Thrombin regulation of synaptic transmission and plasticity: implications for health and disease
 Marina Ben Shimon1†
Marina Ben Shimon1†  Maximilian Lenz1,2†
Maximilian Lenz1,2†  Benno Ikenberg1,2
Benno Ikenberg1,2  Denise Becker2#
Denise Becker2#  Efrat Shavit Stein1
Efrat Shavit Stein1  Joab Chapman1,3
Joab Chapman1,3  David Tanne1,3
David Tanne1,3  Chaim G. Pick4
Chaim G. Pick4  Ilan Blatt1,3 Miri Neufeld1,3,5
Ilan Blatt1,3 Miri Neufeld1,3,5  Andreas Vlachos2‡
Andreas Vlachos2‡  Nicola Maggio1,6*‡
Nicola Maggio1,6*‡- 1Department of Neurology, The J. Sagol Neuroscience Center, The Chaim Sheba Medical Center, Tel HaShomer, Israel
- 2Institute of Clinical Neuroanatomy, Neuroscience Center Frankfurt, Goethe-University Frankfurt, Frankfurt, Germany
- 3Department of Neurology, The Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 4Department of Anatomy and Anthropology, The Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 5Department of Neurology and Epilepsy Unit, The Tel Aviv Sourasky Medical Center, Tel Aviv, Israel
- 6Talpiot Medical Leadership Program, The Chaim Sheba Medical Center, Tel HaShomer, Israel
Thrombin, a serine protease involved in the blood coagulation cascade has been shown to affect neural function following blood-brain barrier breakdown. However, several lines of evidence exist that thrombin is also expressed in the brain under physiological conditions, suggesting an involvement of thrombin in the regulation of normal brain functions. Here, we review ours’ as well as others’ recent work on the role of thrombin in synaptic transmission and plasticity through direct or indirect activation of Protease-Activated Receptor-1 (PAR1). These studies propose a novel role of thrombin in synaptic plasticity, both in physiology as well as in neurological diseases associated with increased brain thrombin/PAR1 levels.
Thrombin in the Blood and in the Brain
Coagulation is a fundamental biological process, by which cellular as well as protein components in the blood form a clot to stop bleeding from injured vessels (Kalz et al., 2014). It consists of a cascade of molecular events leading to the activation of thrombin, which catalyzes the conversion of fibrinogen to fibrin, the building blocks of the hemostatic plug (Figure 1; Siller-Matula et al., 2011; Lippi et al., 2012). Thrombin is a serine protease which is activated by the enzymatic cleavage of two sites on prothrombin by activated Factor X (FXa; Furie and Furie, 2005). The activity of FXa is enhanced by binding to activated Factor V (FVa, which forms the prothrombinase complex with FXa; Figure 1; Furie and Furie, 2005).
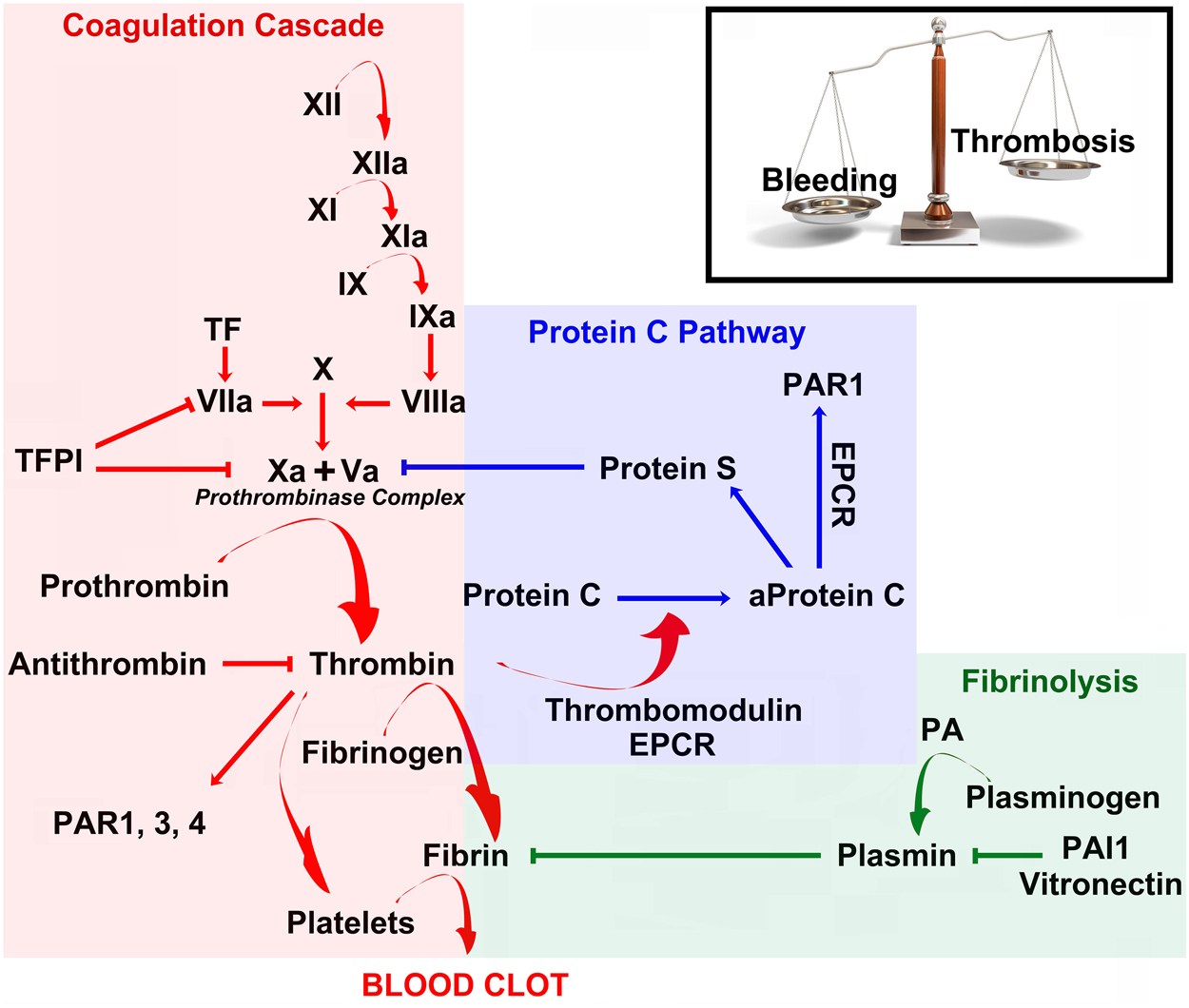
Figure 1. Coagulation, anticoagulation and fibrinolysis maintain a delicate physiological balance between bleeding and thrombosis. Schematic diagram of the coagulation cascade (red), Protein C pathway (blue) and fibrinolysis (green). See main text for more detailed explanation. Abbreviations: TFPI, Tissue Factor Pathway Inhibitor; PAR, Proteases activated receptors; EPCR, Endothelial Protein C Receptor; PA, Plasminogen Activator; PAI, Plasminogen Activator Inhibitor.
