A Case for Microtubule Vulnerability in Amyotrophic Lateral Sclerosis: Altered Dynamics During Disease
 Jayden A. Clark
Jayden A. Clark Elise J. Yeaman
Elise J. Yeaman Catherine A. Blizzard
Catherine A. Blizzard Jyoti A. Chuckowree
Jyoti A. Chuckowree Tracey C. Dickson
Tracey C. Dickson- Menzies Institute for Medical Research, University of Tasmania, Hobart, TAS, Australia
Amyotrophic lateral sclerosis (ALS) is an aggressive multifactorial disease converging on a common pathology: the degeneration of motor neurons (MNs), their axons and neuromuscular synapses. This vulnerability and dysfunction of MNs highlights the dependency of these large cells on their intracellular machinery. Neuronal microtubules (MTs) are intracellular structures that facilitate a myriad of vital neuronal functions, including activity dependent axonal transport. In ALS, it is becoming increasingly apparent that MTs are likely to be a critical component of this disease. Not only are disruptions in this intracellular machinery present in the vast majority of seemingly sporadic cases, recent research has revealed that mutation to a microtubule protein, the tubulin isoform TUBA4A, is sufficient to cause a familial, albeit rare, form of disease. In both sporadic and familial disease, studies have provided evidence that microtubule mediated deficits in axonal transport are the tipping point for MN survivability. Axonal transport deficits would lead to abnormal mitochondrial recycling, decreased vesicle and mRNA transport and limited signaling of key survival factors from the neurons peripheral synapses, causing the characteristic peripheral “die back”. This disruption to microtubule dependant transport in ALS has been shown to result from alterations in the phenomenon of microtubule dynamic instability: the rapid growth and shrinkage of microtubule polymers. This is accomplished primarily due to aberrant alterations to microtubule associated proteins (MAPs) that regulate microtubule stability. Indeed, the current literature would argue that microtubule stability, particularly alterations in their dynamics, may be the initial driving force behind many familial and sporadic insults in ALS. Pharmacological stabilization of the microtubule network offers an attractive therapeutic strategy in ALS; indeed it has shown promise in many neurological disorders, ALS included. However, the pathophysiological involvement of MTs and their functions is still poorly understood in ALS. Future investigations will hopefully uncover further therapeutic targets that may aid in combating this awful disease.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a late onset and ultimately fatal neurodegenerative disease characterized by the loss of upper and lower motor neurons (MNs) from the nervous system. Symptoms can be separated into bulbar (upper MN degeneration), or lumbar (lower MN degeneration) onset, describing the spread of pathology from the initial site. In addition to variance in the spread of pathology, ALS heterogeneity is further exacerbated by its representation as a spectrum disorder, with a range of clinical manifestations from cognitive dysfunction in frontotemporal dementia (FTD), pure motor phenotype in classical ALS, or a combination of both cognitive and motor dysfunctions (Ling et al., 2013). In ALS, cellular loss results in atrophy of cortical and spinal structures, and a loss of muscular innervation and associated muscle wastage (Mezzapesa et al., 2013; Turner and Swash, 2015). Neuronal cell loss is accompanied by reactive gliosis, and characteristic proteinacious intracellular inclusions (Ilieva et al., 2009; Peters et al., 2015).
Neurons are highly refined communicating cells that receive, process and relay information to their target cells, with MNs being particularly vulnerable to ALS-associated pathology. Factors contributing to the selective vulnerability of MNs in ALS include their large cell size and therefore energy dependency, their excitable nature coupled with a lack of buffering capacity, and their intimate relationship with neighboring non-neuronal cells. Many possible disease mechanisms have been proposed to account for the development and progression of ALS (reviewed in Peters et al., 2015). However, one such mechanism, the impairment of the axonal transport system, highlights the significance of the intracellular cytoskeleton, particularly the microtubules (MTs), in the neurodegenerative process.
