Molecular pathophysiology and pharmacology of the voltage-sensing module of neuronal ion channels
 Francesco Miceli1
Francesco Miceli1  Maria Virginia Soldovieri2
Maria Virginia Soldovieri2  Paolo Ambrosino2
Paolo Ambrosino2  Michela De Maria2
Michela De Maria2  Laura Manocchio2
Laura Manocchio2  Alessandro Medoro2
Alessandro Medoro2  Maurizio Taglialatela1,2*
Maurizio Taglialatela1,2*- 1Department of Neuroscience, University of Naples Federico II, Naples, Italy
- 2Department of Medicine and Health Sciences, University of Molise, Campobasso, Italy
Voltage-gated ion channels (VGICs) are membrane proteins that switch from a closed to open state in response to changes in membrane potential, thus enabling ion fluxes across the cell membranes. The mechanism that regulate the structural rearrangements occurring in VGICs in response to changes in membrane potential still remains one of the most challenging topic of modern biophysics. Na+, Ca2+ and K+ voltage-gated channels are structurally formed by the assembly of four similar domains, each comprising six transmembrane segments. Each domain can be divided into two main regions: the Pore Module (PM) and the Voltage-Sensing Module (VSM). The PM (helices S5 and S6 and intervening linker) is responsible for gate opening and ion selectivity; by contrast, the VSM, comprising the first four transmembrane helices (S1–S4), undergoes the first conformational changes in response to membrane voltage variations. In particular, the S4 segment of each domain, which contains several positively charged residues interspersed with hydrophobic amino acids, is located within the membrane electric field and plays an essential role in voltage sensing. In neurons, specific gating properties of each channel subtype underlie a variety of biological events, ranging from the generation and propagation of electrical impulses, to the secretion of neurotransmitters and to the regulation of gene expression. Given the important functional role played by the VSM in neuronal VGICs, it is not surprising that various VSM mutations affecting the gating process of these channels are responsible for human diseases, and that compounds acting on the VSM have emerged as important investigational tools with great therapeutic potential. In the present review we will briefly describe the most recent discoveries concerning how the VSM exerts its function, how genetically inherited diseases caused by mutations occurring in the VSM affects gating in VGICs, and how several classes of drugs and toxins selectively target the VSM.
Introduction
Voltage-dependent changes in ion fluxes are critical for the generation and propagation of electric signals in and between excitable cells. Despite extensive studies performed during the last 50 years, the mechanisms that regulate the voltage sensitivity of Voltage Gated Ion Channels (VGICs) still remain one of the most challenging topic of modern biophysics. As demonstrated by Hodgkin and Huxley (1952), voltage sensitivity is regulated by reorientation of charges (or dipoles) in response to changes in the membrane electric field. The movement of these charges has been experimentally demonstrated upon observation of voltage-activated non linear capacity currents, called gating or sensing currents (Armstrong and Bezanilla, 1973; Bezanilla et al., 1982).
Although these studies first established the biophysical basis of voltage-sensing in VGICs, a major breakthrough allowing to translate into molecular clues such theoretical background was the cloning and sequencing of the first VGIC, namely the voltage-gated Na+ channel (VGNC) from the electroplax of Electrophorus electricus (Noda et al., 1984), followed by the cloning of the first voltage-gated Ca2+ channel (VGCC) from rabbit skeletal muscle (Tanabe et al., 1987), and of a voltage-gated K+ channel (VGKC) from Drosophila (Papazian et al., 1987).
Subsequent studies using different techniques, including mutagenesis, fluorescence spectroscopy, and electrophysiology, have assigned specific functional roles to individual regions of VGICs; during this structure-function era, the “modular” nature of this class of membrane proteins was therefore established. More recently, characterization of the crystal structure of KvAP, Kv1.2 and chimeric Kv1.2/2.1 K+ channels (Jiang et al., 2003; Long et al., 2005a,b, 2007) followed by that of the NavAb VGNC from Arcobacter butzleri (Payandeh et al., 2011) paved the way to a detailed understanding of the intimate molecular architecture of VGIC.
In parallel, the extraordinary advancement of sequencing technologies of the last decade, have allowed the identification of numerous mutations responsible for human diseases in VGICs genes, often revealing previously unexplored functional roles of specific ion channel classes, thereby greatly expanding our understanding of the pathophysiological mechanisms underlying human diseases.
Overall Structure of Voltage-Gated Ion Channels
The VGIC family represents one of the largest group of signal transduction proteins, also acting as fundamental targets for drugs with wide therapeutic applications. Primary sequence analysis indicate VGNCs and VGCCs of eukaryotes are formed by a single peptide (the α subunit) containing four homologous domains (called from I to IV). The membrane core of each of these domains contains six transmembrane helices (from S1 to S6) with an amphipathic loop between the S5 and S6 segments. By contrast, VGKCs are formed upon assembly of four compatible subunits, each showing a high sequence homology with a single domain of VGNCs or VGCCs (Figure 1). Similarly, bacterial VGNCs are composed of homotetramers of single domains whose structure resembles that of each domain of vertebrate VGNCs (Ren et al., 2001; Koishi et al., 2004), likely being the evolutionary ancestors of the larger, four-domain Na+ channels of eukaryotes.
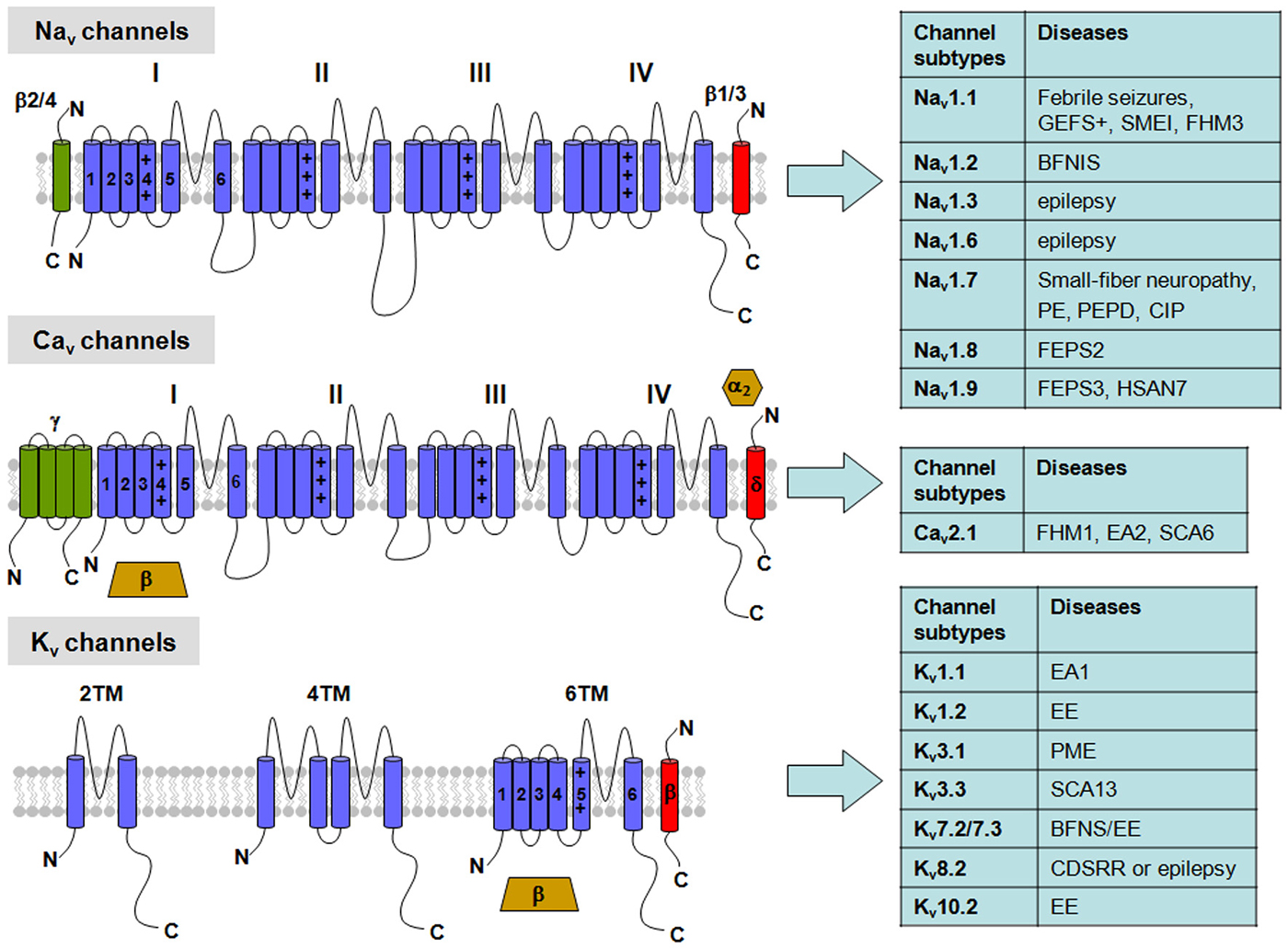
Figure 1. Topological representation of voltage-gated K+ (Kv), Na+ (Nav) and Ca2+ (Cav) channels with related neuronal diseases. Nav channels are formed by a single polypeptide that contains four domains (I-IV), each with six transmembrane segments (S1–S6). β-subunits are single transmembrane proteins that co-assembles with the Nav α-subunit. Cav channels show a similar topology to Nav channels in their α-subunits, but can be associated with four different auxiliary subunits: the α2/δ-complex, linked by disulfide bridges, an intracellular β-subunit, and an occasional γ-subunit with four transmembrane segments. Abbreviations: GEFS+, Generalized Epilepsy with Febrile Seizures plus; SMEI, Severe Myoclonic Epilepsy of Infancy; FHM1-3, Familial Hemiplegic Migraine type 1-3, respectively; BFNS, Benign Familial Neonatal Seizures; BFNIS, Benign Familial Neonatal-Infantile Seizures; EE, Epileptic Encephalopathy; PE, Primary Erythermalgia; PEPD, Paroxysmal Extreme Pain Disorder; CIP, Congenital Insensitivity to Pain; FEPS2-3, Familial Episodic Pain Syndrome type 2-3, respectively; HSAN7, Hereditary Sensory and Autonomic Neuropathy type 7; EA1-2, Episodic Ataxia type 1-2, respectively; SCA6-13, Spinocerebellar Ataxia type 6-13, respectively; PME, Progressive Myoclonus Epilepsy; CDSRR, Cone Dystrophy with Supernormal Rod Electroretinogram.
In all VGICs, each structural domain can be divided into two main regions: the Pore Module (PM) and the Voltage-Sensing Module (VSM). The PM, which allows selective permeability of ion species across the cell membrane, is formed by the fifth and sixth transmembrane segments (S5 and S6, respectively) and the interconnecting loop of all subunits. The VSM of each domain senses the membrane potential variations, switching from a resting to an activated configuration, a necessary prerequisite for subsequent pore opening; the S1–S4 transmembrane segments play a crucial role in this process. Both in VGKCs (Long et al., 2007) as well as in bacterial VGNCs (Payandeh et al., 2011), the VSM of each domain interacts with the PM of a neighboring domain, thus allowing movements of the VSM module to be directly translated into conformational changes of the PM. In detail, while each of the subunits undergo voltage-dependent transitions independently (Zagotta et al., 1994), the final voltage-independent conformational change leading to pore opening appears to occur in a cooperative, all-or-none fashion (Zandany et al., 2008), suggesting only two configuration states of the pore, namely open and closed. However, this binary theory appears to not exhaustively explain the gating behavior of all VGICs: as an example, heteromeric pore conformations have been proposed for Kv2.1 channels, as single-channel analysis revealed the occurrence of discrete subconductance levels, possibly resulting from the pore opening of only one or two subunits (Chapman and VanDongen, 2005). Whether discrete subconductance states occur and can be detected in all VGICs, and whether they result from “partial” pore opening, is still debated.
The VSM is an independent functional module that can be transplanted into other proteins to confer voltage sensitivity; indeed, voltage-sensing phosphatases (VSP) are formed by a VSM linked to a cytoplasmic phosphatase (Murata et al., 2005; Li et al., 2014c). On the other hand, the VSM can also allow ion transport by itself; indeed, subunits forming proton-selective Hv1 channels only contain an isolated VSM, without a canonical S5–S6 pore region (Ramsey et al., 2006; Sasaki et al., 2006).
Structural Basis of Ion Permeation and Inactivation
The PM of VGIC contains the permeation pathway that regulates ion fluxes and selectivity. Structural information regarding the molecular mechanisms of ion permeation, selectivity, and pore opening/closing were obtained from the crystal structure of bacterial “inward rectifier” K+ channels (KIR) KcsA (Doyle et al., 1998), MthK (Jiang et al., 2002a,b), and KirBac1.1 (Kuo et al., 2003), whose membrane core only contains the regions corresponding to the S5–S6 module. In these channels, the PM contains a narrow constriction near its extracellular side, which forms the selectivity filter. Below this region is a relatively large water-filled cavity at the center of the plasma membrane. In K+ channels, the highly conserved GYG sequence forms the selectivity filter that allows ion discrimination via direct interactions of the dehydrated K+ ions and the protein backbone carbonyls. While no eukaryotic VGNC structure is yet available, the crystal structure of bacterial NavAb, recently described at 2.7 Å of resolution (Payandeh et al., 2011), have revealed that a similar region is involved in ion discrimination, although Na+ ions permeate in their partially hydrated configuration across a wider selectivity filter. This region is likely to mediate ion discrimination also in VGNCs and VGCCs of vertebrates; in fact, it has been long known that substitution of positively charged amino acids in the PM of rat Nav1.2 with glutamic acid residues (which occupy equivalent positions in VGCCs), enhances Ca2+ ion permeability (Heinemann et al., 1992). In both VGNCs and VGCCs, a second pore-helix forms an extracellular funnel, a unique feature that could represent a conserved structural element in the outer vestibule of these channels.
Comparing the structural data of KcsA and KirBac1.1 channels, trapped in a closed conformation, with that of MthK channels, trapped in an open conformation, it has been possible to deduce structural changes that underlie pore opening in K+ channels. In KcsA and KirBac1.1 channels, the M2 segments, corresponding to the S6 segments in VGIC, form a four-helix bundle near the intracellular membrane surface that occludes the ion conduction pathway; whereas, in MthK channels the inner region of M2 is bent at a glycine residue; this residue is highly conserved among bacterial K+ channels and in some eukaryotic VGKCs. This glycine residue has been proposed to serve as a gating hinge that allows M2 to oscillate between a close and an open conformation. In eukaryotic K+ channels, it has been proposed that a conserved PVP (Proline-Valine-Proline) motif, not found in bacterial K+ channels and located in S6 downstream of the glycine residue, acts as a flexible hinge that allows channel switching from a closed to an open state (Webster et al., 2004; Long et al., 2005a,b). In particular, this inner part of the S6 segment, also called “bundle crossing” (BC) gate, appears to come close to the S4–S5 linker during channel activation, thus allowing to “sense” VSM movement (Prole and Yellen, 2006); BC gate widening prompts a conformational change in a further gate located at the extracellular entrance of the pore, called the “selectivity filter” gate, leading to pore opening (Labro and Snyders, 2012). The differences in single channel conductance observed between bacterial and mammalian channels might be explained by different degree of movement of the distal S6 regions; indeed, movements seem much wider in bacterial (having larger single channel conductance) when compared to mammalian channels, the latter having a 10-times smaller single channel conductance.
