A single-cross, RNA interference-based genetic tool for examining the long-term maintenance of homeostatic plasticity
 Douglas J. Brusich1†
Douglas J. Brusich1†  Ashlyn M. Spring
Ashlyn M. Spring C. Andrew Frank
C. Andrew Frank- 1Department of Anatomy and Cell Biology, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
- 2Interdisciplinary Graduate Program in Genetics, University of Iowa, Iowa City, IA, USA
- 3Interdisciplinary Programs in Genetics, Neuroscience, and MCB, University of Iowa, Iowa City, IA, USA
Homeostatic synaptic plasticity (HSP) helps neurons and synapses maintain physiologically appropriate levels of output. The fruit fly Drosophila melanogaster larval neuromuscular junction (NMJ) is a valuable model for studying HSP. Here we introduce a genetic tool that allows fruit fly researchers to examine the lifelong maintenance of HSP with a single cross. The tool is a fruit fly stock that combines the GAL4/UAS expression system with RNA interference (RNAi)-based knock down of a glutamate receptor subunit gene. With this stock, we uncover important new information about the maintenance of HSP. We address an open question about the role that presynaptic CaV2-type Ca2+ channels play in NMJ homeostasis. Published experiments have demonstrated that hypomorphic missense mutations in the CaV2 α1a subunit gene cacophony (cac) can impair homeostatic plasticity at the NMJ. Here we report that reducing cac expression levels by RNAi is not sufficient to impair homeostatic plasticity. The presence of wild-type channels appears to support HSP—even when total CaV2 function is severely reduced. We also conduct an RNAi- and electrophysiology-based screen to identify new factors required for sustained homeostatic signaling throughout development. We uncover novel roles in HSP for Drosophila homologs of Cysteine string protein (CSP) and Phospholipase Cβ (Plc21C). We characterize those roles through follow-up genetic tests. We discuss how CSP, Plc21C, and associated factors could modulate presynaptic CaV2 function, presynaptic Ca2+ handling, or other signaling processes crucial for sustained homeostatic regulation of NMJ function throughout development. Our findings expand the scope of signaling pathways and processes that contribute to the durable strength of the NMJ.
Introduction
Animal nervous systems continually face challenges to normal function. When encountering neuronal stress, the outputs of synapses and circuits must be kept within a physiologically appropriate range. This restriction requires the activity of homeostatic regulatory systems. Homeostatic plasticity is a conserved principle across metazoan nervous systems. This fact is demonstrated by studies examining nematode, insect, crustacean, and mammalian synaptic preparations (Perez-Otano and Ehlers, 2005; Marder and Goaillard, 2006; Turrigiano, 2008; Pozo and Goda, 2010; Davis, 2013; Davis and Müller, 2015). Few molecules required for homeostatic synaptic plasticity (HSP) have been categorized into coherent signaling pathways. This is a gap in knowledge that limits our understanding of how neurons and synapses maintain stable function.
The Drosophila melanogaster third instar larval neuromuscular junction (NMJ) is a superb synapse for studying the molecular underpinnings of HSP (Frank, 2014a). At the fruit fly NMJ, genetic and pharmacological manipulations can be used to decrease the sensitivity of postsynaptic glutamate receptors to single vesicles of glutamate (decreased quantal size) (Petersen et al., 1997; Frank et al., 2006; Frank, 2014a). Decreased quantal size triggers retrograde (muscle-to-nerve) signaling that drives increased neurotransmitter release (increased quantal content, QC). As a result of this homeostatic signaling process, normal levels of muscle excitation are maintained.
Robust NMJ regulation has been exploited in genetic screens to uncover molecules required for HSP. One approach employs acute application of the glutamate receptor inhibitor, philanthotoxin-433 (PhTox) on semi-intact NMJ preparations of Drosophila larvae (Frank et al., 2006). Using Drosophila mutants, this approach has uncovered factors required for a short-term induction of synaptic homeostasis at the NMJ (10 min PhTox treatment), including presynaptic CaV2-type Ca2+ channels (Frank et al., 2006; Müller and Davis, 2012), KV potassium channels (Bergquist et al., 2010), epithelial sodium (ENaC) channels (Younger et al., 2013), BLOC-1 complex members (biogenesis of lysosome-related organelles complex-1) (Dickman and Davis, 2009), SNARE complex members (soluble N-ethylmalemide-sensitive factor attachment receptors) (Dickman et al., 2012), Rab3-GAP (Müller et al., 2011), RIM (Rab3 interacting molecule) (Müller et al., 2012), RIM binding protein (Müller et al., 2015), and secreted endostatin (Wang et al., 2014). Some of these proteins gate important presynaptic molecular events such as an increase in Ca2+ influx or an increase in the size of the readily releasable pool of presynaptic vesicles (Weyhersmüller et al., 2011; Müller and Davis, 2012; Müller et al., 2012; Younger et al., 2013). These presynaptic events mirror salient aspects of HSP in mammalian neurons (Murthy et al., 2001; Burrone et al., 2002; Zhao et al., 2011). Therefore, homeostatic processes at the Drosophila NMJ appear to target fundamentally conserved mechanisms that are discoverable by genetic approaches.
The aggregate research at the NMJ suggests overlapping (yet distinct) classes of molecules are required for the acute induction of HSP and the long-term maintenance of HSP (Frank, 2014a). However, acute application of PhTox misses notable factors needed for the continued expression of synaptic homeostasis throughout life, such as the Rho-type guanine exchange factor Ephexin (Frank et al., 2009), the pair-rule transcription factor Gooseberry (Marie et al., 2010), and the protein translation regulator Target of Rapamycin (TOR) (Penney et al., 2012). Alternative approaches are required to identify and elucidate signaling processes the NMJ employs to maintain faithful neurotransmission in response to chronic challenges met throughout development. Signaling processes needed for the prolonged developmental expression of synaptic homeostasis at the Drosophila NMJ could serve a similar function in higher organisms.
A null Drosophila GluRIIA glutamate receptor subunit mutation (Petersen et al., 1997) is valuable for characterizing molecules that work to maintain homeostatic plasticity for extended developmental time (Frank et al., 2009; Marie et al., 2010; Penney et al., 2012; Frank, 2014a). GluRIIA loss decreases quantal size, and the NMJ responds with a homeostatic increase in presynaptic release (Petersen et al., 1997; DiAntonio et al., 1999). Yet Drosophila GluRIIA mutations are not perfectly ideal for large-scale, high-throughput genetic approaches to identify homeostatic factors. Use of these mutations in screens requires generations of genetic crossing, recombination (depending upon the genomic location of the screened mutation to be tested), and the generation of homozygous double mutants. All of this work needs to be completed prior to conducting electrophysiological analyses. Partial impairment of GluRIII (also known as GluRIIC) gene function presents an alternate possibility. GluRIII is an essential glutamate receptor subunit; null GluRIII mutations are embryonic lethal, but GluRIII mutant animals can be rescued to viability with low levels of GluRIII gene expression (Marrus et al., 2004). Third instar larval GluRIII hypomorphs have decreased quantal size and quantal frequency, but evoked excitation is normal because of a homeostatic increase in presynaptic release (Marrus et al., 2004).
For this study, we constructed a Drosophila melanogaster stock that exploits partial GluRIII loss to study the long-term maintenance of HSP in a single genetic cross. The stock takes advantage of the GAL4/UAS expression system and RNA interference (RNAi)-based expression tools (Brand and Perrimon, 1993; Lee and Carthew, 2003; Dietzl et al., 2007; Ni et al., 2008, 2009). Using this stock, we address an open question regarding how CaV2/Cacophony Ca2+ channel function gates synaptic homeostasis at the NMJ. Prior studies (ours included), demonstrate that hypomorphic, missense mutations in cacophony (cac) block homeostatic plasticity (Frank et al., 2006, 2009; Müller and Davis, 2012). Yet here we show that strong knock down of cacophony (cac) gene function throughout life is not sufficient on its own to impair homeostatic plasticity. We also conduct an RNAi- and electrophysiology-based screen to identify new molecules required for the long-term maintenance of homeostatic plasticity. We document novel roles in synaptic homeostasis for proteins previously implicated in intracellular Ca2+ regulation, including Drosophila homologs of Cysteine String Protein (CSP), Phospholipase Cβ (Plc21C), and Gαq.
Materials and Methods
Drosophila Husbandry and Stocks
Fruit flies were reared in chambers with temperature control (29°C for RNAi screen; otherwise 25°C). w1118 (Hazelrigg et al., 1984) is utilized as a wild-type control unless otherwise indicated. Drosophila stocks carrying various mutant alleles, GAL4 drivers, or UAS-driven transgenes were used. Stocks were either obtained from the Bloomington Drosophila Stock Center (BDSC, Bloomington, IN), the Vienna Drosophila RNAi Center (VDRC, Vienna, Austria), or directly from researchers who generated them. UAS-RNAi lines from VDRC (Dietzl et al., 2007) detailed in this manuscript include: UAS-cac[RNAi] (VDRC transformant #104168; cacKK101478), UAS-Csp[RNAi] (VDRC #34168; CspGD10571), and UAS-Plc21C[RNAi] (VDRC #26558; Plc21CGD11359). Plc21C TRiP UAS-RNAi lines (Ni et al., 2008, 2009) obtained from BDSC include: Plc21CHMS0600, Plc21CJF01210, Plc21CJF01211. A transgenic wild-type cac expression construct is UAS-cac-eGFP786C (Kawasaki et al., 2004). Classical mutant alleles and deficiency stocks include: GluRIIASP16 (Petersen et al., 1997), CspDG29203 (Wishart et al., 2012), CspEY22488 (BDGP gene disruption project, Bellen et al., 2004), cacS (Smith et al., 1998), cacHC129 (Kulkarni and Hall, 1987), Plc21Cp60A (Weinkove et al., 1999), Df(2L)BSC4 (J. Deal and K. Cook, BDSC), Gαq1370 (Kain et al., 2008), Gαqf04219 and Gαq221c (Banerjee et al., 2006), and Gαq28 (Yao and Carlson, 2010). GAL4 drivers include elaV(C155)-GAL4 (Lin and Goodman, 1994), Scabrous-GAL4 (Budnik et al., 1996), and BG57-GAL4 (Budnik et al., 1996).
We constructed UAS-GluRIII[RNAi] by PCR amplification from genomic DNA, introducing Xba I restriction sites, and cloning a tandem duplicated fold-back version of the PCR product into the pUAST pWiz vector (Lee and Carthew, 2003). For the PCR amplification step, the GluRIII gene PCR primers were as follows: CAF2L: 5′-TCGATCTAGAGATCCTCGAGCGAGGATGGACAGCGGA-3′ and CAF2R: 5′-TTATTCTAGATGATTATCTCGCCAATGATGC-3′. Standard injection procedures were used to generate UAS-GluRIII[RNAi] transgenic stocks. A UAS-GluRIII[RNAi] insertion on chromosome III was built into a screening stock with the full genotype: elaV(C155)-GAL4; Scabrous-GAL4/CyO-GFP; BG57-GAL4, UAS-GluRIII[RNAi]/TM6B. For simplicity, this screening stock is termed T15 in the manuscript, and balancer chromosomes are not included in its description in the Results Section of the text.
Electrophysiology and Analysis
Wandering third instar larvae were selected for electrophysiological recordings. Sharp electrode recordings were taken from muscle 6 of abdominal segments 2 and 3, as previously described (Davis et al., 1998; Frank et al., 2006). Larvae were dissected in a modified HL3 saline: NaCl (70 mM), KCl (5 mM), MgCl2 (10 mM), NaHCO3 (10 mM), sucrose (115 mM = 3.9%), trehalose (4.2 mM = 0.16%), HEPES (5.0 mM = 0.12%), and CaCl2 (0.5 mM, unless otherwise indicated). Data were collected using Axopatch 200B or Axoclamp 900A amplifiers (Molecular Devices, Sunnyvale, CA), digitized using a Digidata 1440A data acquisition system (Molecular Devices), and recorded with pCLAMP 10 acquisition software (Molecular Devices). For presynaptic nerve stimulation, a Master-8 pulse stimulator (A.M.P. Instruments, Jerusalem, Israel) and an ISO-Flex isolation unit (A.M.P. Instruments) were utilized to deliver 1 ms suprathreshold stimuli to the appropriate segmental nerve. The average spontaneous miniature EPSP (mEPSP) amplitude was quantified by measuring the amplitude of ~100–200 individual spontaneous release events per NMJ (unless mEPSP frequency was too low for ~1 min continuous recording, in which case all mEPSPs available were measured). The average per-NMJ mEPSP amplitudes were then averaged for each genotype. The average evoked EPSP amplitude was calculated for each NMJ. Quantal content (QC) was determined for each recording by calculating the ratio of average EPSP and average mEPSP amplitudes. Quantal contents were calculated for each recording and then averaged across NMJs of the indicated genotypes. Where indicated, quantal content was corrected for non-linear summation (Martin, 1955). Selected raw electrophysiological data are included as Supplementary Table 1.