Microtubules Are Integral to Neuronal Function
Microtubules are structural cytoskeletal elements expressed in all eukaryotic cells. Their composition and general function are conserved between different cell types and organisms, and is essential for cell division and motility. MTs are of particular importance to neurons and are involved in a great number of additional functions including the development of neuronal cell polarity, the generation of neuronal compartments, growth cone mechanics, neurite remodeling and intracellular transport (Chen et al., 2006; Baas and Lin, 2011; Sakakibara et al., 2013). Within neurons, MTs form protofilaments from heterodimerized tubulin. These cylindrical structures are vitally important for the function of long extending axons, which have a high demand for intracellular transport of organelles, proteins and RNA granules. Therefore, it can be said MTs are essential to both the development and maintenance of the neuronal circuitry.
MT protofilaments are comprised of dimers of α and β-tubulin, which through lateral interactions, form the characteristic MT structure (Desai and Mitchison, 1997). MTs undergo bouts of assembly and disassembly from their ends, a process termed dynamic instability (Mitchison and Kirschner, 1984; Figure 1A). A slow growing α-tubulin “minus end” and fast growing β-tubulin “plus end” (Allen and Borisy, 1974; Van Beuningen et al., 2015; Yau et al., 2016) can be accounted for by the guanosine triphosphate (GTP) cap model. GTP bound to β-tubulin confers plus end stability, and upon hydrolysis, leads to MT depolymerization (Desai and Mitchison, 1997; Nogales and Wang, 2006). This directionality in MT structure allows the formation of neuronal compartments and therefore confers neuronal polarity. The axonal compartment has exclusively distal facing positive polarity, or “plus end out” orientating MTs (Heidemann et al., 1981). Contrastingly, the somatodendritic compartment contains a mixed orientation of MT directions, with equal quantities of plus and minus end facing MTs (Yau et al., 2016).
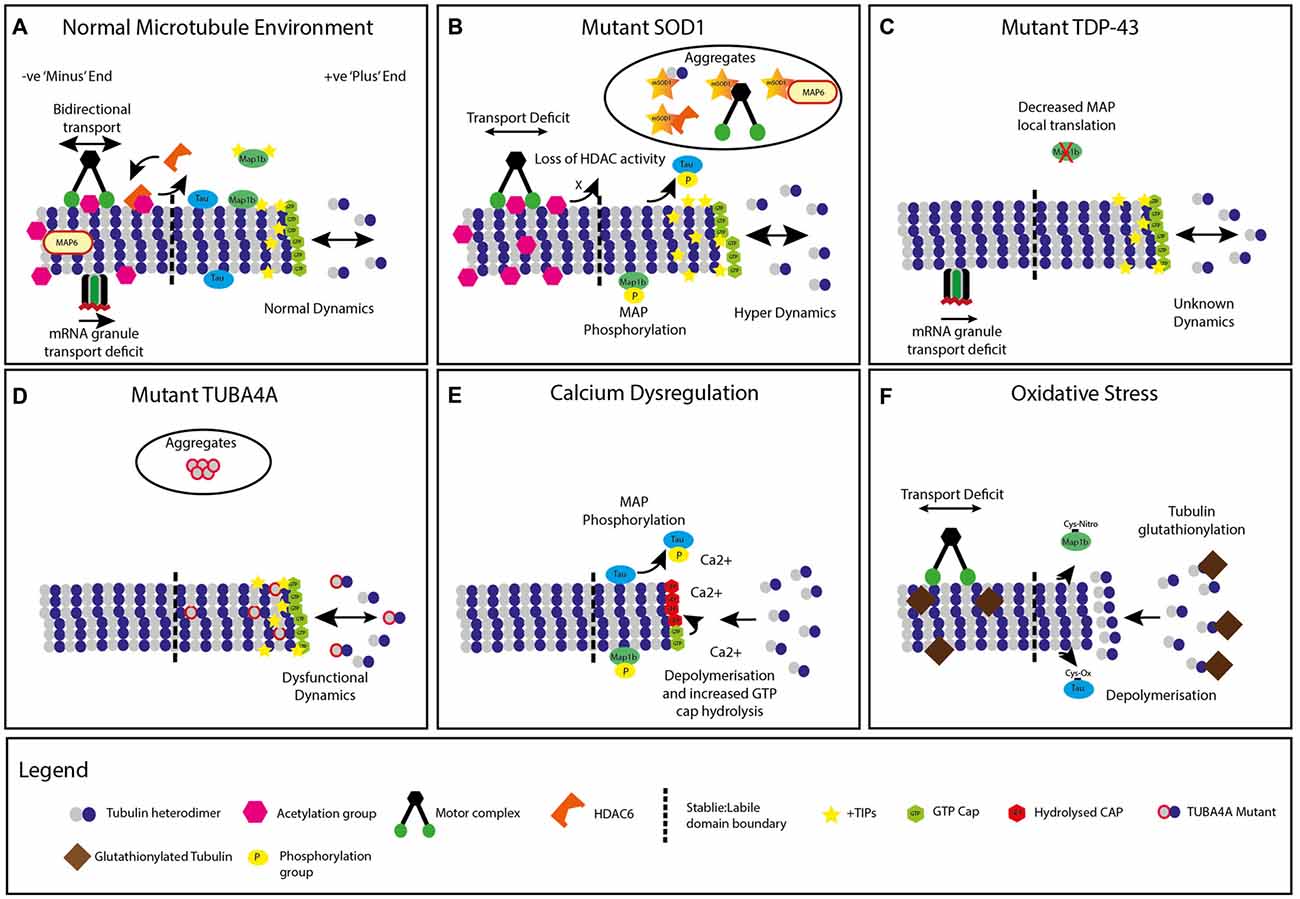
