Additional mechanisms conferring genetic susceptibility to Alzheimer’s disease
 Miguel Calero1,2,3
Miguel Calero1,2,3  Alberto Gómez-Ramos1,4
Alberto Gómez-Ramos1,4  Olga Calero1,2
Olga Calero1,2  Eduardo Soriano1,5
Eduardo Soriano1,5  Jesús Avila1,4*
Jesús Avila1,4*  Miguel Medina1,3*
Miguel Medina1,3*
- 1Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Madrid, Spain
- 2Chronic Disease Programme, Instituto de Salud Carlos III, Madrid, Spain
- 3Alzheimer Disease Research Unit, CIEN Foundation, Queen Sofia Foundation Alzheimer Center, Madrid, Spain
- 4Centro de Biología Molecular Severo Ochoa CSIC-UAM, Madrid, Spain
- 5University of Barcelona, Barcelona, Spain
Familial Alzheimer’s disease (AD), mostly associated with early onset, is caused by mutations in three genes (APP, PSEN1, and PSEN2) involved in the production of the amyloid β peptide. In contrast, the molecular mechanisms that trigger the most common late onset sporadic AD remain largely unknown. With the implementation of an increasing number of case-control studies and the upcoming of large-scale genome-wide association studies there is a mounting list of genetic risk factors associated with common genetic variants that have been associated with sporadic AD. Besides apolipoprotein E, that presents a strong association with the disease (OR∼4), the rest of these genes have moderate or low degrees of association, with OR ranging from 0.88 to 1.23. Taking together, these genes may account only for a fraction of the attributable AD risk and therefore, rare variants and epistastic gene interactions should be taken into account in order to get the full picture of the genetic risks associated with AD. Here, we review recent whole-exome studies looking for rare variants, somatic brain mutations with a strong association to the disease, and several studies dealing with epistasis as additional mechanisms conferring genetic susceptibility to AD. Altogether, recent evidence underlines the importance of defining molecular and genetic pathways, and networks rather than the contribution of specific genes.
Introduction
Alzheimer’s disease (AD) is characterized clinically by a gradual decline in memory and other cognitive functions and neuropathologically by gross brain atrophy and accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles. AD is the most common cause of dementia in the elderly, still without effective treatment. The disease has a strong genetic component and, in a small number of cases, AD segregates as an autosomal dominant trait in families. Although uncommon, the identification of these mutations in the last two or three decades has been very critical not only for diagnosing presymptomatic individuals from autosomal dominant families, but also for important advances in understanding AD pathobiology.
The inheritance of AD exhibits a dichotomous pattern. On one hand, early genetic linkage family studies led to the identification of dominantly inherited, rare mutations in the genes for the amyloid β precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) that are associated with early-onset AD (EOAD) with a penetrance close to 100%. Mutations in those three genes account for one third to one half of all autosomal dominant cases, which in turn represent less than 1% of total AD cases (reviewed in Guerreiro et al., 2012). On the other hand, the risk for late onset AD (LOAD), the most common form of the disease (>95 of total cases), is influenced by common variants such as the APOE haplotypes, as discussed below (see Table 1).
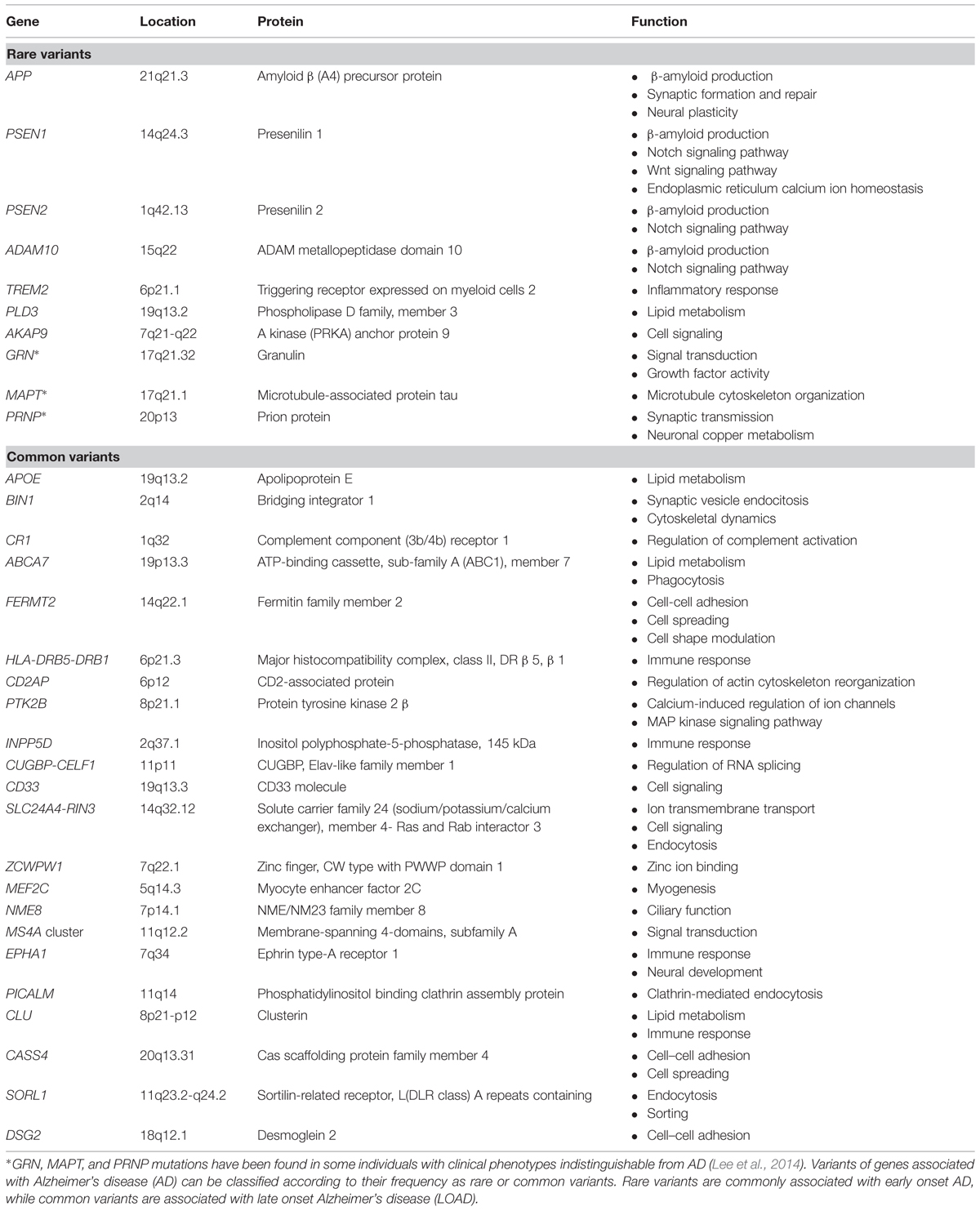
TABLE 1. AD-associated genes.