Immunostaining
Third instar larvae were fileted in HL3 saline. Dissected animals were fixed for 3 min in Bouin's fixative (Ricca Chemical Company, Arlington, TX), washed using standard procedures, and incubated in primary antibodies overnight at 4°C. This was followed by additional washes and a 2-hour incubation in secondary antibody at room temperature. Staining was performed using the following primary antibodies: mouse anti-Synapsin (3C11) 1:50 (Developmental Studies Hybridoma Bank, University of Iowa—DSHB) (Klagges et al., 1996); rabbit anti-Dlg 1:30,000 (Budnik et al., 1996); mouse anti-Brp (nc82) 1:250 (DSHB); rabbit anti-GluRIII 1:4000 (Marrus et al., 2004). The following fluorophore-conjugated antibodies were also used (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA): goat anti-mouse-488 1:1000 (DyLight); goat anti-rabbit-549 1:2000 (DyLight); Alexa-647 goat anti-HRP 1:500. Larval preparations were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and imaged at room temperature using Zen software on a Zeiss 700 LSM mounted on an Axio Observer.Z1 using an EC Plan-Neofluar 40X Oil DIC Objective (aperture 1.30) or an EC Plan-Apochromat 63x Oil DIC Objective (aperture 1.40) (Carl Zeiss Microscopy, Jena, Germany). For each experiment, experimental and control larval preps were stained in the same container, mounted on the same slide, imaged using identical acquisition settings, and analyzed using the same procedure and thresholds.
Image Analyses
Bouton numbers were quantified semi-automatically using the “Spots” function in Imaris x64 v7.6.0 (Bitplane, Zurich Switzerland). Boutons were counted using the anti-Synapsin channel with the XY diameter set at 3 μm. Active Zones were counted using the anti-Brp channel with an XY diameter of 0.3 μm. GluRIII clusters were counted using the anti-GluRIII channel with an XY diameter of 0.5 μm. The threshold was adjusted so that each bouton, active zone, or GluRIII cluster was recognized once. Any errors in automated counting were corrected by hand to arrive at the final value. GluRIII levels and area were assessed using ImageJ 1.48s/Java 1.6.0_24 (64-bit) with Fiji plugins. Z-stack images were compressed using the maximum projection function; an ROI was generated from a mask of the HRP channel and used to define the synapse; a second ROI was hand drawn to exclude any non-synaptic structures in the image; a minimum threshold was set for each channel to eliminate background fluorescence and held consistent within each experiment; the Measure function was used to assess fluorescence intensity and area for each channel [Brp(488), GluRIII(549), HRP(647)]. For each NMJ, total GluRIII fluorescence intensity and synapse coverage was normalized to the synaptic area of the HRP channel and compared between the genotypes analyzed. Cluster area was calculated by dividing the total synaptic area of GluRIII by the number of GluRIII clusters for each synapse imaged.
RNA Extraction and Quantitative RT-PCR
For each genotype, 25–30 wandering third instar, female larvae were sorted in HL3 saline, collected on ice and rinsed in HL3 with excess saline pipetted off before homogenization. Larvae were homogenized in 200 μL TRIzol reagent (Ambion, Life Technologies) using disposable plastic micropestles. Total homogenate was then brought to 1 mL by addition of 800 μL TRIzol, tubes were vigorously inverted, and the resultant homogenate was frozen at −80°C until all genotypes were prepared. Samples were allowed to thaw on ice and incubated for 5 min at room temperature before proceeding with RNA isolation using the manufacturer's instructions with the following changes: Following isopropanol addition, samples were placed at −20°C for precipitation, and two ethanol wash cycles were used. Samples were quantified using a Nanodrop (Thermo Scientific). Reverse transcription was done using iScript (Bio-Rad) according to manufacturer's instructions using 1 μg of total input. Quantitative PCR (Bio-Rad CFX96) was done using SYBR Green reagent (Bio-Rad) with a 10 μL final reaction volume and technical replicates in triplicate. Final primer concentrations were 250 nM. For cac, exon spanning primers were designed with the following sequences: cac-F 5′-cgggaacgagagttgtacg-3′ and cac-R 5′-actggagatggcagtacacg-3′ (this study), and RpL32-F 5′-atgaccatccgcccagcatac-3′ and RpL32-R 5′-ctgcatgagcaggacctccag-3′ (PK Geyer Lab, University of Iowa). Primer piloting was done using stepwise dilutions of iScript product to verify PCR efficiency. Final quantification was done using iScript product diluted to 1:444.4 in the final PCR. Melt curve analysis verified presence of a single product for all reactions.
Statistical Analyses
Statistical significance was assessed either by Student's T-Test, comparing an experimental data set and a control data set, or One-Way ANOVA with Tukey's post-hoc test across multiple data sets, as appropriate. Specific p value ranges are given in the text, figures, and figure legends, with p < 0.05 marked as significant (#p = 0.05; *p < 0.05; **p < 0.01; ***p < 0.001). For some p-values that trend toward statistical significance (0.05 < p < 0.1), specific values are given as indicated. The values reported or plotted on bar graphs are mean ± SEM.
Results
Homeostatic Challenge: GluRIII Gene Knock Down by RNAi
Partial knock down of GluRIII gene function should induce a significant decrease in NMJ quantal size and a homeostatic increase in NMJ quantal content (Marrus et al., 2004). Using the Drosophila pWiz vector (Lee and Carthew, 2003), we created a transgene to target the GluRIII gene for functional knock down by RNAi (see Materials and Methods). From a pWiz-UAS-GluRIII[RNAi] clone, we generated UAS-GluRIII[RNAi] transgenic Drosophila stocks and crossed them to stocks harboring the muscle-specific GAL4 driver, BG57-GAL4 (Budnik et al., 1996) (Figure 1A). We then analyzed the larval cross progeny by electrophysiology to assess the efficiency of glutamate receptor subunit knock down.
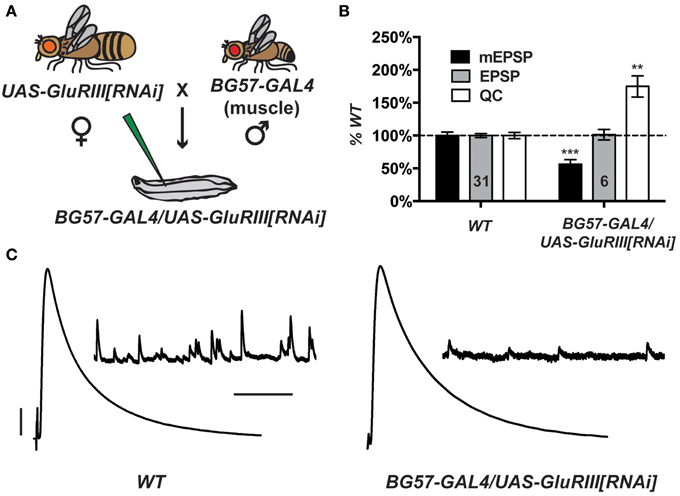
Figure 1. Postsynaptic GluRIII gene knock down induces robust homeostatic compensation. (A) Crossing scheme. NMJs from F1 larvae (genotype BG57-GAL4/UAS-GluRIII[RNAi]) are subjected to electrophysiological analyses. (B) Quantal size (miniature excitatory postsynaptic potentials, mEPSP) is decreased for BG57-GAL4/UAS-GluRIII[RNAi] larvae (***p < 0.001, Student's T-Test). Evoked potentials (excitatory postsynaptic potentials, EPSP) are normal because of a homeostatic increase in quantal content (QC) (**p < 0.01). (C) Representative electrophysiological traces. Scale bars for EPSPs (mEPSPs): 5 mV (1 mV); 50 ms (1000 ms).
Postsynaptically driven BG57-GAL4 >> UAS-GluRIII[RNAi] yields a robust homeostatic challenge to NMJ function (Figures 1B,C) (See Supplementary Table 1 for selected raw electrophysiological data throughout the manuscript). NMJ effects are similar to the published GluRIII hypomorphic condition (Marrus et al., 2004). Compared to controls, BG57-GAL4/UAS-GluRIII[RNAi] NMJs show drastically decreased quantal amplitude (mEPSP = 0.81 ± 0.04 mV for wild-type control vs. 0.46 ± 0.05 mV for GluRIII knock down, p < 0.001, T-Test) and frequency (4.2 ± 0.2 Hz for control vs. 0.8 ± 0.1 Hz for GluRIII knock down, p < 0.001, T-Test). Despite these decreases in spontaneous miniature neurotransmission, evoked neurotransmission is normal (EPSP = 33.2 ± 1.0 mV for control vs. 33.6 ± 2.7 mV for GluRIII knock down) because of a homeostatic enhancement of quantal content (QC = 43.8 ± 2.1 for control vs. 76.6 ± 7.1 for GluRIII knock down, p < 0.01, T-Test) (Figures 1B,C).
By meiotic recombination, we placed a UAS-GluRIII[RNAi] transgene on chromosome III in cis with the BG57-GAL4 driver. To increase the potential of this stock as a genetic tool to study NMJ homeostasis, we augmented it with two presynaptic GAL4 drivers. We chose neuronal drivers elaV(C155)-GAL4 (Lin and Goodman, 1994) and Scabrous-GAL4 (Budnik et al., 1996). Multiple presynaptic drivers were used to enhance the efficiency of RNAi in neurons. For simplicity, we refer to the new stock incorporating all drivers as T15 (trans-synaptic). The genotype of T15 is elaV(C155)-GAL4; Sca-GAL4; BG57-GAL4, UAS-GluRIII[RNAi] (chromosomes X; II; III, balancer chromosomes omitted from this genotype). We also generated a control stock, containing the same GAL4 drivers, but not the UAS-GluRIII[RNAi] transgene.
When T15 females are crossed to wild-type males (herein T15 × WT, Figure 2A), the larval progeny exhibit starkly diminished NMJ quantal size and frequency, and a homeostatic increase in NMJ quantal content (Figures 2B,C). By contrast, when GAL4 driver control females are crossed to wild-type males (herein GAL4 Cont × WT), the NMJ electrophysiological profile of larval progeny is largely similar to that of wild-type NMJs (Figures 2B,C). The GAL4 drivers or genetic background may induce a slight increase in quantal size (Figure 2B). However, this increase is not statistically significant, and the data show that the presence of presynaptic GAL4 drivers exerts no adverse effects on evoked NMJ excitation (Figures 2B,C).
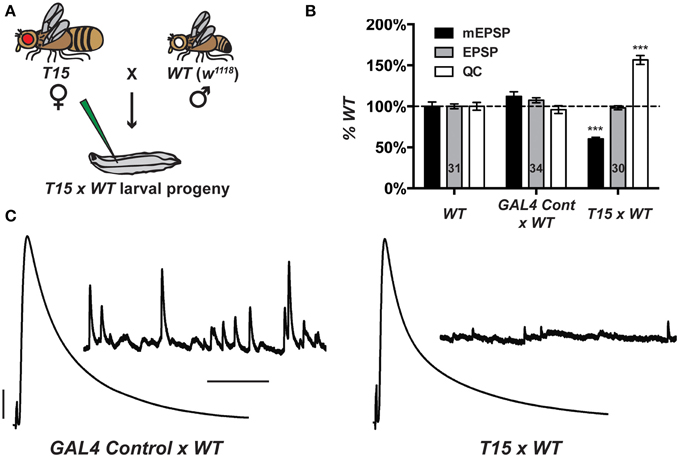
Figure 2. The T15 stock induces robust homeostatic compensation. (A) T15 × WT crossing scheme. The genotype for T15 is (chromosomes X; II; III): elaV(C155)-GAL4; Scabrous-GAL4; BG57-GAL4, UAS-GluRIII[RNAi]. (B) T15 × WT larval NMJs have decreased quantal size (***p < 0.001, T-Test vs. WT). Evoked potentials are normal because of a homeostatic increase in QC (***p < 0.001). A control stock with only GAL4 drivers behaves similarly to WT. (C) Representative electrophysiological traces. Scale bars for EPSPs (mEPSPs): 5 mV (1 mV); 50 ms (1000 ms).
