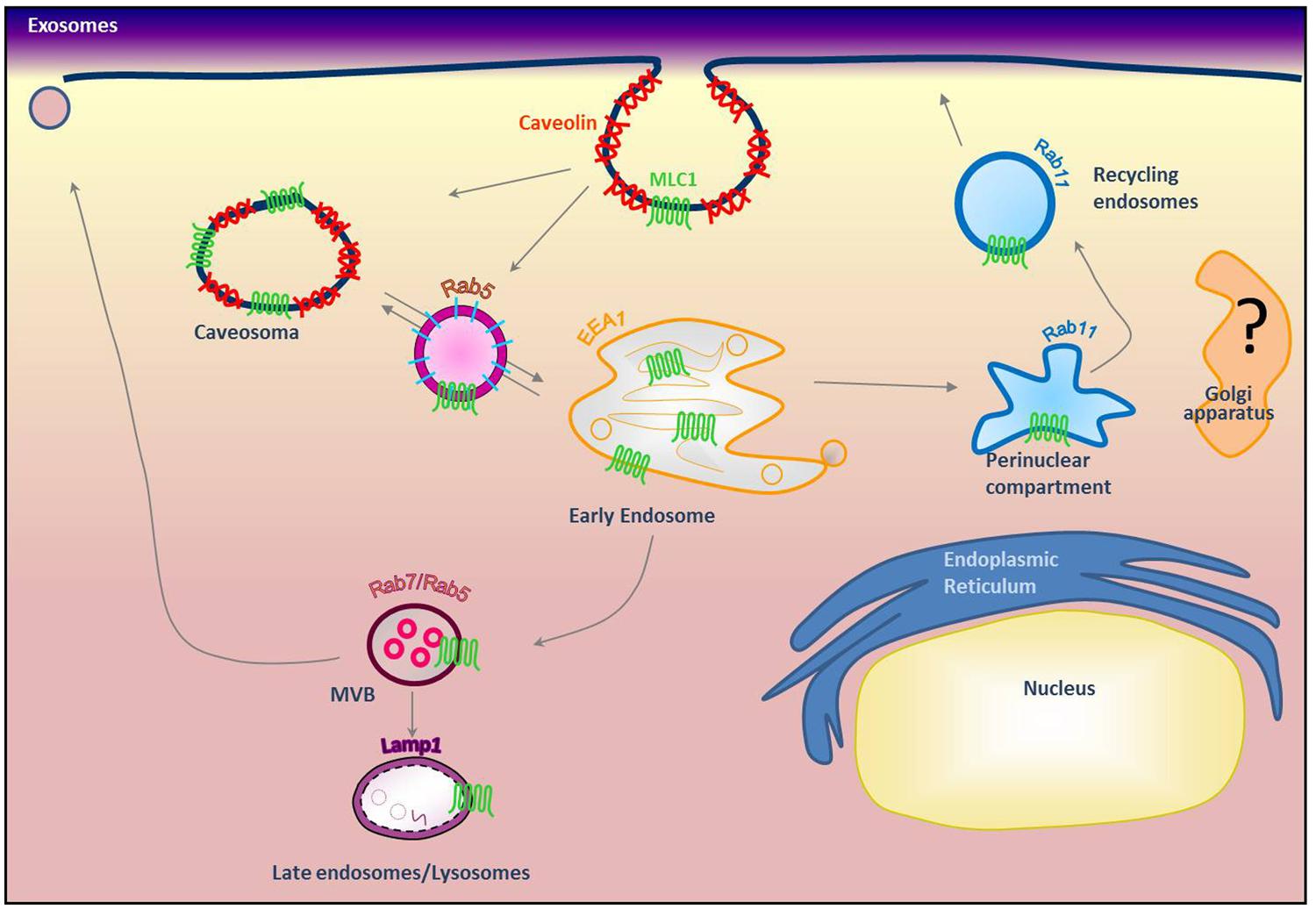
FIGURE 3. Modeling of MLC1 intracellular trafficking. Our previous results indicated that MLC1 is internalized via caveolae-mediated endocytosis and traffics through Rab5+ and EEA1+ early endosomes where it is sorted to the recycling (Rab11+) or degradative pathway (Lamp-1+; Lanciotti et al., 2010; Brignone et al., 2014) (modified from Brignone et al., 2014).
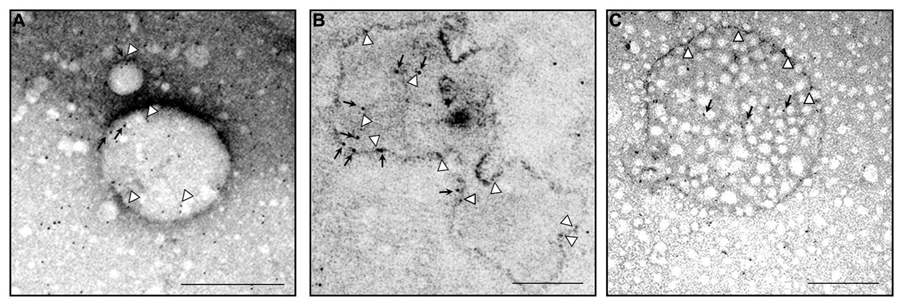
FIGURE 4. Ultrastructural analysis of endogenous MLC1 localization in early endosome compartment (EEA1+ and Rab5+ organelles) and in a multivesicular body. Electron microscopy analysis of ultracentrifuged cytosolic protein fractions obtained from cultured rat astrocytes was performed using anti-MLC1 Ab (10 nm gold particles) in combination with anti-EEA1 or anti-Rab5 Abs (5 nm gold particles). (A) MLC1 (black arrows) is localized in Rab5+ organelles (arrowheads) (B) MLC1 is localized in the membrane and intraluminal areas (black arrows) of EEA1+ organelles (arrowheads). (C) MLC1 immunostaining in the membrane (arrowheads) and intracellular vesicles (black arrows) of a multivesicular body-like structure. Scale Bars: 200 μm.
MLC1 Molecular Interactors
One of the first steps to disclose the function of a novel protein is the identification of its molecular interactors, which can provide information on the biochemical pathways in which the protein of interest is involved (Table 2). By using this approach, GlialCAM has been identified as the second MLC disease-associated gene and the molecular chaperon that targets MLC1 to astrocyte cell–cell junctions (López-Hernández et al., 2011a,b; Capdevila-Nortes et al., 2013).
Recently, new findings obtained in knock-out (KO) animal models have allowed to better understand the relationship between GlialCAM and MLC1 in vivo (see below). As this article focuses on the role of MLC1 protein, we refer the reader to specific publications for detailed studies on GlialCAM involvement in MLC disease (López-Hernández et al., 2011a,b; Capdevila-Nortes et al., 2013).
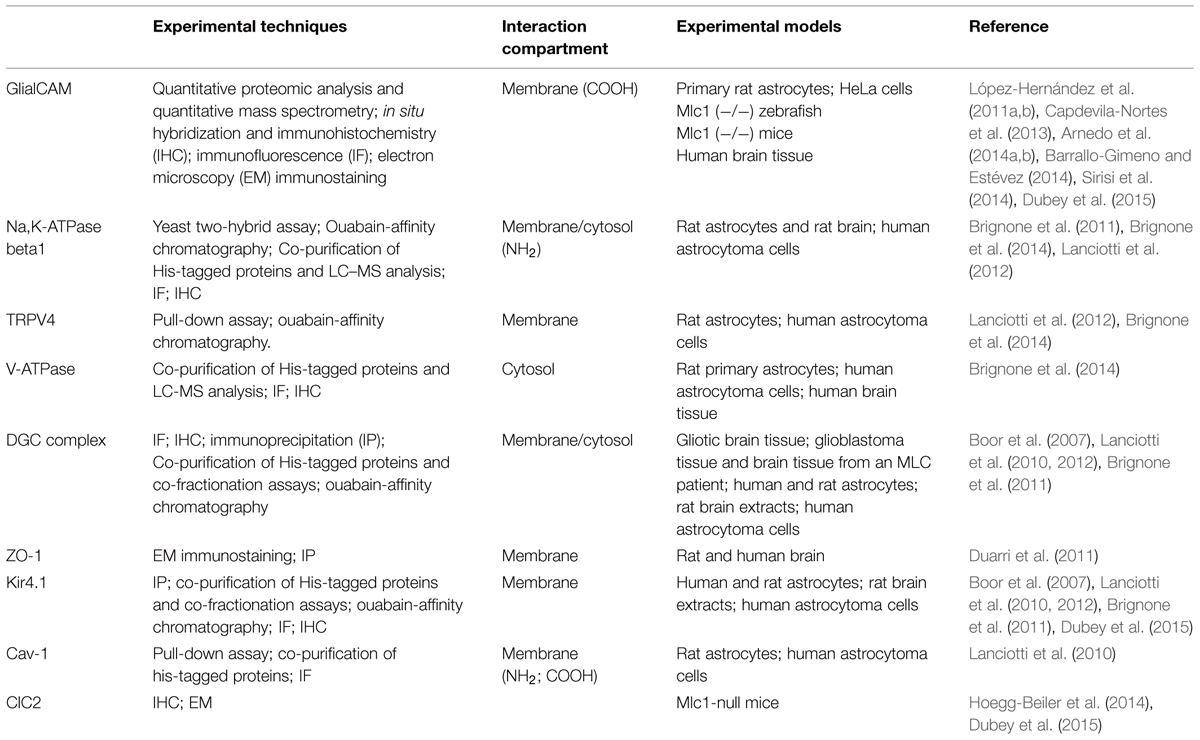
TABLE 2. List of described MLC1 molecular interactors in astrocytes.