Over 30 dominant mutations in the APP gene (located at chromosome 21q21) account for about 15% of early-onset autosomal dominant cases of AD. Interestingly, two recessive APP mutations, A673V, and E693D, also reportedly cause EOAD (Tomiyama et al., 2008; Di Fede et al., 2009). APP encodes a ubiquitously expressed transmembrane protein and most mutations cluster around or within the Aβ domain. Duplications of APP and neighboring sequences are also linked to EOAD. Families carrying these duplications exhibit classic AD and cerebral amyloid angiopathy (reviewed in Guerreiro et al., 2012). These copy number mutations display different frequencies depending on the geographic population studied, being much more frequent in Japanese than in Europeans early-onset familial cases (Raux et al., 2005; Sleegers et al., 2006; Blom et al., 2008; Kasuga et al., 2009). Furthermore, patients with Down syndrome, which results from a chromosome 21 trisomy, develop AD neuropathology (Hartley et al., 2014).
Structurally similar integral membrane proteins PSEN1 and PSEN2 are part of the γ-secretase protein complex responsible for APP cleavage and Aβ generation (Medina and Dotti, 2003). About 80% of EOAD cases have been reported to carry dominant, pathogenic mutations in PSEN1 (located at chromosome 14q24.3), whereas approximately 5% have been identified in PSEN2, which is located at chromosome 1q31–q42 (Bird, 1999) Mutations in PSEN1 and PSEN2 appears scattered throughout the protein, although some clustering can be observed in the transmembrane domains (Guerreiro et al., 2012).
On the other hand, in non-familial, sporadic cases of AD mostly associated with late onset (LOAD), the ε4 allele of the apolipoprotein E (APOE) gene has been identified as a major genetic risk factor contributing to the pathogenesis of AD (Corder et al., 1993; Strittmatter et al., 1993). The APOE gene is located at chromosome 19q13.2 and encodes a highly pleiotropic glycoprotein (Siest et al., 1995) involved in the transport of cholesterol and other lipids in the periphery and brain (Mahley, 1988). There are three polymorphic alleles (ε2, ε3, and ε4) encoding three isoforms that differ on two amino acid residues (112 and 158): ApoE2, ApoE3 (the most common form), and ApoE4. Presence of the ApoE4 allele increases risk in familial and sporadic EOAD and LOAD, increasing risk threefold when in heterozygosis and up to 15-fold when in homozygosis (Ashford, 2004). The ε4 allele has also a dose-dependent effect on the age at onset. Conversely, the allele ε2 appears to lowers the risk for LOAD and delays age at onset (Corder et al., 1994).
Overall, these four genes account for 30–50% of the inheritability of AD (Jonsson et al., 2012). The advent of genome-wide association studies (GWAS) in recent years has allowed the identification of novel genetic associations. Hence, over 20 genetic loci have been reported to associate with increased susceptibility for LOAD, essentially common variants with a small individual effect on risk (unlike APOE), but also recently identified rare susceptibility variants in LOAD with larger effects (see below).
Thus, besides APOE, successive GWAS analyses have generated replicable associations with LOAD for several genes: CLU, PICALM, CR1, BIN1, CD33, MS4A cluster, CD2AP, EPHA1, and ABCA7 (reviewed in Karch et al., 2014). A recent meta-analysis of GWAS data from four large consortia confirmed these previous associations (except for CD33) and reported 12 new susceptibility loci for AD: CASS4, CUGBP-CELF1, DSG2, FERMT2, HLA-DRB5-DRB1, INPP5D, MEF2C, NME8, PTK2B, SLC24A4-RIN3, SORL1, ZCWPW1 (Lambert et al., 2013; Table 1). These genes increase AD risk in a non-Mendelian fashion, but first-degree relatives of LOAD patients have twice the expected risk of AD and LOAD is more frequent in monozygotic than in dizygotic co-twins (Reitz and Mayeux, 2014). However, the observed risk or protective effects of all the single nucleotide polymorphisms (SNPs) tagging these 21 loci are rather small, with odds ratio (OR) ranging from 1.22 to 0.77 (Karch et al., 2014), and the genetic effect attributable to each of these associated loci had population-attributable fractions or preventive fractions between 1.0 and 8.0% (Lambert et al., 2013). Although these data are of great value to delineate the fundamental physiopathological avenues of the disease, they only explain a small proportion of familial clustering (Manolio et al., 2009). Therefore, strong additional efforts in sequencing and post-GWAS analyses have to be put forward to find the remaining missing heritability in order to completely undercover the genetics of AD, utterly aiming at enabling effective prevention, prediction and treatment of the disease (Manolio et al., 2009; Lambert et al., 2013).
A number of recent excellent reviews have focused on the analysis of common genetic risk factors for LOAD and their role in pathogenesis (Guerreiro et al., 2013a; Lambert et al., 2013; Karch et al., 2014; Reitz and Mayeux, 2014). The genes identified can be classified in a few pathways, mainly lipid metabolism, immune response, endocytosis (Karch and Goate, 2015). Despite the identification of all the above-mentioned loci associated with AD, a large proportion of the genetic component of the disorder remains unexplained (Lord et al., 2014). Using alternative AD phenotypes may serve as a tool to unveil additional genes that could modify particular aspects of the disease (Karch and Goate, 2015). In here, we review additional mechanisms that may at least partially explain this missing heritability such as epistasis, rare variants, or the presence of somatic mutations. In this sense, this review does not intend to comprehend an exhaustive analysis of the literature on these subjects, but to put forward important concepts and the general evolution of the field. The analysis of epigenetic modifications or microRNA studies are out of the scope of the present review.