Prothrombin is produced in the liver and is post-translationally modified in a vitamin K-dependent reaction that converts 10 glutamic acids on prothrombin to gamma-carboxyglutamic acid (Gla; Huang et al., 2003). In the presence of Ca2+, the Gla residues promote the binding of prothrombin and other coagulation factors to exposed phospholipid bilayers, which accelerates but also restricts clotting procedure in a dose-dependent manner to injured sites (Huang et al., 2003). Deficiency of vitamin K or administration of the anticoagulant warfarin, an inhibitor of the vitamin k epoxide reductase, inhibits the production of Gla residues and slows the activation of coagulation (Huang et al., 2003). As part of its activity in the coagulation cascade, thrombin also promotes platelet-activation and aggregation (Figure 1; Borissoff et al., 2011).
Conversely, thrombin initiates a feedback mechanism which leads to its own inhibition. Once bound to thrombomodulin, an integral membrane protein expressed by endothelial cells, it increases its affinity to and activates Protein C. Activated Protein C (aPC) in turn leads to the inactivation of the prothrombinase complex (= FXa + FVa) at sites with an intact endothelium (Figure 1; Mosnier et al., 2014). In addition, freely circulating thrombin is blocked by antithrombin-III (Figure 1), a serine protease inhibitor, which is enhanced in its activity by the anticoagulant heparin (Jo et al., 2014). Taken together, blood thrombin activity is tightly regulated by a set of positive- and negative-feedback mechanisms, which promote clotting at injured sites and prevent coagulation at healthy sites. Considering its key role in the coagulation cascade it is not surprising that novel oral anticoagulants (NOACs) have been developed that act as direct (i.e., dabigatran) or indirect thrombin inhibitors (i.e., rivaroxaban and apixaban, via FXa inhibition) (Levy et al., 2014).
Beyond its role in the dynamic process of blood clot formation, thrombin has pronounced pro-inflammatory effects (Esmon, 2014). Acting via specific cell membrane receptors, the Protease-Activated Receptors (PARs), which are abundantly expressed in all arterial vessel wall constituents, thrombin has the potential to exert pro-atherogenic actions, such as leukocyte migration, cellular proliferation, regulation of vascular permeability and tone, platelet-activation, and edema formation (Coughlin, 2000, 2001; Sambrano et al., 2001; Chen and Dorling, 2009; Schuepbach et al., 2009; Spiel et al., 2011). PARs belong to a unique family of G protein-coupled receptors (Luo et al., 2007). Their activation is initiated by an irreversible, site-specific proteolytic cleavage in the N-terminal extracellular region, which uncovers a tethered ligand activating Gαq/11, Gαi/o, or Gα12/13 -proteins (Coughlin, 2000; Macfarlane et al., 2001; Traynelis and Trejo, 2007). Activation of PARs can recruit multiple intracellular signaling pathways depending on the activating ligand (Russo et al., 2009). This agonist-biased signal transduction and the resulting diversity of intracellular signaling pathways appear to be crucial for the multiple actions of PARs (Russo et al., 2009; Bourgognon et al., 2013).
Interestingly, PARs are also expressed in the brain and while PAR2 represents a class of trypsin/tryptase-activated receptors, PAR1, PAR3, and PAR4 are most effectively activated by thrombin (Gingrich and Traynelis, 2000). In the brain, PAR1 has been detected in both neurons and astrocytes, with the latter demonstrating stronger immunohistochemical signal in human brain tissue (Junge et al., 2004). High levels of PAR1 are detected in the hippocampus, cerebral cortex, and striatum of humans (Junge et al., 2004). While the precise molecular pathways activated by neural PAR1 are yet under investigation, in the brain PAR1-activation has been shown to modulate synaptic transmission and plasticity through the enhancement of N-methyl-D-aspartate receptor (NMDAR) currents (Gingrich et al., 2000; Lee et al., 2007; Maggio et al., 2008; Becker et al., 2014; Vance et al., 2015). In addition, PAR1-deficient animals have been reported to have alterations in hippocampus-dependent learning and memory processes (Almonte et al., 2007, 2013), indicating that PAR1 plays a critical role in memory formation and synaptic plasticity under physiological conditions.
A variety of neurological conditions have been associated with changes in the expression of PAR1 in the brain. In Parkinson’s disease, a significant increase in the number of astrocytes expressing PAR1 has been reported in the substantia nigra pars compacta (Ishida et al., 2006). In addition, upregulation of PAR1 in astrocytes has been observed in HIV encephalitis (Boven et al., 2003), implicating this receptor in neuroinflammatory responses. This idea is supported by the evidence of elevated levels of thrombin in an experimental model of multiple sclerosis (Beilin et al., 2005) and other inflammatory brain diseases (Chapman, 2006). Stimulation of PAR1 by thrombin causes proliferation of glia and produces reactive gliosis, infiltration of inflammatory cells, and angiogenesis (Striggow et al., 2001). Finally, expression of PAR1 is increased in experimental models of Alzheimer’s disease (Pompili et al., 2004) and brain ischemia (Striggow et al., 2001).
Both thrombin and its inactive precursor prothrombin have been also detected in the brain (Dihanich et al., 1991; Xi et al., 2003). Prothrombin mRNA shows the highest expression in the cerebral cortex and a moderate expression in the hippocampus and cerebellum (Figure 2A; Dihanich et al., 1991). In the hippocampus pronounced immunohistochemical labeling of thrombin is observed in pyramidal cell layers, while lower (clustered) thrombin signal is seen in the fiber-layers (Figure 2B). Although the precise cellular source of thrombin in the brain and the molecular mechanisms responsible for its formation and release warrant further investigation, experimental evidence has been provided that neural prothrombin expression and thrombin activity are highly regulated under physiological and pathological conditions (Xi et al., 2003; Stein et al., 2015). Hence, the molecular machinery of thrombin/PAR1 signaling is detected not only in the vascular system but also present in brain tissue, where it seems to act as a modulator of neural plasticity.