Dystrophin–Dystroglycan Complex and Junctional Proteins
The observation that MLC disease shares some pathological features with congenital muscular dystrophies with brain involvement caused by mutations in proteins of the dystroglycan (DG) associated protein complex dystrophin–dystroglycan complex (DGC; Boor et al., 2007) prompted us and other research groups to investigate the relationship between MLC1 and DGC. The DGC is a multiprotein complex highly expressed in astrocyte end-feet where it stabilizes ion and water channels, like the inward rectifier potassium 4.1 (Kir4.1) channel and the water channel aquaporin-4 (AQP4; Noell et al., 2011). It has been shown that in the human brain MLC1 co-localizes with DGC proteins, like α/β-DG, syntrophin and agrin, in astrocyte end-feet contacting blood vessels (Boor et al., 2007; Ambrosini et al., 2008). Moreover, in brain tissue from a patient with MLC some components of the DGC (Kir4.1, agrin, and α-DG) are mislocalized while others (merosin, β-DG and AQP4) retain their normal perivascular localization (Boor et al., 2007). These results appeared of interest since agrin and α-DG are specifically involved in Kir4.1 clustering in the glial cell plasma membrane (Noël et al., 2005) and deletion of the Kir4.1 gene in mice causes myelin vacuolation and cyst formation that are reminiscent of the MLC phenotype (Neusch et al., 2001). The MLC1/DGC protein interaction was also supported by biochemical experiments showing that Kir4.1 and MLC1 co-immunoprecipitate in human brain (Boor et al., 2007) and that MLC1 is detected among DGC complex proteins in cultured rat and human astrocytes (Boor et al., 2007; Ambrosini et al., 2008; Lanciotti et al., 2010). In contrast with these data, electron microscopy showed that MLC1 co-localizes with the zonula occludens protein-1 (ZO-1), a protein present in astrocyte–astrocyte junctions, but not with β-DG in astrocyte end-feet contacting cerebrovascular endothelial cells in the normal human adult cerebellum (Duarri et al., 2011; Figure 5). The same study reports that MLC1 does not co-immunoprecipitate with DGC components in cultured rat astrocytes acquiring a differentiated morphology upon prolonged treatment with cytosine arabinoside (Duarri et al., 2011). Differences in MLC1 molecular interactions in different brain regions (cerebral cortex versus cerebellum) and/or astrocyte developmental stages (proliferating neonatal rat astrocytes versus mature differentiated astrocytes) could account for these discrepancies. Interestingly, the DGC itself is also involved in the organization and maintenance of junctional complexes during brain development (Nico et al., 2003, 2004; Sjö et al., 2005). Further studies are needed to clarify the issue of MLC1/DGC relationship.
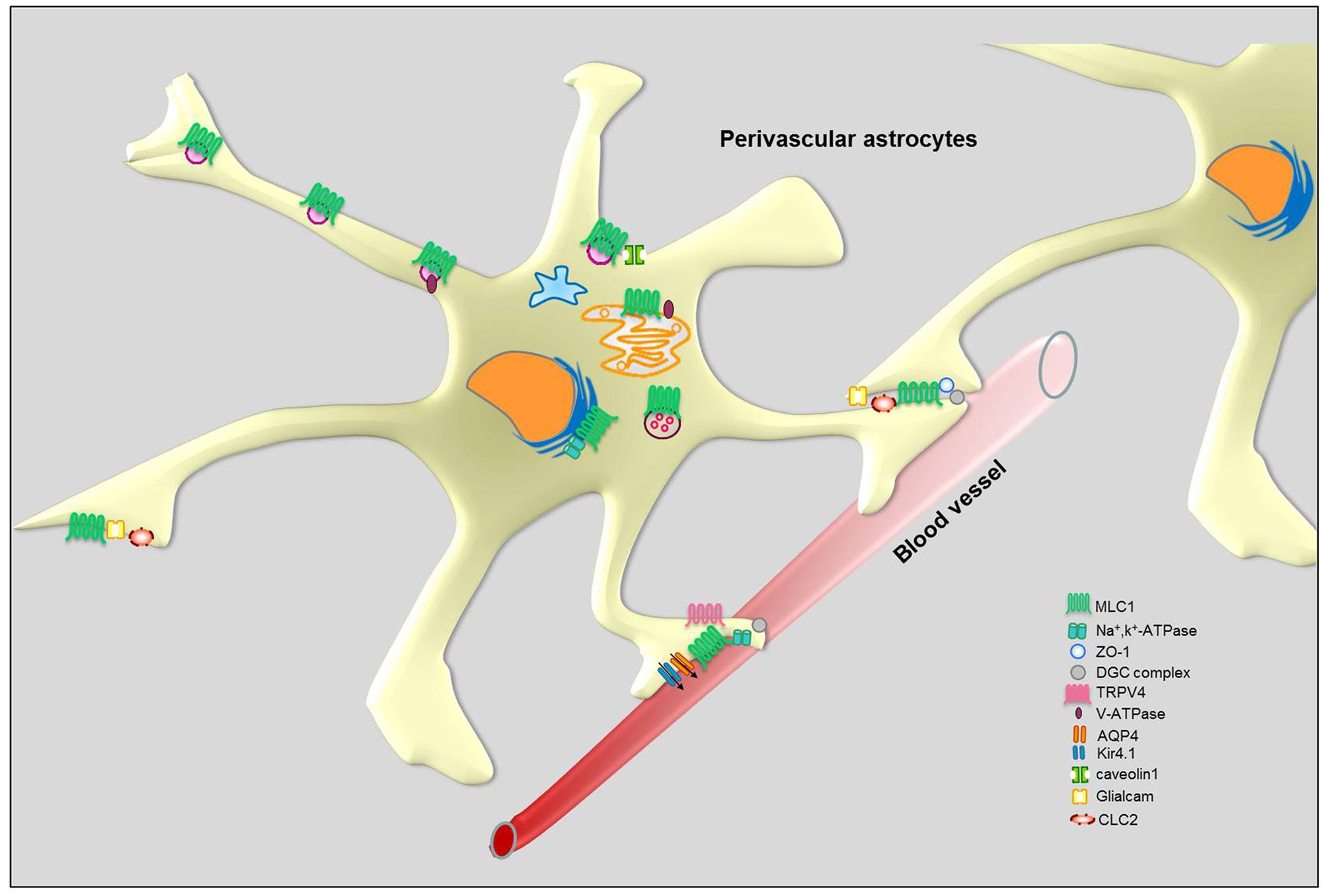
FIGURE 5. Schematic representation of MLC1 molecular interactions in perivascular astrocytes.