Rare Variants
After a decade of intense efforts on GWAS analysis, lately followed by meta-analysis of studies performed on very large cohorts, a number of common variants have been identified to have replicable, although small effects on LOAD with no clear functionality in some cases (Cruchaga et al., 2014); and it is unlikely that many common variants with moderate-large effects remain to be identified (Ridge et al., 2013). However, new rare functional variants, with larger effects are expected to be associated with AD. Thus, researchers from the AD Genetics Consortium argue that more than 25% of phenotypic variance remains unexplained by known markers, but it is tagged by common SNPs, hence suggesting that novel AD markers that account for large amounts of phenotypic variance are likely to be rare (Ridge et al., 2013). Thus, both rare and common variants contribute to AD risk (Guerreiro et al., 2013a; see Table 1). However, GWAS are not well suited for the discovery of these rare variants, and the use of new techniques such as exome analysis or whole genome sequencing will be required. Genome and exome sequencing studies in large data sets are likely to add new genes (with a moderate or low association). However, it remains to be seen whether additional pathways can be identified (Karch and Goate, 2015).
Besides the well-characterized mutations on PSEN1, PSEN2, and APP found in pedigrees of familiar EOAD, the advent of new powerful tools for genome analysis is already delivering a more comprehensive identification of rare variants potentially associated with the disease. For example, whole-genome sequence analysis from 1,795 Icelanders has led to the identification for the first time of a coding mutation (A673T) in APP that protects against AD and cognitive decline (Jonsson et al., 2012). In a different study, exome sequencing analysis has identified two novel pathogenic PSEN1 mutations (p.L166V and p.S230R) in British EOAD (Sassi et al., 2014a), although a similar study did not identify novel variants in AD in an Asian population (Chung et al., 2014).
Additionally, rare variants in other genes previously not associated with familiar EOAD have been found to have a high to moderate effect size. Thus, two rare, highly penetrant mutations in ADAM10 (p.Q170H and p.R181G) for LOAD have been reported (Kim et al., 2009), emphasizing the importance of whole genome or whole exome sequencing approaches to find rare variants causing LOAD, in addition to common variants (Jonsson et al., 2012).
Following whole-exome sequencing and whole-genome sequencing strategies, a rare variant in the TREM2 gene (p.R47H) has also been associated with an increased risk of AD with an OR of 3.4 (Guerreiro et al., 2013b; Guerreiro and Hardy, 2013; Jonsson et al., 2013). Recently, two more TREM2 variants have been associated with either increased (p.R62H) or decreased (p.S144G ) risk in AD (Benitez et al., 2014; Cuyvers et al., 2014). Interestingly, TREM2 also appears to be associated with Parkinson’s disease, frontotemporal dementia (Rayaprolu et al., 2013; Le Ber et al., 2014), and amyotrophic lateral sclerosis (Guerreiro and Hardy, 2013), although this remains controversial (Slattery et al., 2014).
Applying whole exome sequencing with a family-based design aimed at detecting novel AD risk genes, several rare, missense, and synonymous variants in phospholipase D3 (PLD3) have been reported to be associated with AD risk (Cruchaga et al., 2014). One variant in PLD3 (p.V232M), which segregated with some LOAD families appears to increase two- to threefold the risk for AD, perhaps through its influence on APP processing (Cruchaga et al., 2014).
Whole exome sequencing analysis performed on seven African American AD cases, (Logue et al., 2014) has led to find two new rare variants in AKAP9 (A-kinase anchor protein nine gene) potentially associated with AD, that in the replica population from the AD Genetics Consortium showed a strong association (rs144662445, OR = 2.75; rs149979685, OR = 3.61).
Recently, the Ibero-American Alzheimer Disease Genetics Group Researchers analyzed the coding region and flanking sequences of APP, PSEN1, PSEN2, MAPT, and GRN by pooled-DNA exon sequencing in 167 clinical and five autopsy-confirmed, mainly EOAD cases (Jin et al., 2012). Interestingly, pathogenic mutations in PSEN1, GRN, and MAPT genes were found in 2.3% of the screened cases, suggesting that pathogenic mutations or risk variants in MAPT and in GRN are as frequent in clinical AD cases as mutations in APP, PSEN1, and PSEN2 (Jin et al., 2012). These findings underscore the pleotropic role of genes such as MAPT and GRN that can influence both frontotemporal dementia and AD (Jin et al., 2012; Lee et al., 2014; Table 1). Likewise, missense or nonsense haplotypes in PRNP (p.I215V or p.Q160) further highlight how very similar genotypes in PRNP result in strikingly different clinical phenotypes such as prion diseases and AD (Muñoz-Nieto et al., 2013; Guerreiro et al., 2014).
In contrast, the role of rare coding variability in Mendelian inherited dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) has also been investigated in LOAD. Results indicated that rare coding variability in PSEN1 and PSEN2 may influence the susceptibility for LOAD, while GRN, MAPT, and PRNP do not appear to be major contributors to LOAD (Sassi et al., 2014b).
Somatic Mutations
Advanced age is the single most important risk factor for AD. Randomly acquired DNA damage in the nuclear genome is also associated with aging, and post-mitotic neuronal tissue is at special risk of DNA damage due to the elevated production of DNA-damaging reactive oxygen species (ROS) associated with their high metabolism (Barja, 2004; Kennedy et al., 2012). Moreover, the mutation rate of somatic cells is roughly an order of magnitude higher than mutation rates for germ-line cells (Lynch, 2010). On the other hand, evolution cannot put selective pressure on those deleterious mutations that produce defects long after the age of reproduction as it is the case of AD (Medawar, 1952; Kennedy et al., 2012). According to the so called somatic mutation theory of aging, the accumulation of mutations in the genetic material of somatic cells as a function of time results in a decrease in cellular function. A significant amount of research has shown that somatic mutations play an important role in aging and a number of age-related pathologies (Jeppesen et al., 2011; Kennedy et al., 2012). Additionally, some genetic traits may present antagonistic pleiotropy as in the case of APOE ε4 variant that appears to be associated with improved perinatal health and survival, favoring the selection of the ε4 allele despite its later life deleterious effects (reviewed in Eisenberg et al., 2010). With this perspective, it is not surprising that somatic cells, especially in the brain, may accumulate a much higher proportion of mutations in certain DNA hot spots (e.g., CpG dinucleotides) that correlate (at a lower scale) with the reported occurrence of pathogenic mutations in germinal-cell lines. The effect of these somatic mutations will depend on stochastic processes that allow the generation of cellular damage that may also propagate to other cells (Aguzzi and Rajendran, 2009). Therefore, in a wide sense, somatic mutations could be considered as rare variants locally generated.