In some VGICs, currents show prominent inactivation following activation. Two main types of inactivation have been described in VGKCs: the N-type and the C-type inactivation. The N-type inactivation, which is usually more rapid than the C-type, occurs by a ball peptide tethered to the N-terminus of the channels that enters into the pore cavity blocking ion flow (Hoshi et al., 1990). By contrast, the C-type inactivation gate is located in the selectivity filter, and current inactivation is due to the pore collapse (Cuello et al., 2010a,b); structural data from bacterial KCsA confirm such a hypothesis (Bhate and McDermott, 2012). In VGNCs of vertebrates, the short intracellular loop between domains III and IV is responsible for fast inactivation by folding into the intracellular mouth of the pore and blocking it (Vassilev et al., 1988). In particular, the amino acid motif isoleucine, phenylalanine, and methionine (IFM) is required to stabilize the inactivated state of the channels (West et al., 1992). Finally, inactivation processes in VGCCs are more complex, including both voltage- and/or calcium-dependent mechanisms (Simms and Zamponi, 2014).
Structural Basis of Voltage-Sensing
As above mentioned, voltage-sensitivity of VGICs is conferred by the VSM. In particular, the S4 segments are believed to play a key role in the voltage-sensitivity. Indeed, S4 primary sequence shows the presence of 4–8 positively-charged arginines (Rs) or lysine (Ks) residues separated by 2–3 uncharged residues; this configuration would allow positive charges to be localized on the same side of an α-helix. These basic residues are located within the electrical field existing across the two sides of the plasma membrane, thus directly sensing changes in membrane potential. The first four Rs in S4 are considered the most important voltage-sensing elements and are believed to be the major component of gating currents (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). In Kv1.1 channels, it has been calculated that approximately 13 elementary charges cross the electric field in response to membrane depolarization, corresponding to about three elementary charges per subunit (Schoppa et al., 1992).
The molecular mechanisms underlying VSM movement during gating are highly debated topics; nevertheless, results obtained with molecular, biochemical, electrophysiological, optic and crystallographic techniques have enormously expanded our knowledge in this field. In particular, the crystal structures of bacterial KvAP (Jiang et al., 2003), followed by that of mammalian Kv1.2 and Kv1.2/2.1 chimeric VGKCs (Long et al., 2005a, 2007), provide important information on the activated configurations of the VSM. Although no crystal structure is available for the resting state of the VSM, several hypotheses (helical-screw/sliding-helix, transporter and paddle models; see Tombola et al., 2006; Catterall, 2010; Miceli et al., 2011 for reviews) have been formulated to explain how the voltage sensor reaches its activated configuration during channel gating. Currently, these models converge toward an unified hypothesis (Khalili-Araghi et al., 2010; Vargas et al., 2011; Jensen et al., 2012; Yarov-Yarovoy et al., 2012), in which the resting and activated positions of the VSM would be stabilized by ionized hydrogen bonds between the S4 positive charges and two clusters of negative charges: one facing the extracellular side of the membrane and provided by the S1 and S2 helices, and another closer to the intracellular membrane surface, involving S2 and the proximal part of S3 (S3a) helices. At rest, S4 is drawn inwardly by the electrostatic forces of the negative resting membrane potential (RMP); this would allow the first S4 Rs (gating charges) to preferentially interact with the inner cluster of negative charges. During the activation process, S4 would rotate around its helical axis and translate outwardly, reaching a final configuration stabilized by the interaction of the same positively-charged residues with the negative charges provided by the extracellular cluster. This relatively small movement would allow the displacement of the gating charges from an intracellular-facing crevice to one directly connected to the extracellular milieu. A conserved phenylalanine located in the middle of S2 has been proposed to form the charge-transfer center that catalyzes the gating charges movement (Tao et al., 2010) and separates the extracellular and the intracellular crevices of the VSM.
In several VGKCs including Kv1.1, the replacement of specific Rs in S4 with smaller amino acids widens this gating charge pathway, generating cation-permeable non-selective currents (also known as gating pore currents, or ω currents). Interestingly, gating-pore currents generated by mutations in S4 of Nav1.4 or Cav1.1 channels seem to provide a pathophysiological-relevant mechanism in hypokalemic (Sokolov et al., 2007; Struyk and Cannon, 2007) or normokalemic (Sokolov et al., 2008) periodic paralysis in skeletal muscle. Similar mechanisms seem implicated in a mixed arrhythmias and the dilated cardiomyopathy caused by the R219H mutation in domain I of Nav1.5, which induces a proton-specific gating pore current (Gosselin-Badaroudine et al., 2012), and in peripheral nerve hyperexcitability caused by the R207Q in Kv7.2 channels (Miceli et al., 2012). Noteworthy, sequence analysis has revealed that several other channelopathies (including some affecting neuronal channels) might be attributable to the formation of gating pore currents, however more studies are required to confirm the presence of gating pores (Moreau et al., 2014).
Structural Basis of Electromechanical Coupling
Electromechanical coupling describes the process of transferring the energy generated by the VSM movement upon changes in membrane potential to the PM. To simplify a complex process, at resting state the S4–S5 linker directly interacts with the distal part of S6, making the BC gate the main voltage-controllable activation gate. Upon depolarization, S4 movement pulls the S4–S5 linker outwardly leading to a widening of the helical BC gate, which triggers a further conformational change at the level of the selectivity filter (SF) gate that allows ions flow through the pore. The crystal structures of Kv1.2 and of chimeric Kv1.2/2.1 channels suggest at least two non-covalent interactions between VSM and PM: one between the S4–S5 linker and the distal S6 segment at the cytoplasmic side of the membrane, and the other between the C-terminal region of S1 and the C-terminal region of S5 at extracellular site of the membrane (Lu et al., 2002; Long et al., 2005b, 2007). The interaction between S4–S5 linker and the bottom part of S6 seems to play an important role for transmission of conformational changes during channel gating; while the interaction between S1 and S5 acts as an anchor point between the VSM and the PM, thus allowing efficient transmission of conformational changes to the pore’s gate (Lee et al., 2009). Other interactions between VSM and PM include: the bottom part of S4 with S5 segments of neighboring subunits (Soler-Llavina et al., 2006) and the upper part of S5 with the upper part of S4 of the neighboring subunit (Lainé et al., 2003). Based on the available molecular dynamic simulations and experimental data, Blunck and Batulan (2012) proposed two different mechanisms to describe the electromechanical coupling in Shaker-like Kv channels. In one model S4 and S4–S5 linker act as a spring that is relaxed or compressed by the activation/deactivation of the VSM. In the second model, S4 and S4–S5 linker act as a bolt that keeps the pore closed; only when all four S4–S5 linkers have moved out the pore passively follows the opening.
The general mechanism of the electromechanical coupling seems to be conserved among the different K+ channels, although changes in the type of interactions and in the tightness of the coupling may occur in specific channels. Moreover, this general mechanism also seems to apply to skeletal VGNCs (Muroi et al., 2010) and VGCCs (Wall-Lacelle et al., 2011). By contrast, in hyperpolarization-activated cyclic nucleotide gated channels (HCN) decoupling between VSM and PM triggers pore closure (Blunck and Batulan, 2012).
Neuronal Channelopathies Caused by Mutations in the VSM
Potassium Channelopathies
Structure and Function of Voltage-Gated Potassium Channels (VGKCs)
K+ channels are the largest and the most functionally heterogeneous class of ion channels expressed in all eukaryotic cells and in prokaryotes. K+ channels mostly inhibit neuronal excitability by setting the membrane potential closer to the K+ equilibrium potential. Moreover, activation of K+ channels shortens the duration of the action potential (AP), terminates periods of intense electrical activity, reduces neuronal firing frequency, and, in general terms, decreases the efficacy of cell excitatory inputs. Beside these roles, K+ channels participate in solute transport across epithelial membranes, and K+ clearance from brain interstitial spaces from glial cells. In humans, more than 70 genes encode for K+ channels. On the basis of their presumed topology, K+ channels can be classified into three groups: (1) the classical family with 6 transmembrane segments (6TM) including the voltage-gated K+ channels (Kv channels; Kv1-Kv12; Figure 1); (2) a family with only 2 transmembrane segment (2TM), homologous to the S5–S6 segments of Kv channels. To this group belong the inward-rectifier channels (both constitutively-active and G-protein-gated), which include at least seven gene families (KIR1-KIR7); (3) the 4 transmembrane segment family (4TM), formed by subunits encoded by at least 15 different genes (K2P1-K2P17; Yu et al., 2005). While channels formed by subunits of the first two groups are tetrameric, those of the third group are dimers. However, these three groups do not account for all K+ channel structural heterogeneity; in fact, large conductance Ca2+-dependent K+ channels (BK channels) assemble as tetramers of subunits containing seven transmembrane segments, which differ from Kv subunits for the presence of an extra transmembrane segment (S0) at the N-terminus.
Several VGKCs interact with accessory β-subunits that influence a wide range of channel properties such as gating, assembly, trafficking, and targeting of channels to different cellular compartments. Accessory VGKC subunits include different protein structures reflecting their divergent biological roles. Indeed, some β-subunits are integral membrane proteins such as β-subunits of both BK and Kv7 channels; while, others are cytosolic proteins and bind to cytoplasmic modules of VGKC such as Kvβ-subunits for Shaker Kv channels and KChIPs for Kv4 channels (Pongs and Schwarz, 2010 for review).
Kv1.1 (KCNA1) and Kv1.2 (KCNA2) Channel Mutations Cause Episodic Ataxia Type 1 (EA1) or Epileptic Encephalopathy (EE), Respectively
Kv1.1 and Kv1.2 subunits, belonging to the Kv1 family of VGKCs, generate delayed rectifier channels active at or below the RMP, with fast activation/deactivation kinetics and slow inactivation, mainly contributing to transient, slowly inactivating current (ID). Kv1 family members are expressed throughout many regions of the central nervous system (CNS), including hippocampus, cerebellum, and brainstem nuclei. In several brain regions, co-assembling of Kv1.1 with Kv1.4 subunits, which contain N-terminal Inactivation Modules, confers N-type inactivating properties to heteromeric channels. At neuronal subcellular level, Kv1.1, Kv1.2 and Kv1.4 are expressed in the axon initial segment (AIS) where they control AP threshold and firing (Figure 2), in juxtaparanodal regions next to Ranvier’s nodes, in synaptic terminals and in proximal dendrites (Trimmer and Rhodes, 2004). The important pathophysiological role of Kv1.1 channels is underlined by the occurrence of mutations in this channel in patients affected with Episodic Ataxia type 1 [EA1; Online Mendelian Inheritance in Man (OMIM) 160120]. EA1 is a rare, disabling condition with autosomal-dominant inheritance characterized by constant myokymia and dramatic episodes of spastic contractions of head, arms and leg muscles, together with the loss of motor coordination and balance. Moreover, EA1 is associated with an increased incidence of epilepsy. Other features include delayed motor development, cognitive disability, choreoathetosis, and carpal spasms. Disease onset is in childhood or early adolescence (D’Adamo et al., 2014). An altered function of Kv1.1 channels expressed in the cerebellum may be responsible for ataxia, whereas seizures and cognitive dysfunctions associated with EA1 may be caused by impaired Kv1.1 channels in the hippocampus. The first description of a family affected by EA1 was in 1975 (VanDyke et al., 1975); subsequent genetic analysis revealed a heterozygous point mutation in the coding sequence of the human gene KCNA1, encoding for Kv1.1 channels, leading to the F184C substitution in the S1 segment. Electrophysiological studies demonstrated that this mutation shifts the activation curves of Kv1.1 channels to more positive voltages, therefore leading to loss-of-function effects (Adelman et al., 1995). Several other Kv1.1 mutations causing EA1 have been described and they appear to be localized in relevant functional regions of the channels, particularly in the VSM (Figure 3) or in the PM. Overall, functional studies performed on numerous mutant channels revealed that EA1-causing mutations may alter membrane expression or gating processes, such as opening and closure kinetics, voltage-dependence of activation, N- and C-type inactivation. An atypical mechanism by which the V408A pore-mutation affects gating of Kv1.1 channel has been described by means of voltage-clamp fluorometry and single-channel recordings. This mutation accelerates the outward current decay during depolarization by promoting an “activated-not-open” conformation rather than by increasing C-type inactivation (Peters et al., 2011). Moreover, mutations within the VSM cause EA1 mainly by a reduction of maximal currents, in some cases with dominant-negative effects (as in the case of R167M, C185W, F249I, I262T or R307C mutations; Adelman et al., 1995; Zerr et al., 1998; Graves et al., 2010; Zhu et al., 2012; Tomlinson et al., 2013). These results suggest that EA1-causing mutations induce loss-of-function effects on Kv1.1 channels, with haploinsufficiency appearing as the most likely pathogenetic mechanism for the disease. Accordingly, the loss of Kv1.1 channels, obtained either by genetic or pharmacological manipulations, caused alterations in the hippocampal network, which showed more frequent and longer spontaneous sharp waves and high frequency oscillations; moreover, in the CA3 region from Kv1.1−/− mice, mossy fibers and medial perforant path axons were hyperexcitable and produce greater pre- and post-synaptic responses, also showing a reduced paired-pulse ratios suggestive of an increased neurotransmitter release at these terminals (Simeone et al., 2013).
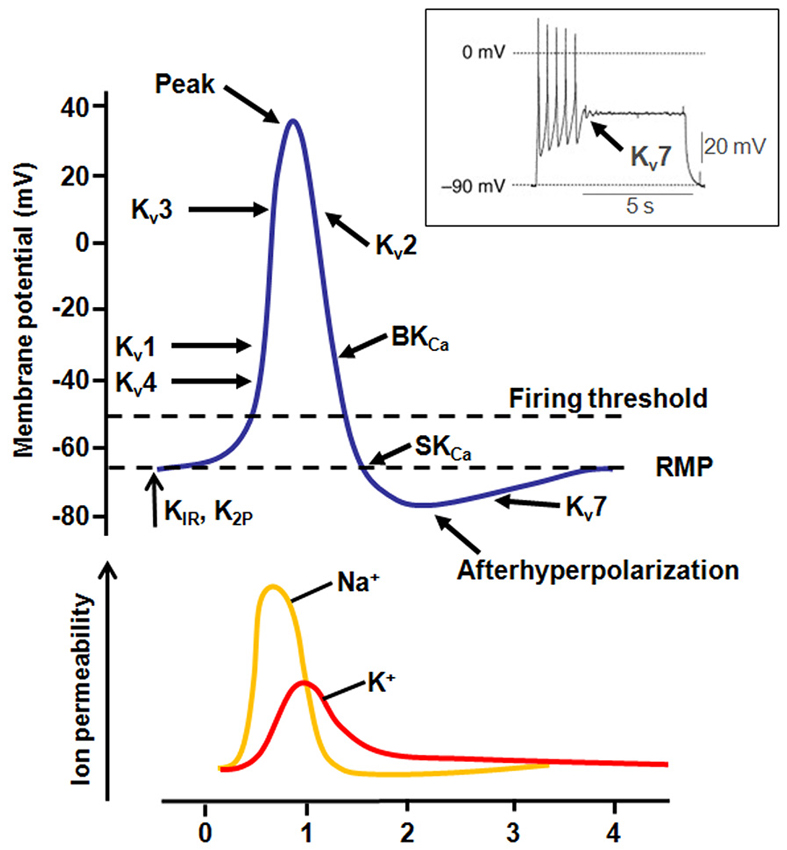
Figure 2. Contribution of K+ currents during AP firing. Representation of the different K+ channel subtypes activated during an AP. The inset shows the effect of Kv7 channel activation during AP firing (modified from Tsantoulas and McMahon, 2014).