MLC1 Molecular Interactors Involved in Osmoregulation and pH Control
Consistent with the hypothesis that MLC1 plays a role in astrocyte-mediated osmoregulation, the MLC1 molecular interactors we identified in preliminary studies included ion channels and accessory proteins that are enriched in astrocyte end-feet (Table 2). Using complementary biochemical, proteomic and in vivo protein interaction assays (like pull-down, yeast two-hybrid system assays, His co-fractionation associated to LC-MS analysis and ouabain affinity chromatography) in cultured rat astrocytes and human astrocytoma cells, we found that MLC1 binds the β1 subunit of the Na, K-ATPase enzyme (Brignone et al., 2011). We also showed that MLC1 is part of a multiprotein complex comprising Kir4.1, cav-1, TRPV4, AQP4, syntrophin, and the vacuolar ATPase (V-ATPase) and is involved in the regulation of ion transport, cellular volume changes and intraorganelle pH control (Brignone et al., 2011, 2014; Lanciotti et al., 2012). We have found that other ion channels/transporters interact with MLC1, such as the chloride intracellular channel 4 (CLIC4), the voltage-dependent mitochondrial anion channels (VDAC1-3) and the excitatory amino acid transporter 1 (EAAT1; Table 1). The functional relationship of these molecules with MLC1 is under investigation. Cell fractionation and immunolocalization experiments revealed that in astrocytes MLC1 interaction with Na, K-ATPase and V-ATPase occurs mainly in the membrane of intracellular organelles, like early and recycling endosomes, in basal culture conditions and also in the plasma membrane after exposure to osmotic stress (Brignone et al., 2011, 2014). These results indicate that macromolecular complexes comprising MLC1 may modulate the astrocyte response to osmotic imbalance by spatially operating in the plasma membrane and in intracellular membranes, similarly to what observed for protein complexes linked to Ca2+, K+, and Cl- channels (Babcock and Li, 2013; Venkatachalam et al., 2014). A more systematic evaluation of the function and mutation-induced alterations of MLC1 in different membrane compartments should be pursued also in view of possible therapeutic applications. In fact, there is increasing evidence that pharmacological treatments can differently target ion channel and transporter protein complexes in the plasma membrane or intracellular compartments (Babcock and Li, 2013).
Deciphering MLC Pathogenesis: From Cellular to Animal Models
To shed light on MLC1 function and its role in MLC pathogenesis, in the last years different in vitro and in vivo pathological models have been developed. In vitro MLC disease models were set up by overexpressing WT and mutated MLC1 in heterologous cells and astrocytes or by knocking-down MLC1 in astrocytes. Earlier studies performed in heterologous cells (HeLa cells, HEK293 cells, Xenopus oocytes) expressing mutated MLC1 protein showed that many missense pathological mutations impaired MLC1 translocation to the plasma membrane. MLC1 mutations also affected the protein half-life by causing an increase in protein degradation (Teijido et al., 2004; Duarri et al., 2008). This effect was mainly ascribed to protein misfolding problems and activation of the endoplasmic reticulum associated degradation pathway (ERAD) since ERAD inhibitors could block MLC1 degradation. After the discovery that cultured astrocytes express MLC1 protein (Ambrosini et al., 2008), these cells became the most relevant in vitro model to study MLC1 pathophysiology. Interestingly, some MLC1 mutations behave differently when overexpressed in rat astrocytes compared to heterologous cells (Duarri et al., 2008), highlighting the need to investigate MLC1 mutation-induced phenotypes in an astrocyte background. To this end, we selected human astrocytoma cells (U251 cells), since these cells express almost undetectable levels of endogenous MLC1 protein. We therefore generated U251 cell lines stably expressing MLC1 WT or carrying some of the several missense mutations found in MLC patients (Brignone et al., 2011). The obtained astrocytoma cell lines expressed MLC1 at levels comparable to those detected in primary rat astrocytes.
Using U251 cell lines expressing different MLC1 pathological mutations (i.e., S426R, S280L, and C125R) we found that not all the mutated proteins were retained in the ER. For example, MLC1 protein carrying the S426R mutation was able to reach the plasma membrane, similarly to WT MLC1 (Brignone et al., 2011). This result suggested that in vitro modeling of MLC1 pathological mutations could unravel MLC1 mutation-specific functional defects and eventually prove useful for testing patient-tailored therapies. Using this system, we also found that the analyzed missense mutations altered the interaction of MLC1 with some previously identified interactors, such as GlialCAM, Kir4.1, Na, K-ATPase, V-ATPase, and TRPV4 channel (Brignone et al., 2011; Lanciotti et al., 2012), supporting further the functional involvement of MLC1 in this macromolecular complex. Interestingly, downregulation of MLC1 in primary rat astrocytes by small interfering RNA (siRNA) silencing induced astrocyte vacuolation, a pathological feature found in the MLC-affected brain (Duarri et al., 2011) and in mlc1-null mice (Dubey et al., 2015), but did not alter the localization of the MLC1 interactor ZO-1 (Duarri et al., 2011). Collectively, these results indicate that in vitro modeling of MLC disease can, at least in part, recapitulate MLC1 dysfunction.
Two different mouse models have been recently developed in which MLC1 has been constitutively knocked-down in all tissues (Hoegg-Beiler et al., 2014; Dubey et al., 2015). A zebrafish model has also been generated by deleting the MLC1 ortholog gene (Sirisi et al., 2014). MLC1-null mice recapitulate some features of the human MLC disease, such as early-onset megalencephaly and augmented brain water content, with appearance of aberrant astrocytic processes adjacent to fluid/brain barriers and progressive white matter vacuolization. However, both KO mice do not develop motor disabilities (even at later stages of life) or brain cysts that are characteristic of the human disease. In general, when neuropathological hallmarks were compared, brain lesions in MLC1-null mice were less severe than those in MLC-affected brain and zebrafish lesions were even less pronounced than mice lesions, predicting interspecies differences, or occurrence of compensatory mechanisms in KO animals. Analysis of mlc1-null mice revealed that, contrary to what was found in vitro (López-Hernández et al., 2011b), MLC1 is important for the correct localization of GlialCAM and ClC-2 in astrocytes. It has been shown that MLC1 also acts in trans, by influencing GlialCAM expression in oligodendrocytes which are devoid of MLC1 (Hoegg-Beiler et al., 2014). On the contrary, lack of MLC1 does not affect the distribution of α/β-DG, agrin, Na, K-ATPase, TRPV4, and ZO-1 in astrocyte perivascular end-feet (Dubey et al., 2015). Evidence supporting the importance of MLC1 for the correct localization of GlialCAM and ClC-2 was also obtained in cultured astrocytes derived from mlc1-null mice that were exposed to a high extracellular K+ concentration mimicking neuronal activity (Hoegg-Beiler et al., 2014; Sirisi et al., 2014). However, immunohistochemical analysis of brain sections from a MLC patient carrying a missense mutation (S69L) leading to MLC1 downregulation showed no apparent abnormalities in GlialCAM localization in perivascular astrocytes (López-Hernández et al., 2011b); in the same patient GlialCAM mislocalization was found restricted to the Bergman glia in the cerebellum (Sirisi et al., 2014).
Although KO animal models do not completely recapitulate the human disease, they confirm that astrocyte-mediated defects in water fluxes and cell volume regulation are involved in MLC disease pathogenesis, as suggested by results in in vitro models. Overall these findings indicate that the experimental models used to study MLC1 function and MLC disease pathogenesis can provide different (sometimes contradictory) results. Some discrepancies may reflect the inability of the in vitro systems to reproduce the complexity of astrocyte functional and structural connections in vivo. It is generally accepted that gene inactivation in vivo represents an essential approach to study the physiological roles of several proteins. However, KO mice, while useful for a rough characterization of gene function, have some disadvantages for deciphering precisely pathogenetic mechanisms, including gene redundancy, and developmental issues. Furthermore, not all the MLC1 mutations analyzed in vitro lead to complete protein degradation, a phenotype recapitulated by null-mouse models. Noteworthy, astrocytes display heterogeneous characteristics depending on species of origin, brain region, developmental stage, environmental factors, and disease states, all of which may render experimental results highly variable. The complexity of the glia themselves as well as the marked increase in the ratio of glia to neurons in higher mammals (Pfrieger, 2009) suggest that some roles of glia studied in mouse models might be non conserved or even more critical in primates and humans (Molofsky et al., 2012). For these reasons, advantages and disadvantages of each experimental system should be taken into account for a correct evaluation of the results obtained.