Recent technological advances in the field of genomics and the emergence of next generation sequencing (NGS) techniques have aggravated the difficulties of interpreting GWAS to reveal the genetic basis of brain disorders. Despite a wealth of data on candidate genes affecting the susceptibility to AD and other neurological disorders, their inherent contribution to the pathogenesis and their relationship with non-genetic environmental risk factors is not yet fully understood. Evidence has accumulated showing that somatic variations can affect neuronal populations and may play a role in brain pathogenic processes. Thus, it has been suggested that, at least in some cases copy number variations (CNV) or SNP linked to major AD and other neurological disorders may lead to genetic dysregulation resulting in somatic mosaicism (Iourov et al., 2013). Moreover, environmental factors may affect cellular repair mechanisms and trigger a series of events that end up generating genomic instability.
Increasing evidence has also shown that, even at low levels of mosaicism, somatic mutations can cause neuropsychiatric, and pediatric disorders such as epilepsy, autism, lissencephaly, and intellectual disability. Interestingly, at least some of these cases appears to have been caused by somatic mutations limited to the brain (Poduri et al., 2013). In addition, a number of reports have shown that a significant population of neurons exhibit aneuploidy in both mice and humans and the level of aneuploidy could be as high as 1–3% in adult brain (Rehen et al., 2005). Furthermore, brain tissue from both AD and other disorders exhibits increased levels of aneuploidy when compared to unaffected brain (Iourov et al., 2009; Frade and López-Sánchez, 2010).
Furthermore, a recent study has investigated the presence of tissue-specific exonic single nucleotide variations (SNV), taking blood exome as a control (Gómez-Ramos et al., 2014). Interestingly, the number of SNV per chromosome was independent of chromosome size, but it was suggested to mainly relate to the number of protein-coding genes per chromosome. Although similar patterns of chromosomal distribution of tissue-specific SNVs were found, clear differences were also detected, supporting the notion that each individual tissue has a specific SNV exome signature.
Some neurodegenerative disorders have been associated with or can be affected by somatic mutations. Thus, EOAD has been attributed to a somatic mosaic PSEN1 mutation present in the brain (Beck et al., 2004). Similarly, a case of sporadic Creutzfeldt–Jakob disease has been reported to be caused by an early embryonic somatic mutation in the PRNP gene (Alzualde et al., 2010). Also, patients of disorders caused by the expansion of highly mutable repeats such as Friedrich’s ataxia or Huntington’s disease, can exhibit notable somatic heterogeneity in repeat lengths across different brain regions and tissues (Kahlem and Djian, 2000; Hellenbroich et al., 2001; Møllersen et al., 2010). Age-related somatic mutations, known to play a role in cancer (Jacobs et al., 2012), have also been proposed to be involved in normal aging processes as well as in neurodegeneration (Kennedy et al., 2012). Interestingly, very recent whole-exome sequencing analyses of various tissues from sporadic AD patients have found a remarkably high number of brain-specific SNV in AD hippocampal samples when compared with blood (Parcerisas et al., 2014), suggesting that somatic genetic mosaicism and brain-specific genome reshaping may contribute to the pathogenesis of sporadic AD.
As NGS techniques become more efficient and affordable in the coming years, somatic mosaic mutations could be more readily detected, and somatic mutation rates could be systematically determined across different regions, cell types, and time points of the human brain development. Although the limitation of brain tissue will still limit studies on brain-specific somatic mutations, novel techniques, and improved bioinformatic analyses will allow us to address the role of somatic mosaicism in neurological conditions. Determining whether brain somatic mutation are at least partially responsible for AD and other neurodegenerative disorders will be one of the next major challenges in the field.
Epistasis
The functional expression of certain genes may be determined by the interaction with other genes, as well as with environment factors. Therefore, some of the missing heritability may be conditioned by epistasis, a crucial factor to understand and interpret genetic pathways and their functional role in complex systems. Epistasis may refer to different phenomena, including the functional interaction between genes or the statistical deviation from additive gene action, among others (Phillips, 2008), yet in the context of this review, we will use the term epistasis as synonym of functional gene–gene interaction. However, GWAS, whole genome and exome sequencing analyses have been mainly focused on standard single-locus tests with different level of precision. With the increasing availability of genetic data delivered by the new genomic technologies, it is becoming clearer the need to address the problems from a holistic position, taking into account the interaction among different players at different levels.
The analysis of epistasis, even if limited to the interaction between two genes, is technically demanding and methodologically limited at present. Part of the challenge for epistasis analysis in GWAS is the magnitude of the search and the computational complexity associated with it (Ritchie, 2015). The methods and related software packages used to detect the interactions between genetic loci that contribute to human genetic disease have been recently re-examined (Cordell, 2009). Lately, several studies have reviewed novel advances in the methodology for detecting epistasis (Ma et al., 2013; Wei et al., 2014) and discussed its relevance in the context of GWAS (Wei et al., 2014). A protocol for exhaustive genome-wide association interaction analysis and applied to AD has been proposed, finding replicable epistasis between the KHDRBS2 and CRYL1 gene loci (Gusareva et al., 2014).
A powerful tool to disentangle the complexity of a disorder such as AD is the use of the concept of endophenotypes to define genetic association elements with more elementary phenomena rather than with the whole spectrum of complex diagnostic entities. Therefore, markers of disease (at any level) that correlate with a genetic trait may be useful to explore the pathophysiological mechanisms related to specific aspects of the syndrome (Cannon and Keller, 2006).