Figure 3. Alignment of the VSM of different Kv, Nav and Cav channel subtypes. The VSM have been aligned by using ClustalW2 software; highlighted in yellow are the missense mutations causing EA1 (in Kv1.1); PME (in Kv3.1); SCA13 (in Kv3.3); BFNS (in Kv7.2 and Kv7.3); EEIE7 (in Kv7.2); RCD3B (in Kv8.2); FHM3, SMEI, and GEFS+ (in Nav1.1); and FHM1, EA2 and SCA6 (in Cav2.1). Mutations in SCN1A are from a database available at http://www.molgen.ua.ac.be/SCN1AMutations/home/Default.cfm.
More recently, four different de novo mutations in the KCNA2 gene, encoding for Kv1.2 K+ channel subunits, have been identified in patients with Epileptic Encephalopathy (EE). Three of these mutations affect conserved residues in the VSM (I263T in S3, R297Q and L298F in S4) and one in the PM (P405L). Functional studies in heterologous expression systems revealed that channels formed by subunits carrying the I263T or P405L mutations almost completely lost their function, with also dominant-negative effects on wild-type channels; by contrast, the two S4 mutations caused a gain of function effect (Syrbe et al., 2015). A Kv1.2 mutation has been also described in a mouse model of cerebral ataxia (I402T substitution in the S6 segment), called Pingu: patch-clamp recordings from cerebellar slices revealed an increased frequency and amplitude of spontaneous GABAergic inhibitory postsynaptic currents and reduced AP firing frequency in Purkinje cells, suggesting that Kv1.2 mutation-dependent increase in GABA release from basket cells is involved in the loss of motor coordination in Pingu mice (Xie et al., 2010).
Kv3.1 (KCNC1) and Kv3.3 (KCNC3) Channel Mutations Cause Progressive Myoclonus Epilepsy (PME) or Spinocerebellar Ataxia Type 13 (SCA13), Respectively
The Kv3 family of VGKCs comprises four members (from Kv3.1 to Kv3.4, encoded by the KCNC1-KCNC4 genes) that differ from others VGKCs because of a more positive activation threshold (thus, they are identified as “high-threshold channels”) and their fast rate of deactivation upon repolarization. These unique properties specifically enable AP fast repolarization without affecting spike initiation and AP duration (Rudy and McBain, 2001; Figure 2). The expression of Kv3.1, Kv3.2 and Kv3.3 is limited to the CNS, whereas Kv3.4 is mainly expressed in skeletal muscles and sympathetic neurons, and only weakly expressed in a few neuronal types. In the CNS, Kv3 channels are mainly expressed in fast-spiking neurons, such as GABAergic interneurons. Accordingly, suppression of Kv3 currents by pharmacological tools (Erisir et al., 1999) or in knockout mice (Lau et al., 2000) impairs firing properties of fast-spiking neurons, affects neurotransmitter release (Sabatini and Regehr, 1997), and induces cell death (Irie et al., 2014).
Spinocerebellar Ataxias (SCAs) are a very heterogeneous group of autosomal dominant neurological disorders caused by degeneration of the cerebellum and spinal cord. SCAs show a wide range of phenotypes, including cerebellar ataxia, dysarthria, extrapyramidal symptoms, oculomotor disturbance, cognitive impairment, and epilepsy. Most SCAs are caused by trinucleotide repeat expansions, while other forms are caused by point mutations in protein kinases, cytoskeletal and mitochondrial proteins, and proteins regulating intracellular calcium stores, such as beta 3 Spectrin (SPTBN2), Tau Tubulin Kinase 2 (TTBK2), Protein Kinase C Gamma type (PRKCG), Inositol 1,4,5-Trisphosphate Receptor type 1 (ITPR1), ATPase Family Gene 3 Like 2 (AFG3L2). In particular, SCA13 (OMIM 605259) is caused by point mutations in the KCNC3 gene, encoding for Kv3.3 channels. So far, three mutations have been identified in Kv3.3, two of which affect residues in the VSM (R420H and R423H both in S4; Figure 3) and one in the PM (F448L; Waters et al., 2006; Figueroa et al., 2010). Functional studies performed in heterologous expression systems revealed that both VSM mutations caused a complete abolishment of channel function when expressed homomerically, whereas the co-expression of mutant subunits with wild-type subunits caused a significant suppression of Kv3.3 activity, consistent with dominant-negative effects. By contrast, the pore mutant F448L channels did not affect current amplitude, but causes a hyperpolarization shift of the current activation and slows the rate of deactivation, suggesting that this mutation increases the stability of the open state (Waters et al., 2006). Taken together, these data suggest that mutations in KCNC3 may cause SCA both by gain- and loss-of-function mechanisms (Figueroa et al., 2010).
More recently, a recurrent mutation in the KCNC1 gene, encoding for Kv3.1 channels, has been identified in patients with Progressive Myoclonus Epilepsy (PME7; OMIM 616187; Muona et al., 2015), one of the most devastating form of epilepsy, characterized by a very wide clinical and genetic heterogeneity (Berkovic et al., 1986). Most of PME cases are autosomal recessive, although in few cases an autosomal dominat transmission occurs. Clinically, the most common features of this disease are myoclonus, tonic-clonic seizures and progressive neurological decline. The mutation occurring in Kv3.1 channel causing PME affects a highly conserved residue in the S4 segment (R320H; Figure 3). Functional experiments in heterologous expression systems showed that the R320H substitution caused a prominent loss of function, with dominant-negative effects on wild-type Kv3.1 channels.
Kv7.2/7.3 (KCNQ2/KCNQ3) and Benign Familial Neonatal Seizures (BFNS)/Epileptic Encephalopathy (EE)
Kv7.2 and Kv7.3 channels belong to the KCNQ gene family that comprises five members (KCNQ1-5) encoding for K+ channel subunits showing different tissue distribution and physiological roles (Soldovieri et al., 2011). When heterologously expressed, homomeric Kv7.2 channels generate K+-selective currents activated by depolarization at membrane potentials around −50 mV with slow activation and deactivation kinetics and lacking significant inactivation. Currents carried by Kv7.3 homomultimers are rather small and activate around −60 mV; among Kv7 members, Kv7.3 channels show the highest opening probability and unitary conductance at the single-channel level (Li et al., 2005). Co-expression of Kv7.2 and Kv7.3 channels generate currents 10-times larger than those obtained by the simple sum of the currents produced by Kv7.2 or Kv7.3 homomultimers (Wang et al., 1998; Yang et al., 1998). This effect is mainly mediated by a higher opening probability of Kv7.2/Kv7.3 heteromers compared to Kv7.2 homomers, together with a two- to threefold increase in the number of channel-forming subunits expressed at the plasma membrane (Schwake et al., 2000). In the brain, heteromeric assembly of Kv7.2 and Kv7.3 subunits underlies the M-current (IKM), a slowly activating and deactivating K+ current that regulates neuronal excitability in the sub-threshold range for AP generation (Soldovieri et al., 2011; Figure 2). Mutations in KCNQ2 (Kv7.2) and KCNQ3 (Kv7.3) genes are responsible for neonatal-onset epileptic diseases with widely-diverging clinical manifestations, ranging from benign to very dramatic phenotypes. Indeed, mutations in these two genes have been identified in patients affected with Benign Familial Neonatal Seizures (BFNS1; OMIM 121200), an autosomal dominant epilepsy of newborns (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998), characterized by recurrent seizures that begin in the first days of life and remit after a few weeks or months, with mostly normal interictal EEG, neuroimaging, and psychomotor development (Bellini et al., 2010). In addition, de novo missense Kv7.2 mutations have been recently found in neonates affected with pharmacoresistant seizures, distinct EEG and neuroradiological features, and various degrees of developmental delay, defining a “Kv7.2 encephalopathy” (Weckhuysen et al., 2012; EIEE7; OMIM 613720). Subsequently, de novo missense Kv7.2 mutations have been also shown as one of the most common cause of early-onset EEs, including the Ohtahara syndrome (Saitsu et al., 2012; Kato et al., 2013), the most severe and earliest developing age-related EE. Most disease-causing Kv7.2 mutations cluster in the VSM (Figure 3), in the PM or in the long C-terminus of the channel. Functional studies for a significant fraction of the described Kv7.2 or Kv7.3 mutations have been carried out, with consequences ranging from slight changes in channel behavior to a complete ablation of channel function (Castaldo et al., 2002; Soldovieri et al., 2006; Miceli et al., 2009). A slight (about 25%) decrease of IKM is believed to be sufficient to cause BFNS, and haploinsufficiency seems to be the primary pathogenetic mechanism for BFNS. Moreover, two independent studies suggested that the clinical disease severity may be related to the extent of mutation-induced functional K+ channel impairment (Miceli et al., 2013; Orhan et al., 2014).
BFNS-causing mutations have been also found to neutralize some of the positively-charged residues in the S4 segment (R207W, R213W, or R214W; Miceli et al., 2008; Bellini et al., 2010): the functional characterization of Kv7.2 channels carrying each of these mutations suggests a reduced channel sensitivity to depolarization; accordingly, homology model of Kv7.2 based on the crystal structure of Kv1.2/Kv2.1 chimeric channels revealed that these residues form intrasubunit ionized hydrogen bonds with negatively-charged residues present in the outer cluster which stabilize the activated state of the voltage sensor. Thus, it seems plausible to hypothesize that in mutant channels the activated configuration of the VSM is destabilized, explaining the positive shift in steady-state voltage dependence of activations and possibly the neuronal hyperexcitability in individuals carrying these mutations (Miceli et al., 2008, 2013).
Notably, although most epilepsy-causing mutations in Kv7.2 or Kv7.3 so far indentified induced loss-of-function effects, electrophysiological experiments performed on Kv7 channels carrying de novo mutations recently found in individuals affected with various forms of early-onset EEs (R144Q, R201C, or R201H in Kv7.2, and R230C in Kv7.3; Rauch et al., 2012; Allen et al., 2013; Carvill et al., 2013; Weckhuysen et al., 2013) have demonstrated that all four mutations cause hyperpolarization shifts of the current activation process by destabilizing the resting state of the VSM (Miceli et al., 2015). Accordingly, computational modeling of a feedforward inhibitory microcircuit formed by a principal hyppocampal CA1 neuron and an inhibitory interneuron suggest that a gain-of-function mutation in Kv7.2/3-IKM can increase the (apparent) excitability of hippocampal CA1 pyramidal neurons by selective suppression of interneuron activity. These data suggest that Kv7.2/3 mutations may cause human epilepsy by both loss-of-function and gain-of function mechanisms; whether these two mechanisms underlie different clinical entities (in terms of age of onset, severity, prognosis, and pharmacosensitivity) is yet unknown.
Kv8.2 (KCNV2) Mutations Cause Cone Dystrophy with Supernormal Rod Electroretinogram (CDSRR) or Epilepsy
Kv8.2 subunits are structurally similar to others VGKCs, although they do not form functional channels in homomeric configuration: for this reason, they are also called “silent modulators”. Conversely, they are functional upon assembly in heteromeric channels with other subunits (e.g., Kv2). The KCNV2 gene encoding for Kv8.2 subunits was first cloned from human testis cDNA, but its expression has been also detected in other tissues, such as the photoreceptor layer of the human retina (Wu et al., 2006) and in the hippocampus, where it co-localizes with Kv2.1 (Allen Institute for Brain Science1), the major contributor for delayed rectifier K+ currents in neurons (Murakoshi and Trimmer, 1999). Co-assembling of silent Kv8.2 with Kv2.1 subunits causes a decrease in the maximal currents as well as modifications in the activation, inactivation, and deactivation kinetics (Bocksteins and Snyders, 2012). Mutations in Kv8.2 channels, some of them located within the VSM (Figure 3), cause cone dystrophy with supernormal rod response (RCD3B; OMIM 610356), a very rare autosomal recessive retinal disorder, characterized by reduced visual acuity, photoaversion, night blindness, and abnormal color vision. At an early age, the retina shows subtle depigmentation of the macula and, later, more obvious areas of atrophy. Electroretinographic features are characteristic and essential for diagnostic purposes. In addition to RCD3B, two mutations in Kv8.2 channels have been identified in two unrelated children affected by epilepsy: one mutation is located in the N-terminal region (R7K), and the other in the S1 segment of the VSM (M285R; Figure 3). Functional studies demonstrated that both mutant channels increased Kv8.2-mediated suppression of Kv2.1 currents. Moreover, heteromeric channels incorporating M285R mutant subunits exhibited additional gating defects, including a +10 mV shift in the voltage dependence of activation and slower activation kinetics. Taken together, these data suggest that both variants may decrease delayed rectifier K+ currents in neurons, possibly leading to an increased excitability under conditions of repetitive stimulation (Jorge et al., 2011).
Kv10.2 (KCNH5) and Epileptic Encephalopathy (EE)
The KCNH5 gene encodes for Kv10.1 potassium channels belonging to the ether-a-go-go (EAG) family (Saganich et al., 1999). EAG family comprises two channel subtypes (Kv10.1 and Kv10.2), that are over-expressed in cancer cells (Camacho, 2006). Kv10.2 is also expressed in the CNS (Saganich et al., 1999), although its function is still unclear (Wulff et al., 2009). Recently, by using whole-exome sequencing in children with sporadic cases of EE of unknown etiology, a novel variant in KCNH5 has been identified (R327H in S4). Structural and functional analysis showed that this pathogenic variant destabilized the resting state of the VSM, therefore leading to a hyperpolarizing shift of the activation curve, suggesting a gain-of-function pathogenetic mechanism associated to this mutation (Yang et al., 2013).
KCa4.1 (KCNT1) Mutations Cause Malignant Migrating Partial Seizures of Infancy (MMPSI) and Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE)
The KCNT1 gene encodes for KCa4.1, a sodium-activated potassium channel, also known as “sequence like a calcium activated potassium channel” (Slack) or Slo2.2. Structurally, this channel is similar to the classic VGKCs except for two properties: no charged residues in S4 and a very large cytoplasmic C-terminal region. KCa4.1 is highly expressed in different regions of the mammalian brain where it generates a delayed outward current termed IKNa that regulates neuronal excitability. Mutations in KCNT1 gene have been identified in patients with two different type of epilepsy occurring in infancy or childhood: Malignant Migrating Partial Seizures of Infancy (MMPSI; also called Epilepsy of Infancy with Migrating Focal Seizures, EIMFS) and Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE; Barcia et al., 2012; Heron et al., 2012). MMPSI is a severe early onset epileptic encephalopathy beginning before of 6 months of age. It is characterized by heterogeneous focal seizures, with seizures foci migrating from one brain region and hemisphere to another, and is associated with arrest or regression of development, resulting in profound disability. By contrast, ADNFLE is characterized by nocturnal frontal lobe seizures beginning in mid childhood, with psychiatric, behavioral, and cognitive disabilities in some cases. All the described mutations causing MMPSI and ADNFLE are not located into the VSM; however, they dramatically affect channel function, with a striking gain-of-function phenotype in vitro (Milligan et al., 2014).
Sodium Channelopathies
Structure and Function of Voltage-Gated Sodium Channels (VGNCs)
VGNCs contribute to the generation of both somatodendritic and axonal AP and conduct subthreshold persistent Na+ currents following AP. Structurally, they are complexes of a pore-forming α subunit of 260 kDa, associated to auxiliary β subunits (β1–β4) of 33-36 kDa (Figure 1; Catterall, 2000). Auxiliary β subunits can assemble with α-subunits and modify their subcellular localization and biophysical properties. In particular, β2 and β4 subunits form disulfide bonds with α subunits, whereas β1 and β3 subunits interact with α-subunits non covalently. Structurally, β subunits are formed by an N-terminal extracellular immunoglobulin-like fold, a single transmembrane α-helix and a short intracellular segment (Isom et al., 1992, 1995).