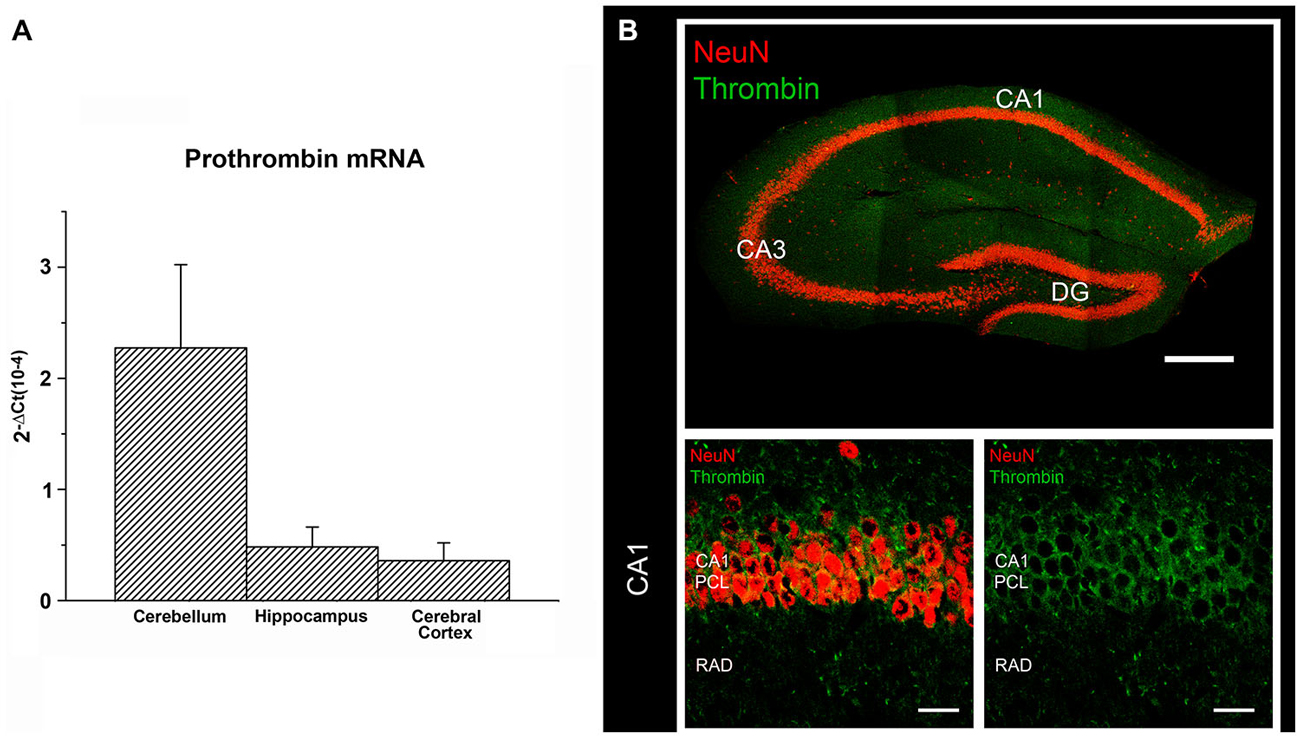
Figure 2. Prothrombin and Thrombin are expressed in the brain. (A) Prothrombin mRNA measured by (RT)qPCR in cerebellum, hippocampus and cerebral cortex of C57BL/6J mice (primers: 5′ CCGAAAGGGCAACCTAGAGC, 5′ GGCCCAGAACACGTCTGTG). The results were normalized to HPRT gene expression within the same cDNA sample and calculated using the ΔCT method with values being expressed as 2−ΔCt. (B) Immunostaining for thrombin (Santa Cruz Biotech, goat polyclonal IgG, cleaved thrombin HC (m361), sc-23335; dilution 1:100 in PBS-based solution) and NeuN (Millipore, mouse monoclonal, MAB377, dilution 1:1000 in PBS-based solution) in the dorsal hippocampus of adult C57BL/6J-mice. Thrombin labeling is predominantly detected in the pyramidal cell layer (PCL) of CA1. The fiber-layers (e.g., stratum radiatum, rad) display weaker but more clustered immunohistochemical signal for thrombin. Scale bar at low magnification = 300 μm; scale bar at higher magnification = 20 μm.
Concentration-Dependent Effects of Thrombin on Synaptic Plasticity
Long term potentiation (LTP) or depression (LTD) of synaptic strength, homeostatic plasticity and metaplasticity are considered to play important roles for the ability of the brain to effectively learn and adapt to novel challenges (Malenka, 2003; Turrigiano, 2012; Hulme et al., 2013). Consistent with the crucial role of synaptic plasticity in brain function (and similar to the blood coagulation cascade), plasticity is a highly regulated process composed of multiple feed-forward and feed-back mechanisms. This situation tunes neural networks in a way that promotes stability, but at the same time allows for rapid and site-specific responses; including changes in the threshold, direction and duration of synaptic events. Conceptually this stable dynamic state of synaptic plasticity is comparable to the blood coagulation cascade, which is kept in a delicate balance between bleeding and site-specific thrombosis. Therefore, it has been hypothesized that similar signaling pathways are employed in blood coagulation and synaptic plasticity.
Indeed, robust experimental evidence exists that thrombin and other coagulation factors are involved in the regulation of LTP in the hippocampus (Gingrich et al., 2000; Almonte et al., 2007; Maggio et al., 2008, 2013b,c; Mannaioni et al., 2008; Hamill et al., 2009). Furthermore, it has been shown that PAR1-signaling mediates thrombin-induced synaptic plasticity, which requires the activation of NMDARs (Lee et al., 2007; Han et al., 2011; Oh et al., 2012; Park et al., 2013).
In a recent attempt to shed more light on the physiological vs. pathological functions of thrombin and PAR1 in the brain, we studied the effects of different concentrations of thrombin and PAR1 activating peptide (PAR1-AP) on hippocampal LTP. This set of experiments disclosed that thrombin regulates the threshold for synaptic plasticity in a concentration-dependent manner (Figure 3; Maggio et al., 2013c). Specifically, [Thrombin]high induced an NMDAR-dependent slow onset LTP, which occluded the ability of neurons to express further LTP (Maggio et al., 2008, 2013c). Conversely, [Thrombin]low promoted L-type voltage gated calcium channel (L-VGCC), metabotropic glutamate receptor 5 (mGluR-5), and intracellular Ca2+ store-dependent LTP, which required aPC and the activation of the Endothelial Protein C Receptor (EPCR, detected on astrocytes and neurons) (Maggio et al., 2013c). In a follow-up study, we further clarified the role of aPC and EPCR in regulating [Thrombin]low-mediated synaptic plasticity (Maggio et al., 2014). We demonstrated that aPC induces metaplasticity and thereby enhances the ability to induce LTP (Maggio et al., 2014). This effect of aPC is mediated by EPCR-dependent PAR1-activation that triggers the production of Sphingosine-1-Phosphate (S1P). In turn, the activation of S1P-receptor 1 and intracellular Ca2+ stores, ultimately lead to a reduction in the LTP threshold (Maggio et al., 2014). Hence, high concentrations of thrombin saturate the ability of neurons to express further LTP via direct PAR1-activation (Maggio et al., 2008), while at low concentration thrombin mediates metaplasticity by enhancing LTP via aPC-EPCR mediated PAR1-signaling (Figure 3; Maggio et al., 2013c).