Synergy factor (SF) analysis has been used to assess over 100 claims of epistasis in sporadic LOAD, finding 27 gene–gene interactions that were significantly associated with AD (Combarros et al., 2009). Additionally, the authors demonstrated by meta-analysis the interaction of APOE ε4 with specific variation in four different genes, namely ACT, BACE1, IL6, and BCHE (Combarros et al., 2009). More recently, a systematic review of genetic studies published between 2009 and 2012 by the Genetics Core of the AD Neuroimaging Initiative (ADNI) focused on genetic associations with disease status or quantitative disease endophenotypes including structural and functional neuroimaging, fluid biomarker assays, and cognitive performance. In this review, the authors summarize the association of several AD risk genes with imaging, fluid and cognitive phenotypes, and indicated the association with multiple ADNI phenotypes of several other genes (i.e., APOC1, FTO, GRIN2B, MAGI2, and TOMM40). The authors suggested the combination of genetic data and phenotypes for targeting future studies employing NGS and convergent multi-omics approaches, and for clinical drug and biomarker development (Shen et al., 2014). Likewise, the use of neuroimaging measures to define powerful quantitative endophenotypes for exploring epistatic relationships in order to explain some of the missing heritability in AD has been proposed (Hohman et al., 2013). Moreover, the analysis of epistasis has also been shown to be a key factor for genomic prediction and its application in preventive strategies through the identification of population strata at increased risk of disease and clinical decision making (Wray et al., 2013).
Many studies are now focused on finding how genetic variants influence specific traits of AD pathology, such as amyloid burden (Kauwe et al., 2011, 2014; Bali et al., 2012; Hohman et al., 2013; Shen et al., 2014), neurofibrilar pathology (Kauwe et al., 2011; Shen et al., 2014), brain atrophy (Shen et al., 2014), cognitive decline (Pedraza et al., 2014), or inflammation (Kauwe et al., 2014). However, the complexity and heterogeneity of AD requires further analysis going beyond the study of single markers in isolation by taking into account interactions between genes. For example, many of the LOAD risk genes do not show single marker associations with amyloid pathology, but only through interaction with other genes (e.g., BIN1 ×PICALM; Hohman et al., 2013).
Therefore, it is important to note that epistasis may be a barrier to uncover the genetic basis of complex disorders, since the effects of quantitative trait loci can be masked by interactions with other loci (Phillips, 2008). As demonstrated for GSTM3 gene and the HHEX/IDE/KIF11 locus, the association with AD could be purely epistatic with neither polymorphism showing an independent effect (Bullock et al., 2013). The same group, involved in the Epistasis Project, has also reported an interaction between transferrin and HFE genes, confirming previous findings (Robson et al., 2004) and suggesting that iron overload may be involved in the development of AD (Lehmann et al., 2012).
Arguably, APOE is the main genetic risk factor in AD and it is likely to interact with other genes. The epistatic effect for APOE has been recognized both in familial EOAD and LOAD modifying the age at onset (Sorbi et al., 1995; Combarros et al., 2009; Hohman et al., 2013). Using whole-trascriptome analysis of brain gene expression, It has been demonstrated that APOE ε4 carrier status was associated with a consistent transcriptomic shift that resembled the LOAD profile (Rhinn et al., 2013). However, unlike the genes underlying familial AD, LOAD susceptibility genes do not specifically alter the Aβ42/40 ratios, suggesting that these genes probably contribute to AD through distinct mechanisms (Bali et al., 2012). Interestingly, a recent study (Naj et al., 2014) has also demonstrated an association of APOE variants with age at onset among affected individuals with LOAD and observed novel associations of CR1, BIN1, and PICALM with age at onset.
Along the same lines, we have explored epistasis and pleiotropic effects of various genes and neurodegenerative disorders. AD and Creutzfeldt-Jakob disease (CJD) are now considered both as part of a wider group generically named as conformational disorders, and more specifically brain amyloidosis. Under the assumption that these two disorders share common pathophysiologic mechanisms involving protein aggregation in the brain leading to fatal degeneration, we investigated a possible genetic interaction between APOE ε4 allele and the polymorphic codon 129 of the PRNP gene in both AD and CJD populations compared to a common control population (Calero et al., 2011). Interestingly, we found a synergistic age-dependent interaction between the two genes (APOE × PRNP) in both disorders (SF = 3.59, p = 0.027 for AD; and SF = 7.26, p = 0.005 for CJD). Our data suggest the involvement of common pathways involved in the generation, clearance, and neurotoxic signal transduction of Aβ peptides and PrP in AD and CJD. The finding of an age-dependent interaction underscores the importance of genetic risk analysis stratified according to other potential interacting/confounding factors such age, sex, or comorbidities.
Finally, the comprehensive analysis of high order interactions is limited by the exponential number of potential interactions and our technical capacity; therefore, we need to focus on particular subsets of the interaction space by using additional functional information (Phillips, 2008). However, the analysis of epistasis should not be limited only to candidate genes.
Conclusion
Over 20 loci have been associated with LOAD, defining three main routes altered in AD: lipid metabolism, immune response, and endocytosis (see Table 1). Altogether, these association analyses to different genes by GWAS underscore the importance of defining pathways and networks rather the contribution of specific genes. Future research should be focused on defining shared and specific mechanisms among distinct neurodegenerative and other chronic disorders including cardiovascular disease, metabolic disorders, or even cancer. Further insights into complex disorders such as AD are expected from the integration of different -omics with detailed high-quality clinico-physiological characterization of cases and controls allowing the development of new disease biomarkers and therapeutic avenues, and will enable the implementation of personalized medicine (Ramanan and Saykin, 2013). The identification by GWAS of multiple disease-associated loci of small effect size emphasizes the polygenic nature of the heritability of complex traits and common disorders such as AD, giving rise to approach the disorder as of quantitative dimensions instead of as a qualitative disorder (Plomin et al., 2009).
However, a large proportion of genetic component of the disorder remains unexplained (Lord et al., 2014). As reviewed above, epistasis, rare variants, and the presence of somatic mutations are additional genetic mechanism that may explain in part this missing heritability. Future research should emphasize: (i) the complete characterization of the genetic risk factors including common-low effect size and rare-high effect size variants, (ii) the use of endophenotypes to associate specific traits of the disease to genetic factors and use this information for risk prediction and definition of treatment response groups, (iii) the analysis of epistasis among different risk genes and the interaction with other factors such as age and sex, and (iv) the potential pleiotropism associated with certain loci that may confer either risk or protection to AD and may transversally regulate different neurodegenerative disorders. From a practical point of view, it is important to acknowledge that most AD patients usually present a combined pathology with other chronic disorders including cardiovascular disease or metabolic disorders such as diabetes mellitus type 2; and therefore we should investigate how these pathologies interact with each other, in order to properly treat each individual patient.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by CIBERNED, CIEN and Reina Sofia Foundations and the Carlos III Health Institute (PI12/00045).