Each α subunit contains four internally repeated domains (called from DI to DIV), each including a VSM and a PM. In the VSM, S4 segments in all four domains have previously-described primary sequence and functional role (Catterall, 1986; Guy and Seetharamulu, 1986; Yarov-Yarovoy et al., 2006, 2012). Furthermore, the DIII-DIV linker and particularly the key IFM motif, plays a crucial role in channel fast inactivation, a process that contributes to the neuronal excitability control.
In humans, 10 genes encode for VGNC α subunits; these are expressed in different excitable tissues and give rise to currents with peculiar biophysical properties (Goldin, 2001). Nav1.1, Nav1.2, Nav1.3 and Nav1.6 channel subtypes are the primary Na+ channels in the CNS; by contrast, Nav1.7, Nav1.8 and Nav1.9 channel subtypes are mainly expressed in the peripheral nervous system (PNS); finally, Nav1.4 channels are expressed in skeletal muscle and Nav1.5 in the heart. The tenth sodium channel protein (Nav1.10) is not voltage-gated and is involved in salt sensing (Watanabe et al., 2000). At the subcellular level, Nav1.1 and Nav1.3 channels are primarily localized in cell bodies (Westenbroek et al., 1989, 1992), whereas Nav1.2 channels are positioned in unmyelinated or pre-myelinated axons and dendrites, and Nav1.6 channels in myelinated axons and dendrites (Caldwell et al., 2000). In the developing neocortex, Nav1.1 channels appear to be abundantly expressed in the proximal part of the AIS of parvalbumin- (Ogiwara et al., 2007; Li et al., 2014b) and, at lower levels, somatostatin-positive GABAergic interneurons (Li et al., 2014b). By contrast, in the same region, Nav1.2 channels are preferentially distributed to the somatostatin-positive population of GABAergic neurons, whereas Nav1.6 channels are mainly located at the distal part of the AIS of both types of interneurons (Li et al., 2014b). Such differences in the distribution of specific Nav channel subtypes appear to explain their differential role in controlling intrinsic excitability of distinct neuronal populations.
A large number of genetic diseases are caused by mutations in VGNCs, including inherited forms of periodic paralysis, cardiac arrhythmia, epilepsy, and chronic pain (Lehmann-Horn and Jurkat-Rott, 1999; Catterall et al., 2008). Among these, the genes most frequently associated with inherited forms of neurological disorders encode for brain Na+ channels Nav1.1 (SCN1A) and Nav1.2 (SCN2A).
Nav1.1 and Epilepsy
Mutations in SCN1A (Nav1.1) are responsible for genetic epilepsy syndromes with a wide range of severity. In fact, SCN1A-associated epilepsies range from simple febrile seizures, to Generalized Epilepsy with Febrile Seizures (GEFS+; OMIM 604233; Escayg et al., 2000), an autosomal dominant epilepsy disorder associated to missense mutations, to Severe Myoclonic Epilepsy of Infancy (SMEI or Dravet syndrome, OMIM 607208; Dravet et al., 1992; Engel and International League Against Epilepsy [ILAE], 2001), a much more severe form of epilepsy often caused by mutations causing truncation or deletions of Nav1.1 channels. SMEI is a drug-resistant epilepsy occurring in the first year of life with seizures often associated with elevated body temperature and progressing to prolonged, clustered, or continuous seizures and to status epilepticus (Dravet et al., 1992; Engel and International League Against Epilepsy [ILAE], 2001).
Although only few of the Nav1.1 mutations responsible for epileptogenic diseases in humans have been functionally characterized, several evidence point to a loss-of-function as their common effect (Bechi et al., 2015), with a correlation between the increasing functional severity of Nav1.1 channel alteration and the worsening of the clinical phenotype (Catterall et al., 2010). In other words, missense mutations found in patients with more benign clinical courses cause milder channel dysfunction, whereas more dramatic functional consequences are caused by truncation or loss-of-function mutations described in more severe phenotypes. Disease-causing Nav1.1 mutations appear to be spread along the α subunit sequence, with 25% of those (157/675) affecting the VSM.2 However, other studies (Kanai et al., 2004) have reported the occurrence of Nav1.1 missense mutations in SMEI affecting the PM, suggesting that, beside the type of mutations, also their localization plays a part in the expressed phenotype. Finally, in some cases mutations of the same residue, but introducing different amino acids, have been identified in patients with SMEI (R1648C) or GEFS+ (R1648H; see below), suggesting that the specific structural and functional changes prompted by each mutation are strictly related to the severity of the disease. One of the possible molecular mechanism responsible for the loss-of-function occurring in Nav1.1 disease-causing mutations is a folding defect, whereby the quality control system of the endoplasmic reticulum recognizes the mutation-induced structural defect and targets the abnormal protein to degradation, severely impeding its membrane trafficking. Therefore, rescuing defective proteins from degradation appears as an attractive intervention strategy in Nav1.1-related disease. Among the various strategies to reverse Nav1.1 folding defects, co-expression of β1 subunits (Sugiura et al., 2012), exposure to (lower) temperatures which facilitate correct folding (Bernier et al., 2004; Rusconi et al., 2007), co-expression with modulatory proteins (such as calmodulin; Rusconi et al., 2007), as well as the exposure to Nav blockers (such as phenytoin; Bechi et al., 2015), are the most studied. In fact, the function of the epileptogenic missense mutations, including the R859C affecting the first arginine residue in the DII-S4, can be rescued by co-expression of the β1 subunit, of the interacting protein ankyrin, and of a modified scorpion toxin binding to the VSM of these channels specifically engineered to target the endoplasmic reticulum (Bechi et al., 2015). Notably, some Nav1.1 mutations identified in patients affected with GEFS+ or SMEI introduce different substitutions at the same residue; in these cases, the extent of the folding defect appears to correlate with disease severity.
As previously indicated, more than half of the SMEI mutations cause loss-of-function effects due to stop codons or deletions, demonstrating that haploinsufficiency of SCN1A is pathogenic. To understand this counterintuitive result, as reduced Na+ currents should lead to hypoexcitability rather than hyperexcitability, mouse models were generated by targeted deletion in SCN1A gene: notably, the loss of Nav1.1 channels substantially reduced Na+ currents only in hippocampal inhibitory interneurons of both Nav1.1+/− and Nav1.1−/− mice, but not in excitatory pyramidal neurons (Yu et al., 2006; Ogiwara et al., 2007), therefore causing a loss of sustained high-frequency firing in hippocampal and cortical interneurons, possibly explaining the hyperexcitability observed in patients affected by SMEI.
Alterations of GABAergic interneurons in Nav1.1−/− mice could also contribute to explain additional clinical manifestations occurring in SMEI patients: in fact, Nav1.1+/− mice have a reduced non-REM sleep, impaired circadian behavior, cognitive impairment and autistic-like traits, reminiscent of those observed in children with SMEI (Han et al., 2012).
Although the functional consequences of SMEI-associated mutations are generally recapitulated in SCN1A knockout mice, functional studies in heterologous expression systems of Nav1.1 channels incorporating GEFS+-associated mutations suggest that both loss- and gain-of-function effects may contribute to the pathogenesis of this disease. In particular, functional studies in an animal model of GEFS+ incorporating the R1648H mutation affecting the S4 in domain IV of Nav1.1 channels (Figure 3; Tang et al., 2009; Martin et al., 2010) have suggested that this variant reduced currents in both excitatory and inhibitory neurons, although in a different way: indeed, this variant reduced peak Na+ currents and enhanced slow inactivation in inhibitory neurons (possibly leading to hypoexcitability of inhibitory neurons), whereas it negatively shifted the voltage dependence of fast inactivation in excitatory neurons (possibly leading to hyperexcitability of excitatory neurons). As similarly observed for SMEI-model mice, the combination of these two mechanisms lead to a predominant reduction in vivo of GABAergic neurons excitability, therefore explaining the hyperexcitability observed in GEFS+-affected patients. More recently, a comprehensive neurophysiological analysis of this mouse model has confirmed that the R1648H mutation reduces firing in inhibitory neurons from various brain regions, but does not affect excitability of excitatory neurons (Hedrich et al., 2014).
Nav1.1 Channels and Familial Hemiplegic Migraine (FHM3)
Familial Hemiplegic Migraine (FHM) is a rare, monogenic subtype of migraine with aura, associated to mutations in three genes. In particular, FHM1 (OMIM 141500) is caused by mutations in the CACNA1A calcium channel subunit (see below), FHM2 is caused by mutations in the α2 subunit of the Na, K-ATPase pump (ATP1A2; OMIM 602481), and FHM3 has been more recently associated to mutations in Nav1.1 channels (de Vries et al., 2009; OMIM 609634). All these proteins are crucial regulators of ion fluxes across neuronal and glial cell membranes, suggesting that FHM, and possibly other forms of migraine, should be considered as cerebral ionopathies.
So far, only six FHM3-causing mutations have been identified: one of them (L1649Q) affects the DIV-S4 segment at the level of a residue following R1648, which is mutated both in SMEI- or GEFS+-affected patients. Other FHM3-causing mutations are present in the pore (L263V) or in the DIII-DIV linker (Q1489K/H and F1499L). Finally, one mutation in the intracellular DII-DIII linker (T1174S) has been recently found in a family affected by FHM and epilepsy. Complex alterations prompted by this mutation on the functional properties of Nav1.1 channels, including loss- (such as a rightward shift in the activation curve and a slower recovery from fast inactivation) and gain- (increased persistent sodium currents, INaP) of-function mechanisms each appear to be involved in the proepiletogenic and pro-FHM manifestations of the disease, respectively (Cestèle et al., 2013a).
The functional consequences prompted by the VSM-affecting mutation L1649Q were first assessed by introducing this mutation in the highly homologous human Nav1.5 (encoded by the SCN5A gene), revealing a slower inactivation and a faster recovery from inactivation, predicting enhanced neuronal excitation (Vanmolkot et al., 2007). However, a subsequent study, in which this mutation was introduced in Nav1.1 channels and co-expressed with both human β1 and β2 accessory subunits (Kahlig et al., 2008), revealed a complete loss of function, possibly because of a markedly reduced membrane expression of Nav1.1-L1649Q mutant channels. Notably, a subsequent paper has demonstrated that the exposure of transfected cells to permissive temperature (30°C for 36–48 h) before recordings, as well as the expression of this mutant in neurons at physiological temperature (37°C), was able to partially restore the function of Nav1.1-L1649Q channels, which therefore behaved like folding defective mutants; strikingly, rescued currents showed altered biophysical properties consistent with gain-of-function effects on neuronal hyperexcitability (Cestèle et al., 2013b).
Gain-of-function effects are also caused by the pore-affecting mutation L263V (Kahlig et al., 2008); by contrast, the Q1489K mutation appears to be associated to complex functional alterations, particularly in the inactivation process, leading to predominant loss-of-function effects when expressed in Nav1.5 (Vanmolkot et al., 2007) or in the long isoform of Nav1.1 (Kahlig et al., 2008) channels, rather than to predominant gain-of-function when introduced in the shorter isoform of Nav1.1 (the predominant variant expressed in the brain; Schaller et al., 1992) and studied by transfections of heterologous cells or cultured neurons (Cestèle et al., 2008).
Two additional Nav1.1 mutations not affecting the VSM but rather the DIII-DIV linker (Q1498H and F1499L) can also cause Elicited Repetitive Daily Blindness (ERDB), suggesting that SCN1A mutations can cause a complex spectrum of human disease, including epilepsy, FHM3 and/or retinal cell excitability (Vahedi et al., 2009).
Nav1.2 and Benign Familial Neonatal-Infantile Seizures (BFNIS)
Benign Familial Neonatal-Infantile Seizures (BFNIS or BFIS3; OMIM 607745) is a mild seizure syndrome caused by dominant mutations in SCN2A encoding for Nav1.2 channels (Heron et al., 2002). Affected individuals have seizures starting in early infancy (Berkovic et al., 2004). Typically, ictal episodes begin as partial seizures, which often become generalized. Febrile seizures are rare. Fortunately, these seizures favorably respond to treatment with anti-epileptic drugs, and they generally remit by 1 year of age. Nav1.2 channels are closely related to Nav1.1 channels in amino acid sequence and in their CNS expression pattern (Goldin et al., 2000). They are primarily expressed in unmyelinated axons in adult rodent brain (Westenbroek et al., 1989), being replaced by Nav1.6 channels during axon myelination (Rasband, 2010). Nav1.2 expression precedes that of Nav1.1, also increasing during the first 4 weeks of postnatal life in rodents (Gordon et al., 1987; Beckh et al., 1989). The relationship between the functional alterations in Nav1.2 channels and the hyperexcitability of BFNIS is not yet clear, also because not all BFNIS-causing mutations have been functionally characterized and because contrasting results have been obtained in some cases upon expression of the same mutant channels in different expression systems. However, for many VSM-affecting mutations, such as R223Q (in the DI-S4) and R1319Q (DIII-S4) gain-of-function effects have been reported (Sugawara et al., 2001; Scalmani et al., 2006), through complex biophysical alterations combining a hyperpolarized shift in the voltage-dependence of activation and a positive shift in the voltage-dependence of inactivation: these results suggest an increase in Na+ channels availability, leading to hyperexcitability. In addition, a further missense mutation (L1563V, within DIV-S2) in a specific Nav1.2 splice variant expressed at the neonatal stage caused a gain-of-function effect (Xu et al., 2007). Notably, this neonatal isoform has a more positive voltage-dependence of activation than the adult one, which may contribute to the relative hypoexcitability of the neonatal brain; moreover, since the effect of the mutation was specific for this neonatal variant, it appears likely that seizure disappearance after the neonatal period in BFNIS patients carrying this mutation is due to the switch between the two Nav1.2 isoforms or to the developmental switch between Nav1.2 and Nav1.6 channels (Liao et al., 2010b).
In addition to BFNIS, de novo dominant mutations in SCN2A can cause also more severe seizure syndromes (Kamiya et al., 2004; Ogiwara et al., 2009; Liao et al., 2010a; EIEE11; OMIM 613721): as recently reviewed (Baasch et al., 2014), 14 mutations in Nav1.2 appear to relate with BFNIS and 21 mutations with more severe phenotypes, including developmental delay and intractable seizures. One of the functionally characterized mutations affecting the VSM, namely the de novo E1211K mutation, affecting a highly-conserved residue in the DIII-S1 and found in a patient affected with intractable seizures and developmental delay, revealed complex alterations in the biophysical properties of Nav1.2 channels, including a hyperpolarization of both the half-activation and half-inactivation potentials and a delay in the recovery from inactivation, therefore leading to unpredictable results on mutation-dependent pathogenetic mechanisms. Intriguingly, computational modeling approaches such as those in Cestèle et al. (2013a) could predict the predominant mutation-induced alteration, therefore allowing a better understanding of the pathogenetic mechanisms associated to specific disease-causing mutations.
These results suggest that the spectrum of epilepsies associated to mutations in Nav1.2 channels may be similar to that observed for Nav1.1 mutations.
Nav1.3-1.6 Channels and Epilepsy
Recently, pediatric epileptic phenotypes have been also associated to mutations in other two isoforms of the Nav1 channel family, namely Nav1.3 (SCN3A) and Nav1.6 (SCN8A). Five missense Nav1.3 mutations have been identified in patients affected by cryptogenic focal epilepsy, mainly localized in the pore region, except for the D766N mutation, affecting a highly-conserved residue in the S2 segment of domain II (Holland et al., 2008; Vanoye et al., 2013). The characterization of the functional consequences prompted by these mutations in Nav1.3 channels revealed heterogeneous effects, including smaller current density, slower activation kinetics, depolarized voltage-dependence of activation and inactivation (particularly in the case of the R357Q mutation), or increased persistent currents (as in the case of the E1111K mutation). However, the presence of each of these mutations induces a common functional alteration in Nav1.3 currents, consisting in an increased current activation in response to depolarizing voltage ramps, which could contribute to neuronal hyperexcitability (Vanoye et al., 2013).