GluRIII Knock Down Does Not Grossly Alter Synapse Development
We wished to examine the effects of these NMJ drivers and the UAS-GluRIII[RNAi] transgene on synapse development. We imaged larval NMJs by immunofluorescence microscopy. We utilized an anti-GluRIII antibody to examine glutamate receptors (Marrus et al., 2004). As expected, T15 × WT NMJs show a marked decrease in anti-GluRIII NMJ staining compared to wild-type controls or GAL4 × WT controls (Figures 3A–N). We note several aspects of anti-GluRIII staining that are diminished at T15 × WT NMJs. First, there is a 50% decrease in the number of anti-GluRIII puncta compared to wild-type control NMJs for muscles 6 and 7 (Figure 3M, p < 0.05, T-Test for both segments A2 and A3). In addition to this reduction in cluster number, we observed that individual GluRIII cluster size was greatly reduced (41.6 ± 3.5% compared to wild-type controls, p < 0.001, T-Test). Further analysis of digital immunofluorescence images reveals that the average intensity of each anti-GluRIII pixel is also decreased (82.3 ± 3.9% for the T15 × WT condition compared to wild-type controls, p = 0.03, T-Test). This intensity decrease is small. However, when considered in combination with the reductions in cluster number and size, we estimate an 88% decrease in GluRIII NMJ protein per unit of synapse area in T15 × WT larvae compared to wild-type larvae (Figure 3N, p < 0.001, T-Test; total fluorescence intensity, normalized for synapse area, see Materials and Methods). As expected with RNAi-mediated knock down, this is not a complete loss of GluRIII protein. Importantly these changes are consistent with our electrophysiological data for T15 × WT showing significantly decreased quantal size (reduced intensity/pixel and cluster size) and frequency (reduced cluster number).
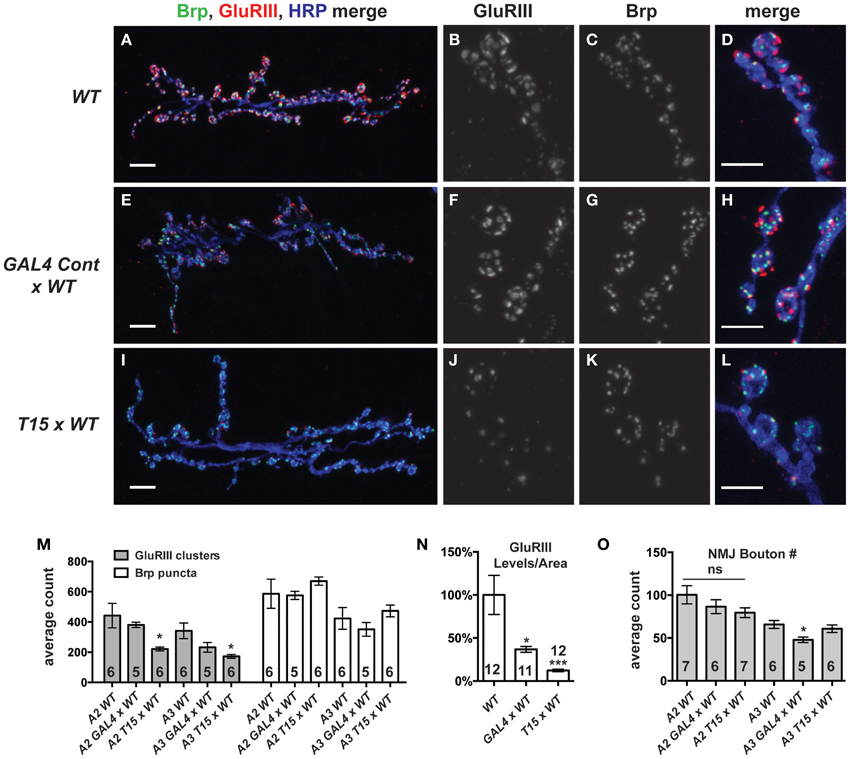
Figure 3. GluRIII glutamate receptor subunits are dramatically decreased in the T15 line. (A-L) Immunostaining of wild type (WT), GAL4 Cont × WT, and T15 × WT NMJs with antibodies against GluRIII (red), Bruchpilot (Brp, green), and HRP (blue). A, E, and I show 40X images (scale bars, 10 μm) of muscle 6/7 NMJs from wandering third instar larvae. (B–D,F–H,J–L) Panels show various channels of 60X images (scale bars, 5 μm) of NMJs. (M) Quantification of the number of presynaptic active zones (marked by Brp) and GluRIII clusters at the muscle 6/7 synapse of segments A2 and A3 (*p < 0.05). (N) Calculation of total GluRIII levels per unit of synapse area. This measure takes into account both GluRIII cluster size and GluRIII intensity (see text for individual values; see Materials and Methods for details; *p < 0.05 compared to WT; ***p < 0.001). (O) Quantification of the number of boutons at segment A2 and A3 muscle 6/7 NMJs. n ≥ 6 NMJs stained for each condition.
Surprisingly, the presence of the GAL4 drivers (or the genetic background of the driver line stock) also appears to diminish GluRIII protein level (per unit of synapse area) in GAL4 × WT compared to wild-type controls (Figure 3N, p < 0.05). We are not certain why GluRIII levels are down in GAL4 × WT. Nevertheless, by electrophysiology, this depression is clearly not severe enough to diminish quantal size or evoked excitation (Figure 2B)—though it could explain a partial decrease in quantal frequency compared to WT (Supplementary Table 1). Nevertheless, the amount of GluRIII protein per unit synapse area in GAL4 × WT is about three times greater than the amount measured in T15 × WT (Figure 3N, p < 0.001).
We wished to examine other basic elements of synapse structure. To do this, we stained NMJs with an anti-Bruchpilot (Brp) antibody to count the number of presynaptic active zones (Wagh et al., 2006) or with anti-Discs Large (Dlg) (Budnik et al., 1996) and anti-Synapsin (Syn) (Klagges et al., 1996) antibodies to count the number of synaptic boutons. For synapses on both segments A2 and A3, neither active zone counts (Figure 3M, Brp puncta) nor bouton counts (Figure 3O) are significantly different when comparing T15 × WT NMJs vs. wild-type NMJs. These findings are interesting, considering the fact that T15 × WT NMJs have fewer GluRIII clusters apposed to the presynaptic active zone (Figure 3M). Collectively, our data show that T15 × WT NMJs have starkly decreased GluRIII levels, but otherwise grossly normal synapse growth and morphology.
RNAi- and Electrophysiology-Based Screening
The prior electrophysiological data illustrate the potential utility of T15 as a genetic tool to study HSP. If T15 animals are crossed to animals bearing an effective UAS-RNAi transgene, the target gene should be knocked down in larval progeny both presynaptically (due to elaV(C155)-GAL4 and Sca-GAL4 driving UAS-RNAi of the target gene in neurons) and postsynaptically (due to BG57-GAL4 driving UAS-RNAi of the target gene in muscle)—all in the context of a homeostatic challenge to NMJ function due to GluRIII gene knock down. To employ T15 for this purpose, we executed a small-scale RNAi- and electrophysiology-based screen. We chose 62 UAS-RNAi stocks on chromosomes II and III (Dietzl et al., 2007; Ni et al., 2008, 2009). The screen was biased: we targeted genes encoding factors potentially required for proper presynaptic Ca2+ entry and handling, G-protein signals (which could impinge upon calcium channel function), factors known to regulate general synaptic functions, and possible trans-synaptic signaling molecules. To conduct the screen, we crossed T15 females with UAS-RNAi males and recorded from the NMJs of male larval progeny (Figure 4A). Male progeny were chosen because they should have a stronger dose of X-linked elaV(C155)-GAL4 than female progeny.
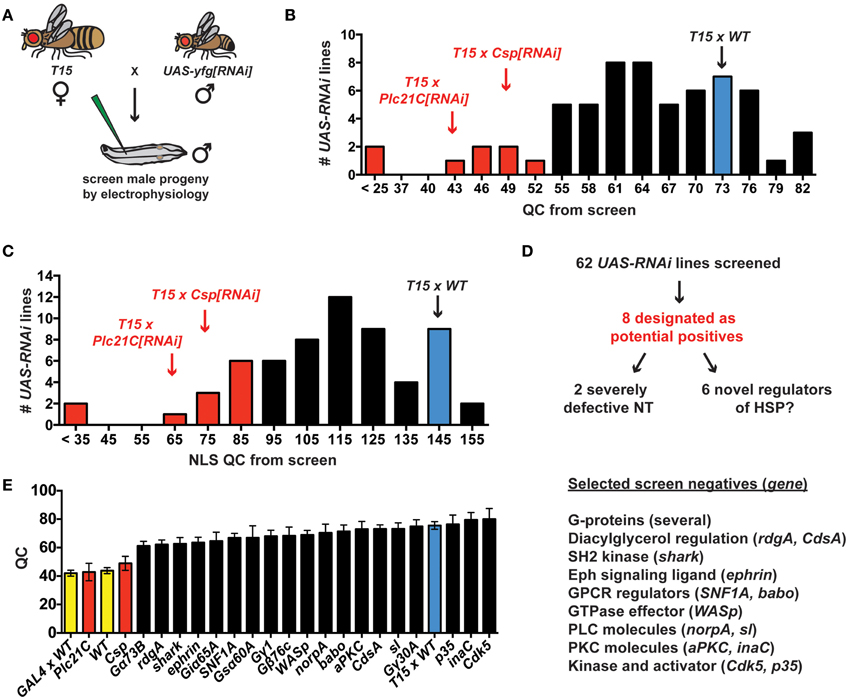
Figure 4. An RNAi- and electrophysiology-based screen for homeostatic factors. (A) Crossing scheme for screen T15 × UAS-yfg[RNAi] (“your favorite gene”). For UAS-RNAi lines on chromosomes II or III, male progeny are examined by electrophysiology because dosage compensated elaV(C155)-GAL4/Y male progeny should have a higher dose of presynaptic GAL4 than elaV(C155)-GAL4/+ female siblings. (B) Distribution of QC values from screened larvae. Eight T15 × UAS-RNAi crosses yield a QC smaller than two standard deviations below T15 × WT (red bars). (C) When QC is corrected for non-linear summation (NLS), twelve T15 × UAS-RNAi line crosses yield an NLS corrected QC (NLS QC) smaller than two standard deviations below WT (red bars). (D) Schematic to sort potential positives for follow-up analyses. (E) Negative data for some genes in the screen. Where a specific gene name is listed, the data represent the QC for T15 × UAS-yfg[RNAi]. Underlying data for displayed screen negatives has average evoked potentials >30 mV (not shown, but WT control EPSP = 33.2 ± 1.0 mV) and QC > 60.
T15 × WT NMJs have significantly elevated quantal content compared to non-transgenic wild-type controls (Figures 2B, 4B). In analyzing the T15 × UAS-RNAi screen data, we considered quantal content as a measure of presynaptic release. We identified eight T15 × UAS-RNAi crosses that yielded progeny with a low NMJ quantal content (QC < 52, Figure 4B). For all eight, the level of presynaptic release is indistinguishable (or lower) than that of unchallenged, wild-type NMJs, potentially indicative of a block in homeostatic compensation. Considering the distribution of QC for all of the UAS-RNAi crosses from the screen, this level of presynaptic release is also about two standard deviations smaller than T15 × WT controls. We wished to ensure that our synaptic release analysis was accurate and restrictive. Therefore, we also applied a correction to our calculations of quantal content to account for effects of non-linear summation (NLS) when recording synaptic voltages (Martin, 1955). Considering the distribution of NLS corrected QC for all of the UAS-RNAi crosses from the screen, an NLS corrected QC < 85 is two standard deviations smaller than T15 × WT controls. Twelve T15 × UAS-RNAi crosses fall below this NLS QC threshold, including all eight identified by the prior cutoff (Figure 4C). We chose to focus initial follow-up efforts on the eight potential positives that showed a low quantal content by both criteria (Figure 4D).