Astrocyte Functional Pathways Affected by MLC1 Mutations
Regulation of Ca2+ Influx
Intracellular Ca2+ changes participate in the regulation of regulatory volume decrease (RVD) that occurs after cell swelling in astrocytes. It has been shown that the cation channel TRPV4 co-localizes with AQP4 in the astrocyte end-feet and is responsible for an outward-rectifying Ca2+ conductance and Ca2+ influx following cell swelling induced by mechanical and osmotic stress (Benfenati et al., 2007, 2011; Figure 5). The TRPV4-mediated Ca2+ influx is an essential step in the activation of RVD in astrocytes which is needed to rescue the temporary cell swelling induced by hyposmosis. Using the U251 astrocytoma cell model, we found that MLC1 mutations causing retention of the protein in the cytoplasmic perinuclear area affect TRPV4-mediated Ca2+ influx induced by hyposmotic stress (Lanciotti et al., 2012). This finding supports the hypothesis that MLC1 localized in the plasma membrane can functionally cooperate with TRPV4 to regulate Ca2+ influx and RVD after osmotic imbalance. Moreover, as in primary astrocytes (Benfenati et al., 2007, 2011), also in astrocytoma cells hyposmotic stress induces heterogeneous Ca2+ transients that differ for lag time of onset, profile of the transient (i.e., peak shaped, sustained, or a combination of the two) and maximal amplitude of the Ca2+ changes (Lanciotti et al., 2012). The high variability in the Ca2+ response is hardly compatible with a direct effect of swelling on TRPV4 channels, but more likely is the result of an indirect effect mediated by a different molecular sensor (Christensen and Corey, 2007). The TRPV4 cation channel is known as a polymodal transducer of mechanical stretch, osmotic pressure, and warmth perception (Nilius and Voets, 2013). However, it is debated whether TRPV4 is itself sensitive to such stimuli or lies downstream of an osmotic sensor and only mediates the transduction of the stimulus. The available evidence indicates that TRPV4 can be activated by second messengers and phosphorylations (Xu et al., 2003; Poole et al., 2013). It has been shown that activation of TRPV4 depends on phosphorylation on tyrosine 253 by Src kinase or on 5,6′-epoxyeicosatrienoic acid (5,6′-EET) resulting from the breakdown of arachidonic acid by cytochrome P450 epoxygenase (Xu et al., 2003; Poole et al., 2013). Notably, our recent unpublished data indicate that MLC1 can bind to a number of proteins with enzymatic activity, like kinases and phosphatases (Table 1). Further studies are needed to understand if MLC1 affects TRPV4 function by acting as a docking site for mechanical sensors. We recently demonstrated that MLC1, by modulating endosome acidity (see below), can influence TRPV4 trafficking favoring channel recycling versus degradation. We cannot exclude that this effect might mechanistically explain the functional cooperation between MLC1 and TRPV4. Accordingly, defects in calcium influx during hyposmosis were also observed in monocyte-derived macrophages from MLC patients (Petrini et al., 2013).
Regulation of Cl- Channel Function
Among the processes involved in cell volume regulation chloride currents are the object of intense research aimed at understanding their involvement in the pathophysiology of MLC1. Chloride is largely used as co-ion together with cations for electrical neutralization during membrane ion fluxes or as a counter-ion in anion-exchange mechanisms. According to Walz (2002) the cytoplasmic chloride concentration in cultured astrocytes is around 40 mM. In vivo studies generated conflicting results. Depending on the brain area, the intracellular chloride concentration ranged between 36 and 46 mM (Smith et al., 1981), while a lower concentration (6 mM) was found in unspecified glial cells in slice preparations (Ballanyi et al., 1987). It seems that oligodendrocytes have a lower intracellular chloride concentration compared to astrocytes, in which the chloride intracellular concentration is actively kept around 40 mM. In astrocytes the resulting reversal potential, which is more positive than the resting potential, gives rise to an efflux of the ion upon opening of chloride channels during volume regulatory events (Walz, 2002).
Based on the available evidence, two different chloride currents are potentially involved in MLC pathology. Defects in a RVD-induced chloride current have been noted in rat astrocytes following siRNA-mediated MLC1 downregulation and in MLC patient-derived lymphoblasts (Ridder et al., 2011). Based on activation by hypotonic shock, ion selectivity, pharmacological sensitivity, and outward rectifying property, the MLC1-dependent current was identified as a volume regulated anion current (VRAC). VRAC currents were also studied in astrocytes derived from mlc1-null mice (Sirisi et al., 2014; Dubey et al., 2015). In one of these studies, a VRAC current was identified as the Cl- current sensitive to the VRAC inhibitor DCPIB (Sirisi et al., 2014). The hypotonicity-induced VRAC current amplitude in MLC1-/- astrocytes differed slightly, but not significantly, from that recorded in WT astrocytes. However, the slightly smaller VRAC current in MLC1-/- astrocytes was associated with more pronounced vacuolation in brain tissue, consistent with a functional decay of water/ion regulatory mechanisms (Sirisi et al., 2014). In another study performed in astrocytes derived from mlc1-null mice where the MLC1 gene has been replaced by the green fluorescent protein encoding sequence, astrocytes exhibited a hypotonic challenge-induced Cl- current that was identified as due to VRAC channels by cadmium insensitivity and tamoxifen sensitivity (Dubey et al., 2015). Such a current had a lower amplitude in astrocytes from MLC1-/- mice compared to those from wild-type mice, indicating a VRAC current functional decay due to MLC1 ablation. Consistent with the role of VRAC in RVD, astrocytes from MLC1-/- mice reacted to a hypotonic challenge with a slower RVD (Dubey et al., 2015). It is not clear whether differences in the detection of the VRAC-induced chloride current in the two mlc1-null mice models are due to technical problems or to the influence of the genetic background (C57BL/6 versus C57BL/6-129/Svj mixed genetic background). VRAC is not only involved in the RVD, but also in pathological events like apoptotic volume decrease, necrotic volume increase following lactacidosis and excitotoxicity (Okada et al., 2009; Pedersen et al., 2015). Notably, VRAC is the target of several regulatory mechanisms activated by osmotic cell swelling; among these, are EGFR activation and phosphorylation by PI3K, src and ERK (Pedersen et al., 2015). Data supporting the possibility that MLC1 protein itself acts as VRAC are not available. Recently, LRRC8A, a member of the four-transmembrane protein family, was recognized as a component of the VRAC channel in non-neural cells (Qiu et al., 2014; Voss et al., 2014) and also in glial cells were LRRC8A is an indispensable component of a permeability pathway that mediates both swelling-activated and agonist-induced amino acid release (Hyzinski-García et al., 2014). However, the relationship between MLC1 and LRRC8A is still unknown.
Based on the presence of white matter vacuolation in mice lacking ClC-2 (Blanz et al., 2007), this chloride channel was considered as potentially involved in MLC1 disease but no ClC-2 mutations were found in MLC patients (Scheper et al., 2010). However, although initially in humans ClC-2 loss- and gain-of function mutations were linked to idiopathic generalized epilepsy with no apparent leukodystrophy (Haug et al., 2003), a recent study has revealed that autosomal-recessive CLCN2 mutations cause a leukoencephalopathy characterized by intramyelinic oedema (Depienne et al., 2013).
ClC-2 channels are almost ubiquitous and their whole-cell current is characterized by hyperpolarization-induced slow activation, slow deactivating tail currents at more positive potentials, strong inward rectification, and blockade by iodide but not by classical Cl- channel inhibitors and tamoxifen (Jentsch et al., 2002). In the first in vitro study aimed at elucidating the role of ClC-2 channels in MLC, no correlations between MLC1 protein and ClC-2 were found, MLC1 mRNA interference did not change ClC-2 level, and the two proteins did not co-precipitate (Jeworutzki et al., 2012). In the same study, attention was also focused on GlialCAM, whose mutations were found in a subpopulation of MLC1 patients (López-Hernández et al., 2011a,b; Capdevila-Nortes et al., 2013). Overexpression of GlialCAM together with ClC-2 in Xenopus oocytes caused remarkable changes of current features, such as ohmic conductance instead of inward-rectification, instantaneous activation instead of slow activation, and large increase in the current amplitude (Jeworutzki et al., 2012). However, the current recorded in primary astrocytes and identified as carried by ClC-2 because of lack of permeability to iodide and insensitivity to tamoxifen, did not show the above features even if cultured astrocytes are known to express GlialCAM. It cannot be ruled out that the abrupt changes recorded in the heterologous system are the result of GlialCAM protein overexpression (Jeworutzki et al., 2012). A recent study performed in parallel in three different KO mouse models, MLC1-/-, GlialCAM-/- and ClC-2-/-, addressed this point with a detailed analysis of whole-cell recordings from slice preparations (Hoegg-Beiler et al., 2014). As in primary cortical astrocytes obtained from the previously characterized mouse models (Sirisi et al., 2014), also in WT Bergman glia, known to express GlialCAM, Cl- currents showed features expected in the absence of GlialCAM (i.e., inward rectification and slow activation). Moreover, ablation of GlialCAM did not result in the expected changes (i.e., large decrease of the amplitude and stronger inward rectification) and caused a mild decrease of the current, compatible with the decrease in ClC-2 expression observed in MLC1-/- and GlialCAM-/- mice by immunohistochemistry. So far, the most likely interpretation of these results is that MLC1 plays a role as a chaperone contributing to the correct localization of ClC-2 proteins, but is not involved in the modulation of the biophysical features of ClC-2 channel.
pH Regulation in the Endosomal Compartment
Both the V-ATPase and the Na, K-ATPase pumps are known to regulate acidity of the endosomal compartment. Aiming at investigating further the functional relationship between MLC1 and V-ATPase/Na, K-ATPase in the context of the MLC1-associated macromolecular complex described in in vitro experiments (Brignone et al., 2011, 2014), we studied the effects of MLC1 mutations on organelle pH.