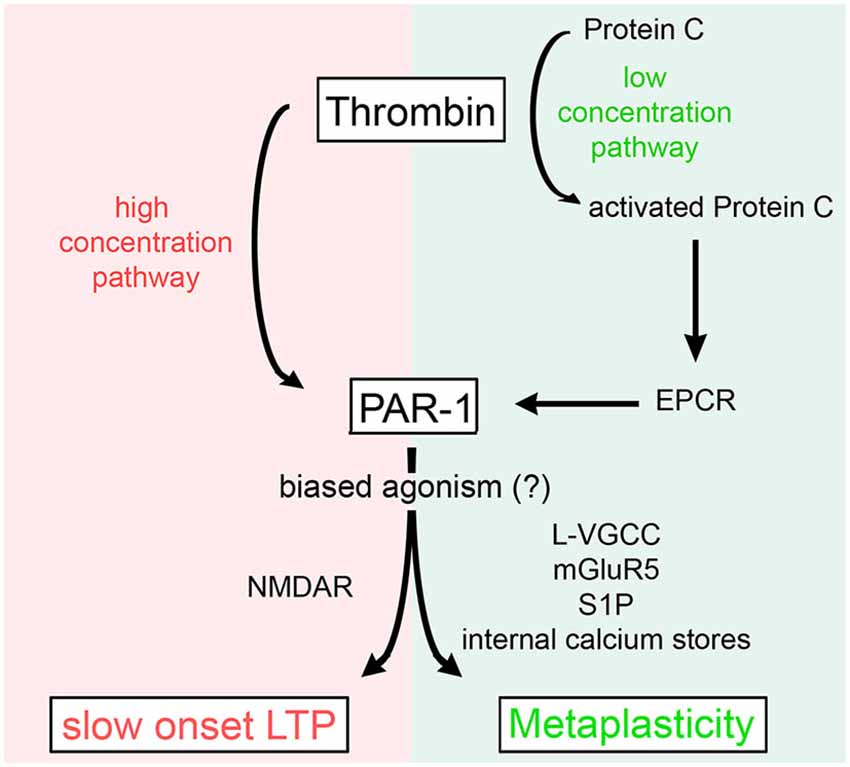
Figure 3. Concentration-dependent effects of thrombin on synaptic plasticity. High levels of thrombin cause a slow onset NMDAR-dependent LTP by direct activation of PAR1. Low concentrations of thrombin activate Protein C which binds to EPCR and activates PAR1 to induce metaplasticity, i.e., a reduction in the LTP threshold [by recruitment of L-type voltage gated calcium channels (L-VGCC), metabotropic glutamate receptors 5 (mGluR5), sphingosine-1-phosphate (S1P), and internal calcium stores]. For further details refer to the main text and Maggio et al. (2013c).
This finding is of considerable interest in clinical settings of cerebrovascular events: upon blood-brain barrier (BBB) opening, exposure to blood-derived thrombin and aPC could interfere with endogenous, i.e., neural thrombin/aPC mediated synaptic plasticity. On the one hand high thrombin concentrations may affect the ability of the brain to use LTP-like plastic processes for acquisition of new “memories”, due to the [Thrombin]high-mediated saturation of LTP. On the other hand aPC/PAR1 mediated synaptic plasticity (Maggio et al., 2013c, 2014), could facilitate LTP through other mechanisms and may counteract the [Thrombin]high effect (Maggio et al., 2008). The precise role and interplay of thrombin vs. aPC mediated synaptic plasticity under physiological and pathological conditions and the biological consequences for the course of diseases is currently under investigation.
PAR1-Activation Affects Homeostatic Synaptic Plasticity
To learn more about the role of thrombin/PAR1-signaling under pathological conditions and to study its role in other forms of synaptic plasticity, we used the entorhinal denervation in vitro model (Del Turco and Deller, 2007) and assessed the role of PAR1 in denervation-induced homeostatic synaptic plasticity (Vlachos et al., 2012). This model takes advantage of the highly organized entorhino-hippocampal projection, which originates in the entorhinal cortex, forms the perforant pathway, and terminates in the hippocampus. Lesioning this fiber tract results in the partial denervation of neurons in the hippocampus, without directly damaging the dendritic tree of target neurons (Müller et al., 2010). In turn, neurons respond to the loss of entorhinal input with a slow adaptive, i.e., compensatory increase in excitatory synaptic strength (Vlachos et al., 2012, 2013a,b), which is considered to play an important role in stabilizing the activity of neuronal networks under physiological and pathological conditions (Marder and Goaillard, 2006; Maffei and Fontanini, 2009; Turrigiano, 2012; Vitureira et al., 2012). While the presence of PAR1 was not required for homeostatic plasticity of excitatory synapses to occur, our study revealed that PAR1-activation occludes the ability of partially denervated neurons to increase their excitatory synaptic strength in a compensatory manner (Becker et al., 2014). Thus our results provided first experimental evidence that PAR1 activity affects neural homeostasis, which could be of relevance for neurological diseases associated with increased brain thrombin concentrations, neuronal cell loss and denervation of connected brain regions (see also Maggio and Vlachos, 2014).
The Role of Thrombin/PAR1-Signaling in Ischemic LTP
Finally, in a recent study we demonstrated that thrombin/PAR1-signaling has a fundamental role in oxygen glucose deprivation (OGD)-mediated alterations in synaptic plasticity (Stein et al., 2015). A brief (i.e., a few minutes) deprivation of oxygen and glucose, as seen for example in the early stage of acute ischemia or transient ischemic attack, potentiates synaptic transmission at hippocampal CA3-CA1 synapses in an NMDAR-dependent manner and induces ischemic LTP (iLTP) without causing neuronal cell death (Crépel et al., 1993; Hsu and Huang, 1997; Calabresi et al., 2003). It has been proposed that iLTP could have a major impact on the functional reorganization of neuronal networks during the early phase of ischemic stroke (Calabresi et al., 2003). In this context, we were able to demonstrate that OGD increases thrombin activity in neural tissue, i.e., in acute hippocampal slices, and triggers the induction of iLTP. Moreover, we showed that the induction of iLTP impairs the ability of neurons to express further LTP in a thrombin/PAR1-dependent fashion. Indeed, iLTP was blocked when thrombin activity or PAR1 signaling were inhibited and the ability of neurons to express LTP was rescued (Stein et al., 2015). We concluded from this series of experiments that iLTP resembles a [Thrombin]high-mediated LTP and hypothesized that counteracting thrombin signaling in the brain during the acute phase of stroke may improve synaptic plasticity (Stein et al., 2015).
Together, these studies provided robust experimental evidence that PAR1-signaling (which is activated, e.g., by thrombin or aPC) is involved in the regulation of distinct forms of synaptic plasticity under physiological and pathological conditions.