In addition, mutations in SCN8A have been recently identified in patients affected with epilepsy and/or intellectual disability (Larsen et al., 2015; Epileptic Encephalopathy-13, EIEE13; OMIM 614558). The role of this channel in neuronal excitability has been firmly assessed by numerous studies in animal models expressing diverse null SCN8 variants and revealing that a reduced expression of SCN8A-encoded Nav1.6 channels protects against seizures by decreasing neuronal excitability (O’Brien and Meisler, 2013). After the identification of the first mutation in the pore of Nav1.6 channels in a patient affected with a severe EE (N1768D; Veeramah et al., 2012), other mutations have been identified also in the VSM (R223G in the DI-S4, T767I in DII-S1, and others; Estacion et al., 2014; de Kovel et al., 2014; Blanchard et al., 2015; Kong et al., 2015). The functional properties of these mutant channels include both loss- and gain-of-function characteristics, revealing also in this case complex pathogenetic mechanisms underlying these severe phenotypes.
Nav1.7, 1.8, and 1.9 and Pain Disorders
Human genetic studies have clearly revealed a central role for Nav1.7, Nav1.8, and Nav1.9 Na+ channels in nociception. In fact, mutations in SCN9A (encoding for Nav1.7), SCN10A (encoding for Nav1.8) or SCN11A (encoding for Nav1.9) occur in many human disorders characterized by an altered pain sensation.
SCN9A mutations have been found in families affected by excruciating pain syndromes including Primary Erythermalgia (PE; OMIM 133020), Paroxysmal Extreme Pain Disorder (PEPD; OMIM 167400), or small-fiber neuropathy (OMIM 133020), as well as by Congenital Insensitivity to Pain (CIP; OMIM 243000), an autosomal recessive disorder in which patients are insensitive to pain caused by fractures, burns, dental extractions, and childbirth. Functional characterization of PE-, PEPD- or neuropathy-causing mutations (often occurring in the VSM, as in the case of W1538R in DIV-S2 and L823R in DII-S4; Lampert et al., 2009; Cregg et al., 2013) revealed gain-of-function effects through different mechanisms (including hyperpolarization in the activation voltage, slowing of the deactivation, increased current size, or impairment of channel inactivation). On the other hand, loss-of-function coding or splicing mutations in SCN9A leading to a substantial impairment in dorsal root ganglion (DRG) AP firing have been found in CIP families (Cox et al., 2006).
A wide genetic screen in patients with small-fiber neuropathy and negative for SCN9A mutations identified seven disease-causing variants in SCN10A (Familial Episodic Pain Syndrome, FEPS2; OMIM 615551). The functional consequences prompted by these mutations have been assessed only for some of them, revealing gain-of-function effects on Nav1.8 currents, and hyperexcitability of DRG neurons. None of these mutations has been found to affect the VSM (Faber et al., 2012; Huang et al., 2013; Han et al., 2014).
Other cohorts of patients affected by episodic pain or by small-fiber neuropathy have been also reported to carry mutations in the SCN11A gene (Familial Episodic Pain Syndrome type 3, FEPS3; OMIM 615552). Some of these mutations, which also affect the voltage sensing modules, increased Nav1.9 current density and enhanced excitability of DRG neurons (Zhang et al., 2013; Huang et al., 2014). On the other hand, a different mutation in SCN11A has been also identified in individuals with congenital inability to experience pain who suffer from recurrent tissue damage and severe mutilations (Leipold et al., 2013; Hereditary Sensory and Autonomic Neuropathy type 7, HSAN7; OMIM 615548). This mutation strongly hyperpolarized the voltage-dependence of Nav1.9 current activation (a gain-of-function change), while reducing the excitability of DRG neurons expressing the mutant channel (a loss-of-function at the cellular level). The enhanced activity of Nav1.9 channels is believed to lead to an inhibition of neuronal firing of DRGs because of a strong depolarization of the RMP which impairs transmission from presynaptic afferents to postsynaptic cells within the spinal cord, thereby producing pain insensitivity (Waxman et al., 2014).
Calcium Channelopathies
Structure and Function of Voltage-Gated Calcium Channels (VGCCs)
VGCCs are involved in a wide range of physiological processes, such as muscle contraction, neurotransmitter release, hormone secretion and gene expression. According to their threshold of activation, VGCCs are classified in two major categories: low voltage-activated (LVA) and high voltage-activated (HVA). Structurally, HVA are heteromultimeric protein complexes formed by co-assembling of a pore forming α1 subunit and α2, δ, β, γ ancillary subunits (Figure 1); by contrast, LVA channels appear to lack ancillary subunits (Catterall et al., 2005). There are three major families of Cav α1 subunits, namely Cav1, Cav2 and Cav3, each including several members: based on their biophysical and pharmacological features, HVA channels include Cav1 (encoding for L-type channels) and Cav2 (encoding for P/Q-, N- or R-type channels) families, whereas LVA channels comprise the Cav3 family (encoding for T-type channels). The Cav1 channel family encodes three different neuronal L-type channels (called Cav1.2, Cav1.3, and Cav1.4), in addition to a skeletal muscle-specific isoform Cav1.1 channel; the Cav2 channel family includes three members (Cav2.1, Cav2.2, and Cav2.3); among them, Cav2.1 channels gives rise to P- or Q-type channels by alternative splicing and assembly with specific ancillary subunits; Cav2.2 encodes for N-type channels and Cav2.3 for R-type channels. Cav3 channel family includes three members (Cav3.1, Cav3.2 and Cav3.3), corresponding to T-type calcium channels (Simms and Zamponi, 2014).
So far, several human disorders have been linked to mutations in VGCCs affecting the skeletal muscle and the nervous system. In particular, mutations in Cav1.2 are associated to Timothy Syndrome (TS), characterized by severe arrhythmia and multiple organ systems dysfunctions, including neurological disorders such as autism and mental retardation. By contrast, mutations in Cav3.2 channels have been found in various forms of idiopathic generalized epilepsies: disease-associated mutations prompted gain-of-function effects, leading to an increased neuronal excitability and changes in gene transcription (Eckle et al., 2014). To the best of our knowledge, no TS- or epilepsy-causing mutations in Cav1.2 or Cav3.2 genes, respectively, has been found to affect the VSM; thus, these mutations will not be further addressed in the present manuscript. By contrast, mutations in Cav2.1 are associated to migraine (FHM1) and ataxias (EA2 and SCA6); since mutations in Cav2.1 often affect the VSM, these diseases will be discussed in the following section.
Cav2.1 Mutations Linked to Familial Hemiplegic Migraine Type 1 (FHM1), Episodic Ataxia Type 2 (EA2) and Spinocerebellar Ataxia Type 6 (SCA6)
Cav2.1 channels are predominantly expressed in neuronal and neuroendocrine cells, where they play a crucial role in neurotransmitter release, particularly at excitatory synapses (Westenbroek et al., 1995). Based on their expression in areas involved in migraine and/or migraine pain pathogenesis, including the cerebral cortex, the trigeminal ganglia, and brainstem nuclei, as well as the cerebellum, it is not surprising that mutations in the CACNA1A gene encoding for Cav2.1 have been linked to neurological disorders, such as Familial Hemiplegic Migraine type 1 (FHM1; OMIM 141500), Episodic Ataxia type 2 (EA2; OMIM 108500) and Spinocerebellar Ataxia type 6 (SCA6; OMIM 183086); patients carrying Cav2.1 mutations often present significant overlapping symptoms, suggesting shared pathophysiological mechanisms (Pietrobon, 2013). As previously described for FHM3, FHM1 is a rare, autosomal dominant form of migraine with aura, characterized by recurrent attacks of disabling headache and, in some cases, progressive cerebellar atrophy. So far, 25 mutations in Cav2.1 have been found in FHM1-affected patients (de Vries et al., 2009; Pietrobon, 2013), mostly affecting the pore and the VSM (Figure 3). Functional expression in heterologous systems revealed that a common functional effect of FHM1 mutations is a shift in the activation curve toward hyperpolarized voltages, thus increasing Cav2.1 open probability and leading to gain-of-function effects (Tottene et al., 2002). In particular, the functional effects of the VSM-affecting R192Q mutation have been studied in a knock-in mouse model: electrophysiological recordings from cerebellar granule cells of this mouse showed an increase in Cav2.1 current density, a hyperpolarizing shift in the voltage-dependence of current activation, an increase in calcitonin gene-related peptide (CGRP) release and increased susceptibility to cortical spreading depression (Tottene et al., 2009), a mechanism that is believed to cause the migraine-preceding aura (Lauritzen, 1994). R192Q-induced gain-of-function of Cav2.1 channels appears also responsible for a larger AP-evoked calcium current and a longer AP duration in Capsaicin-Insensitive Trigeminal ganglion neurons (CI-T) when compared to CI-T neurons from WT mice (Fioretti et al., 2011). These effects appear rather specific for glutamatergic neurons, as also the probability of glutamate release appears enhanced in Cav2.1-R192Q expressing neurons (Tottene et al., 2009) when compared to wild-type neurons, whereas gating properties of the Cav2.1 channels expressed in GABAergic interneurons are barely affected (Vecchia et al., 2014).
EA2 is an autosomal dominant, paroxysmal cerebellar disorder, characterized by ataxia, migraine-like symptoms, interictal nystagmus and cerebellar atrophy. More than 20 CACNA1A mutations have been linked to EA2: most of them are nonsense (Jen et al., 1999) or missense (Guida et al., 2001; Wappl et al., 2002) mutations. Most nonsense mutations are predicted to truncate Cav2.1 subunits, resulting in loss-of-function effects; furthermore, their expression in heterologous systems together with wild-type channels produces dominant-negative effects (Jeng et al., 2006). In some cases, generalized epilepsy can also be found associated with EA2 (Jouvenceau et al., 2001), as reported for the R1820X nonsense mutation having both loss-of-function and dominant-negative effects on co-expressed wild-type Cav2.1. No EA2-causing mutation has been identified in the VSM.
SCA6 is an autosomal dominant, paroxysmal cerebellar disorder characterized by late-onset, slowly-progressive ataxia and Purkinje neuron degeneration. This disease is associated with a different degree of a CAG repeat expansion (20–33 repeats) at the distal C-terminus of Cav2.1 channels (Ishikawa et al., 1997; Zhuchenko et al., 1997): therefore, also in this case the VSM is not a hot-spot region for disease-causing mutations.
The VSM as a Molecular Target for Toxins and Syntetic Compounds
VGICs are molecular targets for several classes of drugs, including anticonvulsants, antiarrhythmics, and local anesthetic molecules; most of them recognize a binding site located within the pore region. More recently, the VSM also emerged as a novel pharmacological target for some classes of toxins and synthetic compounds. In VGNCs, three binding sites for neurotoxins have been identified in the VSM (Catterall et al., 2007). Site 3, located in the transmembrane loop between DIV-S3 and DIV-S4, is targeted by several groups of polypeptide toxins: α-scorpion toxins, sea-anemone toxins, and some spider toxins. These molecules slow or block Na+ channel inactivation by inhibiting the conformational changes coupling channel activation to fast inactivation (Rogers et al., 1996). This mechanism, called “voltage-sensor trapping” (Cestèle et al., 1998), may also describe the mode of action of other gating modifier toxins. Site 4, located in the S1–S2 and S3–S4 loops of domain II, is recognized by β-scorpion toxins. These toxins induce both a negative shift in the voltage-dependence of Na+ channel activation and a reduction in the maximal current. Finally, site 6, probably located near site 3, is occupied by δ-conotoxins that cause a slowing in Na+ channel inactivation. Toxin-induced sensor-trapping can also occur in VGCCs (agatoxins from spiders; McDonough, 2007), and in VGKCs of the Kv2 and Kv4 (hanatoxins from tarantula; Swartz and MacKinnon, 1997) or Kv3 and Kv11 (sea anemone toxins; Diochot and Lazdunski, 2009) subclasses. More recently, a novel molecular variant of AaTXKβ, namely AaTXKβ(2–64), from Androctonus australis scorpion venom has been shown to cause a hyperpolarization shift in the voltage-dependence of activation of Kv7.4 channels (Landoulsi et al., 2013) with a mechanism reminiscent of that described for β-scorpion toxins acting at site 4 of VGNCs (Dutertre and Lewis, 2010).
Besides toxins, also synthetic compounds may recognize a binding site located within the VSM; this pharmacological action has been exploited in some VGNCs and, among VGKCs, in the Kv7 subfamily. In fact, two compounds (ICA-121431 and PF-04856264) selectively inhibit currents from Nav1.1/Nav1.3 and Nav1.7 channels, respectively, by interacting with a site located at the extracellular ends of the S2 and S3 transmembrane segments of domain IV, a region that overlaps with the interaction sites for some scorpion and anemone toxins (McCormack et al., 2013). An analog of the analgesic drug diclofenac, called NH29, has been demonstrated to interact with an external groove formed by the interface of S1, S2, and S4 helices of the VSM in Kv7.2 channels, where it stabilizes the interaction between two conserved residues in S2 (E130) and S4 (R207). This compound shifts toward the left the activation curve and increases the maximal current of Kv7.2 channels, thereby acting as a non-toxin gating modifier (Peretz et al., 2010). By contrast, another anolog of diclofenac, called NH17, is able to block Kv7.2 channels recognizing a binding site in the VSM. Intriguingly, both compounds, NH17 and NH29, bind to the VSM of transient receptor potential vanilloid 1 (TRPV1) channels exerting opposite effects: indeed, they act as an activator and a blocker of TRPV1 currents, respectively. These two compounds also recognize a binding site in the VSM of the proton channel mVSOP (Kornilov et al., 2014). In addition, also another Kv7.2/3 channel opener, referred as ICA-27243, seems to interact with the VSM (Wickenden et al., 2008), although the specific amino acids involved in drug binding have not been yet identified (Padilla et al., 2009). The compound ztz240, discovered by a screening of 20,000 compounds using rubidium flux combined with atomic absorption spectrometry, increases Kv7.2 currents by interacting with the F137 residue in the gating charge pathway of Kv7.2 channels (Gao et al., 2010; Li et al., 2013). Interestingly, the same phenylalanine (F137) in the S2 of Kv7.2 forms the binding site for a previously reported Kv11.1 activator, NS1643, which also causes a leftward shift in current activation and slows current deactivation of Kv7.2 channels (Li et al., 2014a).