Using a video-imaging approach with the pH sensitive dye FITC-dextran, we found that in astrocytoma cells expressing WT, but not mutated MLC1, early endosomes (i.e., EEA1+ and RAB5+) showed a higher pH compared to early endosomes from the parental U251 cells (Brignone et al., 2014). This result is compatible with a possible role for MLC1 in the modulation of V-ATPase and Na, K-ATPase function. Being the early endosomal compartment a check-point station where proteins are sorted toward recycling or degradation depending on further acidification of the organelles, these results suggest that MLC1 can influence the trafficking of proteins co-localizing in the same early endosomes. Indeed, we found that WT but not mutated MLC1 favored TRPV4 channel recycling (Brignone et al., 2014). These results are consistent with a functional role of MLC1 in endosome pH regulation, since recycling vesicles are characterized by a slightly less acidic pH compared to vesicles sorted toward the lysosomal degradative pathway (Grant and Donaldson, 2009; Scott and Gruenberg, 2011). From a pathogenic point of view, it is interesting to note that alterations in organelle pH regulation induce endosome enlargement and formation of intracellular vacuoles similar to those observed in MLC1 KO astrocytes (Duarri et al., 2011) and that some neurodegenerative diseases are characterized by abnormal giant endosomes (Gouras, 2013). It has been hypothesized that disturbance of the endoplasmic compartment pH might be associated with the development of autistic behavior resulting from an irregular arrangement of membrane components like channels and transporters in the plasmalemma of neurons and astrocytes during development (Kondapalli et al., 2014). Notably, autism is part of the symptomatology of MLC disease (López-Hernández et al., 2011a).
MLC1 Functions: Astrocyte Specific Ion Channel or Ion Channel Regulator?
The almost exclusive expression of MLC1 in astrocytes together with the neuropathological alterations observed in MLC patients and the results obtained in cellular models have led to hypothesize a role for MLC1 in the regulation of ion/water fluxes controlling cell volume in specific astrocytic districts such as those facing the brain barriers (blood–brain and cerebrospinal fluid–brain barriers).
Does MLC1 directly function as an ion/water permeation site or as a transport protein? Ion channels herein referred to are crucial for the transepithelial transport of salt and water, the regulation of cellular volume and pH, the acidification of intracellular organelles, and chemical signaling. They may be regulated by calcium, pH, phosphorylation, and lipids. Many channels are oligomers of identical or homologous pore-forming α subunits and assemble in complexes with β/γ subunits which may be essential for their function or modulate their properties. Most of these features have been described for MLC1 (see above). Experimental studies revealed that several different (also redundant) mechanisms control MLC1 membrane localization (protein–protein, protein–lipid interactions, post-translational modifications) suggesting that a strict temporal and spatial regulation of MLC1 is necessary in astrocytes. Although at present conclusive results ruling out MLC1 direct involvement in ion/water permeation/transport in astrocytes are missing, the ensemble of the data collected so far converge to delineate a dual role for MLC1 ranging from a chaperoning to a regulatory role of functional events.
The observation that several of the pathological MLC1 mutations cause a mislocalization of the protein together with that of some of its functional interactors suggests a chaperoning-like role of MLC1 in the correct assembly of those specific proteins. On the other hand, the variety of events affected by MLC1 ablation or mutations, together with the recognition of several putative binding sites that could act as docking sites for regulatory enzymes, prompt to consider its regulatory role in the complex scenario of events taking place in specific intracellular domains and involving water/ion movements and pH control.
Besides the exact definition of the multifaceted role played by MLC1, the advent of MLC1 in animals developing myelin prompts to consider in a phylogenetic perspective the importance of this protein for the new needs of such a complex functional structure comprising astrocytes, oligodendrocytes, and neurons. In MLC disease myelin degeneration is not attributable to directmyelin or oligodendrocyte protein alterations but to an indirect effect mediated by astrocytic dysfunction. Although at present the molecular mechanisms causing myelin degeneration in MLC disease are not known it is established that defects in astrocyte maturation, astrocyte functional impairment causing accumulation of toxic substrates, and/or failure of specific astrocyte-mediated homeostatic pathways can affect myelin formation and maintenance leading to cystic or vacuolating myelin degeneration (reviewed in Lanciotti et al., 2013 and references therein). Due to the important role played by astrocytes in regulating oligodendrocyte progenitor cell proliferation, migration, and differentiation during CNS development (Clemente et al., 2013), we cannot exclude that dysfunctional astrocytes could affect myelination also by acting on oligodendrocyte development.
Concluding Remarks
Channelopathies are a diverse set of disorders associated with defects in ion channel (and transporter) function that are mostly linked with inherited mutations impairing the biophysical properties of the channels. The rapid progress made in molecular genetics and electrophysiology has allowed to expand the field of channelopathies revealing that many disorders reflect dysfunctions in regulatory proteins that alter ion channel synthesis, membrane trafficking and/or posttranslational modifications.
Moreover, associated proteins can define tissue/organ functional specificity, making ion channels at a specific location more susceptible to a pathological condition even if their expression is not tissue-specific. This could be the case of MLC1 which is highly and exclusively expressed by astrocytes in the CNS. The results obtained so far demonstrate MLC1 involvement in astrocyte volume control. Astrocytes not only undergo dramatic volume changes as a result of the ionic dysregulation that occurs in pathological conditions such as ischemia, trauma, and epilepsy but also during neuronal activity in conjunction with extracellular K+ buffering (Florence et al., 2012 and references therein). These volume regulatory mechanisms may thus be fundamental for the maintenance of the homeostatic conditions allowing optimal CNS function. A deeper understanding of the structure and function of MLC1 protein and its relationship with ion channels and ion channel-related proteins may contribute not only to identify novel therapeutic approaches for the rare MLC genetic disease, but may also provide insights into the mechanisms underlying more common brain disorders such as migraines, epilepsies, ischemia, and autism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The financial support of ELA Foundation (grants ELA 2006-001/4 and ELA 2009-002C5A to EA) and Telethon Italy (grants GGP1118 and GEP14134 to EA) is gratefully acknowledged. AL is the recipient of an ELA foundation fellowship (grant no. 2012-021F2). We thank Dr. Francesca Aloisi for helpful comments and critical reading of the manuscript and Dr. Marco Crescenzi for support with the protein mass spectrometry facility at ISS, Rome.