A Role for Thrombin/PAR1 in Epileptogenesis
Although serine proteases are expressed at very low concentration in neural tissue (Luo et al., 2007), their levels increase significantly in the brain following BBB breakdown. In this condition, a large, non-selective increase in the permeability of brain capillaries and tight junctions takes place, such that high molecular weight proteins (Ballabh et al., 2004) and other blood components gain access to the brain. This particular event occurs in several neurological conditions (Ballabh et al., 2004; Tomkins et al., 2008), including hemorrhagic/ischemic stroke (Hjort et al., 2008; Bang et al., 2009) and traumatic brain injury (Barzó et al., 1997; Tomkins et al., 2008; Itzekson et al., 2014). Even though there is a paucity of information concerning the amount of thrombin crossing the BBB, it has been reported that thrombin levels increase more than 200 fold (from 100 pM to 25 nM) in the cerebrospinal fluid of patients with subarachnoid hemorrhage (Suzuki et al., 1992). In addition, if bleeding occurs directly within the brain tissue, active thrombin and other proteases freely diffuse into the brain parenchyma until blood coagulation closes off the injured vessels (Suzuki et al., 1992). A direct consequence of the contact of thrombin with the brain tissue is the onset of seizures (Lee et al., 1997). In 1997, Lee et al. reported that intracerebral injections of thrombin results in focal motor seizures. Interestingly, the injection of thrombin together with its inhibitor alpha-(2-naphthylsulfonyl-glycyl)-4-amidinophenylalaninepiperidide (alpha-NAPAP) prevented both clinical and electrographic seizures (Lee et al., 1997). Similarly, mice engineered to lack Nexin-1, an endogenous thrombin inhibitor, have an increased susceptibility for kainic acid-induced seizures (Luthi et al., 1997).
Following this line of evidence we were able to demonstrate that thrombin-induced seizures are mediated by activation of PAR1 (Maggio et al., 2008). In hippocampal slices, thrombin at a concentration of 5 nM (1 U/ml) increased spontaneous firing of CA3 pyramidal cells (Maggio et al., 2008). In order to examine whether thrombin facilitates the onset of epileptic discharges in conditions mimicking BBB breakdown in the slice (Chen and Swanson, 2003; Beart and O’Shea, 2007), we exposed neurons to thrombin in presence of elevated [K+]o or glutamate. In normal slices, addition of 4 mM [K+]o did not produce any noticeable spontaneous seizures, which were clearly seen when [K+]o were increased to 15 mM. Similarly, 500 μM but not 100 μM glutamate produced spontaneous seizure-like activity in the slice. Remarkably, thrombin facilitated the response to the lower concentration of [K+]o (4 mM) and glutamate (100 μM) to produce seizure-like activity. This facilitatory action of thrombin on the production of seizure-like activity was mediated by PAR1, since it was mimicked by PAR1-AP and blocked by the PAR1-inhibitor SCH79797. However, it did not depend on NMDARs as it was not affected by the selective NMDAR-inhibitors ifenprodil or by 2R-amino-5-phosphonovalericacid (APV), indicating that [Thrombin]high-mediated LTP does not explain the acute effects of thrombin on seizure-induction (Maggio et al., 2008). While thrombin induced epileptogenesis seems to be NMDAR-independent, it is possible that thrombin could also indirectly modulate synaptic transmission, e.g., through thrombin-mediated proteolysis and remodeling of the extracellular matrix (e.g., Dityatev, 2010).
More recently, Isaeva et al. (2012) exposed hippocampal slices prepared from young postnatal rats (P6 to P15) to large concentrations of thrombin (10 U/ml), to find that thrombin depolarizes membrane potential of neurons and produces a hyperpolarizing shift of tetrodotoxin-sensitive INap through a PAR1-mediated mechanism (Isaeva et al., 2012). In addition, we have reported that thrombin affects synaptic transmission in hippocampal CA3 neurons by enhancing both frequency and amplitude of mEPSCs, while reducing frequency and amplitude of mIPSCs (Maggio et al., 2013b). Together, these studies showed a pro-epileptic effect of thrombin, which induces membrane and synaptic changes leading to seizures via PAR1-activation. However, whether the thrombin-induced increase in neuronal activity leads to the development of epilepsy at a later stage remains unclear. Nevertheless, it has been demonstrated that high concentrations of thrombin induce apoptosis (Xi et al., 2003; Luo et al., 2007), which may lead to maladaptive circuit reorganization and to the development of epilepsy. Additional work is required to test this intriguing possibility and to assess whether thrombin could play a crucial role in epileptogenesis.
Open Questions and Conclusions
While robust experimental evidence exists that thrombin affects brain functioning through the concentration-dependent regulation of distinct forms of synaptic plasticity, it remains unclear how thrombin exerts such diverse effects by acting on the same receptor, i.e., via PAR1. Conceptually, several possibilities, or a combination of different mechanisms, need to be considered. In endothelial cells for example the recruitment of distinct PAR1-mediated downstream signaling pathways has been reported: while thrombin mediated PAR1-activation recruits Gq/G12/13, aPC seems to act via PAR1-mediated Gi (Riewald and Ruf, 2005). This observation is consistent with a study showing that PAR1 interacts with various G proteins (Russo et al., 2009; McCoy et al., 2012). Also, precise location of PAR1 in neuronal or astrocytic membrane compartments and the current state of neuronal networks seem to influence selectivity of PAR1 to distinct ligands and the recruitment of diverse intracellular signaling pathways by PAR1 (Russo et al., 2009; Bourgognon et al., 2013). Such agonist-biased signal transduction may explain the involvement of PAR1 in mediating and influencing various forms of plasticity, depending on the actual thrombin concentration (Figure 3). However, whether distinct subtypes of PAR1 exist, which recruit specific downstream signals, and even show different affinities for thrombin and aPC needs to be tested. The role of simultaneous or selective activation of PAR1 in distinct cell types and/or cellular compartments remains unclear in this context as well. While some evidence for astrocytic PAR1 signaling has been provided (e.g., Vance et al., 2015), the contribution of PAR1 signaling in pre- vs. postsynaptic compartments, in distinct types of interneurons, microglia cells, oligodendrocytes or endothelial cells warrants further investigation. Thrombin may also exert its function through other receptors and proteolysis of specific components in the extracellular matrix (e.g., fibrinogen) under physiological and pathological conditions (e.g., epileptogenesis, see Dityatev, 2010). Indeed, evidence exists that fibrinogen plays an important role in microglia recruitment (Davalos et al., 2012). Hence, it is interesting to speculate that thrombin could modulate neuroinflammatory responses by acting on PAR1 and fibrinogen (Tripathy et al., 2013). Whether thrombin-induced, fibrin-mediated pathways also act on synaptic transmission and plasticity, and the role of PAR1 in this context has not been addressed so far.