We identified eight potential hits out of 62 UAS-RNAi stocks, which is a very high positive rate for a screen (12.9%). However, the screen is biased, and it is not surprising that several pre-selected factors could impair synapse function. Nevertheless, most T15 × UAS-RNAi crosses do not yield progeny with diminished NMJ QC. Several screen negatives with EPSP > 30 mV and QC > 60 are shown (Figure 4E). It is possible the corresponding target genes are not involved in synaptic homeostasis. However, it is also possible that conditions in this experiment do not yield sufficient gene knock down to reveal a homeostatic phenotype. One interesting example to consider in this regard is Drosophila ephrin. Prior work demonstrates that a presynaptic signaling system consisting of the Eph receptor tyrosine kinase and the cytoplasmic guanine exchange factor Ephexin is needed for the long-term maintenance of HSP (Frank et al., 2009). Drosophila Ephrin is a top candidate ligand for Drosophila Eph in this process, but the RNAi data from our screen do not support this conclusion (Figure 4E).
A low QC value from a screened T15 × UAS-RNAi line could reflect a genuine defect in synaptic homeostasis. It could also simply reflect a decrease in neurotransmission when the targeted gene is knocked down in NMJ tissues. We note that two of the eight UAS-RNAi lines identified appear to cause severe neurotransmission defects (Figures 4B–D), suggesting synapse dysfunction independent of possible defects in homeostatic plasticity (QC < 25 when crossed to T15; NLS QC < 35 when crossed to T15). One of these two RNAi lines targets Drosophila cacophony (cac)—a positive control RNAi line included in the screen that targets the α1a pore-forming subunit of CaV2 channels. Given previous studies examining CaV2 in neurotransmission and NMJ homeostasis (Frank et al., 2006, 2009; Tsurudome et al., 2010; Müller and Davis, 2012), this result is not surprising. However, using T15, we are able to garner new information about Cac/CaV2 and its role in synaptic homeostasis (see below). Finally, we also report initial characterizations of two novel homeostatic genes, Drosophila Csp and Plc21C (Figures 4B,C). Our screening and characterizations of other genes are ongoing, and we will report further characterizations elsewhere.
Cacophony Knock Down by RNAi Impairs Neurotransmission but Not Homeostatic Plasticity
CaV2-type voltage-gated Ca2+ channels are critical for presynaptic neurotransmission. For many vertebrate and invertebrate synapses, CaV2 channels also gate homeostatic modulations of neurotransmission (Frank, 2014b). In Drosophila, homozygous, partial loss-of-function missense mutations like cacS block the homeostatic potentiation of transmitter release at the NMJ after glutamate receptor impairment (Frank et al., 2006, 2009; Müller and Davis, 2012). By contrast, cac null mutants arrest as embryos (Kulkarni and Hall, 1987; Kurshan et al., 2009). Therefore, it is unknown if cac mutant impairment of HSP is due to partial loss of channel function—or if amino-acid substitutions such as the one encoded by cacS (F1029I, transmembrane domain III S6) (Smith et al., 1998) impair specific functions or interactions critical to homeostatic signaling. Intriguingly, cacS/+ heterozygotes display partial defects in courtship song behavior (Smith et al., 1998) and the execution of synaptic homeostasis (Frank et al., 2006, 2009). These data are consistent with the possibility that missense cac mutations may exert antimorphic effects on synaptic function.
Knocking down cac gene activity by RNAi allows one to test whether a simple reduction of CaV2 function suffices to impair synaptic homeostasis. Efficient RNAi-mediated cac knock down also circumvents possible antimorphic effects associated with genetic mutations (but not potential effects due to haploinsufficiency). For the screen and subsequent analyses here, we used a UAS-cac[RNAi] transgene to knock down cac gene function (cacKK101478, see Materials and Methods) (Dietzl et al., 2007). By primary target sequence, cacKK101478 is not predicted to induce off-target gene knock down. By quantitative RT-PCR (qPCR), we find that driving this transgene under simultaneous control of the neuronal drivers elaV(C155)-GAL4 and Sca-GAL4 diminishes total levels of cac mRNA by ~50% vs. a control utilizing just the drivers (See Materials and Methods).
We prepared mRNA for qPCR using whole larvae. Therefore, Cac protein loss in motor neurons and at synapses could be even greater than qPCR measurements. We tested the effectiveness of UAS-cac[RNAi] on cac gene function in two different ways. First, we crossed transgenically rescued cac null females elaV(C155)-GAL4, cacHC129;; UAS-cac-eGFP786c × UAS-cac[RNAi] males (or WT males for control). This UAS-cac[RNAi] cross (but not the WT control cross) kills all progeny, with only a small number of adults reaching the pupal eclosion stage and then either arresting during the process of eclosion or getting stuck in the food shortly after eclosing (not shown). Second, we find that presynaptic expression of cacKK101478 starkly diminishes evoked neurotransmission (elaV(C155)-GAL4, Sca-GAL4 >> UAS-cac[RNAi]: EPSP = 8.5 ± 1.1 mV; QC = 8.4 ± 1.5, n = 8) compared to a wild-type control (WT: EPSP = 33.2 ± 1.0 mV, QC = 43.8 ± 2.1, n = 31). This diminishment of evoked neurotransmission is similar in severity to a cacS mutant (Frank et al., 2006), and it suggests a significant loss of Cac protein at the NMJ. By contrast, muscle expression of cacKK101478 (BG57-GAL4 >> UAS-cac[RNAi]) has no adverse effect on evoked neurotransmission (EPSP = 37.5 ± 2.0 mV; QC = 53.2 ± 2.9, n = 8).
We crossed either the T15 stock or the aforementioned pre- and post-synaptic GAL4 driver control stock (Figure 2) to UAS-cac[RNAi]. For GAL4 Cont × UAS-cac[RNAi] progeny, NMJ evoked excitation and quantal content are markedly decreased compared to GAL4 Cont × WT controls (Figure 5A). This is true across a range of extracellular Ca2+ concentrations, though perhaps a bit less pronounced at physiological [Ca2+] (1.5 mM) (Figure 5A). As expected, for T15 × UAS-cac[RNAi] larval progeny, there is a significant decrease in NMJ quantal size (mEPSP) compared to GAL4 Cont × UAS-cac[RNAi] larval progeny (Figure 5B). However, T15 × UAS-cac[RNAi] NMJ EPSP amplitude is no different than control cross UAS-cac[RNAi] progeny (Figure 5C). This is due to a homeostatic enhancement in presynaptic quantal content (Figure 5B). This result holds for extracellular Ca2+ concentrations that permit a range of presynaptic release: 0.5, 1.0, and 1.5 mM. For each condition, homeostatic compensation of presynaptic release remains robust (Figure 5B).
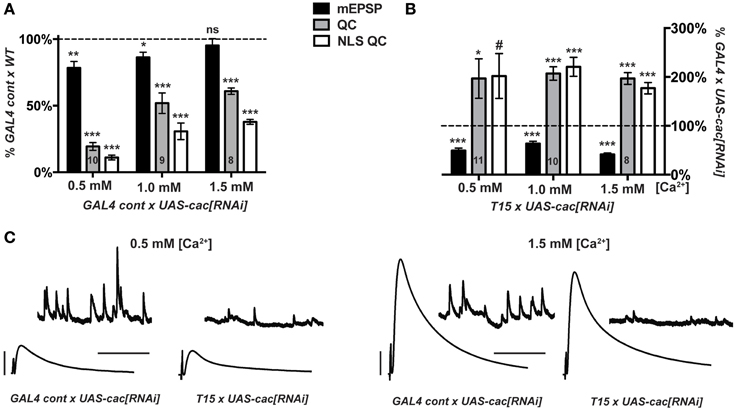
Figure 5. Knock down of cacophony gene function does not impair synaptic homeostasis. (A,B) mEPSP (black), QC (gray), NLS QC (white). (#p = 0.05; *p < 0.05; **p < 0.01; ***p < 0.001, T-test compared to control at 100% dotted line) (A) Diminished baseline neurotransmission for GAL4 control × UAS-cac[RNAi] larvae is consistent with cac gene knock down. Diminished release occurs across a range of extracellular [Ca2+]. (B) T15 × UAS-cac[RNAi] larvae show robust homeostatic compensation compared to GAL4 control × UAS-cac[RNAi] larvae. As expected, the T15 line induces a marked diminishment of quantal size (mEPSP). The NMJ responds with a robust increase in release. This response is observed across a range of extracellular calcium concentrations, and the same result holds whether or not QC is corrected for non-linear summation. (C) Representative electrophysiological traces. Scale bars for EPSPs (mEPSPs): 5 mV (1 mV); 50 ms (1000 ms).
Our data suggest diminishment of cac gene function itself is not sufficient to block homeostatic potentiation of function. Nor is extracellular calcium concentration a factor. If Cac/CaV2 levels are simply diminished—but the copies of Cac α1a subunits present are wild-type copies—homeostatic plasticity is intact. By contrast, the presence of function-impairing CaV2 α1a subunits due to amino-acid substitution appears sufficient to block the induction and maintenance of homeostatic plasticity (Frank et al., 2006, 2009; Müller and Davis, 2012).
Cysteine String Protein Mutations Block the Long-Term Maintenance of Synaptic Homeostasis
CSP is a conserved synaptic protein in the DnaJ family of chaperones. For both vertebrates and invertebrates, CSP executes functions that promote viability, coordinated locomotion, neuroprotection, and evoked neurotransmitter release (Zinsmaier, 2010). At the Drosophila NMJ, synaptic functions of CSP appear related to Ca2+ regulation. In Csp loss-of-function mutants, resting intra-terminal Ca2+ levels are altered (Dawson-Scully et al., 2000; Bronk et al., 2001; Dawson-Scully et al., 2007), and neurotransmission defects can be suppressed by high [Ca2+]e or repetitive nerve stimulation (Dawson-Scully et al., 2000; Bronk et al., 2001). The fact that a UAS-Csp[RNAi] line emerged from our screen was somewhat surprising because a prior genetic screen demonstrated that a Csp mutation does not impair homeostatic plasticity when the NMJ is challenged with PhTox application (Goold and Davis, 2007; Dickman and Davis, 2009). However, this previous screen specifically examined factors for a role in the short-term induction of homeostatic plasticity, not its long-term maintenance.
Our screen found no elevation of NMJ quantal content when comparing T15 × UAS-Csp[RNAi] larval progeny vs. wild-type larvae (Figure 4). This result indicates a possible role for CSP in homeostatic plasticity, although it could also be consistent with expected Csp defects in neurotransmission. In a follow-up test, we found that T15 × UAS-Csp[RNAi] NMJ quantal content is not significantly different than GAL4 driver control × UAS-Csp[RNAi] NMJ quantal content (Figure 6A). This result suggests a bona fide defect in synaptic homeostasis (Figures 6A,B).