References
Ambrosini, E., Serafini, B., Lanciotti, A., Tosini, F., Scialpi, F., Psaila, R.,et al. (2008). Biochemical characterization of MLC1 protein in astrocytes and its association with the dystrophin-glycoprotein complex. Mol. Cell Neurosci. 37, 480–493. doi: 10.1016/j.mcn.2007.11.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arnedo, T., Aiello, C., Jeworutzki, E., Dentici, M. L., Uziel, G., Simonati, A.,et al. (2014a). Expanding the spectrum of megalencephalic leukoencephalopathy with subcortical cysts in two patients with GLIALCAM mutations. Neurogenetics 15, 41–48. doi: 10.1007/s10048-013-0381-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arnedo, T., López-Hernández, T., Jeworutzki, E., Capdevila-Nortes, X., Sirisi, S., Pusch, M.,et al. (2014b). Functional analyses of mutations in HEPACAM causing megalencephalic leukoencephalopathy. Hum. Mutat. 35, 1175–1178. doi: 10.1002/humu.22622
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Babcock, J. J., and Li, M. (2013). Inside job: ligand-receptor pharmacology beneath the plasma membrane. Acta Pharmacol. Sin. 34, 859–869. doi: 10.1038/aps.2013.51
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ballanyi, K., Grafe, P., and Ten Bruggencate, G. (1987). Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J. Physiol. 382, 159–174. doi: 10.1113/jphysiol.1987.sp016361
Barlowe, C. (2003). Signals for COPII-dependent export from the ER: what’s the ticket out? Trends Cell Biol. 13, 295–300. doi: 10.1016/S0962-8924(03)00082-5
Barrallo-Gimeno, A., and Estévez, R. (2014). GlialCAM, a glial cell adhesion molecule implicated in neurological disease. Adv. Neurobiol. 8, 47–59. doi: 10.1007/978-1-4614-8090-7_3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benfenati, V., Amiry-Moghaddam, M., Caprini, M., Mylonakou, M. N., Rapisarda, C., Ottersen, O. P.,et al. (2007). Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience 148, 876–892. doi: 10.1016/j.neuroscience.2007.06.039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benfenati, V., Caprini, M., Dovizio, M., Mylonakou, M. N., Ferroni, S., Ottersen, O. P.,et al. (2011). An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cellvolume control in astrocytes, Proc. Natl. Acad. Sci. U.S.A. 108, 2563–2568. doi: 10.1073/pnas.1012867108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ben-Zeev, B., Levy-Nissenbaum, E., Lahat, H., Anikster, Y., Shinar, Y., Brand, N.,et al. (2002). Megalencephalic leukoencephalopathy with subcortical cysts; a founder effect in Israeli patients and a higher than expected carrier rate among. Libyan Jews. Hum. Genet. 111, 214–218. doi: 10.1007/s00439-002-0770-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blanz, J., Schweizer, M., Auberson, M., Maier, H., Muenscher, A., Hübner, C. A.,et al. (2007). Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 27, 6581–6589. doi: 10.1523/JNEUROSCI.0338-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bonkowsky, J. L., Nelson, C., Kingston, J. L., Filloux, F. M., Mundorff, M. B., and Srivastava, R. (2010). The burden of inherited leukodystrophies in children. Neurology 75, 718–725. doi: 10.1212/WNL.0b013e3181eee46b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boor, I., Nagtegaal, M., Kamphorst, W., van der Valk, P., Pronk, J. C., van Horssen, J.,et al. (2007). MLC1 is associated with the dystrophin–glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 114, 403–410. doi: 10.1007/s00401-007-0247-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boor, I. P. K., de Groot, K., Mejaski-Bosnjak, V., Brenner, C., van der Knaap, M. S., Scheper, G. C.,et al. (2006). Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum. Mutat. 27, 505–512. doi: 10.1002/humu.20332
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boor, P. K., de Groot, K., Waisfisz, Q., Kamphorst, W., Oudejans, C. B., Powers, J. M.,et al. (2005). MLC1 a novel protein in distal astroglial processes, J. Neuropathol. Exp. Neurol. 64, 412–419.
Brignone, M. S., Lanciotti, A., Macioce, P., Macchia, G., Gaetani, M., Aloisi, F.,et al. (2011). The beta1 subunit of the Na,K-ATPase pump interacts with megalencephalic leukoencephalopathy with subcortical cysts protein 1 in brain astrocytes: new insights into MLC pathogenesis, Hum. Mol. Gen. 20, 90–103. doi: 10.1093/hmg/ddq435
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brignone, M. S., Lanciotti, A., Visentin, S., De Nuccio, C., Molinari, P., Camerini, S.,et al. (2014). Megalencephalic leukoencephalopathy with subcortical cysts protein-1 modulates endosomal pH and protein trafficking in astrocytes: relevance to MLC disease pathogenesis. Neurobiol. Dis. 66, 1–18. doi: 10.1016/j.nbd.2014.02.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brown, D. A. (2006). Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology (Bethesda) 21, 430–439. doi: 10.1152/physiol.00032.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bugiani, M., Moroni, I., Bizzi, A., Nardocci, N., Bettecken, T., Gartner, J.,et al. (2003). Consciousness disturbances in megalencephalic leukoencephalopathy with subcortical cysts. Neuropediatrics 34, 211–212. doi: 10.1055/s-2003-42209
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Capdevila-Nortes, X., López-Hernández, T., Apaja, P. M., López de Heredia, M., Sirisi, S., Callejo, G.,et al. (2013). Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum. Mol. Genet. 22, 4405–4416. doi: 10.1093/hmg/ddt290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, S. Y., Di, A., Naren, A. P., Palfrey, H. C., Kirk, K. L., and Nelson, D. J. (2002). Mechanisms of CFTR regulation by syntaxin 1A and PKA. J. Cell Sci. 115(Pt 4), 783–791.
Christensen, A. P., and Corey, D. P. (2007). TRP channels in mechanosensation: direct or indirect activation? Nat. Rev. Neurosci. 8, 510–521. doi: 10.1038/nrn2149
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Curran, J., and Mohler, P. J. (2014). Alternative paradigms for ion channelopathies: disorders of ion channel membrane trafficking and posttranslational modification. Annu. Rev. Physiol. 77, 505–524. doi: 10.1146/annurev-physiol-021014-071838
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clemente, D., Ortega, M. C., Melero-Jerez, C., and de Castro, F. (2013). The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell Neurosci. 7:268. doi: 10.3389/fncel.2013.00268
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dart, C. (2010). Lipid microdomains and the regulation of ion channel function. J. Physiol. 588(Pt 17), 3169–3178. doi: 10.1113/jphysiol.2010.191585
Depienne, C., Bugiani, M., Dupuits, C., Galanaud, D., Touitou, V., Postma, N.,et al. (2013). Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 12, 659–668. doi: 10.1016/S1474-4422(13)70053-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duarri, A., Lopez de Heredia, M., Capdevila-Nortes, X., Ridder, M. C., Montolio, M., López-Hernández, T.,et al. (2011). Knockdown of MLC1 in primary astrocytes causes cell vacuolation: a MLC disease cell model. Neurobiol. Dis. 43, 228–238. doi: 10.1016/j.nbd.2011.03.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duarri, A., Teijido, O., López-Hernández, T., Scheper, G. C., Barriere, H., Boor, I.,et al. (2008). Molecular pathogenesis of megalencephalic leukoencephalopathy with subcortical cysts: mutations in MLC1 cause folding defects. Hum. Mol. Genet. 17, 3728–3739. doi: 10.1093/hmg/ddn269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dubey, M., Bugiani, M., Ridder, M. C., Postma, N. L., Brouwers, E., Polder, E.,et al. (2015). Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann. Neurol. 77, 114–131. doi: 10.1002/ana.24307
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farhan, H., Wendeler, M. W., Mitrovic, S., Fava, E., Silberberg, Y., Sharan, R.,et al. (2010). MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J. Cell Biol. 189, 997–1011. doi: 10.1083/jcb.200912082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Florence, C. M., Baillie, L. D., and Mulligan, S. J. (2012). Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS ONE 7:e51124. doi: 10.1371/journal.pone.0051124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González, M. I., Krizman-Genda, E., and Robinson, M. B. (2007). Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J. Biol. Chem. 282, 29855–29865. doi: 10.1074/jbc.M704738200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gouras, G. K. (2013). Convergence of synapses, endosomes, and prions in the biology of neurodegenerative diseases. Int. J. Cell Biol. 2013, 141083. doi: 10.1155/2013/141083
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grant, B. D., and Donaldson, J. G. (2009). Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608. doi: 10.1038/nrm2755
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haug, K., Warnstedt, M., Alekov, A. K., Sander, T., Ramírez, A., Poser, B.,et al. (2003). Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat. Genet. 33, 527–532. doi: 10.1038/ng1121
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoegg-Beiler, M. B., Sirisi, S., Orozco, I. J., Ferrer, I., Hohensee, S., Auberson, M.,et al. (2014). Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 5, 3475. doi: 10.1038/ncomms4475
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hyzinski-García, M. C., Rudkouskaya, A., and Mongin, A. A. (2014). LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J. Physiol. 592(Pt 22), 4855–4862. doi: 10.1113/jphysiol.2014.278887
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jentsch, T. J., Stein, V., Weinreich, F., and Zdebik, A. A. (2002). Molecular structure and physiological function of chloride channels. Physiol. Rev. 82, 503–68.