Considering the suggested role of thrombin as a major factor for seizures, the cellular source of thrombin in the brain and the molecular machinery responsible for its formation and release needs to be further clarified. Nevertheless, it is interesting to speculate that new oral anticoagulants, which target thrombin could have an unexpected antiepileptic effect (Maggio et al., 2013a), and may restore the ability of neurons to express plasticity under conditions of increased brain thrombin concentrations and PAR1-activity, as seen for example upon BBB breakdown. Its central role in neural plasticity classifies PAR1 as an interesting therapeutic target in thrombin associated pathologies. We are confident that future studies will provide new important insights on the role of thrombin/aPC/PAR1-mediated neural plasticity and excitability and will shed more light on concentration-, ligand-, source-, target-, and state-dependent recruitment of PAR1 signaling cascades. These information may contribute to the development of new strategies to restore brain function under pathological conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Almonte, A. G., Hamill, C. E., Chhatwal, J. P., Wingo, T. S., Barber, J. A., Lyuboslavsky, P. N., et al. (2007). Learning and memory deficits in mice lacking protease activated receptor-1. Neurobiol. Learn. Mem. 88, 295–304. doi: 10.1016/j.nlm.2007.04.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Almonte, A. G., Qadri, L. H., Sultan, F. A., Watson, J. A., Mount, D. J., Rumbaugh, G., et al. (2013). Protease-activated receptor-1 modulates hippocampal memory formation and synaptic plasticity. J. Neurochem. 124, 109–122. doi: 10.1111/jnc.12075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ballabh, P., Braun, A., and Nedergaard, M. (2004). The blood-brain barrier: an overview: structure, regulation and clinical implications. Neurobiol. Dis. 16, 1–13. doi: 10.1016/j.nbd.2003.12.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bang, O. Y., Saver, J. L., Alger, J. R., Shah, S. H., Buck, B. H., Starkman, S., et al. (2009). Patterns and predictors of blood-brain barrier permeability derangements in acute ischemic stroke. Stroke 40, 454–461. doi: 10.1161/STROKEAHA.108.522847
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barzó, P., Marmarou, A., Fatouros, P., Corwin, F., and Dunbar, J. G. (1997). Acute blood-brain barrier changes in experimental closed head injury as measured by MRI and Gd-DTPA. Acta Neurochir. Suppl. 70, 243–246. doi: 10.1007/978-3-7091-6837-0_75
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beart, P. M., and O’Shea, R. D. (2007). Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br. J. Pharmacol. 150, 5–17. doi: 10.1038/sj.bjp.0706949
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Becker, D., Ikenberg, B., Schiener, S., Maggio, N., and Vlachos, A. (2014). NMDA-receptor inhibition restores Protease-Activated Receptor 1 (PAR1) mediated alterations in homeostatic synaptic plasticity of denervated mouse dentate granule cells. Neuropharmacology 86, 212–218. doi: 10.1016/j.neuropharm.2014.07.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beilin, O., Karussis, D. M., Korczyn, A. D., Gurwitz, D., Aronovich, R., Hantai, D., et al. (2005). Increased thrombin inhibition in experimental autoimmune encephalomyelitis. J. Neurosci. Res. 79, 351–359. doi: 10.1002/jnr.20270
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Borissoff, J. I., Spronk, H. M., and ten Cate, H. (2011). The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 364, 1746–1760. doi: 10.1056/NEJMra1011670
Bourgognon, J. M., Schiavon, E., Salah-Uddin, H., Skrzypiec, A. E., Attwood, B. K., Shah, R. S., et al. (2013). Regulation of neuronal plasticity and fear by a dynamic change in PAR1-G protein coupling in the amygdala. Mol. Psychiatry 18, 1136–1145. doi: 10.1038/mp.2012.133
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boven, L. A., Vergnolle, N., Henry, S. D., Silva, C., Imai, Y., Holden, J., et al. (2003). Up-regulation of proteinase-activated receptor 1 expression in astrocytes during HIV encephalitis. J. Immunol. 170, 2638–2646. doi: 10.4049/jimmunol.170.5.2638
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Calabresi, P., Centonze, D., Pisani, A., Cupini, L., and Bernardi, G. (2003). Synaptic plasticity in the ischaemic brain. Lancet Neurol. 2, 622–629. doi: 10.1016/s1474-4422(03)00532-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chapman, J. (2006). Thrombin in inflammatory brain diseases. Autoimmun. Rev. 5, 528–531. doi: 10.1016/j.autrev.2006.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, D., and Dorling, A. (2009). Critical roles for thrombin in acute and chronic inflammation. J. Thromb. Haemost. 7(Suppl. 1), 122–126. doi: 10.1111/j.1538-7836.2009.03413.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Y., and Swanson, R. A. (2003). Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 23, 137–149. doi: 10.1097/00004647-200302000-00001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coughlin, S. R. (2000). Thrombin signalling and protease-activated receptors. Nature 407, 258–264. doi: 10.1038/35025229
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coughlin, S. R. (2001). Protease-activated receptors in vascular biology. Thromb. Haemost. 86, 298–307.
Crépel, V., Hammond, C., Chinestra, P., Diabira, D., and Ben-Ari, Y. (1993). A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J. Neurophysiol. 70, 2045–2055.
Davalos, D., Ryu, J. K., Merlini, M., Baeten, K. M., Le Moan, N., and Petersen, M. A. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3:1227. doi: 10.1038/ncomms2230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Del Turco, D., and Deller, T. (2007). Organotypic entorhino-hippocampal slice cultures–a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol. Biol. 399, 55–66. doi: 10.1007/978-1-59745-504-6_5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dihanich, M., Kaser, M., Reinhard, E., Cunningham, D., and Monard, D. (1991). Prothrombin mRNA is expressed by cells of the nervous system. Neuron 6, 575–581. doi: 10.1016/0896-6273(91)90060-d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dityatev, A. (2010). Remodelling of extracellular matrix and epileptogenesis. Epilepsia 51(Suppl. 3), 61–65. doi: 10.1111/j.1528-1167.2010.02612.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Esmon, C. T. (2014). Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb. Haemost. 111, 625–633. doi: 10.1160/TH13-09-0730
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Furie, B., and Furie, B. C. (2005). Thrombus formation in vivo. J. Clin. Invest. 115, 3355–3362. doi: 10.1172/jci26987
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gingrich, M. B., Junge, C. E., Lyuboslavsky, P., and Traynelis, S. F. (2000). Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 20, 4582–4595.