References
Aguzzi, A., and Rajendran, L. (2009). The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790. doi: 10.1016/j.neuron.2009.12.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alzualde, A., Moreno, F., Martínez-Lage, P., Ferrer, I., Gorostidi, A., Otaegui, D., et al. (2010). Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178N mutation in the PRNP gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 1283–1291. doi: 10.1002/ajmg.b.31099
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ashford, J. W. (2004). APOE genotype effects on Alzheimer’s disease onset and epidemiology. J. Mol. Neurosci. 23, 157–165. doi: 10.1385/JMN:23:3:157
Bali, J., Gheinani, A. H., Zurbriggen, S., and Rajendran, L. (2012). Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 109, 15307–15311. doi: 10.1073/pnas.1201632109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barja, G. (2004). Free radicals and aging. Trends Neurosci. 27, 595–600. doi: 10.1016/j.tins.2004.07.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beck, J. A., Poulter, M., Campbell, T. A., Uphill, J. B., Adamson, G., Geddes, J. F., et al. (2004). Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum. Mol. Genet. 13, 1219–1224. doi: 10.1093/hmg/ddh134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benitez, B. A., Jin, S. C., Guerreiro, R., Graham, R., Lord, J., Harold, D., et al. (2014). Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol. Aging 35, 1510. doi: 10.1016/j.neurobiolaging.2013.12.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bird, T. D. (1999). “Early-Onset Familial Alzheimer Disease,” in GeneReviews® [Internet], eds R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, and C. T. Fong (Seattle, WA; University of Washington, Seattle), 1993–2014.
Blom, E. S., Viswanathan, J., Kilander, L., Helisalmi, S., Soininen, H., Lannfelt, L., et al. (2008). Low prevalence of APP duplications in Swedish and Finnish patients with early-onset Alzheimer’s disease. Eur. J. Hum. Genet. 16, 171–175. doi: 10.1038/sj.ejhg.5201966
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bullock, J. M., Medway, C., Cortina-Borja, M., Turton, J. C., Prince, J. A., Ibrahim-Verbaas, C. A., et al. (2013). Discovery by the Epistasis Project of an epistatic interaction between the GSTM3 gene and the HHEX/IDE/KIF11 locus in the risk of Alzheimer’s disease. Neurobiol. Aging 34, 1309.e1–1309.e7. doi: 10.1016/j.neurobiolaging.2012.08.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Calero, O., Bullido, M. J., Clarimón, J., Frank-García, A., Martínez-Martín, P., Lleó, A., et al. (2011). Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer’s and Creutzfeldt-Jakob diseases. PLoS ONE 6:e22090. doi: 10.1371/journal.pone.0022090
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cannon, T. D., and Keller, M. C. (2006). Endophenotypes in the genetic analyses of mental disorders. Annu. Rev. Clin. Psychol. 2, 267–290. doi: 10.1146/annurev.clinpsy.2.022305.095232
Chung, S. J., Kim, M. J., Kim, J., Kim, Y. J., You, S., Koh, J., et al. (2014). Exome array study did not identify novel variants in Alzheimer’s disease. Neurobiol. Aging 35, 1958.e13–1958.e14. doi: 10.1016/j.neurobiolaging.2014.03.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Combarros, O., Cortina-Borja, M., Smith, A. D., and Lehmann, D. J. (2009). Epistasis in sporadic Alzheimer’s disease. Neurobiol. Aging 30, 1333–1349. doi: 10.1016/j.neurobiolaging.2007.11.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cordell, H. J. (2009). Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 10, 392–404. doi: 10.1038/nrg2579
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C. Jr., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Cruchaga, C., Karch, C. M., Jin, S. C., Benitez, B. A., Cai, Y., Guerreiro, R., et al. (2014). Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505, 550–554. doi: 10.1038/nature12825
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cuyvers, E., Bettens, K., Philtjens, S., Van Langenhove, T., Gijselinck, I., van der Zee, J., et al. (2014). Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e11–726.e19. doi: 10.1016/j.neurobiolaging.2013.09.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Di Fede, G., Catania, M., Morbin, M., Rossi, G., Suardi, S., Mazzoleni, G., et al. (2009). A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477. doi: 10.1126/science.1168979
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eisenberg, D. T., Kuzawa, C. W., and Hayes, M. G. (2010). Worldwide allele frequencies of the human apolipoprotein E gene: climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 143, 100–111. doi: 10.1002/ajpa.21298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frade, J. M., and López-Sánchez, N. (2010). A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75 (NTR). Cell Cycle 9, 1934–1941. doi: 10.4161/cc.9.10.11582
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gómez-Ramos, A., Sanchez-Sanchez, R., Muhaisen, A., Rábano, A., Soriano, E., and Avila, J. (2014). Similarities and differences between exome sequences found in a variety of tissues from the same individual. PLoS ONE 9:e101412. doi: 10.1371/journal.pone.0101412
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., Brás, J., and Hardy, J. (2013a). SnapShot: genetics of Alzheimer’s disease. Cell 155, 968–968. doi: 10.1016/j.cell.2013.10.037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013b). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., Brás, J., Wojtas, A., Rademakers, R., Hardy, J., and Graff-Radford, N. (2014). A nonsense mutation in PRNP associated with clinical Alzheimer’s disease. Neurobiol. Aging 35, 2656.e13–2656.e16. doi: 10.1016/j.neurobiolaging.2014.05.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R. J., Gustafson, D. R., and Hardy, J. (2012). The genetic architecture of Alzheimer’s disease: beyond APP, PSENs and APOE. Neurobiol. Aging 33, 437–456. doi: 10.1016/j.neurobiolaging.2010.03.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., and Hardy, J. (2013). TREM2 and neurodegenerative disease. N. Engl. J. Med. 369, 1569–1570. doi: 10.1056/NEJMc1306509#SA1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gusareva, E. S., Carrasquillo, M. M., Bellenguez, C., Cuyvers, E., Colon, S., Graff-Radford, N. R., et al. (2014). Genome-wide association interaction analysis for Alzheimer’s disease. Neurobiol. Aging 35, 2436–2443. doi: 10.1016/j.neurobiolaging.2014.05.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hartley, D., Blumenthal, T., Carrillo, M., DiPaolo, G., Esralew, L., Gardiner, K.et al. (2014). Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement. doi: 10.1016/j.jalz.2014.10.007 [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hellenbroich, Y., Schwinger, E., and Zühlke, C. (2001). Limited somatic mosaicism for Friedreich’s ataxia GAA triplet repeat expansions identified by small pool PCR in blood leukocytes. Acta Neurol. Scand. 103, 188–192. doi: 10.1034/j.1600-0404.2001.103003188.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hohman, T. J., Koran, M. E., Thornton-Wells, T., and Alzheimer’s Neuroimaging Initiative. (2013). Epistatic genetic effects among Alzheimer’s candidate genes. PLoS ONE 8:e80839. doi: 10.1371/journal.pone.0080839