Jeworutzki, E., Lagostena, L., Elorza-Vidal, X., Lopez-Hernandez, T., Estévez, R., and Pusch, M. (2014). GlialCAM, a CLC-2 Cl(-) channel subunit, activates the slow gate of CLC chloride channels. Biophys. J. 107, 1105–1116. doi: 10.1016/j.bpj.2014.07.040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeworutzki, E., López-Hernández, T., Capdevila-Nortes, X., Sirisi, S., Bengtsson, L., Montolio, M.,et al. (2012). GlialCAM A protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron 73, 951–961. doi: 10.1016/j.neuron.2011.12.039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. B. (2014). Channelopathies. Korean J. Pediatr. 57, 1–18. doi: 10.3345/kjp.2014.57.1.1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kondapalli, K. C., Prasad, H., and Rao, R. (2014). An inside job: how endosomal Na(+)/H(+) exchangers link to autism and neurological disease. Front. Cell Neurosci. 8:172. doi: 10.3389/fncel.2014.00172
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kristensen, B., Birkelund, S., and Jorgensen, P. L. (2003). Trafficking of Na, K-ATPase fused to enhanced green fluorescent protein is mediated by protein kinase A or C. J. Membr. Biol. 191, 25–36. doi: 10.1007/s00232-002-1043-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lanciotti, A., Brignone, M. S., Bertini, E., Petrucci, T. C., Aloisi, F., and Ambrosini, E. (2013). Astrocytes: emerging stars in leukodystrophy pathogenesis. Transl. Neurosci. 4, 12. doi: 10.2478/s13380-013-0118-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lanciotti, A., Brignone, M. S., Camerini, S., Serafini, B., Macchia, G., Raggi, C.,et al. (2010). MLC1 trafficking and membrane expression in astrocytes: role of caveolin-1 and phosphorylation. Neurobiol. Dis. 37, 581–595. doi: 10.1016/j.nbd.2009.11.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lanciotti, A., Brignone, M. S., Molinari, P., Visentin, S., De Nuccio, C., Macchia, G.,et al. (2012). Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum. Mol. Genet. 21, 2166–2180. doi: 10.1093/hmg/dds032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leegwater, P. A., Yuan, B. Q., Van Der Steen, J., Mulders, J., Konst, A. A., Boor, P. K.,et al. (2001). Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 68, 831–838. doi: 10.1086/319519
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
López-Hernández, T., Ridder, M. C., Montolio, M., Capdevila-Nortes, X., Polder, E., Sirisi, S.,et al. (2011a). Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88, 422–432. doi: 10.1016/j.ajhg.2011.02.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
López-Hernández, T., Sirisi, S., Capdevila-Nortes, X., Montolio, M., Fernández-Dueñas, V., Scheper, G. C.,et al. (2011b). Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 20, 3266–3277. doi: 10.1093/hmg/ddr238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mamenko, M., Zaika, O. L., Boukelmoune, N., Berrout, J., O’Neil, R. G., and Pochynyuk, O. (2013). Discrete control of TRPV4 channel function in the distal nephron by protein kinases A and C. J. Biol. Chem. 88, 20306–20314. doi: 10.1074/jbc.M113.466797
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Masaki, K., Suzuki, S. O., Matsushita, T., Yonekawa, T., Matsuoka, T., Isobe, N.,et al. (2012). Extensive loss of connexins in Baló’s disease: evidence for an auto-antibody-independent astrocytopathy via impaired astrocyte-oligodendrocyte/myelin interaction. Acta Neuropathol. 123, 887–900. doi: 10.1007/s00401-012-0972-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mejaski-Bosnjak, V., Besenski, N., Brockmann, K., Pouwels, P. J., Frahm, J., and Hanefeld, F. A. (1997). Cystic leukoencephalopathy in a megalencephalic child: clinical and magnetic resonance imaging/magnetic resonance spectroscopy findings. Pediatr. Neurol. 16, 347–350. doi: 10.1016/S0887-8994(97)00044-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meyer, J., Huberth, A., Ortega, G., Syagailo, Y. V., Jatzke, S., Mössner, R.,et al. (2001). A missense mutation in a novel gene encoding a putative cation channel is associated with catatonic schizophrenia in a large pedigree. Mol. Psychiatry 6, 302–306. doi: 10.1038/sj.mp.4000869
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Michelsen, K., Yuan, H., and Schwappach, B. (2005). Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 6, 717–22. doi: 10.1038/sj.embor.7400480
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Molofsky, A. V., Krencik, R., Ullian, E. M., Tsai, H. H., Deneen, B., Richardson, W. D.,et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Montagna, G., Teijido, O., Eymard-Pierre, E., Muraki, K., Cohen, B., Loizzo, A.,et al. (2006). Vacuolating megalencephalic leukoencephalopathy with subcortical cysts: functional studies of novel variants in MLC1. Hum. Mutat. 27, 292. doi: 10.1002/humu.9407
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neusch, C., Rozengurt, N., Jacobs, R. E., Lester, H. A., and Kofuji, P. (2001). Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 21, 5429–5438.
Nico, B., Frigeri, A., Nicchia, G. P., Corsi, P., Ribatti, D., Quondamatteo, F.,et al. (2003). Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 42, 235–251. doi: 10.1002/glia.10216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nico, B., Paola Nicchia, G., Frigeri, A., Corsi, P., Mangieri, D., Ribatti, D.,et al. (2004). Altered blood-brain barrier development in dystrophic MDX mice. Neuroscience 125, 921–935. doi: 10.1016/j.neuroscience.2004.02.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nilius, B., and Voets, T. (2013). The puzzle of TRPV4 channelopathies. EMBO Rep. 14, 152–163. doi: 10.1038/embor.2012.219
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noël, G., Belda, M., Guadagno, E., Micoud, J., Klöcker, N., and Moukhles, H. (2005). Dystroglycan and Kir4.1 coclustering in retinal Müller glia is regulated by laminin-1 and requires the PDZ-ligand domain of Kir4.1. J Neurochem. 94, 691–702. doi: 10.1111/j.1471-4159.2005.03191.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noell, S., Wolburg-Buchholz, K., Mack, A. F., Beedle, A. M., Satz, J. S., Campbell, K. P.,et al. (2011). Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur. J. Neurosci. 33, 2179–2186. doi: 10.1111/j.1460-9568.2011.07688.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Okada, Y., Sato, K., and Numata, T. (2009). Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 587(Pt 10), 2141–2149. doi: 10.1113/jphysiol.2008.165076
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pascual-Castroviejo, I., van der Knaap, M. S., Pronk, J. C., García-Segura, J. M., Gutiérrez-Molina, M., and Pascual-Pascual, S. I. (2005). Vacuolating megalencephalic leukoencephalopathy: 24 year follow-up of two siblings. Neurologia 20, 33–40.
Patrono, C., Di Giacinto, G., Eymard-Pierre, E., Santorelli, F. M., Rodriguez, D., De Stefano, N.,et al. (2003). Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts. Neurology 61, 534–537. doi: 10.1212/01.WNL.0000076184.21183.CA
Pedersen, S. F., Klausen, T. K., and Nilius, B. (2015). The identification of VRAC (Volume Regulated Anion Channel): an amazing Odyssey. Acta Physiol. (Oxf) doi: 10.1111/apha.12450 [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petrini, S., Minnone, G., Coccetti, M., Frank, C., Aiello, C., Cutarelli, A.,et al. (2013). Monocytes and macrophages as biomarkers for the diagnosis of megalencephalic leukoencephalopathy with subcortical cysts. Mol. Cell Neurosci. 56, 307–321. doi: 10.1016/j.mcn.2013.07.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pfrieger, F. W. (2009). Roles of glial cells in synapse development. Cell Mol. Life Sci. 66, 662037–2047. doi: 10.1007/s00018-009-0005-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poole, D. P., Amadesi, S., Veldhuis, N. A., Abogadie, F. C., Lieu, T., Darby, W.,et al. (2013). Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J. Biol. Chem. 288, 5790–5802. doi: 10.1074/jbc.M112.438184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qiu, Z., Dubin, A. E., Mathur, J., Tu, B., Reddy, K., Miraglia, L. J.,et al. (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458. doi: 10.1016/j.cell.2014.03.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ridder, M. C., Boor, I., Lodder, J. C., Postma, N. L., Capdevila-Nortes, X., Duarri, A.,et al. (2011). Megalencephalic leucoencephalopathy with cysts: defect in chloride currents and cell volume regulation. Brain 134, 3342–3354. doi: 10.1093/brain/awr255
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scheper, G. C., van Berkel, C. G., Leisle, L., de Groot, K. E., Errami, A., Jentsch, T. J.,et al. (2010). Analysis of CLCN2 as candidate gene for megalencephalic leukoencephalopathy with subcortical cysts. Genet. Test Mol. Biomarkers 14, 225–257. doi: 10.1089/gtmb.2009.0148
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmitt, A., Gofferje, V., Weber, M., Meyer, J., Mössner, R., and Lesch, K. P. (2003). The brainspecific protein MLC1 implicated in megalencephalic leukoencephalopathy with subcortical cysts is expressed in glial cells in the murine brain. Glia 44, 283–295. doi: 10.1002/glia.10304
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schutze, M. P., Peterson, P. A., and Jackson, M. R. (1994). An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J. 13, 1696–1705.