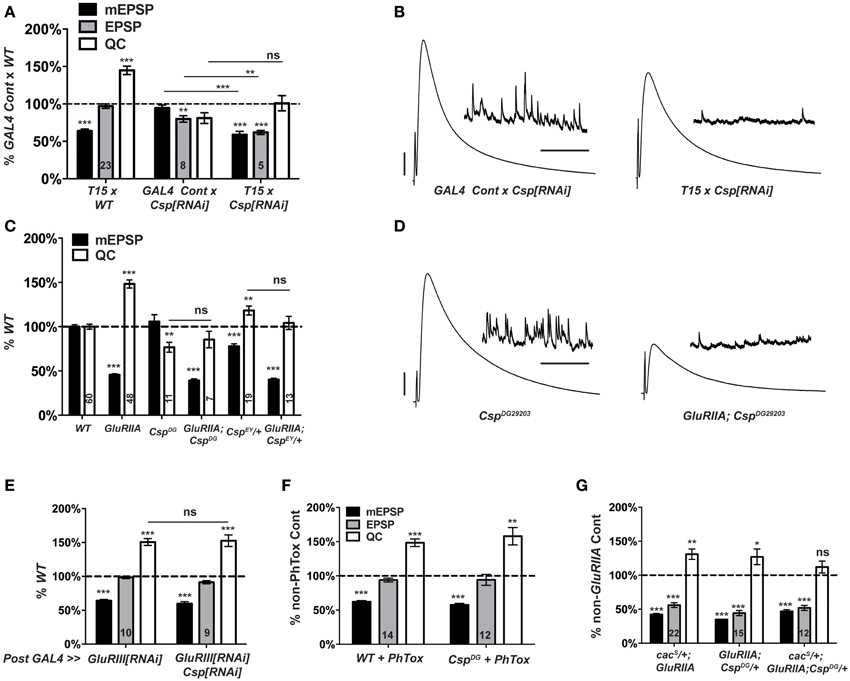
Figure 6. Csp is required for long-term homeostatic compensation. (A) T15 × UAS-Csp[RNAi] shows a failure to upregulate quantal content compared to its GAL4-driven UAS-Csp[RNAi] control (ns, p = 0.15). Knock down of Csp shows a slight impairment in evoked neurotransmission (EPSP) compared to control (**p < 0.01). (B) Representative electrophysiological traces show the failure of T15 × UAS-Csp[RNAi] larvae to maintain evoked potentials at control levels. (C) Homozygosity for the CspDG29203 allele or heterozygosity for the CspEY22488 allele block homeostatic upregulation of quantal content compared to their respective non-GluRIIASP16genetic controls (ns, p = 0.44 and p = 0.14, respectively). (D) Representative traces show a failure of homeostatic compensation for GluRIIASP16, CspDG29203. (E) Postsynaptic knock down of Csp function leaves homeostatic plasticity intact. (F) Acute homeostatic compensation is intact in CspDG29203 as evidenced by the elevated quantal content in response to philanthotoxin-433 (PhTox) application (**p < 0.01). (G) A doubly heterozygous combination of CspDG29203/+ and cacS/+ shows a homeostatic block in the GluRIIASP16 background because of a failure to increase quantal content over cacS/+; CspDG29203/+ controls (ns, p = 0.24). By contrast, the single heterozygous mutations retain partial homeostatic compensatory capacity. Scale bars for EPSPs (mEPSPs): 5 mV (1 mV); 50 ms (2000 ms). (ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001).
To follow up on the RNAi screen with genetic mutations, we examined two transposon insertion-induced Csp alleles, CspDG29203 and CspEY22488. CspDG29203 disrupts the 5′ UTR of Csp, and genetic evidence suggests that it is hypomorphic (Wishart et al., 2012). CspEY22488 is inserted in an intronic region of Csp. It causes significant homozygous lethality and appears to be a strong mutation genetically (data not shown). We generated two double mutant combinations: GluRIIASP16; CspDG29203, and GluRIIA; CspEY22488/+. The GluRIIASP16 deletion mutation induces a marked decrease in quantal size and a homeostatic increase in quantal content compared to wild-type controls (Figure 6C) (Petersen et al., 1997). By contrast, for the double mutant combinations, synaptic homeostasis is impaired: there is no significant increase in release compared to Csp mutants alone (Figure 6C). As a result, evoked postsynaptic excitation is stunted (Figure 6D). Interestingly, the CspEY22488/+ and CspDG29203 genetic conditions display somewhat different baseline electrophysiology, in terms of both quantal size and quantal content (Figure 6C). These differences could be due to different genetic backgrounds or Csp allelic strength. Nevertheless, both alleles block homeostatic compensation in response to GluRIIA gene loss (Figures 6C,D).
The vast majority of research published about Drosophila CSP demonstrates that it localizes to presynaptic terminals. However, it has also been reported that Drosophila muscles can express low levels of CSP (Eberle et al., 1998). To test whether CSP's function in synaptic homeostasis could reside in the postsynaptic compartment, we crossed the muscle driver stock BG57-GAL4, UAS-GluRIII[RNAi] to UAS-Csp[RNAi]. Knocking down Csp gene function in the muscle leaves synaptic homeostasis intact (Figure 6E); this result is consistent with a presynaptic role for CSP in synaptic homeostasis.
Collectively, our data demonstrate that Csp function is needed for the long-term maintenance of homeostatic plasticity. To double check whether Csp could also be required for the short-term induction of homeostatic plasticity, we applied PhTox to CspDG29203 mutant NMJs. By this assay, the rapid induction of homeostatic plasticity remains intact (Figure 6F), consistent with the previously reported results (Goold and Davis, 2007; Dickman and Davis, 2009). Therefore, CSP appears to be needed for the sustained expression of homeostatic plasticity throughout development, not its short-term induction.
A Csp and cac Mutant Combination Blocks HSP
cacS/+; GluRIIA double mutant NMJs have partially impaired synaptic homeostasis (Frank et al., 2006) (and Figure 6G). As discussed above, this partial impairment could be due to an antimorphic allelic effect. The cacS/+ genetic condition has been previously exploited to characterize second-site factors that could potentially execute homeostatic plasticity in conjunction with CaV2 by searching for genetic interactions with cacS (Frank et al., 2009; Wang et al., 2014). In the sensitized cacS/+ genetic background, we find that cacS/+; GluRIIA; CspDG29203/+ NMJs have completely blocked synaptic homeostasis—i.e., there is no increase in quantal content compared to the cacS/+;; CspDG29203/+ genetic control condition (Figure 6G). A strong double heterozygous phenotype could be consistent with Cac and CSP both functioning within synaptic homeostasis, either in a linear signaling pathway or in parallel processes. However, it could also reflect a summation of separate, partial defects in HSP for both cacS/+; GluRIIA NMJs and GluRIIA; CspDG29203/+ NMJs (Figure 6G).
Loss of PLCβ Function Impairs Synaptic Homeostasis
In designing our screen, we postulated that lipid signaling at the synapse could affect NMJ homeostasis. Canonically, Gαq-GTP (together with Gβγ G-proteins) activates PLCβ. In turn, PLCβ cleaves phosphatidylinositol 4,5-bisphosphase (PIP2) into diacylglycerol (DAG) and inositol triphosphate (IP3) (Tedford and Zamponi, 2006; Philip et al., 2010; Kadamur and Ross, 2013). These molecules are known to influence neurotransmission in several ways at many synapses, including the NMJ (Goni and Alonso, 1999; Cremona and De Camilli, 2001; Peters et al., 2001; Wu et al., 2002; Rohrbough and Broadie, 2005; Huang et al., 2006). To examine this pathway in the context of NMJ homeostasis, we considered a Drosophila PLCβ homolog gene, Plc21C—among other genes, some of which showed no phenotype (Figure 4E).
When T15 is crossed to a UAS-Plc21C[RNAi] transgenic line (Dietzl et al., 2007), larval progeny have significantly lower NMJ quantal content than T15 × WT controls (Figures 4B,C, 7A). By contrast, when the muscle driver stock BG57-GAL4, UAS-GluRIII[RNAi] is crossed to the same UAS-Plc21C[RNAi] transgenic line, full homeostatic compensation occurs (Figures 7B,C). This latter result argues against Plc21C acting in the muscle to control synaptic homeostasis. A prior study demonstrated that presynaptic expression of the same UAS-Plc21C[RNAi] construct utilized in our screen (Plc21CGD11359) diminishes Plc21C mRNA levels by more than 50% (Dahdal et al., 2010). Moreover, Plc21C mRNA is found in the larval nervous system (Shortridge et al., 1991). Collectively, these data are consistent with a neuronal role for Plc21C in the homeostatic control of NMJ function.
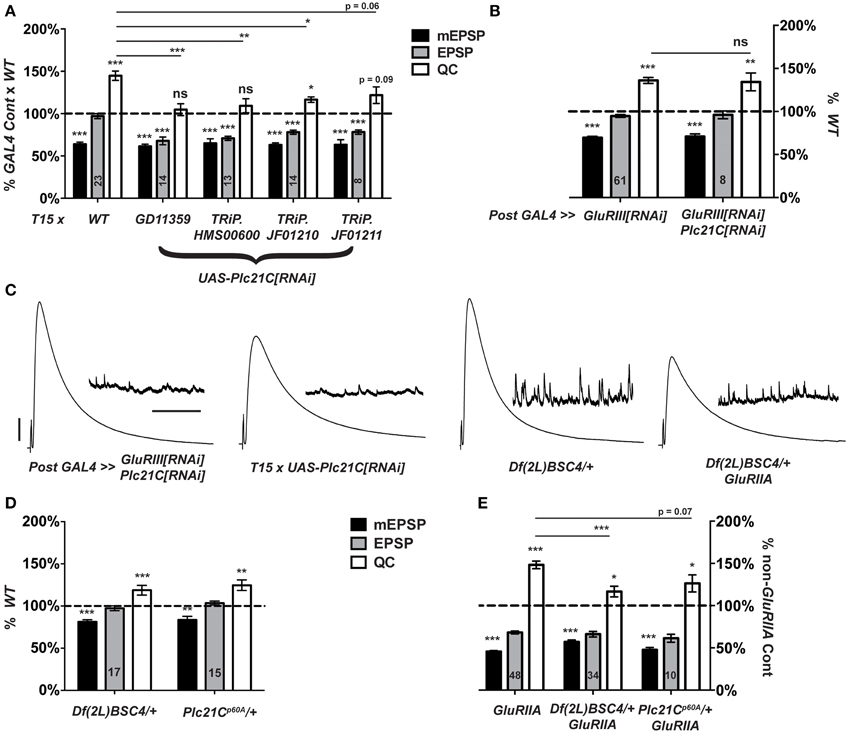
Figure 7. Presynaptic Plc21C is required for homeostatic compensation. (A) T15-mediated RNAi knock down of Plc21C results in a failure to maintain the EPSP at control levels (***p < 0.001). Concerning homeostatic compensation, T15 × TRiP.JF01210 is the only cross to show increased quantal content compared to its GAL4 cross control (*p = 0.04), but even this quantal content was still significantly depressed compared to the T15 × WT control (*p < 0.05, One-Way ANOVA including all crosses in dataset, with Tukey's post-hoc). (B) Postsynaptic knock down of Plc21C does not affect homeostatic compensation: the EPSP is maintained (ns, p = 0.45) at control levels because the quantal content is upregulated (**p < 0.01). (C) Representative traces show normal neurotransmission upon postsynaptic knock down of GluRIII and Plc21C or a heterozygous loss of Plc21C via use of the deficiency chromosome Df(2L)BSC4. However, evoked release is not maintained upon T15-mediated knock down of Plc21C or the heterozygous loss of Plc21C in a GluRIIASP16 mutant background. (D) Evoked amplitudes (EPSPs) are unaltered upon heterozygous loss of Plc21C by use of the deficiency Df(2L)BSC4 or Plc21Cp60A allele though both do show a small, yet significant decrease in the amplitude of mEPSP events (***p < 0.001 and **p < 0.01, respectively). (E) Quantal content is minimally increased in the GluRIIASP16 background upon heterozygous loss of Plc21C compared to their respective genetic controls (*p < 0.05). For the Df(2L)BSC4 deficiency, the increased quantal content does not reach the level of increase found in GluRIIASP16 (***p < 0.001, One-Way ANOVA, Tukey's post-hoc). This is indicative of a partial impairment in homeostatic compensation. Scale bars for EPSPs (mEPSPs): 5 mV (1 mV); 50 ms (2000 ms).
To address possible off-target effects of UAS-Plc21C[RNAi] expression, we acquired three additional UAS-Plc21C[RNAi] lines—Drosophila TRiP constructs (Transgenic RNAi Project) (Ni et al., 2009, 2008). Each TRiP line fails to robustly increase quantal content when crossed to T15, (Figure 7A). This result is consistent with an impairment of HSP. Additionally, we examined two heterozygous Plc21C deletion mutations: a chromosomal deficiency that removes one copy of Plc21C from the genome, Df(2L)BSC4, and a 5′ UTR and first exon deletion called Plc21Cp60A (Weinkove et al., 1999). Alone, neither heterozygous deletion diminishes evoked neurotransmission (Figures 7C,D). Heterozygous Plc21C/+ animals in a GluRIIASP16 mutant background do have a mild increase in quantal content compared to Plc21C/+ genetic controls (Figure 7E). However, this increase is not nearly as robust as one would expect for a GluRIIA condition (Figure 7E). This result indicates a partial impairment of NMJ homeostatic compensation (Figures 7C,E). Collectively, the data utilizing hypomorphic Plc21C conditions support the idea that presynaptic Plc21C gates the sustained expression of homeostatic plasticity.