Figure 1. Modifications to neuronal microtubules (MT) in amyotrophic lateral sclerosis (ALS). (A) Normal microtubule +TIP dynamics and kinesin/dynein transport, mRNA granule transport and chemical modifications. (B) Mutant superoxide dismutase 1 (SOD1) expression leading to microtubule hyperdynamics, increased +TIP protein density, decreased transport, increased acetylation, phosphorylation of microtubule associated proteins (MAPs) and accumulation of microtubule protein containing aggregates. A global decrease in Histone Deacetylase (HDAC) activity is also present. (C) Mutant TDP-43 (TARDBP) expression causes dysfunction in mRNA granule transport. Decreased local translation of MAP mRNA is also implicated in TDP-43 mutants. (D) Mutant TUBA4A expression alters microtubule dynamics and network stability, with unknown impact on +TIP proteins, transport or chemical modifications. Select mutations are incorporated into intracellular aggregates. (E) Energy depletion and calcium dysregulation generates increased microtubule depolymerization, tubulin guanosine triphosphate (GTP) cap hydrolysis, and increased MAP phosphorylation. (F) Neuronal oxidative stress leads to tubulin glutathionylation, increased microtubule depolymerization, decreased axonal transport and alterations to MAPs, with unknown impact on classical chemical modifications or +TIP proteins.
Neurons, being relatively long-lived and stable cells, require a stable cytoskeleton. Neuronal MTs can be separated into two domains: labile and stable (Baas and Black, 1990; Baas, 2013). While mature neurons have an MT network consisting of both domains, the majority of the MTs are stable (Ferreira and Cáceres, 1989; Lim et al., 1989). These domains cannot be fully explained by the myriad of tubulin isoforms available in the genome (Tischfield and Engle, 2010). This functional diversity has instead been associated with domain-specific chemical modification to tubulin subunits, which aid in increasing the functionality of MTs, and in some cases, can impact upon their stability (Baas, 2013; Janke, 2014). These modifications to neuronal MTs include, but are not limited to, tubulin tyrosination, acetylation, polyamination, glutathyonilation, glutamylation and glycylation (reviewed previously in Janke and Kneussel, 2010; Janke, 2014). These post-translational modifications may confer stability to MT structure and alter binding affinity of MT-associated proteins, thereby altering their function. Although not being fully understood, these chemical modifications can also be utilized as molecular markers of MT network stability, and alterations to chemical modifications are thought to be attractive therapeutic targets for neurodegenerative disorders (d’Ydewalle et al., 2011; Taes et al., 2013).