Conclusion
Although the general principles of voltage-sensing have been described by purely functional experiments well over 50 years ago, recent progress in apparently distinct fields of investigation ranging from molecular genetics to X-ray crystallography have contributed to the identification of human diseases caused by altered gating of a large number of VGICs, and to the description of the intimate molecular mechanisms of this process at nearly atomic level resolution. In the present work, we have described the diseases associated with mutations in the VSM of neuronal voltage-dependent K+, Na+, and Ca2+ channels, at the same time reporting on the described functional consequences of some of these disease-causing mutations and on the potential pathogenetic mechanisms. In addition, it should be underlined that the study of the functional consequences of naturally-occurring mutations, beside unraveling the potential disease pathogenetic mechanisms, provide unique opportunities to refine our current view of the role played by specific amino acids in channel function; as an example, the opposite functional behavior shown by Kv7.2 mutations affecting the distal end of S4 (causing a marked rightward shift in gating) when compared to those in the proximal end of the same transmembrane segment (which, instead, caused a negative gating shift), provides strong support to the current view of the molecular motions occurring in S4 during voltage-dependent gating. By this effort, we took the opportunity to highlight some of the major controversies and pressing issues in the field. Most channelopathies herein described have quite high penetrance and recognize the genetic mutations as a critical event in disease etiology; therefore, whenever possible, we have attempted to highlight genotype-phenotype correlations emerging from the detailed study of the extent of functional derangements found in most commonly used experimental systems. In an era of fast-advancing genomic techniques which allow the identification of a large number of variants in ion channel genes in several human diseases, such studies may contribute to a better definition of genotype-phenotype correlations, assisting in the definition of disease-based criteria to score variant severity, predict their pathogenetic role, and formulate prognostic hypotheses. Such efforts are likely to provide critical data which will be fed into current efforts to standardize and provide guidelines for the interpretation of sequence variants such as those recently provided by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015). Curation tools such as ClinGen3 or ClinVar,4 will be instrumental in the interpretation of the association between variants and genetic diseases, and are also likely to standardize phenotyping routines which may result in a more in-depth disease classification. Another crucial issue is the need for better experimental tools (cellular and animal models) to investigate the mechanism(s) by which each specific variant contributes to disease pathogenesis; similar clinical courses are apparently associated with both loss- and gain-of-function effects by specific ion channel variants, highlighting our rudimentary understanding of the molecular steps involved. Finally, given that the subtle structural transitions occurring during gating are likely specific and distinct for each ion channel family and subfamily, we also believe that the VSM is an underestimated target for drug action; we envision that an improved structural knowledge of channel gating may lead to the design of drugs effective on specific VSM states, thereby allowing patient-tailored therapies for rare diseases caused by each VSM mutation. Such drugs could be also of great therapeutic benefit for more common disorders involving changes in ion channel gating.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Telethon Grant GGP15113, the Fondazione San Paolo-IMI (Project Neuroscience), Regione Molise (Convenzione AIFA/Regione Molise), the Science and Technology Council of the Province of Avellino, Grant PRIN 2009 to MT, and the Scientific Independence of Young Researchers (SIR) Program to FM. In addition, postdoctoral grants from Fondazione Umberto Veronesi to FM and from Italian Society of Pharmacology to MVS supported this work.
Footnotes
- ^ http://mouse.brain-map.org
- ^ http://www.molgen.ua.ac.be/SCN1AMutations/home/Default.cfm
- ^ http://clinicalgenome.org/
- ^ http://www.ncbi.nlm.nih.gov/clinvar/
References
Adelman, J. P., Bond, C. T., Pessia, M., and Maylie, J. (1995). Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15, 1449–1454. doi: 10.1016/0896-6273(95)90022-5
Aggarwal, S. K., and MacKinnon, R. (1996). Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 16, 1169–1177. doi: 10.1016/s0896-6273(00)80143-9
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Armstrong, C. M., and Bezanilla, F. (1973). Currents related to movement of gating particles of sodium channels. Nature 242, 459–461. doi: 10.1038/242459a0
Baasch, A. L., Hüning, I., Gilissen, C., Klepper, J., Veltman, J. A., Gillessen-Kaesbach, G., et al. (2014). Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia and brain abnormalities. Epilepsia 55, e25–29. doi: 10.1111/epi.12554
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., and Langouet, M. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259. doi: 10.1038/ng.2441
Bechi, G., Rusconi, R., Cestèle, S., Striano, P., Franceschetti, S., and Mantegazza, M. (2015). Rescuable folding defective Nav1.1 (SCN1A) mutants in epilepsy: properties, occurrence and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol. Dis. 75, 100–114. doi: 10.1016/j.nbd.2014.12.028
Beckh, S., Noda, M., Lübbert, H., and Numa, S. (1989). Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 8, 3611–3616.
Bellini, G., Miceli, F., Soldovieri, M. V., Miraglia del Giudice, E., Coppola, G., and Taglialatela, M. (2010). “KCNQ2-related disorders,” in GeneReviews® [Internet], eds. R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, C. T. Fong, et al. (Seattle, WA: University of Washington).
Berkovic, S. F., Andermann, F., Carpenter, S., and Wolfe, L. S. (1986). Progressive myoclonus epilepsies: specific causes and diagnosis. N. Engl. J. Med. 315, 296–305. doi: 10.1056/nejm198607313150506
Berkovic, S. F., Heron, S. E., Giordano, L., Marini, C., Guerrini, R., Kaplan, R. E., et al. (2004). Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann. Neurol. 55, 550–557. doi: 10.1002/ana.20029
Bernier, V., Lagacé, M., Bichet, D. G., and Bouvier, M. (2004). Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol. Metab. 15, 222–228. doi: 10.1016/j.tem.2004.05.003
Bezanilla, F., White, M. M., and Taylor, R. E. (1982). Gating currents associated with potassium channel activation. Nature 296, 657–659. doi: 10.1038/296657a0
Bhate, M. P., and McDermott, A. E. (2012). Protonation state of E71 in KcsA and its role for channel collapse and inactivation. Proc. Natl. Acad. Sci. U S A 109, 15265–15270. doi: 10.1073/pnas.1211900109
Biervert, C., Schroeder, B. C., Kubisch, C., Berkovic, S. F., Propping, P., Jentsch, T. J., et al. (1998). A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406. doi: 10.1126/science.279.5349.403
Blanchard, M. G., Willemsen, M. H., Walker, J. B., Dib-Hajj, S. D., Waxman, S. G., Jongmans, M. C., et al. (2015). De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J. Med. Genet. 52, 330–337. doi: 10.1136/jmedgenet-2014-102813
Blunck, R., and Batulan, Z. (2012). Mechanism of electromechanical coupling in voltage-gated potassium channels. Front. Pharmacol. 3:166. doi: 10.3389/fphar.2012.00166
Bocksteins, E., and Snyders, D. J. (2012). Electrically silent Kv subunits: their molecular and functional characteristics. Physiology (Bethesda) 27, 73–84. doi: 10.1152/physiol.00023.2011
Caldwell, J. H., Schaller, K. L., Lasher, R. S., Peles, E., and Levinson, S. R. (2000). Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites and synapses. Proc. Natl. Acad. Sci. U S A 97, 5616–5620. doi: 10.1073/pnas.090034797
Camacho, J. (2006). Ether a go-go potassium channels and cancer. Cancer Lett. 233, 1–9. doi: 10.1016/j.canlet.2005.02.016
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Castaldo, P., del Giudice, E. M., Coppola, G., Pascotto, A., Annunziato, L., and Taglialatela, M. (2002). Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J. Neurosci. 22:RC199.
Catterall, W. A. (1986). Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55, 953–985. doi: 10.1146/annurev.biochem.55.1.953
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi: 10.1016/S0896-6273(00)81133-2
Catterall, W. A. (2010). Ion channel voltage sensors: structure, function and pathophysiology. Neuron 67, 915–928. Review. doi: 10.1016/j.neuron.2010.08.021
Catterall, W. A., Cestèle, S., Yarov-Yarovoy, V., Yu, F. H., Konoki, K., and Scheuer, T. (2007). Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141. doi: 10.1016/j.toxicon.2006.09.022
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 28, 11768–11777. doi: 10.1523/JNEUROSCI.3901-08.2008
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). Nav1.1 channels and epilepsy. J. Physiol. 588, 1849–1859. doi: 10.1113/jphysiol.2010.187484
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005). International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425. doi: 10.1124/pr.57.4.5
Cestèle, S., Labate, A., Rusconi, R., Tarantino, P., Mumoli, L., Franceschetti, S., et al. (2013a). Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 54, 927–935. doi: 10.1111/epi.12123
Cestèle, S., Schiavon, E., Rusconi, R., Franceschetti, S., and Mantegazza, M. (2013b). Nonfunctional Nav1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc. Natl. Acad. Sci. U S A 110, 17546–17551. doi: 10.1073/pnas.1309827110
Cestèle, S., Qu, Y., Rogers, J. C., Rochat, H., Scheuer, T., and Catterall, W. A. (1998). Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3–S4 loop in domain II. Neuron 21, 919–931. doi: 10.1016/S0896-6273(00)80606-6
Cestèle, S., Scalmani, P., Rusconi, R., Terragni, B., Franceschetti, S., and Mantegazza, M. (2008). Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J. Neurosci. 28, 7273–7283. doi: 10.1523/JNEUROSCI.4453-07.2008
Chapman, M. L., and VanDongen, A. M. (2005). K channel subconductance levels result from heteromeric pore conformations. J. Gen. Physiol. 126, 87–103. doi: 10.1085/jgp.200509253
Charlier, C., Singh, N. A., Ryan, S. G., Lewis, T. B., Reus, B. E., Leach, R. J., et al. (1998). A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 18, 53–55. doi: 10.1038/ng0198-53
Cox, J. J., Reimann, F., Nicholas, A. K., Thornton, G., Roberts, E., Springell, K., et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. doi: 10.1038/nature05413
Cregg, R., Laguda, B., Werdehausen, R., Cox, J. J., Linley, J. E., Ramirez, J. D., et al. (2013). Novel mutations mapping to the fourth sodium channel domain of Nav1.7 result in variable clinical manifestations of primary erythromelalgia. Neuromolecular Med. 15, 265–278. doi: 10.1007/s12017-012-8216-8
Cuello, L. G., Jogini, V., Cortes, D. M., Pan, A. C., Gagnon, D. G., Dalmas, O., et al. (2010a). Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature 466, 272–275. doi: 10.1038/nature09136
Cuello, L. G., Jogini, V., Cortes, D. M., and Perozo, E. (2010b). Structural mechanism of C-type inactivation in K(+) channels. Nature 466, 203–208. doi: 10.1038/nature09153
D’Adamo, M. C., Hanna, M. G., Di Giovanni, G., and Pessia, M. (2014). “Episodic ataxia type 1,” in Source GeneReviews® [Internet], eds R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, C. T. Fong, et al. (Seattle, WA: University of Washington), 1993–2014.
de Kovel, C. G., Meisler, M. H., Brilstra, E. H., van Berkestijn, F. M., van ’t Slot, R., van Lieshout, S., et al. (2014). Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res. 108, 1511–1518. doi: 10.1016/j.eplepsyres.2014.08.020
de Vries, B., Frants, R. R., Ferrari, M. D., and van den Maagdenberg, A. M. (2009). Molecular genetics of migraine. Hum. Genet. 126, 115–132. doi: 10.1007/s00439-009-0684-z
Diochot, S., and Lazdunski, M. (2009). Sea anemone toxins affecting potassium channels. Prog. Mol. Subcell. Biol. 46, 99–122. doi: 10.1007/978-3-540-87895-7_4
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. doi: 10.1126/science.280.5360.69
Dravet, C., Bureau, M., Guerrini, R., Giraud, N., and Roger, J. (1992). “Severe myoclonic epilepsy in infants,” in Epileptic Syndromes in Infancy, Childhood and Adolescence, eds Roger, J., Dravet, C., Bureau, M., Dreifus, F. E., Perret, A., and Wolf, P. 2nd Edn. (London: John Libbey), 75–102.
Dutertre, S., and Lewis, R. J. (2010). Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 285, 13315–13320. doi: 10.1074/jbc.R109.076596
Eckle, V. S., Shcheglovitov, A., Vitko, I., Dey, D., Yap, C. C., Winckler, B., et al. (2014). Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J. Physiol. 592, 795–809. doi: 10.1113/jphysiol.2013.264176
Engel, J. Jr., and International League Against Epilepsy [ILAE]. (2001). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE task force on classification and terminology. Epilepsia 42, 796–803. doi: 10.1046/j.1528-1157.2001.10401.x
Erisir, A., Lau, D., Rudy, B., and Leonard, C. S. (1999). Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J. Neurophysiol. 82, 2476–2489.
Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., et al. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345. doi: 10.1038/74159
Estacion, M., O’Brien, J. E., Conravey, A., Hammer, M. F., Waxman, S. G., Dib-Hajj, S. D., et al. (2014). A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 69, 117–123. doi: 10.1016/j.nbd.2014.05.017
Faber, C. G., Lauria, G., Merkies, I. S., Cheng, X., Han, C., Ahn, H. S., et al. (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U S A 109, 19444–19449. doi: 10.1073/pnas.1216080109
Figueroa, K. P., Minassian, N. A., Stevanin, G., Waters, M., Garibyan, V., Forlani, S., et al. (2010). KCNC3: phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum. Mutat. 31, 191–196. doi: 10.1002/humu.21165
Fioretti, B., Catacuzzeno, L., Sforna, L., Gerke-Duncan, M. B., van den Maagdenberg, A. M., Franciolini, F., et al. (2011). Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 589(Pt. 23), 5879–5895. doi: 10.1113/jphysiol.2011.220533
Gao, Z., Zhang, T., Wu, M., Xiong, Q., Sun, H., Zhang, Y., et al. (2010). Isoform-specific prolongation of Kv7 (KCNQ) potassium channel opening mediated by new molecular determinants for drug-channel interactions. J. Biol. Chem. 285, 28322–28332. doi: 10.1074/jbc.M110.116392
Goldin, A. L. (2001). Resurgence of sodium channel research. Annu. Rev. Physiol. 63, 871–894. doi: 10.1146/annurev.physiol.63.1.871
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. doi: 10.1016/S0896-6273(00)00116-1
Gordon, D., Merrick, D., Auld, V., Dunn, R., Goldin, A. L., Davidson, N., et al. (1987). Tissue-specific expression of the RI and RII sodium channel subtypes. Proc. Natl. Acad. Sci. U S A 84, 8682–8686. doi: 10.1073/pnas.84.23.8682
Gosselin-Badaroudine, P., Keller, D. I., Huang, H., Pouliot, V., Chatelier, A., Osswald, S., et al. (2012). A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One 7:e38331. doi: 10.1371/journal.pone.0038331
Graves, T. D., Rajakulendran, S., Zuberi, S. M., Morris, H. R., Schorge, S., Hanna, M. G., et al. (2010). Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 75, 367–372. doi: 10.1212/WNL.0b013e3181ea9ee3
Guida, S., Trettel, F., Pagnutti, S., Mantuano, E., Tottene, A., Veneziano, L., et al. (2001). Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am. J. Hum. Genet. 68, 759–764. doi: 10.1086/318804
Guy, H. R., and Seetharamulu, P. (1986). Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U S A 83, 508–512. doi: 10.1073/pnas.83.2.508
Han, C., Vasylyev, D., Macala, L. J., Gerrits, M. M., Hoeijmakers, J. G., Bekelaar, K. J., et al. (2014). The G1662S Nav1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 85, 499–505. doi: 10.1136/jnnp-2013-306095
Han, S., Yu, F. H., Schwartz, M. D., Linton, J. D., Bosma, M. M., Hurley, J. B., et al. (2012). Nav1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc. Natl. Acad. Sci. U S A 109, E368–E377. doi: 10.1073/pnas.1115729109
Hedrich, U. B., Liautard, C., Kirschenbaum, D., Pofahl, M., Lavigne, J., Liu, Y., et al. (2014). Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Nav1.1 mutation. J. Neurosci. 34, 14874–14889. doi: 10.1523/JNEUROSCI.0721-14.2014
Heinemann, S. H., Terlau, H., Stühmer, W., Imoto, K., and Numa, S. (1992). Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 356, 441–443. doi: 10.1038/356441a0
Heron, S. E., Crossland, K. M., Andermann, E., Phillips, H. A., Hall, A. J., Bleasel, A., et al. (2002). Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360, 851–852. doi: 10.1016/s0140-6736(02)09968-3
Heron, S. E., Smith, K. R., Bahlo, M., Nobili, L., Kahana, E., and Licchetta, L. (2012). Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190. doi: 10.1038/ng.2440
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544. doi: 10.1113/jphysiol.1952.sp004764
Holland, K. D., Kearney, J. A., Glauser, T. A., Buck, G., Keddache, M., Blankston, J. R., et al. (2008). Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci. Lett. 433, 65–70. doi: 10.1016/j.neulet.2007.12.064
Hoshi, T., Zagotta, W. N., and Aldrich, R. W. (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538. doi: 10.1126/science.2122519
Huang, J., Han, C., Estacion, M., Vasylyev, D., Hoeijmakers, J. G., Gerrits, M. M., et al. (2014). Gain-of-function sodium channel Nav1.9 mutations in painful neuropathy. Brain 137, 1627–1642. doi: 10.1093/brain/awu079
Huang, J., Yang, Y., Zhao, P., Gerrits, M. M., Hoeijmakers, J. G., Bekelaar, K., et al. (2013). Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J. Neurosci. 33, 14087–14097. doi: 10.1523/JNEUROSCI.2710-13.2013
Irie, T., Matsuzaki, Y., Sekino, Y., and Hirai, H. (2014). Kv3.3 channels harbouring a mutation of spinocerebellar ataxia type 13 alter excitability and induce cell death in cultured cerebellar Purkinje cells. J. Physiol. 592, 229–247. doi: 10.1113/jphysiol.2013.264309
Ishikawa, K., Tanaka, H., Saito, M., Ohkoshi, N., Fujita, T., Yoshizawa, K., et al. (1997). Japanese families with autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1–p13.2 and are strongly associated with mild CAG expansions in the spinocerebellar ataxia type 6 gene in chromosome 19p13.1. Am. J. Hum. Genet. 61, 336–346. doi: 10.1086/514867
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F., Offord, J., Charbonneau, H., et al. (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. doi: 10.1126/science.1375395
Isom, L. L., Ragsdale, D. S., De Jongh, K. S., Westenbroek, R. E., Reber, B. F., Scheuer, T., et al. (1995). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442. doi: 10.1016/0092-8674(95)90121-3
Jen, J., Yue, Q., Nelson, S. F., Yu, H., Litt, M., Nutt, J., et al. (1999). A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia. Neurology 53, 34–37. doi: 10.1212/WNL.53.1.34
Jeng, C. J., Chen, Y. T., Chen, Y. W., and Tang, C. Y. (2006). Dominant-negative effects of human P/Q-type Ca2+ channel mutations associated with episodic ataxia type 2. Am. J. Physiol. Cell Physiol. 290, C1209–C1220. doi: 10.1152/ajpcell.00247.2005
Jensen, M. Ø., Jogini, V., Borhani, D. W., Leffler, A. E., Dror, R. O., and Shaw, D. E. (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. doi: 10.1126/science.1216533
Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T., and MacKinnon, R. (2002a). Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–522. doi: 10.1038/417515a
Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T., and MacKinnon, R. (2002b). The open pore conformation of potassium channels. Nature 417, 523–526. doi: 10.1038/417523a
Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T., et al. (2003). X-ray structure of a voltage dependent K+ channel. Nature 423, 33–41. doi: 10.1038/nature01580
Jorge, B. S., Campbell, C. M., Miller, A. R., Rutter, E. D., Gurnett, C. A., Vanoye, C. G., et al. (2011). Voltage gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc. Natl. Acad. Sci. U S A 108, 5443–5448. doi: 10.1073/pnas.1017539108
Jouvenceau, A., Eunson, L. H., Spauschus, A., Ramesh, V., Zuberi, S. M., Kullmann, D. M., et al. (2001). Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 358, 801–807. doi: 10.1016/s0140-6736(01)05971-2
Kahlig, K. M., Rhodes, T. H., Pusch, M., Freilinger, T., Pereira-Monteiro, J. M., Ferrari, M. D., et al. (2008). Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. U S A 105, 9799–9804. doi: 10.1073/pnas.0711717105
Kamiya, K., Kaneda, M., Sugawara, T., Mazaki, E., Okamura, N., Montal, M., et al. (2004). A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J. Neurosci. 24, 2690–2698. doi: 10.1523/jneurosci.3089-03.2004
Kanai, K., Hirose, S., Oguni, H., Fukuma, G., Shirasaka, Y., Miyajima, T., et al. (2004). Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology 63, 329–334. doi: 10.1212/01.WNL.0000129829.31179.5B
Kato, M., Yamagata, T., Kubota, M., Arai, H., Yamashita, S., Nakagawa, T., et al. (2013). Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287. doi: 10.1111/epi.12200
Khalili-Araghi, F., Jogini, V., Yarov-Yarovoy, V., Tajkhorshid, E., Roux, B., and Schulten, K. (2010). Calculation of the gating charge for the Kv1.2 voltage-activated potassium channel. Biophys. J. 98, 2189–2198. doi: 10.1016/j.bpj.2010.02.056
Koishi, R., Xu, H., Ren, D., Navarro, B., Spiller, B. W., Shi, Q., et al. (2004). A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. 279, 9532–9538. doi: 10.1074/jbc.m313100200
Kong, W., Zhang, Y., Gao, Y., Liu, X., Gao, K., Xie, H., et al. (2015). SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia 56, 431–438. doi: 10.1111/epi.12925
Kornilov, P., Peretz, A., Lee, Y., Son, K., Lee, J. H., Refaeli, B., et al. (2014). Promiscuous gating modifiers target the voltage sensor of K(v)7.2, TRPV1 and H(v)1 cation channels. FASEB J. 28, 2591–2602. doi: 10.1096/fj.14-250647
Kuo, A., Gulbis, J. M., Antcliff, J. F., Rahman, T., Lowe, E. D., Zimmer, J., et al. (2003). Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926. doi: 10.1126/science.1085028
Labro, A. J., and Snyders, D. J. (2012). Being flexible: the voltage-controllable activation gate of Kv channels. Front. Pharmacol. 3:168. doi: 10.3389/fphar.2012.00168
Lainé, M., Lin, M. C., Bannister, J. P., Silverman, W. R., Mock, A. F., Roux, B., et al. (2003). Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron 39, 467–481. doi: 10.1016/s0896-6273(03)00468-9
Lampert, A., Dib-Hajj, S. D., Eastman, E. M., Tyrrell, L., Lin, Z., Yang, Y., et al. (2009). Erythromelalgia mutation L823R shifts activation and inactivation of threshold sodium channel Nav1.7 to hyperpolarized potentials. Biochem. Biophys. Res. Commun. 390, 319–324. doi: 10.1016/j.bbrc.2009.09.121
Landoulsi, Z., Miceli, F., Palmese, A., Amoresano, A., Marino, G., El Ayeb, M., et al. (2013). Subtype-selective activation of K(v)7 channels by AaTXKβ(2–64), a novel toxin variant from the Androctonus australis scorpion venom. Mol. Pharmacol. 84, 763–773. doi: 10.1124/mol.113.088971
Larsen, J., Carvill, G. L., Gardella, E., Kluger, G., Schmiedel, G., Barisic, N., et al. (2015). The phenotypic spectrum of SCN8A encephalopathy. Neurology 84, 480–489. doi: 10.1212/wnl.0000000000001211
Lau, D., Vega-Saenz de Miera, E. C., Contreras, D., Ozaita, A., Harvey, M., Chow, A., et al. (2000). Impaired fast-spiking, suppressed cortical inhibition and increased susceptibility to seizures in mice lacking Kv3.2 K+ channel proteins. J. Neurosci. 20, 9071–9085.
Lauritzen, M. (1994). Pathophysiology of the migraine aura. The spreading depression theory. Brain 117, 199–210. doi: 10.1093/brain/117.1.199
Lee, S. Y., Banerjee, A., and MacKinnon, R. (2009). Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS Biol. 7:e47. doi: 10.1371/journal.pbio.1000047
Lehmann-Horn, F., and Jurkat-Rott, K. (1999). Voltage-gated ion channels and hereditary disease. Physiol. Rev. 79, 1317–1372.
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Giesselmann, S., Baets, J., et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404. doi: 10.1038/ng.2767
Li, P., Chen, Z., Xu, H., Sun, H., Li, H., Liu, H., et al. (2013). The gating charge pathway of an epilepsy-associated potassium channel accommodates chemical ligands. Cell Res. 23, 1106–1118. doi: 10.1038/cr.2013.82
Li, P., Chen, X., Zhang, Q., Zheng, Y., Jiang, H., Yang, H., et al. (2014a). The human ether-a-go-go-related gene activator NS1643 enhances epilepsy-associated KCNQ channels. J. Pharmacol. Exp. Ther. 351, 596–604. doi: 10.1124/jpet.114.217703
Li, T., Tian, C., Scalmani, P., Frassoni, C., Mantegazza, M., Wang, Y., et al. (2014b). Action potential initiation in neocortical inhibitory interneurons. PLoS Biol. 12:e1001944. doi: 10.1371/journal.pbio.1001944
Li, Q., Wanderling, S., Paduch, M., Medovoy, D., Singharoy, A., McGreevy, R., et al. (2014c). Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat. Struct. Mol. Biol. 21, 244–252. doi: 10.1038/nsmb.2768
Li, Y., Gamper, N., Hilgemann, D. W., and Shapiro, M. S. (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835. doi: 10.1523/jneurosci.2597-05.2005
Liao, Y., Anttonen, A. K., Liukkonen, E., Gaily, E., Maljevic, S., Schubert, S., et al. (2010a). SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus and pain. Neurology 75, 1454–1458. doi: 10.1212/wnl.0b013e3181f8812e
Liao, Y., Deprez, L., Maljevic, S., Pitsch, J., Claes, L., Hristova, D., et al. (2010b). Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133(Pt. 5), 1403–1414. doi: 10.1093/brain/awq057
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005a). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903. doi: 10.1126/science.1116269
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005b). Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908. doi: 10.1126/science.1116270
Long, S. B., Tao, X., Campbell, E. B., and MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. doi: 10.1038/nature06265
Lu, Z., Klem, A. M., and Ramu, Y. (2002). Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120, 663–676. doi: 10.1085/jgp.20028696
Martin, M. S., Dutt, K., Papale, L. A., Dubé, C. M., Dutton, S. B., de Haan, G., et al. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834. doi: 10.1074/jbc.m109.078568
McCormack, K., Santos, S., Chapman, M. L., Krafte, D. S., Marron, B. E., West, C. W., et al. (2013). Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. U S A 110, E2724–E2732. doi: 10.1073/pnas.1220844110
McDonough, S. I. (2007). Gating modifier toxins of voltage-gated calcium channels. Toxicon 49, 202–212. doi: 10.1016/j.toxicon.2006.09.018
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc. Natl. Acad. Sci. U S A 110, 4386–4391. doi: 10.1073/pnas.1216867110
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 35, 3782–3793. doi: 10.1523/jneurosci.4423-14.2015
Miceli, F., Soldovieri, M. V., Hernandez, C. C., Shapiro, M. S., Annunziato, L., and Taglialatela, M. (2008). Gating consequences of charge neutralization of arginine residues in the S4 segment of Kv7.2, an epilepsy-linked K+ channel subunit. Biophys. J. 95, 2254–2264. doi: 10.1529/biophysj.107.128371
Miceli, F., Soldovieri, M. V., Iannotti, F. A., Barrese, V., Ambrosino, P., Martire, M., et al. (2011). The voltage-sensing domain of K(v)7.2 channels as a molecular target for epilepsy-causing mutations and Anticonvulsants. Front. Pharmacol. 2:2. doi: 10.3389/fphar.2011.00002
Miceli, F., Soldovieri, M. V., Lugli, L., Bellini, G., Ambrosino, P., Migliore, M., et al. (2009). Neutralization of a unique, negatively-charged residue in the voltage sensor of Kv7.2 subunits in a sporadic case of benign familial neonatal seizures. Neurobiol. Dis. 34, 501–510. doi: 10.1016/j.nbd.2009.03.009
Miceli, F., Vargas, E., Bezanilla, F., and Taglialatela, M. (2012). Gating currents from Kv7 channels carrying neuronal hyperexcitability mutations in the voltage-sensing domain. Biophys. J. 102, 1372–1382. doi: 10.1016/j.bpj.2012.02.004
Milligan, C. J., Li, M., Gazina, E. V., Heron, S. E., Nair, U., and Trager, C. (2014). KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75, 581–590. doi: 10.1002/ana.24128
Moreau, A., Gosselin-Badaroudine, P., and Chahine, M. (2014). Biophysics, pathophysiology and pharmacology of ion channel gating pores. Front. Pharmacol. 5:53. doi: 10.3389/fphar.2014.00053
Muona, M., Berkovic, S. F., Dibbens, L. M., Oliver, K. L., Maljevic, S., Bayly, M. A., et al. (2015). A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet. 47, 39–46. doi: 10.1038/ng.3144
Murakoshi, H., and Trimmer, J. S. (1999). Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J. Neurosci. 19, 1728–1735.
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K., and Okamura, Y. (2005). Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. doi: 10.1038/nature03650
Muroi, Y., Arcisio-Miranda, M., Chowdhury, S., and Chanda, B. (2010). Molecular determinants of coupling between the domain III voltage sensor and pore of a sodium channel. Nat. Struct. Mol. Biol. 17, 230–237. doi: 10.1038/nsmb.1749
Noda, M., Shimizu, S., Tanabe, T., Takai, T., Kayano, T., Ikeda, T., et al. (1984). Primary structure of electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127. doi: 10.1038/312121a0
O’Brien, J. E., and Meisler, M. H. (2013). Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 4:213. doi: 10.3389/fgene.2013.00213
Ogiwara, I., Ito, K., Sawaishi, Y., Osaka, H., Mazaki, E., Inoue, I., et al. (2009). De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 73, 1046–14053. doi: 10.1212/wnl.0b013e3181b9cebc
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. doi: 10.1523/jneurosci.5270-06.2007
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Löffler, H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. doi: 10.1002/ana.24080
Padilla, K., Wickenden, A. D., Gerlach, A. C., and McCormack, K. (2009). The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci. Lett. 465, 138–142. doi: 10.1016/j.neulet.2009.08.071
Papazian, D. M., Schwarz, T. L., Tempel, B. L., Jan, Y. N., and Jan, L. Y. (1987). Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from drosophila. Science 237, 749–753. doi: 10.1126/science.2441470
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi: 10.1038/nature10238
Peretz, A., Pell, L., Gofman, Y., Haitin, Y., Shamgar, L., Patrich, E., et al. (2010). Targeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifier. Proc. Natl. Acad. Sci. U S A 107, 15637–15642. doi: 10.1073/pnas.0911294107
Peters, C. J., Werry, D., Gill, H. S., Accili, E. A., and Fedida, D. (2011). Mechanism of accelerated current decay caused by an episodic ataxia type-1-associated mutant in a potassium channel pore. J. Neurosci. 31, 17449–17459. doi: 10.1523/jneurosci.2940-11.2011
Pietrobon, D. (2013). Calcium channels and migraine. Biochim. Biophys. Acta 1828, 1655–1665. doi: 10.1016/j.bbamem.2012.11.012
Pongs, O., and Schwarz, J. R. (2010). Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796. doi: 10.1152/physrev.00020.2009
Prole, D. L., and Yellen, G. (2006). Reversal of HCN channel voltage dependence via bridging of the S4-S5 linker and post-S6. J. Gen. Physiol. 128, 273–282. doi: 10.1085/jgp.200609590
Ramsey, I. S., Moran, M. M., Chong, J. A., and Clapham, D. E. (2006). A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216. doi: 10.1038/nature04700
Rasband, M. N. (2010). The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562. doi: 10.1038/nrn2852
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. doi: 10.1016/S0140-6736(12)61480-9
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., and Clapham, D. E. (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. doi: 10.1126/science.1065635
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Rogers, J. C., Qu, Y., Tanada, T. N., Scheuer, T., and Catterall, W. A. (1996). Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 271, 15950–15962. doi: 10.1074/jbc.271.27.15950
Rudy, B., and McBain, C. J. (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526. doi: 10.1016/s0166-2236(00)01892-0
Rusconi, R., Scalmani, P., Cassulini, R. R., Giunti, G., Gambardella, A., Franceschetti, S., et al. (2007). Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J. Neurosci. 27, 11037–11046. doi: 10.1523/jneurosci.3515-07.2007
Sabatini, B. L., and Regehr, W. G. (1997). Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J. Neurosci. 17, 3425–3435.