A Role for Gαq in Synaptic Homeostasis?
Gαq-GTP and Gβγ G-proteins are classically known to activate PLCβ function (Tedford and Zamponi, 2006). A positive Gαq-Plc21C regulatory relationship appears conserved for Drosophila flight behavior (Banerjee et al., 2006). Therefore, we postulated that Plc21C could be playing a role in synaptic homeostasis downstream of G-protein signals. In the course of our screen, we discovered that genetic knock down of a Gαq-encoding gene only mildly impairs synaptic release compared to wild-type: T15 × UAS-Gαq[RNAi] (EPSP = 29.8 ± 1.6 mV; QC = 64.0 ± 3.7). This effect was not strong (possibly due to partial gene knock down by RNAi) and was not identified as a positive hit from the screen. However, given the results with Plc21C, we wished to probe Gαq with genetic mutants. We examined four strong Gαq loss-of-function alleles: Gαq1370, Gαqf04219, Gαq221c, and Gαq28. Each Gαq null allele is homozygous lethal (well before the third instar larval stage), but Gαq/+ heterozygotes are viable. For baseline neurotransmission, Gαq/+ mutant NMJs show normal levels of postsynaptic excitation (EPSPs, Figure 8A). Interestingly, however, heterozygous Gαq/+ NMJs show partially impaired homeostatic plasticity in a GluRIIASP16 mutant background (Figure 8B).
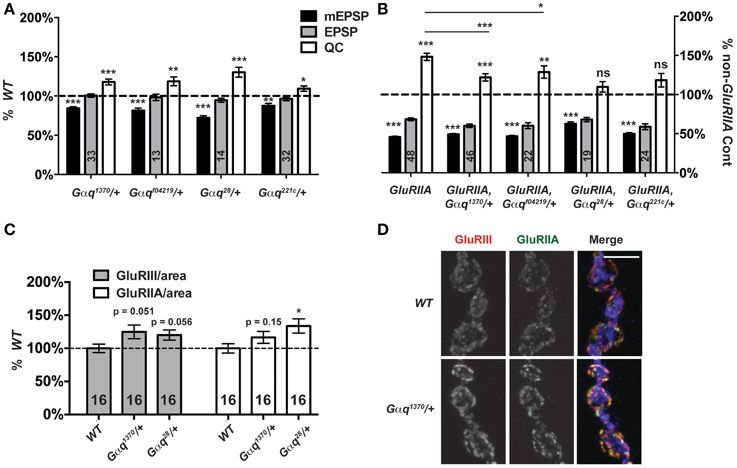
Figure 8. Partial Gαq loss impairs homeostatic compensation. (A) Heterozygous loss of Gαq does not affect levels of evoked neurotransmission (ns, p > 0.05 for each allele compared to WT). However, as with loss of Plc21C, there is a small decrease in mEPSP amplitude (**p < 0.01 for each allele) and a small increase in QC (p-values vary for each allele, but all < 0.05). (B) When challenged with a loss of GluRIIA, Gαq28/+ and Gαq221c/+ NMJs show a complete block in homeostatic compensation (QC—ns, p > 0.05) while Gαq1370/+ and Gαqf04219/+ show partial compensation with quantal content elevated slightly compared to genetic control (***p < 0.001 and **p < 0.01, respectively, Student's T-Test) but not to the full extent seen in the GluRIIASP16 control (***p < 0.001 and *p < 0.05). (C,D) NMJ glutamate receptor subunit levels per unit of synapse area are not decreased in Gαq/+ mutants; if anything, they may be slightly enhanced (scale bar, 5 μm). Merged images include anti-GluRIII (red), -GluRIIA (green), and -HRP (blue) staining.
Each Gαq/+ mutant condition shows a small, but significant decrease in NMJ quantal size compared to wild-type controls (Figure 8A). We wondered if this decrease in quantal size could reflect a decrease in glutamate receptor expression levels and a postsynaptic role for Gαq. This possibility could potentially confound our analyses of homeostatic plasticity. Therefore, we stained Gαq/+ mutant NMJs with anti-GluRIII and anti-GluRIIA antibodies and quantified stained receptor levels per unit synapse area, as before (Figure 3). We find no significant decrease in glutamate receptor levels in Gαq/+ mutant NMJs (Figures 8C,D). In fact, for most measures, there appears to be a slight increase or a statistical trend toward a slight increase of glutamate receptor subunit expression (Figure 8C). This single experiment does not rule out possible postsynaptic functions for Gαq. However, our collective findings are consistent with the possibility that Gαq and Plc21C play concerted roles in the execution of homeostatic plasticity. Further experiments will be required to test if there is a direct regulatory relationship in the presynaptic nerve between the two proteins.
Discussion
There exists overwhelming evidence that forms of HSP shape how neurons and synapses maintain stable function. Physiologically appropriate levels of synaptic activity must be maintained throughout life while facing numerous endogenous and exogenous challenges to neuronal function. To understand how HSP works on a molecular level, it is important to uncover mechanisms that govern both its initiation and long-term maintenance. Here, we apply prior knowledge and reagents to develop a new genetic stock abbreviated as T15: elaV(C155)-GAL4; Sca-GAL4; BG57-GAL4, UAS-GluRIII[RNAi]. T15 allows examination of the long-term maintenance of synaptic homeostasis at the NMJ in a single genetic cross. We validate the synaptic properties of T15 progeny through a combination of genetics, electrophysiology, and synapse imaging. We demonstrate the utility of T15 as a genetic tool in multiple ways: (1) we address one aspect of how CaV2 function impinges upon synaptic homeostasis throughout development; (2) we identify CSP as a novel regulator of homeostatic plasticity; and (3) we identify a putative presynaptic system consisting of Drosophila Gαq and PLCβ homologs that also regulates homeostatic plasticity.
Flexibility and Uses of the T15 Drosophila Stock
The concept of expressing GAL4-responsive UAS transgenes both pre- and post-synaptically at the NMJ is not novel, but it is useful. In our study we add the UAS-GluRIII[RNAi] transgene to provide a homeostatic challenge to the NMJ. We have utilized T15 by crossing it with UAS-RNAi transgenes and interrogating the synapse electrophysiologically. Variations on this genetic theme beyond RNAi screening are possible. For example, to test whether NMJ overexpression of a particular UAS-driven target gene impairs HSP, a single cross to T15 would be sufficient. In fact, an “overexpression screen” could be conducted by crossing T15 to Drosophila stocks carrying transposons with UAS insertions in orientations that drive gene expression (Rorth, 1996; Beinert et al., 2004; Bellen et al., 2004; Thibault et al., 2004; Staudt et al., 2005). For another example, viable loss-of-function mutations on the X-chromosome could be directly assayed for roles in homeostatic plasticity. This could be done by crossing mutant females to BG57-GAL4, UAS-GluRIII[RNAi] males and examining the mutant male larval progeny by electrophysiology. Chromosomal deficiency or duplication screens are also feasible by employing the BG57-GAL4, UAS-GluRIII[RNAi] chromosome in a similar crossing scheme. With GluRIIA genetic mutations, such screens or approaches require more extensive genetic work. The single-cross utility of a BG57-GAL4, UAS-GluRIII[RNAi] chromosome makes large-scale screens to isolate factors involved in developmental or long-term HSP possible. Finally, T15 does present hurdles. For example, with T15, RNAi-targeted genes are impaired both pre- and post-synaptically. Therefore, the screen itself provides limited information regarding tissue specificity of new homeostatic factors. Follow-up experiments examining positives in the pre- and/or post-synaptic compartments need to be done to garner this information. The advantage of being able to identify both pre- and post-synaptic regulators of HSP in a single-generation crossing scheme outweighs the disadvantage of follow-up work.
CaV2 Channel Expression Levels and Homeostatic Synaptic Plasticity
CaV2 channels help to evoke fast neurotransmission by allowing an influx of calcium into the presynaptic terminal upon neuronal depolarization (for comprehensive reviews on CaV channels, see Catterall et al., 2005; Zamponi, 2005). Considerable data from multiple synaptic preparations suggest that CaV2 channels also gate neurotransmitter release in a homeostatic fashion (Frank, 2014b). At the Drosophila NMJ, increased CaV2 function could be directly responsible for a homeostatic potentiation of presynaptic neurotransmitter release (Frank et al., 2006). Yet it is unclear precisely how this process works. It is not known if increased neurotransmission results from trafficking of new CaV2 channels to the synapse, a modulation of functional properties of CaV2 channels already present at the synapse—or even a modulation of Ca2+-triggered signaling processes downstream of CaV2 function.
Prior data examining cac loss-of-function mutants have not answered this question. Hypomorphic cacS; GluRIIA double mutant NMJs have no significant increase in quantal content compared to cacS mutant NMJs (Frank et al., 2006). Corroborating this finding, presynaptic Ca2+ imaging experiments demonstrate that postsynaptic glutamate receptor impairment correlates with an increase of presynaptic calcium transients (Müller and Davis, 2012)—and cacS mutant NMJs are particularly resistant to potentiation of function after glutamate receptor impairment (Müller and Davis, 2012). A weaker hypomorphic allele, cacTS2 can impair homeostatic compensation induced by GluRIIA deletion, but only if the cacTS2; GluRIIA animals are reared at a high temperature (29°C)—likely lowering effective cac gene function (Frank et al., 2006). Together, these data suggest that absolute amount of Cac/CaV2 function may correlate with the homeostatic capacity of the synapse. Yet additional observations counter this idea. For instance, cacS/+ heterozygous NMJs have nearly normal neurotransmission—significantly better than cacTS2 homozygous NMJs—but cacS/+; GluRIIA NMJs raised at 22°C have partially impaired synaptic homeostasis, while cacTS2; GluRIIA animals reared at 22°C have intact homeostatic compensation (Frank et al., 2006). Additionally, heterozygous null cacHC129/+ animals display normal homeostatic compensation (Frank et al., 2009). Taken altogether, it is possible that the presence of dysfunctional CaV2 channel subunits (e.g., CacS) throughout development—rather than the absolute degree of CaV2 dysfunction—could be the driving factor behind impairments of synaptic homeostasis.
Given these background data, it was important to check if severe diminishment of cac function is sufficient to impair homeostatic plasticity at the NMJ. Our analyses of T15 × UAS-cac[RNAi] NMJs demonstrate that synaptic homeostasis is still intact across multiple concentrations of [Ca2+]e, even when there is severely defective baseline neurotransmission (Figure 5). It appears if the CaV2 channels present at the synapse are wild-type channels, the NMJ retains its homeostatic capacity—even if the number of functional CaV2 channels that successfully make it to the terminal is sharply reduced. This result is inconsistent with a model in which Ca2+-directed signaling downstream of CaV2 activity drives homeostatic plasticity. However, it does not resolve the question of whether the homeostatic potentiation of release normally proceeds through the addition of functional channels to the synapse or through modulation of channel properties. According to a “slot model” of CaV2.1 channel positioning, at presynaptic terminals there may exist only a limited number of channel-type-specific slots (Cao et al., 2004). If this were true at the Drosophila NMJ—and if all slots were normally occupied—it would point to modulation of CaV2 gating properties as a probable mechanism.
A recent paper corroborates this idea. Mutations in two Drosophila genes encoding epithelial sodium (ENaC) channels block both the rapid induction and long-term expression of synaptic homeostasis—importantly, by impairing necessary increases in presynaptic calcium influx (Younger et al., 2013). Interestingly, when the ENaC antagonist benzamil is acutely applied to GluRIIA mutant NMJs, presynaptic calcium influx is dramatically decreased. By contrast, benzamil has no effect on presynaptic calcium influx at wild-type NMJs (Younger et al., 2013). Collectively, these observations are consistent with a model in which CaV2 gating properties are enhanced during synaptic homeostasis, with ENaC channels mediating this enhancement (Younger et al., 2013).