Scott, C. C., and Gruenberg, J. (2011). Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 33, 103–110. doi: 10.1002/bies.201000108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scott, D. B., Blanpied, T. A., and Ehlers, M. D. (2003). Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45, 755–767. doi: 10.1016/S0028-3908(03)00250-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shikano, S., and Li, M. (2003). Membrane receptor trafficking: evidence of proximal and distal zones conferred by two independent endoplasmic reticulum localization signals. Proc. Natl. Acad. Sci. U.S.A. 100, 5783–5788. doi: 10.1073/pnas.1031748100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Simons, K., and Ikonen, E. (1997). Functional rafts in cell membranes. Nature 387, 569–572. doi: 10.1038/42408
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singhal, B. S., Gorospe, J. R., and Naidu, S. (2003). Megalencephalic leukoencephalopathy with subcortical cysts. J. Child Neurol. 18, 646–652. doi: 10.1177/08830738030180091201
Singhal, B. S., Gursahani, R. D., Udani, V. P., and Biniwale, A. A. (1996). Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr. Neurol. 14, 291–296. doi: 10.1016/0887-8994(96)00048-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sirisi, S., Folgueira, M., Lopez-Hernandez, T., Minieri, L., Pérez-Rius, C., Gaitán-Peñas, H.,et al. (2014). Megalencephalic leukoencephalopathy with subcortical Cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum. Mol. Genet. 23, 5069–5086. doi: 10.1093/hmg/ddu231
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sivaprasadarao, A., Taneja, T. K., Mankouri, J., and Smith, A. J. (2007). Trafficking of ATP-sensitive potassium channels in health and disease. Biochem. Soc. Trans. 35, 1055–1059. doi: 10.1042/BST0351055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sjö, A., Magnusson, K. E., and Peterson, K. H. (2005). Association of alpha-dystrobrevin with reorganizing tight junctions. J. Membr. Biol. 203, 21–30. doi: 10.1007/s00232-004-0728-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, Q. R., Johnson, C. E., and Woodbury, D. M. (1981). Uptake of 36Cl_ and 22Na by the brain-cerebrospinal fluid system. J. Neurochem. 37, 117–124. doi: 10.1111/j.1471-4159.1981.tb05298.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Son, Y. K., Park, H., Firth, A. L., and Park, W. S. (2013). Side-effects of protein kinase inhibitors on ion channels. J. Biosci. 38, 937–949. doi: 10.1007/s12038-013-9383-y
Steinke, V., Meyer, J., Syagailo, Y. V., Ortega, G., Hameister, H., Mössner, R.,et al. (2003). The genomic organization of the murine Mlc1 (Wkl1, KIAA0027) gene. J. Neural. Transm. 110, 333–343. doi: 10.1007/s00702-002-0788-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Teijido, O., Martínez, A., Pusch, M., Zorzano, A., Soriano, E., Del Río, J. A.,et al. (2004). Localization and functional analyses of the MLC1 protein involved in megalencephalic leukoencephalopathy with subcortical cysts. Hum. Mol. Genet. 13, 2581–2594. doi: 10.1093/hmg/ddh291
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Topçu, M., Gartioux, C., Ribierre, F., Yalçinkaya, C., Tokus, E., Oztekin, N.,et al. (2000). Vacuoliting megalencephalic leukoencephalopathy with subcortical cysts, mapped to chromosome 22qtel. Am. J. Hum. Genet. 66, 733–739. doi: 10.1086/302758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Topcu, M., Saatci, I., Topcuoglu, M. A., Kose, G., and Kunak, B. (1998). Megalencephaly and leukodystrophy with mild clinical course: a report on 12 new cases. Brain Dev. 20, 142–153. doi: 10.1016/S0387-7604(98)00002-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vaknin-Dembinsky, A., Karussis, D., Avichzer, J., and Abramsky, O. (2014). NMO spectrum of disorders: a paradigm for astrocyte-targeting autoimmunity and its implications for MS and other CNS inflammatory diseases. J. Autoimmun. 54, 93–99. doi: 10.1016/j.jaut.2014.05.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Knaap, M. S., Barth, P. G., Stroink, H., van Nieuwenhuizen, O., Arts, W. F., Hoogenraad, F.,et al. (1995a). Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol. 37, 324–334. doi: 10.1002/ana.410370308
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Knaap, M. S., Valk, J., Barth, P. G., Smit, L. M., van Engelen, B. G., and Tortori Donati, P. (1995b). Leukoencephalopathy with swelling in children and adolescents: MRI patterns and differential diagnosis. Neuroradiology 37, 679–686. doi: 10.1007/BF00593394
Van der Knaap, M. S., Barth, P. G., Vrensen, G. F., and Valk, J. (1996). Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol. 92, 206–212. doi: 10.1007/s004010050510
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Knaap, M. S., Boor, I., and Estévez, R. (2012). Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 11, 973–985. doi: 10.1016/S1474-4422(12)70192-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Venkatachalam, K., Wong, C. O., and Zhu, M. X. (2014). The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium doi: 10.1016/j.ceca.2014.10.008 [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Voss, F. K., Ullrich, F., Münch, J., Lazarow, K., Lutter, D., Mah, N.,et al. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. doi: 10.1126/science.1252826
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walz, W. (2002). Chloride/anion channels in glial cell membranes. Glia 40, 1–10. doi: 10.1002/glia.10125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., Matteson, J., An, Y., Moyer, B., Yoo, J. S., Bannykh, S.,et al. (2004). COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 167, 65–74. doi: 10.1083/jcb.200401035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., and Paller, A. S. (2006). Lipid Rafts: membrane Triage Centers. J. Invest. Dermatol. 126, 951–953. doi: 10.1038/sj.jid.5700282
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wingerchuk, D. M., Lennon, V. A., Lucchinetti, C. F., Pittock, S. J., and Weinshenker, B. G. (2007). The spectrum of neuromyelitis optica. Lancet Neurol. 6, 805–815. doi: 10.1016/S1474-4422(07)70216-8
Xu, H., Zhao, H., Tian, W., Yoshida, K., Roullet, J. B., and Cohen, D. M. (2003). Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase-dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J. Biol. Chem. 278, 11520–11527. doi: 10.1074/jbc.M211061200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Dong, C., Wu, Q. J., Balch, W. E., and Wu, G. (2011). Di-acidic motifs in the membrane-distal C termini modulate the transport of angiotensin II receptors from the endoplasmic reticulum to the cell surface. J. Biol. Chem. 286, 20525–20535. doi: 10.1074/jbc.M111.222034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zschocke, J., Bayatti, N., and Behl, C. (2005). Caveolin and GLT-1 gene expression is reciprocally regulated in primary astrocytes: association of GLT-1 with non-caveolar lipid rafts. Glia 49, 275–287. doi: 10.1002/glia.20116
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: astrocytes, brain edema, calcium, potassium, chloride ions, myelin vacuolation
Citation: Brignone MS, Lanciotti A, Camerini S, De Nuccio C, Petrucci TC, Visentin S and Ambrosini E (2015) MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front. Cell. Neurosci. 9:106. doi: 10.3389/fncel.2015.00106
Received: 03 February 2015; Paper pending published: 18 February 2015
Accepted: 09 March 2015; Published online: 01 April 2015.
Edited by:
Maria Cristina D'Adamo, University of Perugia, ItalyReviewed by:
Alexander A. Mongin, National Academy of Sciences of Belarus, BelarusMario Valentino, University of Malta, Malta
Copyright © 2015 Brignone, Lanciotti, Camerini, De Nuccio, Petrucci, Visentin and Ambrosini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Ambrosini, Department of Cell Biology and Neuroscience, Istituto Superiore di Sanità, Viale Regina Elena, 299, Rome 00161, Italy elena.ambrosini@iss.it
† These authors have contributed equally to this work.