Gingrich, M. B., and Traynelis, S. F. (2000). Serine proteases and brain damage–is there a link? Trends Neurosci. 23, 399–407. doi: 10.1016/s0166-2236(00)01617-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamill, C. E., Mannaioni, G., Lyuboslavsky, P., Sastre, A. A., and Traynelis, S. F. (2009). Protease-activated receptor 1-dependent neuronal damage involves NMDA receptor function. Exp. Neurol. 217, 136–146. doi: 10.1016/j.expneurol.2009.01.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, K. S., Mannaioni, G., Hamill, C. E., Lee, J., Junge, C. E., Lee, C. J., et al. (2011). Activation of protease activated receptor 1 increases the excitability of the dentate granule neurons of hippocampus. Mol. Brain 4:32. doi: 10.1186/1756-6606-4-32
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hjort, N., Wu, O., Ashkanian, M., Sølling, C., Mouridsen, K., Christensen, S., et al. (2008). MRI detection of early blood-brain barrier disruption: parenchymal enhancement predicts focal hemorrhagic transformation after thrombolysis. Stroke 39, 1025–1028. doi: 10.1161/STROKEAHA.107.497719
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hsu, K. S., and Huang, C. C. (1997). Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br. J. Pharmacol. 122, 671–681. doi: 10.1038/sj.bjp.0701409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, M., Rigby, A. C., Morelli, X., Grant, M. A., Huang, G., Furie, B., et al. (2003). Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 10, 751–756. doi: 10.1038/nsb971
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hulme, S. R., Jones, O. D., and Abraham, W. C. (2013). Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci. 36, 353–362. doi: 10.1016/j.tins.2013.03.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Isaeva, E., Hernan, A., Isaev, D., and Holmes, G. L. (2012). Thrombin facilitates seizures through activation of persistent sodium current. Ann. Neurol. 72, 192–198. doi: 10.1002/ana.23587
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishida, Y., Nagai, A., Kobayashi, S., and Kim, S. U. (2006). Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J. Neuropathol. Exp. Neurol. 65, 66–77. doi: 10.1097/01.jnen.0000195941.48033.eb
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Itzekson, Z., Maggio, N., Milman, A., Shavit, E., Pick, C. G., and Chapman, J. (2014). Reversal of trauma-induced amnesia in mice by a thrombin receptor antagonist. J. Mol. Neurosci. 53, 87–95. doi: 10.1007/s12031-013-0200-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jo, J. T., Schiff, D., and Perry, J. R. (2014). Thrombosis in brain tumors. Semin. Thromb. Hemost. 40, 325–331. doi: 10.1055/s-0034-1370791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Junge, C. E., Lee, C. J., Hubbard, K. B., Zhang, Z., Olson, J. J., Hepler, J. R., et al. (2004). Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp. Neurol. 188, 94–103. doi: 10.1016/j.expneurol.2004.02.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kalz, J., ten Cate, H., and Spronk, H. M. (2014). Thrombin generation and atherosclerosis. J. Thromb. Thrombolysis 37, 45–55. doi: 10.1007/s11239-013-1026-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, K. R., Drury, I., Vitarbo, E., and Hoff, J. T. (1997). Seizures induced by intracerebral injection of thrombin: a model of intracerebral hemorrhage. J. Neurosurg. 87, 73–78. doi: 10.3171/jns.1997.87.1.0073
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, C. J., Mannaioni, G., Yuan, H., Woo, D. H., Gingrich, M. B., and Traynelis, S. F. (2007). Astrocytic control of synaptic NMDA receptors. J. Physiol. 581, 1057–1081. doi: 10.1113/jphysiol.2007.130377
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levy, J. H., Spyropoulos, A. C., Samama, C. M., and Douketis, J. (2014). Direct oral anticoagulants: new drugs and new concepts. JACC Cardiovasc. Interv. 7, 1333–1351. doi: 10.1016/j.jcin.2014.06.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lippi, G., Favaloro, E. J., and Franchini, M. (2012). Paradoxical thrombosis, part 2, anticoagulant and antiplatelet therapy. J. Thromb. Thrombolysis 34, 367–373. doi: 10.1007/s11239-012-0748-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, W., Wang, Y., and Reiser, G. (2007). Protease-activated receptors in the brain: receptor expression, activation and functions in neurodegeneration and neuroprotection. Brain Res. Rev. 56, 331–345. doi: 10.1016/j.brainresrev.2007.08.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luthi, A., Van der Putten, H., Botteri, F. M., Mansuy, I. M., Meins, M., Frey, U., et al. (1997). Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J. Neurosci. 17, 4688–4699.
Macfarlane, S. R., Seatter, M. J., Kanke, T., Hunter, G. D., and Plevin, R. (2001). Proteinase-activated receptors. Pharmacol. Rev. 53, 245–282.
Maffei, A., and Fontanini, A. (2009). Network homeostasis: a matter of coordination. Curr. Opin. Neurobiol. 19, 168–173. doi: 10.1016/j.conb.2009.05.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., Blatt, I., Vlachos, A., Tanne, D., Chapman, J., and Segal, M. (2013a). Treating seizures and epilepsy with anticoagulants? Front. Cell. Neurosci. 7:19. doi: 10.3389/fncel.2013.00019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., Cavaliere, C., Papa, M., Blatt, I., Chapman, J., and Segal, M. (2013b). Thrombin regulation of synaptic transmission: implications for seizure onset. Neurobiol. Dis. 50, 171–178. doi: 10.1016/j.nbd.2012.10.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., Itsekson, Z., Dominissini, D., Blatt, I., Amariglio, N., Rechavi, G., et al. (2013c). Thrombin regulation of synaptic plasticity: implications for physiology and pathology. Exp. Neurol. 247, 595–604. doi: 10.1016/j.expneurol.2013.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., Itsekson, Z., Ikenberg, B., Strehl, A., Vlachos, A., Blatt, I., et al. (2014). The anticoagulant activated protein, C. (aPC) promotes metaplasticity in the hippocampus through an EPCR-PAR1–S1P1 receptors dependent mechanism. Hippocampus 24, 1030–1038. doi: 10.1002/hipo.22288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., Shavit, E., Chapman, J., and Segal, M. (2008). Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. J. Neurosci. 28, 732–736. doi: 10.1523/JNEUROSCI.3665-07.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maggio, N., and Vlachos, A. (2014). Synaptic plasticity at the interface of health and disease: new insights on the role of endoplasmic reticulum intracellular calcium stores. Neuroscience 281C, 135–146. doi: 10.1016/j.neuroscience.2014.09.041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Malenka, R. C. (2003). The long-term potential of LTP. Nat. Rev. Neurosci. 4, 923–926. doi: 10.1038/nrn1258
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mannaioni, G., Orr, A. G., Hamill, C. E., Yuan, H., Pedone, K. H., McCoy, K. L., et al. (2008). Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J. Biol. Chem. 283, 20600–20611. doi: 10.1074/jbc.M803015200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marder, E., and Goaillard, J. M. (2006). Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 7, 563–574. doi: 10.1038/nrn1949
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McCoy, K. L., Gyoneva, S., Vellano, C. P., Smrcka, A. V., Traynelis, S. F., and Hepler, J. R. (2012). Protease-activated receptor 1 (PAR1) coupling to G(q/11) but not to G(i/o) or G(12/13) is mediated by discrete amino acids within the receptor second intracellular loop. Cell. Signal. 24, 1351–1360. doi: 10.1016/j.cellsig.2012.01.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mosnier, L. O., Zlokovic, B. V., and Griffin, J. H. (2014). Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb. Haemost. 112, 883–892. doi: 10.1160/TH14-05-0448