CSP and a Sustained Expression of Homeostatic Plasticity
CSP was originally identified in Drosophila from an antibody-based approach as a synapse-specific antigen (Zinsmaier et al., 1990). Follow-up work has demonstrated that CSP executes multiple synaptic functions (Zinsmaier, 2010; Donnelier and Braun, 2014). Interestingly, loss of CSP function appears to exacerbate synaptic problems over developmental time in organisms as varied as Drosophila (Zinsmaier et al., 1994), mice (Fernandez-Chacon et al., 2004; Garcia-Junco-Clemente et al., 2010), and humans (Benitez et al., 2011; Noskova et al., 2011; Velinov et al., 2012). These facts could be in line with data that CSP is required for the long-term maintenance of homeostatic plasticity—even though CSP is not required for its short-term induction (Dickman and Davis, 2009) (Figure 6F).
What is the specific role of CSP during the execution of HSP? This is unclear. Based on the literature, there are several plausible connections. As stated above, modulation of CaV2 functional properties could be key to synaptic homeostasis, and CSP has previously been implicated in the modulation of CaV2-type calcium channel function (Gundersen and Umbach, 1992). We observe a genetic interaction between Csp and cac loss-of-function mutations (Figure 6G)—however, other studies have failed to find evidence for direct CaV regulation by CSP, including studies at Drosophila synapses (Morales et al., 1999; Dawson-Scully et al., 2000). Therefore, other modes of regulation that genetically interact with cac are likely. For example, CSP has been implicated in the regulation of Ca2+-triggered exocytosis (Dawson-Scully et al., 2000; Bronk et al., 2001; Dawson-Scully et al., 2007), and recent studies have investigated possible CSP chaperone interactions with SNARE molecules. SNAP25 (t-SNARE) expression is significantly decreased in mice lacking CSPα (Chandra et al., 2005; Zhang et al., 2012). This represents a possible link to homeostatic plasticity because reduction of SNAP25 levels compromises homeostatic signaling at the Drosophila NMJ (Dickman et al., 2012).
Gαq, PLCβ, and Lipid Signals
Our data demonstrate that partial losses of Gαq and Plc21C gene function partially impair the homeostatic response (Figures 7, 8). The data open several possibilities for presynaptic control of neurotransmitter release and homeostatic plasticity. By one model, diminishment of PLCβ signaling could result in excess PIP2 at the synapse at the expense of IP3 and membrane-bound DAG. In other systems, it is well known that DAG activates protein kinase C (PKC), which in turn, activates CaV2 channels (Tedford and Zamponi, 2006). In this way, DAG-mediated regulation of CaV2 function could be a key determinant in homeostatic plasticity. However, by a second model, it has been reported that Plc21C gene function and DAG regulate the abundance of synaptic DUNC-13 at the Drosophila NMJ (Aravamudan and Broadie, 2003). DUNC-13 is a highly conserved protein critical for proper maintenance of SNARE-mediated vesicle exocytosis (Jahn and Fasshauer, 2012; Kasai et al., 2012; James and Martin, 2013), a process previously implicated in homeostatic plasticity at the NMJ (Dickman et al., 2012). There exists evidence that DUNC-13 family molecules could regulate the dynamics of the readily releasable pools (RRP) of synaptic vesicles (Chen et al., 2013). RRP size enhancement is another process implemented during synaptic homeostasis at the NMJ (Weyhersmüller et al., 2011; Müller et al., 2012). For a third model, depressed Gαq/PLCβ signaling could result in lower levels of synaptic IP3. Canonically, intracellular IP3 binds to IP3 receptors and liberates Ca2+ from intracellular stores. In turn, Ca2+-dependent signaling cascades could permit the expression of homeostatic plasticity. Finally, PIP2 itself is known to bimodally regulate CaV2 channels: CaV2 channels are stabilized by low levels of PIP2, but inhibited by high levels of PIP2 (Wu et al., 2002). Interestingly, an amino-acid substitution on the intracellular side of transmembrane domain III S6 on CaV2.1 alters its affinity for PIP2. This is the same domain containing the amino-acid substitution in cacS (Smith et al., 1998; Zhen et al., 2006). More experiments are required to define a precise mechanism of Gαq/PLCβ signaling in the execution of synaptic homeostasis at the NMJ. Regardless of mechanism, the results implicating Gαq and Plc21C in synaptic homeostasis suggest that a synaptic signal controls presynaptic excitability through a G-Protein Coupled Receptor (GPCR)/Gαq/PLCβ-mediated signaling pathway. This is an exciting possibility that is open to exploration via additional genetic approaches in Drosophila, taking advantage of tools like those we generated for this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank many researchers and Drosophila stock centers cited in the Materials and Methods for providing Drosophila stocks. We thank the CAF, T. Tootle, and F. Lin lab members for helpful discussions. We thank Catherine Neff and Thomas James for comments on an earlier version of this manuscript. This work was supported in part by NIH grant NS062738 to CAF, a Whitehall Foundation Grant to CAF, as well as funds from the Department of Anatomy and Cell Biology at the University of Iowa Carver College of Medicine, and a Carver Trust Medical Research Initiative Grant to CAF. AMS was supported in part by National Institutes of Health Predoctoral Training Grant T32GM008629 (PI Daniel Eberl).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/journal/10.3389/fncel.2015.00107/abstract
References
Aravamudan, B., and Broadie, K. (2003). Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J. Neurobiol. 54, 417–438. doi: 10.1002/neu.10142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Banerjee, S., Joshi, R., Venkiteswaran, G., Agrawal, N., Srikanth, S., Alam, F., et al. (2006). Compensation of inositol 1,4,5-trisphosphate receptor function by altering sarco-endoplasmic reticulum calcium ATPase activity in the Drosophila flight circuit. J. Neurosci. 26, 8278–8288. doi: 10.1523/JNEUROSCI.1231-06.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beinert, N., Werner, M., Dowe, G., Chung, H. R., Jackle, H., and Schafer, U. (2004). Systematic gene targeting on the × chromosome of Drosophila melanogaster. Chromosoma 113, 271–275. doi: 10.1007/s00412-004-0313-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bellen, H. J., Levis, R. W., Liao, G., He, Y., Carlson, J. W., Tsang, G., et al. (2004). The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167, 761–781. doi: 10.1534/genetics.104.026427
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benitez, B. A., Alvarado, D., Cai, Y., Mayo, K., Chakraverty, S., Norton, J., et al. (2011). Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 6:e26741. doi: 10.1371/journal.pone.0026741
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bergquist, S., Dickman, D. K., and Davis, G. W. (2010). A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron 66, 220–234. doi: 10.1016/j.neuron.2010.03.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415.
Bronk, P., Wenniger, J. J., Dawson-Scully, K., Guo, X., Hong, S., Atwood, H. L., et al. (2001). Drosophila Hsc70-4 is critical for neurotransmitter exocytosis in vivo. Neuron 30, 475–488. doi: 10.1016/S0896-6273(01)00292-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Budnik, V., Koh, Y. H., Guan, B., Hartmann, B., Hough, C., Woods, D., et al. (1996). Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron 17, 627–640. doi: 10.1016/S0896-6273(00)80196-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burrone, J., O'Byrne, M., and Murthy, V. N. (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418. doi: 10.1038/nature01242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cao, Y. Q., Piedras-Renteria, E. S., Smith, G. B., Chen, G., Harata, N. C., and Tsien, R. W. (2004). Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 43, 387–400. doi: 10.1016/j.neuron.2004.07.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005). International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425. doi: 10.1124/pr.57.4.5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., and Sudhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Z., Cooper, B., Kalla, S., Varoqueaux, F., and Young, S. M. Jr. (2013). The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci. 33, 8336–8351. doi: 10.1523/JNEUROSCI.5128-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cremona, O., and De Camilli, P. (2001). Phosphoinositides in membrane traffic at the synapse. J. Cell Sci. 114, 1041–1052.
Dahdal, D., Reeves, D. C., Ruben, M., Akabas, M. H., and Blau, J. (2010). Drosophila pacemaker neurons require g protein signaling and GABAergic inputs to generate twenty-four hour behavioral rhythms. Neuron 68, 964–977. doi: 10.1016/j.neuron.2010.11.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, G. W. (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728. doi: 10.1016/j.neuron.2013.09.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, G. W., DiAntonio, A., Petersen, S. A., and Goodman, C. S. (1998). Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron 20, 305–315. doi: 10.1016/S0896-6273(00)80458-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davis, G. W., and Müller, M. (2015). Homeostatic control of presynaptic neurotransmitter release. Annu. Rev. Physiol. 77, 251–270. doi: 10.1146/annurev-physiol-021014-071740
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dawson-Scully, K., Bronk, P., Atwood, H. L., and Zinsmaier, K. E. (2000). Cysteine-string protein increases the calcium sensitivity of neurotransmitter exocytosis in Drosophila. J. Neurosci. 20, 6039–6047.
Dawson-Scully, K., Lin, Y., Imad, M., Zhang, J., Marin, L., Horne, J. A., et al. (2007). Morphological and functional effects of altered cysteine string protein at the Drosophila larval neuromuscular junction. Synapse 61, 1–16. doi: 10.1002/syn.20335
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiAntonio, A., Petersen, S. A., Heckmann, M., and Goodman, C. S. (1999). Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci. 19, 3023–3032.
Dickman, D. K., and Davis, G. W. (2009). The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326, 1127–1130. doi: 10.1126/science.1179685
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dickman, D. K., Tong, A., and Davis, G. W. (2012). Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci. 32, 8716–8724. doi: 10.1523/JNEUROSCI.5465-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dietzl, G., Chen, D., Schnorrer, F., Su, K. C., Barinova, Y., Fellner, M., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. doi: 10.1038/nature05954
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donnelier, J., and Braun, J. E. (2014). CSPalpha-chaperoning presynaptic proteins. Front. Cell. Neurosci. 8:116. doi: 10.3389/fncel.2014.00116
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eberle, K. K., Zinsmaier, K. E., Buchner, S., Gruhn, M., Jenni, M., Arnold, C., et al. (1998). Wide distribution of the cysteine string proteins in Drosophila tissues revealed by targeted mutagenesis. Cell Tissue Res. 294, 203–217. doi: 10.1007/s004410051170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fernandez-Chacon, R., Wolfel, M., Nishimune, H., Tabares, L., Schmitz, F., Castellano-Munoz, M., et al. (2004). The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42, 237–251. doi: 10.1016/S0896-6273(04)00190-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frank, C. A. (2014a). Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74. doi: 10.1016/j.neuropharm.2013.06.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frank, C. A. (2014b). How voltage-gated calcium channels gate forms of homeostatic synaptic plasticity. Front. Cell. Neurosci. 8:40. doi: 10.3389/fncel.2014.00040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frank, C. A., Kennedy, M. J., Goold, C. P., Marek, K. W., and Davis, G. W. (2006). Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52, 663–677. doi: 10.1016/j.neuron.2006.09.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frank, C. A., Pielage, J., and Davis, G. W. (2009). A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron 61, 556–569. doi: 10.1016/j.neuron.2008.12.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia-Junco-Clemente, P., Cantero, G., Gomez-Sanchez, L., Linares-Clemente, P., Martinez-Lopez, J. A., Lujan, R., et al. (2010). Cysteine string protein-alpha prevents activity-dependent degeneration in GABAergic synapses. J. Neurosci. 30, 7377–7391. doi: 10.1523/JNEUROSCI.0924-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goni, F. M., and Alonso, A. (1999). Structure and functional properties of diacylglycerols in membranes. Prog. Lipid Res. 38, 1–48.