Perturbations in MT and microtubule associated protein (MAP) functions have been implicated in a range of neurodegenerative diseases (Dubey et al., 2015), including Alzheimer’s disease (AD; Matsuyama and Jarvik, 1989), Parkinson’s disease (PD; Ren et al., 2003; Cartelli et al., 2010), Huntington’s disease (HD), various congenital developmental disorders (Tischfield and Engle, 2010), schizophrenia (Morris et al., 2003; Andrieux et al., 2006) and also ALS (Baird and Bennett, 2013; Smith et al., 2014). Of note the MT stabilizer, Tau (Drubin and Kirschner, 1986), is involved in the pathophysiology of AD (reviewed in Hanger et al., 2014); Mcmurray (2000); Stancu et al. (2014) as well as in other neurodegenerative diseases (collectively tauopathies) such as supranuclear palsy, corticobasal degeneration and Picks disease (reviewed in Cairns et al., 2004). Alterations to proteins involved in MT stability, dynamics and MT turnover also occur in PD (Alim et al., 2004; Yang et al., 2005; Gillardon, 2009). Similarly, MT involvement has also been established through 1-methyl-4-phenylpiridinium (MPP+; Cappelletti et al., 2005) and Rotenone-mediated PD (Ren et al., 2005) in in vitro models, with these found to destabilize the MT network. Furthermore, it has been highlighted that perturbations to MT dynamics also leads to MT-dependent transport impairment in a model of PD (Cartelli et al., 2010). The gene disrupted-in-schizophrenia-1 (DISC-1), whose mutations are associated with familial forms of schizophrenia, has been suggested to associate and interact with MT components (Morris et al., 2003; Callicott et al., 2005), further implicating MT alterations as a causal factor in multiple neurodegenerative disorders. Conversely, mutations to tubulin genes generally lead to disorders associated with dysregulated neurogenesis, due to abnormalities in neuronal migration, cellular division of progenitor cells, neuronal differentiation and induction of cell polarity. These disorders include lissencephaly, polymicrogyria, mirocephaly, cerebellar dysplasia and some occulomotor disorders (Francis et al., 2006; Guerrini et al., 2008; Tischfield et al., 2011).
In ALS, the consequence of MT dysfunction has classically been hypothesized as being due to the physical length of the axon in affected MNs, with alterations to MTs being thought to impact on axonal transport (De Vos et al., 2007; Millecamps and Julien, 2013). However, evidence for MT dysfunction having a primary role in ALS has significantly increased over the last 15 years. The recent identification of tubulin rare variants and their impact on MT function, specific interactions with mutant and pathological proteins as well as altered function of MT-associated proteins and signaling pathways which affect MT dynamics, have all been implicated in ALS pathogenesis. This review article will consider the role MTs play in disease pathogenesis and potential mechanisms that may have an impact on MT function in ALS.
The Genetic Inheritance of ALS; Recent Insights into Microtubule Function
Familial genetics of ALS are being increasingly understood, in both the context of the mechanisms through which they act, as well as in the clinical phenotype presented (Marangi and Traynor, 2015; Peters et al., 2015; Turner and Swash, 2015). Moreover, the fact that MTs and their functions are a site of pathological convergence for various ALS mutations is becoming increasingly appreciated. The first identified causative mutation for ALS was in the gene coding for superoxide dismutase 1 (SOD1), which was also used to generate the first animal model of the disease, the widely used SOD1G93A transgenic mouse (Rosen, 1993; Gurney et al., 1994). More than 160 different mutations to the gene encoding for SOD1 have been reported (Rosen, 1993; Al-Chalabi et al., 2012), accounting for approximately 20% of all familial forms of ALS (FALS; Andersen and Al-Chalabi, 2011; Robberecht and Philips, 2013). It has long been established that SOD1 mutations can cause axonal transport dysfunction, thus suggesting an interaction with the MT system (Warita et al., 1999; Ligon et al., 2005; Bilsland et al., 2010; Ikenaka et al., 2012). This has been reported to occur as early as embryonic day 13 in the SOD1G93A mutant mouse (Kieran et al., 2005). Recently, evidence has been published supporting the hypothesis that alterations to MT dynamics are driving MN dysfunction in these mice (Fanara et al., 2007; Kleele et al., 2014; Figure 1B). Indeed, mutant SOD1 has been implicated in the alteration of signaling cascades that may affect proper MT functioning, however, this process is still poorly understood (Evans et al., 2000; Nguyen et al., 2001; Vadlamudi et al., 2005; Lopez-Fanarraga et al., 2007; Ikeda et al., 2011; Zyss et al., 2011; Bunton-Stasyshyn et al., 2015).