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Müller, C. M., Vlachos, A., and Deller, T. (2010). Calcium homeostasis of acutely denervated and lesioned dentate gyrus in organotypic entorhino-hippocampal co-cultures. Cell Calcium 47, 242–252. doi: 10.1016/j.ceca.2009.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oh, S. J., Han, K. S., Park, H., Woo, D. H., Kim, H. Y., Traynelis, S. F., et al. (2012). Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by Bestrophin-1 channel but not by vesicular exocytosis. Mol. Brain 5:38. doi: 10.1186/1756-6606-5-38
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, H., Han, K. S., Oh, S. J., Jo, S., Woo, J., Yoon, B. E., et al. (2013). High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol. Brain 6:54. doi: 10.1186/1756-6606-6-54
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pompili, E., Nori, S. L., Geloso, M. C., Guadagni, E., Corvino, V., Michetti, F., et al. (2004). Trimethyltin-induced differential expression of PAR subtypes in reactive astrocytes of the rat hippocampus. Brain Res. Mol. Brain Res. 122, 93–98. doi: 10.1016/j.molbrainres.2003.12.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Riewald, M., and Ruf, W. (2005). Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J. Biol. Chem. 280, 19808–19814. doi: 10.1074/jbc.m500747200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Russo, A., Soh, U. J., and Trejo, J. (2009). Proteases display biased agonism at protease-activated receptors: location matters! Mol. Interv. 9, 87–96. doi: 10.1124/mi.9.2.8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sambrano, G. R., Weiss, E. J., Zheng, Y. W., Huang, W., and Coughlin, S. R. (2001). Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature 413, 74–78. doi: 10.1038/35092573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schuepbach, R. A., Feistritzer, C., Fernández, J. A., Griffin, J. H., and Riewald, M. (2009). Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thromb. Haemost. 101, 724–733. doi: 10.1160/th08-10-0632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Siller-Matula, J. M., Schwameis, M., Blann, A., Mannhalter, C., and Jilma, B. (2011). Thrombin as a multi-functional enzyme. Focus on in vitro and in vivo effects. Thromb. Haemost. 106, 1020–1033. doi: 10.1160/TH10-11-0711
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spiel, A. O., Bartko, J., Schwameis, M., Firbas, C., Siller-Matula, J., Schuetz, M., et al. (2011). Increased platelet aggregation and in vivo platelet activation after granulocyte colony-stimulating factor administration. A randomised controlled trial. Thromb. Haemost. 105, 655–662. doi: 10.1160/TH10-08-0530
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stein, E. S., Itsekson-Hayosh, Z., Aronovich, A., Reisner, Y., Bushi, D., Pick, C. G., et al. (2015). Thrombin induces ischemic LTP (iLTP): implications for synaptic plasticity in the acute phase of ischemic stroke. Sci. Rep. 5:7912. doi: 10.1038/srep07912
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Striggow, F., Riek-Burchardt, M., Kiesel, A., Schmidt, W., Henrich-Noack, P., Breder, J., et al. (2001). Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur. J. Neurosci. 14, 595–608. doi: 10.1046/j.0953-816x.2001.01676.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suzuki, M., Ogawa, A., Sakurai, Y., Nishino, A., Venohara, K., Mizoi, K., et al. (1992). Thrombin activity in cerebrospinal fluid after subarachnoid hemorrhage. Stroke 23, 1181–1182. doi: 10.1161/01.str.23.8.1181
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomkins, O., Shelef, I., Kaizerman, I., Eliushin, A., Afawi, Z., Misk, A., et al. (2008). Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 79, 774–777. doi: 10.1136/jnnp.2007.126425
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Traynelis, S. F., and Trejo, J. (2007). Protease-activated receptor signaling: new roles and regulatory mechanisms. Curr. Opin. Hematol. 14, 230–235. doi: 10.1097/moh.0b013e3280dce568
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tripathy, D., Sanchez, A., Yin, X., Luo, J., Martinez, J., and Grammas, P. (2013). Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 5:19. doi: 10.3389/fnagi.2013.00019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turrigiano, G. (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736. doi: 10.1101/cshperspect.a005736
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vance, K. M., Rogers, R. C., and Hermann, G. E. (2015). PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J. Neurosci. 35, 776–785. doi: 10.1523/JNEUROSCI.3105-14.2015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vitureira, N., Letellier, M., and Goda, Y. (2012). Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr. Opin. Neurobiol. 22, 516–521. doi: 10.1016/j.conb.2011.09.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vlachos, A., Becker, D., Jedlicka, P., Winkels, R., Roeper, J., and Deller, T. (2012). Entorhinal denervation induces homeostatic synaptic scaling of excitatory postsynapses of dentate granule cells in mouse organotypic slice cultures. PLoS One 7:e32883. doi: 10.1371/journal.pone.0032883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vlachos, A., Helias, M., Becker, D., Diesmann, M., and Deller, T. (2013a). NMDA-receptor inhibition increases spine stability of denervated mouse dentate granule cells and accelerates spine density recovery following entorhinal denervation in vitro. Neurobiol. Dis. 59, 267–276. doi: 10.1016/j.nbd.2013.07.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vlachos, A., Ikenberg, B., Lenz, M., Becker, D., Reifenberg, K., Bas-Orth, C., et al. (2013b). Synaptopodin regulates denervation-induced homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. U S A 110, 8242–8247. doi: 10.1073/pnas.1213677110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xi, G., Reiser, G., and Keep, R. F. (2003). The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J. Neurochem. 84, 3–9. doi: 10.1046/j.1471-4159.2003.01268.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: thrombin, proteases activated receptor 1 (PAR1), clotting factors, long term potentiation, synaptic plasticity, hippocampus
Citation: Ben Shimon M, Lenz M, Ikenberg B, Becker D, Shavit Stein E, Chapman J, Tanne D, Pick CG, Blatt I, Neufeld M, Vlachos A and Maggio N (2015) Thrombin regulation of synaptic transmission and plasticity: implications for health and disease. Front. Cell. Neurosci. 9:151. doi: 10.3389/fncel.2015.00151
Received: 26 January 2015; Accepted: 01 April 2015;
Published online: 21 April 2015.
Edited by:
Leszek Kaczmarek, Nencki Institute, PolandReviewed by:
Sadao Shiosaka, Nara Institute of Science and Technology, JapanRobert Pawlak, University of Exeter, UK
Copyright © 2015 Ben Shimon, Lenz, Ikenberg, Becker, Shavit Stein, Chapman, Tanne, Pick, Blatt, Neufeld, Vlachos and Maggio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Maggio, Department of Neurology, The J. Sagol Neuroscience Center, The Chaim Sheba Medical Center, 52621 Tel HaShomer, Israel nicola.maggio@sheba.health.gov.il
#Present address: Denise Becker, Department of Fundamental Neurosciences, University of Lausanne, Rue de Bugnon 9, 1005 Lausanne, Switzerland
† Joint first authors.
‡ These authors have contributed equally to this work.