Goold, C. P., and Davis, G. W. (2007). The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron 56, 109–123. doi: 10.1016/j.neuron.2007.08.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gundersen, C. B., and Umbach, J. A. (1992). Suppression cloning of the cDNA for a candidate subunit of a presynaptic calcium channel. Neuron 9, 527–537. doi: 10.1016/0896-6273(92)90190-O
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazelrigg, T., Levis, R., and Rubin, G. M. (1984). Transformation of white locus DNA in drosophila: dosage compensation, zeste interaction, and position effects. Cell 36, 469–481. doi: 10.1016/0092-8674(84)90240-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, F. D., Woodruff, E., Mohrmann, R., and Broadie, K. (2006). Rolling blackout is required for synaptic vesicle exocytosis. J. Neurosci. 26, 2369–2379. doi: 10.1523/JNEUROSCI.3770-05.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jahn, R., and Fasshauer, D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. doi: 10.1038/nature11320
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
James, D. J., and Martin, T. F. (2013). CAPS and Munc13: CATCHRs that SNARE Vesicles. Front. Endocrinol. 4:187. doi: 10.3389/fendo.2013.00187
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kadamur, G., and Ross, E. M. (2013). Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154. doi: 10.1146/annurev-physiol-030212-183750
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kain, P., Chakraborty, T. S., Sundaram, S., Siddiqi, O., Rodrigues, V., and Hasan, G. (2008). Reduced odor responses from antennal neurons of G(q)alpha, phospholipase Cbeta, and rdgA mutants in Drosophila support a role for a phospholipid intermediate in insect olfactory transduction. J. Neurosci. 28, 4745–4755. doi: 10.1523/JNEUROSCI.5306-07.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kasai, H., Takahashi, N., and Tokumaru, H. (2012). Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol. Rev. 92, 1915–1964. doi: 10.1152/physrev.00007.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawasaki, F., Zou, B., Xu, X., and Ordway, R. W. (2004). Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J. Neurosci. 24, 282–285. doi: 10.1523/JNEUROSCI.3553-03.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klagges, B. R., Heimbeck, G., Godenschwege, T. A., Hofbauer, A., Pflugfelder, G. O., Reifegerste, R., et al. (1996). Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. J. Neurosci. 16, 3154–3165.
Kulkarni, S. J., and Hall, J. C. (1987). Behavioral and cytogenetic analysis of the cacophony courtship song mutant and interacting genetic variants in Drosophila melanogaster. Genetics 115, 461–475.
Kurshan, P. T., Oztan, A., and Schwarz, T. L. (2009). Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat. Neurosci. 12, 1415–1423. doi: 10.1038/nn.2417
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y. S., and Carthew, R. W. (2003). Making a better RNAi vector for Drosophila: use of intron spacers. Methods 30, 322–329. doi: 10.1016/S1046-2023(03)00051-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, D. M., and Goodman, C. S. (1994). Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron 13, 507–523. doi: 10.1016/0896-6273(94)90022-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marder, E., and Goaillard, J. M. (2006). Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 7, 563–574. doi: 10.1038/nrn1949
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marie, B., Pym, E., Bergquist, S., and Davis, G. W. (2010). Synaptic homeostasis is consolidated by the cell fate gene gooseberry, a Drosophila pax3/7 homolog. J. Neurosci. 30, 8071–8082. doi: 10.1523/JNEUROSCI.5467-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marrus, S. B., Portman, S. L., Allen, M. J., Moffat, K. G., and DiAntonio, A. (2004). Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J. Neurosci. 24, 1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin, A. R. (1955). A further study of the statistical composition on the end-plate potential. J. Physiol. 130, 114–122. doi: 10.1113/jphysiol.1955.sp005397
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morales, M., Ferrus, A., and Martinez-Padron, M. (1999). Presynaptic calcium-channel currents in normal and csp mutant Drosophila peptidergic terminals. Eur. J. Neurosci. 11, 1818–1826. doi: 10.1046/j.1460-9568.1999.00604.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Müller, M., and Davis, G. W. (2012). Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr. Biol. 22, 1102–1108. doi: 10.1016/j.cub.2012.04.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Müller, M., Genç, Ö., and Davis, G. W. (2015). RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron 85, 1056–1069. doi: 10.1016/j.neuron.2015.01.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Müller, M., Liu, K. S., Sigrist, S. J., and Davis, G. W. (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 32, 16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Müller, M., Pym, E. C., Tong, A., and Davis, G. W. (2011). Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron 69, 749–762. doi: 10.1016/j.neuron.2011.01.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murthy, V. N., Schikorski, T., Stevens, C. F., and Zhu, Y. (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682. doi: 10.1016/S0896-6273(01)00500-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ni, J. Q., Liu, L. P., Binari, R., Hardy, R., Shim, H. S., Cavallaro, A., et al. (2009). A Drosophila resource of transgenic RNAi lines for neurogenetics. Genetics 182, 1089–1100. doi: 10.1534/genetics.109.103630
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ni, J. Q., Markstein, M., Binari, R., Pfeiffer, B., Liu, L. P., Villalta, C., et al. (2008). Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods 5, 49–51. doi: 10.1038/nmeth1146
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noskova, L., Stranecky, V., Hartmannova, H., Pristoupilova, A., Baresova, V., Ivanek, R., et al. (2011). Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 89, 241–252. doi: 10.1016/j.ajhg.2011.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Penney, J., Tsurudome, K., Liao, E. H., Elazzouzi, F., Livingstone, M., Gonzalez, M., et al. (2012). TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74, 166–178. doi: 10.1016/j.neuron.2012.01.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Perez-Otano, I., and Ehlers, M. D. (2005). Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 28, 229–238. doi: 10.1016/j.tins.2005.03.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peters, C., Bayer, M. J., Buhler, S., Andersen, J. S., Mann, M., and Mayer, A. (2001). Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 409, 581–588. doi: 10.1038/35054500
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petersen, S. A., Fetter, R. D., Noordermeer, J. N., Goodman, C. S., and DiAntonio, A. (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248. doi: 10.1016/S0896-6273(00)80415-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Philip, F., Kadamur, G., Silos, R. G., Woodson, J., and Ross, E. M. (2010). Synergistic activation of phospholipase C-beta3 by Galpha(q) and Gbetagamma describes a simple two-state coincidence detector. Curr. Biol. 20, 1327–1335. doi: 10.1016/j.cub.2010.06.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. doi: 10.1016/j.neuron.2010.04.028
Rohrbough, J., and Broadie, K. (2005). Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 6, 139–150. doi: 10.1038/nrn1608
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rorth, P. (1996). A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc. Natl. Acad. Sci. U.S.A. 93, 12418–12422. doi: 10.1073/pnas.93.22.12418
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shortridge, R. D., Yoon, J., Lending, C. R., Bloomquist, B. T., Perdew, M. H., and Pak, W. L. (1991). A Drosophila phospholipase C gene that is expressed in the central nervous system. J. Biol. Chem. 266, 12474–12480.
Smith, L. A., Peixoto, A. A., Kramer, E. M., Villella, A., and Hall, J. C. (1998). Courtship and visual defects of cacophony mutants reveal functional complexity of a calcium-channel alpha1 subunit in Drosophila. Genetics 149, 1407–1426.
Staudt, N., Molitor, A., Somogyi, K., Mata, J., Curado, S., Eulenberg, K., et al. (2005). Gain-of-function screen for genes that affect Drosophila muscle pattern formation. PLoS Genet. 1:e55. doi: 10.1371/journal.pgen.0010055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tedford, H. W., and Zamponi, G. W. (2006). Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 58, 837–862. doi: 10.1124/pr.58.4.11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thibault, S. T., Singer, M. A., Miyazaki, W. Y., Milash, B., Dompe, N. A., Singh, C. M., et al. (2004). A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat. Genet. 36, 283–287. doi: 10.1038/ng1314
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsurudome, K., Tsang, K., Liao, E. H., Ball, R., Penney, J., Yang, J. S., et al. (2010). The Drosophila miR-310 cluster negatively regulates synaptic strength at the neuromuscular junction. Neuron 68, 879–893. doi: 10.1016/j.neuron.2010.11.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. doi: 10.1016/j.cell.2008.10.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Velinov, M., Dolzhanskaya, N., Gonzalez, M., Powell, E., Konidari, I., Hulme, W., et al. (2012). Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS ONE 7:e29729. doi: 10.1371/annotation/26d7eb64-ccd2-41db-b1aa-7cdc8c1eff95
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wagh, D. A., Rasse, T. M., Asan, E., Hofbauer, A., Schwenkert, I., Durrbeck, H., et al. (2006). Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 49, 833–844. doi: 10.1016/j.neuron.2006.02.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, T., Hauswirth, A. G., Tong, A., Dickman, D. K., and Davis, G. W. (2014). Endostatin is a trans-synaptic signal for homeostatic synaptic plasticity. Neuron 83, 616–629. doi: 10.1016/j.neuron.2014.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weinkove, D., Neufeld, T. P., Twardzik, T., Waterfield, M. D., and Leevers, S. J. (1999). Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr. Biol. 9, 1019–1029. doi: 10.1016/S0960-9822(99)80450-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weyhersmüller, A., Hallermann, S., Wagner, N., and Eilers, J. (2011). Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 31, 6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wishart, T. M., Rooney, T. M., Lamont, D. J., Wright, A. K., Morton, A. J., Jackson, M., et al. (2012). Combining comparative proteomics and molecular genetics uncovers regulators of synaptic and axonal stability and degeneration in vivo. PLoS Genet. 8:e1002936. doi: 10.1371/journal.pgen.1002936
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, L., Bauer, C. S., Zhen, X. G., Xie, C., and Yang, J. (2002). Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 419, 947–952. doi: 10.1038/nature01118
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yao, C. A., and Carlson, J. R. (2010). Role of G-proteins in odor-sensing and CO2-sensing neurons in Drosophila. J. Neurosci. 30, 4562–4572. doi: 10.1523/JNEUROSCI.6357-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Younger, M. A., Müller, M., Tong, A., Pym, E. C., and Davis, G. W. (2013). A presynaptic ENaC channel drives homeostatic plasticity. Neuron 79, 1183–1196. doi: 10.1016/j.neuron.2013.06.048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zamponi, G. W. (2005). Voltage-Gated Calcium Channels. Georgetown; New York: Landes Bioscience/Eurekah.com; Kluwer Academic/Plenum Publishers.
Zhang, Y. Q., Henderson, M. X., Colangelo, C. M., Ginsberg, S. D., Bruce, C., Wu, T., et al. (2012). Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. doi: 10.1016/j.neuron.2012.01.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, C., Dreosti, E., and Lagnado, L. (2011). Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J. Neurosci. 31, 7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhen, X. G., Xie, C., Yamada, Y., Zhang, Y., Doyle, C., and Yang, J. (2006). A single amino acid mutation attenuates rundown of voltage-gated calcium channels. FEBS Lett. 580, 5733–5738. doi: 10.1016/j.febslet.2006.09.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zinsmaier, K. E. (2010). Cysteine-string protein's neuroprotective role. J. Neurogenet. 24, 120–132. doi: 10.3109/01677063.2010.489625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zinsmaier, K. E., Eberle, K. K., Buchner, E., Walter, N., and Benzer, S. (1994). Paralysis and early death in cysteine string protein mutants of Drosophila. Science 263, 977–980. doi: 10.1126/science.8310297
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zinsmaier, K. E., Hofbauer, A., Heimbeck, G., Pflugfelder, G. O., Buchner, S., and Buchner, E. (1990). A cysteine-string protein is expressed in retina and brain of Drosophila. J. Neurogenet. 7, 15–29. doi: 10.3109/01677069009084150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: homeostatic plasticity, Drosophila melanogaster, neuromuscular junction, RNAi screening, synaptic transmission, CaV2 channels, cysteine string protein, phospholipase C beta
Citation: Brusich DJ, Spring AM and Frank CA (2015) A single-cross, RNA interference-based genetic tool for examining the long-term maintenance of homeostatic plasticity. Front. Cell. Neurosci. 9:107. doi: 10.3389/fncel.2015.00107
Received: 16 December 2014; Accepted: 09 March 2015;
Published: 26 March 2015.
Edited by:
Jaichandar Subramanian, Massachusetts Institute of Technology, USAReviewed by:
Konrad Ernst Zinsmaier, University of Arizona, USASusan Tsunoda, Colorado State University, USA
Copyright © 2015 Brusich, Spring and Frank. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Andrew Frank, Department of Anatomy and Cell Biology, University of Iowa, Carver College of Medicine, 51 Newton Road, 1-661A BSB Iowa City, IA 52242, USA andy-frank@uiowa.edu
†These authors have contributed equally to this work.