Similarly, discoveries of mutations in RNA-processing proteins such as TDP-43 (TARDBP) and fused in sarcoma (FUS) were both identified as a cause of FALS and FTD, and subsequently used to generate further models of ALS (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Kwiatkowski et al., 2009; Vance et al., 2009). TDP-43 is associated with transcription repression (Ou et al., 1995), the regulation of splice variants (Buratti and Baralle, 2001), mRNA stability and/or transport (Tollervey et al., 2011). In disease, both mutated and wild-type TDP-43 mislocalize from the nucleus to the cytoplasm and form cytoplasmic inclusions (Neumann et al., 2006). FUS is also a nuclear DNA/RNA binding protein and is involved in various aspects of gene expression through mRNA metabolism and mRNA transport. Mutated FUS, similar to TDP-43, is mislocalized to the cytoplasm due to a loss of nuclear import signaling (Dormann et al., 2010). The similar function and mislocalization of these two proteins, TDP-43 and FUS, indicates that familial insults in neuronal degeneration may have common mechanisms. It has been found that TDP-43 interacts with MTs in the MT-dependant transport of mRNP granules (Kanai et al., 2004; Alami et al., 2014; Figure 1C). This transport is particularly important due to TDP-43’s role in local mRNA translation of proteins in dendrites, axons and synapses, such as the neuromuscular junction (NMJ; Kanai et al., 2004; Belly et al., 2005; Fallini et al., 2012). Similarly, TDP-43 interacts directly with proteins involved in RNA transport (Freibaum et al., 2010). Mutated TDP-43 has recently been identified to impair axonal transport of mRNA in vivo, as well as the induced pluripotent stem cell (iPSC) derived MNs of patients with TDP-43 mutations (Alami et al., 2014). mRNP granule transport defects were independent of other transport reporter defects such as mitochondrial transport. It was hypothesized that diminished transport of mRNA to compartments such as the axon and NMJ may lead to a decrease in proteins particularly important for NMJ maintenance and survival (Polymenidou et al., 2011; Lagier-Tourenne et al., 2012).
In 2009, two independent studies identified the hexanucleotide repeat expansion of the non-coding region of reading frame 72 on chromosome 9 (C9ORF72); a mutation that occurs in both sporadic ALS (SALS) and FALS, also FTD, at greater frequency than other, previously known mutations (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Initially it was postulated that this abnormal repeat could be transcribed and may have some toxic function, however the exact mechanism of pathogenesis remains unclear (DeJesus-Hernandez et al., 2011). More recently it has been proposed that the presence of the repeat expansion may result in “unconventional translation” (Ash et al., 2013; Todd and Petrucelli, 2016). This leads to five dipeptide repeat proteins, which accumulate in neurons (Mori et al., 2013). It is still unknown whether these repeat expansions lead to a gain or loss of function mechanism, however support for C9ORF72 gain of function is growing (Todd and Petrucelli, 2016). While function of this repeat expansion is still under investigation, Droppelmann et al. (2014) have highlighted that possible interaction with MTs may exist, due to the C9ORF72 homology to a guanine nucleotide exchange factor (GEF) that signals Rab-GTPases. Rab-GTPases are involved in membrane trafficking of proteins (Levine et al., 2013; Droppelmann et al., 2014) and hence, may be dependent upon MTs.
Although the majority of FALS cases follow a dominant pattern of inheritance, mutations to Alsin (ALS2) have been found to exhibit a recessive pattern of inheritance (Hadano et al., 2001). Mutations in the ALS2 gene have been associated with the development of juvenile onset ALS, as well as a range of other conditions such as primary lateral sclerosis and hereditary spastic paraplegia (HSP; Eymard-Pierre et al., 2002; Panzeri et al., 2006), with 12 different mutations identified to contribute to the development of such conditions (Chandran et al., 2007). Many of these mutations result in a premature stop codon, therefore rendering the ALS2 protein non-functional (Yamanaka et al., 2006). ALS2 is a GEF, which promotes guanosine diphosphate (GDP) release and GTP binding onto target proteins, as well as the stimulation certain signaling cascades (Tudor et al., 2005). ALS2 mutations have been suggested to contribute to the pathogenicity of ALS through the disruptions of Rab5-dependent exocytosis, endosome trafficking and also glutamate-associated excitotoxicity, which is considered a hallmark of ALS pathology (Devon et al., 2006; Hadano et al., 2006; Lai et al., 2006). Indeed, ALS2 acts through signaling cascades that impact MTs, and whose loss of function may generate MT dysfunction; a process that requires further research (Tudor et al., 2005).