Saganich, M. J., Vega-Saenz de Miera, E., Nadal, M. S., Baker, H., Coetzee, W. A., and Rudy, B. (1999). Cloning of components of a novel subthreshold-activating K(+) channel with a unique pattern of expression in the cerebral cortex. J. Neurosci. 19, 10789–10802.
Saitsu, H., Kato, M., Koide, A., Goto, T., Fujita, T., Nishiyama, K., et al. (2012). Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann. Neurol. 72, 298–300. doi: 10.1002/ana.23620
Sasaki, M., Takagi, M., and Okamura, Y. (2006). A voltage sensor-domain protein is a voltage-gated proton channel. Science 312, 589–592. doi: 10.1126/science.1122352
Scalmani, P., Rusconi, R., Armatura, E., Zara, F., Avanzini, G., Franceschetti, S., et al. (2006). Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci. 26, 10100–10109. doi: 10.1523/jneurosci.2476-06.2006
Schaller, K. L., Krzemien, D. M., McKenna, N. M., and Caldwell, J. H. (1992). Alternatively spliced sodium channel transcripts in brain and muscle. J. Neurosci. 12, 1370–1381.
Schoppa, N. E., McCormack, K., Tanouye, M. A., and Sigworth, F. J. (1992). The size of gating charge in wild-type and mutant Shaker potassium channels. Science 255, 1712–1715. doi: 10.1126/science.1553560
Schwake, M., Pusch, M., Kharkovets, T., and Jentsch, T. J. (2000). Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J. Biol. Chem. 275, 13343–13348. doi: 10.1074/jbc.275.18.13343
Seoh, S. A., Sigg, D., Papazian, D. M., and Bezanilla, F. (1996). Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 16, 1159–1167. doi: 10.1016/s0896-6273(00)80142-7
Simeone, T. A., Simeone, K. A., Samson, K. K., Kim do, Y., and Rho, J. M. (2013). Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol. Dis. 54, 68–81. doi: 10.1016/j.nbd.2013.02.009
Simms, B. A., and Zamponi, G. W. (2014). Neuronal voltage-gated calcium channels: structure, function and dysfunction. Neuron 82, 24–45. doi: 10.1016/j.neuron.2014.03.016
Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., et al. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29. doi: 10.1038/ng0198-25
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78. doi: 10.1038/nature05598
Sokolov, S., Scheuer, T., and Catterall, W. A. (2008). Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl. Acad. Sci. U S A 105, 19980–19985. doi: 10.1073/pnas.0810562105
Soldovieri, M. V., Castaldo, P., Iodice, L., Miceli, F., Barrese, V., Bellini, G., et al. (2006). Decreased subunit stability as a novel mechanism for potassium current impairment by a KCNQ2 C-terminus mutation causing benign familial neonatal convulsions. J. Biol. Chem. 281, 418–428. doi: 10.1074/jbc.m510980200
Soldovieri, M. V., Miceli, F., and Taglialatela, M. (2011). Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology (Bethesda) 26, 365–376. doi: 10.1152/physiol.00009.2011
Soler-Llavina, G. J., Chang, T. H., and Swartz, K. J. (2006). Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron 52, 623–634. doi: 10.1016/j.neuron.2006.10.005
Struyk, A. F., and Cannon, S. C. (2007). A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20. doi: 10.1085/jgp.200709755
Sugawara, T., Tsurubuchi, Y., Agarwala, K. L., Ito, M., Fukuma, G., Mazaki-Miyazaki, E., et al. (2001). A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc. Natl. Acad. Sci. U S A 98, 6384–6389. doi: 10.1073/pnas.111065098
Sugiura, Y., Ogiwara, I., Hoshi, A., Yamakawa, K., and Ugawa, Y. (2012). Different degrees of loss of function between GEFS+ and SMEI Nav1.1 missense mutants at the same residue induced by rescuable folding defects. Epilepsia 53, e111–e114. doi: 10.1111/j.1528-1167.2012.03467.x
Swartz, K. J., and MacKinnon, R. (1997). Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron 18, 665–673. doi: 10.1016/s0896-6273(00)80306-2
Syrbe, S., Hedrich, U. B., Riesch, E., Djémié, T., Müller, S., Møller, R. S., et al. (2015). De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 47, 393–399. doi: 10.1038/ng.3239
Tanabe, T., Takeshima, H., Mikami, A., Flockerzi, V., Takahashi, H., Kangawa, K., et al. (1987). Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature 328, 313–318. doi: 10.1038/328313a0
Tang, B., Dutt, K., Papale, L., Rusconi, R., Shankar, A., Hunter, J., et al. (2009). A BAC transgenic mouse model reveals neuron subtype-specific effects of a generalized epilepsy with febrile seizures plus (GEFS+) mutation. Neurobiol. Dis. 35, 91–102. doi: 10.1016/j.nbd.2009.04.007
Tao, X., Lee, A., Limapichat, W., Dougherty, D. A., and MacKinnon, R. (2010). A gating charge transfer center in voltage sensors. Science 328, 67–73. doi: 10.1126/science.1185954
Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2006). How does voltage open an ion channel? Annu. Rev. Cell Dev. Biol. 22, 23–52. doi: 10.1146/annurev.cellbio.21.020404.145837
Tomlinson, S. E., Rajakulendran, S., Tan, S. V., Graves, T. D., Bamiou, D. E., Labrum, R. W., et al. (2013). Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J. Neurol. Neurosurg. Psychiatry 84, 1107–1112. doi: 10.1136/jnnp-2012-304131
Tottene, A., Conti, R., Fabbro, A., Vecchia, D., Shapovalova, M., Santello, M., et al. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61, 762–773. doi: 10.1016/j.neuron.2009.01.027
Tottene, A., Fellin, T., Pagnutti, S., Luvisetto, S., Striessnig, J., Fletcher, C., et al. (2002). Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal Cav2.1 current density in neurons. Proc. Natl. Acad. Sci. U S A 99, 13284–13289. doi: 10.1073/pnas.192242399
Trimmer, J. S., and Rhodes, K. J. (2004). Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 66, 477–519. doi: 10.1146/annurev.physiol.66.032102.113328
Tsantoulas, C., and McMahon, S. B. (2014). Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 37, 146–158. doi: 10.1016/j.tins.2013.12.002
Vahedi, K., Depienne, C., Le Fort, D., Riant, F., Chaine, P., Trouillard, O., et al. (2009). Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology 72, 1178–1183. doi: 10.1212/01.wnl.0000345393.53132.8c.
VanDyke, D. H., Griggs, R. C., Murphy, M. J., and Goldstein, M. N. (1975). Hereditary myokymia and periodic ataxia. J. Neurol. Sci. 25, 109–118. doi: 10.1016/0022-510x(75)90191-4
Vanmolkot, K. R., Babini, E., de Vries, B., Stam, A. H., Freilinger, T., Terwindt, G. M., et al. (2007). The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Mutation in brief #957. Online. Hum. Mutat. 28:522. doi: 10.1002/humu.9486
Vanoye, C. G., Gurnett, C. A., Holland, K. D., George, A. L. Jr., and Kearney, J. A. (2013). Novel SCN3A variants associated with focal epilepsy in children. Neurobiol. Dis. 62, 313–322. doi: 10.1016/j.nbd.2013.10.015
Vargas, E., Bezanilla, F., and Roux, B. (2011). In search of a consensus model of the resting state of a voltage-sensing domain. Neuron 72, 713–720. doi: 10.1016/j.neuron.2011.09.024
Vassilev, P. M., Scheuer, T., and Catterall, W. A. (1988). Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661. doi: 10.1126/science.2458625
Vecchia, D., Tottene, A., van den Maagdenberg, A. M., and Pietrobon, D. (2014). Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in Cav2.1 knockin migraine mice. Neurobiol. Dis. 69, 225–234. doi: 10.1016/j.nbd.2014.05.035
Veeramah, K. R., O’Brien, J. E., Meisler, M. H., Cheng, X., Dib-Hajj, S. D., Waxman, S. G., et al. (2012). De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 90, 502–510. doi: 10.1016/j.ajhg.2012.01.006
Wall-Lacelle, S., Hossain, M. I., Sauvé, R., Blunck, R., and Parent, L. (2011). Double mutant cycle analysis identified a critical leucine residue in the IIS4S5 linker for the activation of the Ca(V)2.3 calcium channel. J. Biol. Chem. 286, 27197–27205. doi: 10.1074/jbc.m111.237412
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wappl, E., Koschak, A., Poteser, M., Sinnegger, M. J., Walter, D., Eberhart, A., et al. (2002). Functional consequences of P/Q-type Ca2+ channel Cav2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J. Biol. Chem. 277, 6960–6966. doi: 10.1074/jbc.M110948200
Watanabe, E., Fujikawa, A., Matsunaga, H., Yasoshima, Y., Sako, N., Yamamoto, T., et al. (2000). Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J. Neurosci. 20, 7743–7751.
Waters, M. F., Minassian, N. A., Stevanin, G., Figueroa, K. P., Bannister, J. P., Nolte, D., et al. (2006). Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 38, 447–451. doi: 10.1038/ng1758
Waxman, S. G., Merkies, I. S., Gerrits, M. M., Dib-Hajj, S. D., Lauria, G., Cox, J. J., et al. (2014). Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 13, 1152–1160. doi: 10.1016/s1474-4422(14)70150-4
Webster, S. M., Del Camino, D., Dekker, J. P., and Yellen, G. (2004). Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 428, 864–868. doi: 10.1038/nature02468
Weckhuysen, S., Ivanovic, V., Hendrickx, R., Van Coster, R., Hjalgrim, H., Møller, R. S., et al. (2013). Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 81, 1697–1703. doi: 10.1212/01.wnl.0000435296.72400.a1
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R., et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25. doi: 10.1002/ana.22644
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc. Natl. Acad. Sci. U S A 89, 10910–10914. doi: 10.1073/pnas.89.22.10910
Westenbroek, R. E., Merrick, D. K., and Catterall, W. A. (1989). Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3, 695–704. doi: 10.1016/0896-6273(89)90238-9
Westenbroek, R. E., Noebels, J. L., and Catterall, W. A. (1992). Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. J. Neurosci. 12, 2259–2267.
Westenbroek, R. E., Sakurai, T., Elliott, E. M., Hell, J. W., Starr, T. V., Snutch, T. P., et al. (1995). Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 15, 6403–6418.
Wickenden, A. D., Krajewski, J. L., London, B., Wagoner, P. K., Wilson, W. A., Clark, S., et al. (2008). N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol. Pharmacol. 73, 977–986. doi: 10.1124/mol.107.043216
Wu, H., Cowing, J. A., Michaelides, M., Wilkie, S. E., Jeffery, G., Jenkins, S. A., et al. (2006). Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am. J. Hum. Genet. 79, 574–579. doi: 10.1086/507568
Wulff, H., Castle, N. A., and Pardo, L. A. (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001. doi: 10.1038/nrd2983
Xie, G., Harrison, J., Clapcote, S. J., Huang, Y., Zhang, J. Y., Wang, L. Y., et al. (2010). A new Kv1.2 channelopathy underlying cerebellar ataxia. J. Biol. Chem. 285, 32160–32173. doi: 10.1074/jbc.M110.153676
Xu, R., Thomas, E. A., Jenkins, M., Gazina, E. V., Chiu, C., Heron, S. E., et al. (2007). A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol. Cell. Neurosci. 35, 292–301. doi: 10.1016/j.mcn.2007.03.003
Yang, W. P., Levesque, P. C., Little, W. A., Conder, M. L., Ramakrishnan, P., Neubauer, M. G., et al. (1998). Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J. Biol. Chem. 273, 19419–19423. doi: 10.1074/jbc.273.31.19419
Yang, Y., Vasylyev, D. V., Dib-Hajj, F., Veeramah, K. R., Hammer, M. F., Dib-Hajj, S. D., et al. (2013). Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2–R327H demonstrate the role of this residue in stabilizing the channel closed state. J. Neurosci. 33, 16586–16593. doi: 10.1523/JNEUROSCI.2307-13.2013
Yarov-Yarovoy, V., Baker, D., and Catterall, W. A. (2006). Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. U S A 103, 7292–7297. doi: 10.1073/pnas.0602350103
Yarov-Yarovoy, V., DeCaen, P. G., Westenbroek, R. E., Pan, C. Y., Scheuer, T., Baker, D., et al. (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U S A 109, E93–E102. doi: 10.1073/pnas.1118434109
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Yu, F. H., Yarov-Yarovoy, V., Gutman, G. A., and Catterall, W. A. (2005). Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 57, 387–395. doi: 10.1124/pr.57.4.13
Zagotta, W. N., Hoshi, T., and Aldrich, R. W. (1994). Shaker potassium channel gating. III: evaluation of kinetic models for activation. J. Gen. Physiol. 103, 321–362. doi: 10.1085/jgp.103.2.321
Zandany, N., Ovadia, M., Orr, I., and Yifrach, O. (2008). Direct analysis of cooperativity in multisubunit allosteric proteins. Proc. Natl. Acad. Sci. U S A 105, 11697–11702. doi: 10.1073/pnas.0804104105
Zerr, P., Adelman, J. P., and Maylie, J. (1998). Characterization of three episodic ataxia mutations in the human Kv1.1 potassium channel. FEBS Lett. 431, 461–464. doi: 10.1016/s0014-5793(98)00814-x
Zhang, X. Y., Wen, J., Yang, W., Wang, C., Gao, L., Zheng, L. H., et al. (2013). Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 93, 957–966. doi: 10.1016/j.ajhg.2013.09.016
Zhu, J., Alsaber, R., Zhao, J., Ribeiro-Hurley, E., and Thornhill, W. B. (2012). Characterization of the Kv1.1 I262T and S342I mutations associated with episodic ataxia 1 with distinct phenotypes. Arch. Biochem. Biophys. 524, 99–105. doi: 10.1016/j.abb.2012.05.006
Keywords: ion channels, voltage-sensing module, channelopathies, gating modifier, mutations
Citation: Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Manocchio L, Medoro A and Taglialatela M (2015) Molecular pathophysiology and pharmacology of the voltage-sensing module of neuronal ion channels. Front. Cell. Neurosci. 9:259. doi: 10.3389/fncel.2015.00259
Received: 03 April 2015; Accepted: 22 June 2015;
Published: 15 July 2015.
Edited by:
Mauro Pessia, Universtity of Perugia, ItalyReviewed by:
Jianmin Cui, Washington University, USAMassimo Mantegazza, CNRS UMR7275 and University of Nice Sophia Antipolis, France
Copyright © 2015 Miceli, Soldovieri, Ambrosino, De Maria, Manocchio, Medoro and Taglialatela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Taglialatela, Department of Medicine and Health Sciences, University of Molise, Via De Sanctis, Campobasso 86100, Italy, m.taglialatela@unimol.it