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iourov, I. Y., Vorsanova, S. G., Liehr, T., and Yurov, Y. B. (2009). Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol. Dis. 34, 212–220. doi: 10.1016/j.nbd.2009.01.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iourov, I. Y., Vorsanova, S. G., and Yurov, Y. B. (2013). Somatic cell genomics of brain disorders: a new opportunity to clarify genetic-environmental interactions. Cytogenet. Genome. Res. 139, 181–188. doi: 10.1159/000347053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jacobs, K. B., Yeager, M., Zhou, W., Wacholder, S., Wang, Z., Rodriguez-Santiago, B., et al. (2012). Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 44, 651–658. doi: 10.1038/ng.2270
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeppesen, D. K., Bohr, V. A., and Stevnsner, T. (2011). DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 94, 166–200. doi: 10.1016/j.pneurobio.2011.04.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jin, S. C., Pastor, P., Cooper, B., Cervantes, S., Benitez, B. A., Razquin, C., et al. (2012). Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther. 4, 34. doi: 10.1186/alzrt137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kahlem, P., and Djian, P. (2000). The expanded CAG repeat associated with juvenile Huntington disease shows a common origin of most or all neurons and glia in human cerebrum. Neurosci. Lett. 286, 203–207. doi: 10.1016/S0304-3940(00)01029-6
Karch, C. M., Cruchaga, C., and Goate, A. M. (2014). Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26. doi: 10.1016/j.neuron.2014.05.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kasuga, K., Shimohata, T., Nishimura, A., Shiga, A., Mizuguchi, T., Tokunaga, J., et al. (2009). Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 80, 1050–1052. doi: 10.1136/jnnp.2008.161703
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kauwe, J. S., Bailey, M. H., Ridge, P. G., Perry, R., Wadsworth, M. E., Hoyt, K. L., et al. (2014). Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 10:e1004758. doi: 10.1371/journal.pgen.1004758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kauwe, J. S., Cruchaga, C., Karch, C. M., Sadler, B., Lee, M., Mayo, K., et al. (2011). Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer’s disease. PLoS ONE 6:e15918. doi: 10.1371/journal.pone.0015918
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kennedy, S. R., Loeb, L. A., and Herr, A. J. (2012). Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 133, 118–126. doi: 10.1016/j.mad.2011.10.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, M., Suh, J., Romano, D., Truong, M. H., Mullin, K., Hooli, B., et al. (2009). Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum. Mol. Genet. 18, 3987–3996. doi: 10.1093/hmg/ddp323
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Le Ber, I., De Septenville, A., Guerreiro, R., Bras, J., Camuzat, A., Caroppo, P., et al. (2014). Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35, 2419.e23–2419.e25. doi: 10.1016/j.neurobiolaging.2014.04.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, J. H., Kahn, A., Cheng, R., Reitz, C., Vardarajan, B., Lantigua, R., et al. (2014). Disease-related mutations among Caribbean Hispanics with familial dementia. Mol. Genet. Genomic Med. 2, 430–437. doi: 10.1002/mgg3.85
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lehmann, D. J., Schuur, M., Warden, D. R., Hammond, N., Belbin, O., Kölsch, H., et al. (2012). Transferrin and HFE genes interact in Alzheimer’s disease risk: the Epistasis Project. Neurobiol. Aging 33, 202.e1–202.e13. doi: 10.1016/j.neurobiolaging.2010.07.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Logue, M. W., Schu, M., Vardarajan, B. N., Farrell, J., Bennett, D. A., Buxbaum, J. D., et al. (2014). Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement 10, 609–618. doi: 10.1016/j.jalz.2014.06.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lord, J., Lu, A. J., and Cruchaga, C. (2014). Identification of rare variants in Alzheimer’s disease. Front. Genet. 5:369. doi: 10.3389/fgene.2014.00369
Lynch, M. (2010). Evolution of the mutation rate. Trends Genet. 26, 345–352. doi: 10.1016/j.tig.2010.05.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, L., Clark, A. G., and Keinan, A. (2013). Gene-based testing of interactions in association studies of quantitative traits. PLoS Genet. 9:e1003321. doi: 10.1371/journal.pgen.1003321
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Medina, M., and Dotti, C. G. (2003). RIPped out by presenilin-dependent gamma-secretase. Cell. Signal. 15, 829–841. doi: 10.1016/S0898-6568(03)00041-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Møllersen, L., Rowe, A. D., Larsen, E., Rognes, T., and Klungland, A. (2010). Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 6:e1001242. doi: 10.1371/journal.pgen.1001242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muñoz-Nieto, M., Ramonet, N., López-Gastón, J. I., Cuadrado-Corrales, N., Calero, O., Díaz-Hurtado, M., et al. (2013). A novel mutation I215V in the PRNP gene associated with Creutzfeldt-Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J. Neurol. 260, 77–84. doi: 10.1007/s00415-012-6588-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Naj, A. C., Jun, G., Reitz, C., Kunkle, B. W., Perry, W., Park, Y. S., et al. (2014). Effects of multiple genetic Loci on age at onset in late-onset Alzheimer disease: a genome-wide association study. JAMA Neurol. 71, 1394–1404. doi: 10.1001/jamaneurol.2014.1491
Parcerisas, A., Rubio, S. E., Muhaisen, A., Gómez-Ramos, A., Pujadas, L., Puiggros, M., et al. (2014). Somatic signature of brain-specific single nucleotide variations in sporadic Alzheimer’s disease. J. Alzheimers. Dis. 42, 1357–1382. doi: 10.3233/JAD-140891
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedraza, O., Allen, M., Jennette, K., Carrasquillo, M., Crook, J., Serie, D., et al. (2014). Evaluation of memory endophenotypes for association with CLU, CR1, and PICALM variants in black and white subjects. Alzheimers Dement 10, 205–213. doi: 10.1016/j.jalz.2013.01.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phillips, P. C. (2008). Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9, 855–867. doi: 10.1038/nrg2452
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Plomin, R., Haworth, C. M., and Davis, O. S. (2009). Common disorders are quantitative traits. Nat. Rev. Genet. 10, 872–878. doi: 10.1038/nrg2670
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poduri, A., Evrony, G. D., Cai, X., and Walsh, C. A. (2013). Somatic mutation, genomic variation, and neurological disease. Science 341:1237758. doi: 10.1126/science.1237758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramanan, V. K., and Saykin, A. J. (2013). Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2, 145–175.
Raux, G., Guyant-Maréchal, L., Martin, C., Bou, J., Penet, C., Brice, A., et al. (2005). Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J. Med. Genet. 42, 793–795. doi: 10.1136/jmg.2005.033456
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rayaprolu, S., Mullen, B., Baker, M., Lynch, T., Finger, E., Seeley, W. W., et al. (2013). TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 8, 19. doi: 10.1186/1750-1326-8-19
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rehen, S. K., Yung, Y. C., McCreight, M. P., Kaushal, D., Yang, A. H., Almeida, B. S., et al. (2005). Constitutional aneuploidy in the normal human brain. J. Neurosci. 25, 2176–2180. doi: 10.1523/JNEUROSCI.4560-04.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rhinn, H., Fujita, R., Qiang, L., Cheng, R., Lee, J. H., and Abeliovich, A. (2013). Integrative genomics identifies APOE ε4 effectors in Alzheimer’s disease. Nature 500, 45–50. doi: 10.1038/nature12415
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ridge, P. G., Mukherjee, S., Crane, P. K., Kauwe, J. S., and Alzheimer’s Disease Genetics Consortium. (2013). Alzheimer’s disease: analyzing the missing heritability. PLoS ONE 8:e79771. doi: 10.1371/journal.pone.0079771
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ritchie, M. D. (2015). Finding the epistasis needles in the genome-wide haystack. Methods Mol. Biol. 1253, 19–33. doi: 10.1007/978-1-4939-2155-3_2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Robson, K. J., Lehmann, D. J., Wimhurst, V. L., Livesey, K. J., Combrinck, M., and Merryweather-Clarke, A. T.et al. (2004). Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer’s disease. J. Med. Genet. 41, 261–265. doi: 10.1136/jmg.2003.015552
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sassi, C., Guerreiro, R., Gibbs, R., Ding, J., Lupton, M. K., Troakes, C., et al. (2014a). Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 35, 2422.e13–2422.e16. doi: 10.1016/j.neurobiolaging.2014.04.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sassi, C., Guerreiro, R., Gibbs, R., Ding, J., Lupton, M. K., Troakes, C., et al. (2014b). Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol. Aging 35, 2881.e1–2881.e6. doi: 10.1016/j.neurobiolaging.2014.06.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, L., Thompson, P. M., Potkin, S. G., Bertram, L., Farrer, L. A., Foroud, T. M., et al. (2014). Genetic analysis of quantitative phenotypes in AD and MCI: imaging, cognition and biomarkers. Brain Imag. Behav. 8, 183–207. doi: 10.1007/s11682-013-9262-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Siest, G., Pillot, T., Régis-Bailly, A., Leininger-Muller, B., Steinmetz, J., Galteau, M. M., et al. (1995). Apolipoprotein E: an important gene and protein to follow in laboratory medicine. Clin. Chem. 41(Pt 1), 1068–1086.
Slattery, C. F., Beck, J. A., Harper, L., Adamson, G., Abdi, Z., Uphill, J., et al. (2014). R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement 10, 602–608. doi: 10.1016/j.jalz.2014.05.1751
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D., Wauters, J., et al. (2006). APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129(Pt 11), 2977–2983. doi: 10.1093/brain/awl203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sorbi, S., Nacmias, B., Forleo, P., Piacentini, S., Latorraca, S., and Amaducci, L. (1995). Epistatic effect of APP717 mutation and apolipoprotein E genotype in familial Alzheimer’s disease. Ann. Neurol. 38, 124–127. doi: 10.1002/ana.410380120
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Strittmatter, W. J., Saunders, A. M., Schmechel, D., Pericak-Vance, M., Enghild, J., Salvesen, G. S., et al. (1993). Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981. doi: 10.1073/pnas.90.5.1977
Tomiyama, T., Nagata, T., Shimada, H., Teraoka, R., Fukushima, A., Kanemitsu, H., et al. (2008). A new amyloid β variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 63, 377–387. doi: 10.1002/ana.21321
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wei, W. H., Hemani, G., and Haley, C. S. (2014). Detecting epistasis in human complex traits. Nat. Rev. Genet. 15, 722–733. doi: 10.1038/nrg3747
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wray, N. R., Yang, J., Hayes, B. J., Price, A. L., Goddard, M. E., and Visscher, P. M. (2013). Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 14, 507–515. doi: 10.1038/nrg3457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer, epistasis, exome, GWAS, neurodegeneration, rare variants, risk factors, somatic mutations
Citation: Calero M, Gómez-Ramos A, Calero O, Soriano E, Avila J and Medina M (2015) Additional mechanisms conferring genetic susceptibility to Alzheimer’s disease. Front. Cell. Neurosci. 9:138. doi: 10.3389/fncel.2015.00138
Received: 23 December 2014; Accepted: 23 March 2015;
Published online: 09 April 2015.
Edited by:
Victoria Campos-Peña, Instituto Nacional de Neurologia y Neurocirugia, MexicoReviewed by:
Rafael Linden, Federal University of Rio de Janeiro, BrazilTuck Wah Soong, National University of Singapore, Singapore
Copyright © 2015 Calero, Gómez-Ramos, Calero, Soriano, Avila and Medina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Medina and Jesús Avila Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas, Valderrebollo 5, 28031 Madrid, Spain mmedina@ciberned.es