Vascular endothelial growth factor (VEGF) is a polymorphic risk factor for ALS, and its expression is reduced in patients (Brockington et al., 2006, 2010). A mouse model that produces decreased levels of VEGF develops a motor neurodegenerative phenotype, with behavioral deficits and cellular loss (Oosthuyse et al., 2001). VEGF mice show decreased expression of the MAPs tau, MAP1b and MAP6, which has implications for stability of both the stable and labile domains of MTs in ALS (Brockington et al., 2010). Furthermore, genes relating to transport and dynein complex cargo loading are also down regulated. This occurs well before MN loss, but at the time of motor behavior phenotype generation. This may highlight a link between dysfunction of MTs and other identified genes, leading to MN dysfunction, followed by cellular demise (Brockington et al., 2010).
Microtubule Protein Mutations: A Primary Cause of ALS
Recent research has highlighted that mutations to MTs can also initiate adult onset MN dysfunction, suggesting that MTs may be a primary driver for ALS pathophysiology (Al-Chalabi et al., 1999; Puls et al., 2003; Gros-Louis et al., 2004; Wu et al., 2012; Smith et al., 2014). Smith et al. (2014) found dominant negative mutated variants in the TUBA4A gene on chromosome 2 in FALS patients. These mutations were reported to cause classical spinal onset ALS, with upper and lower MN loss (Smith et al., 2014), and in some cases, FTD-like symptoms. The mutated region normally interacts with β-tubulin and the motor domain of kinesins and other MAPs (Liu et al., 2012; Howes et al., 2014). These variants were further shown to ineffectively form tubulin dimers and displayed a decreased incorporation into protofibrils, inhibiting MT network stability (Figure 1D). Similarly, other mutations affecting the conformation of tubulin proteins may subsequently alter the assembly of tubulin, resulting in an unstable MT structure (Tischfield et al., 2011).
TUBA4A is ubiquitously expressed in all cell types, but at high levels in the nervous system (Rustici et al., 2013; Smith et al., 2014). The expression of TUBA4A also increases over time, possibly illuminating why mutations in these genes cause later age disease phenotypes, unlike congenital tubulin mutations, which generate developmental disorders (Tischfield et al., 2011; Hersheson et al., 2013). Further supporting this, and as highlighted by Smith et al. (2014) expression of the β-tubulin subunit, TUBB4A, whose mutations cause adult onset disease Torsion Dystonia Type 4, increases over time, similar to the expression pattern of α-tubulin TUBA4A (Hersheson et al., 2013). Further impacts of tubulin alterations in ALS can be shown in SALS patients, where there is a down regulation of α-tubulin subunit genes (Jiang et al., 2005). However, how MT dynamics and transport are impacted as a result of TUBA4A mutations are still not clear. Future animal models of TUBA4A mutations will allow for the identification of its role in pathogenesis, particularly focusing on its age-dependant expression pattern.
Collectively, disease-causing mutations have shed light on an integral role for MTs in ALS. However, with the majority of ALS cases still seemingly sporadic, it remains unclear if altered MT function is the cause, or consequence, of upstream initiating pathogenic mechanisms.
Pathogenic ALS Mechanisms Impact on Microtubule Structure and Function
Many mechanisms and molecular pathways involved in both the initiation and maintenance of ALS have been identified (Van Damme et al., 2005; Ferraiuolo et al., 2011; Peters et al., 2015). These include, but are not limited to, mitochondrial-dependant energy depletion, excitotoxicity and calcium dysregulation and cellular oxidative stress. The interplay between these disease mechanisms and insults to MTs are not well understood, however, it is becoming increasingly appreciated that MTs may act as a site for mechanistic convergence, as they are impacted by various pathogenic molecular mechanisms associated with ALS.
Motor Neurons are Metabolically Demanding
One such pathological mechanism is mitochondria-dependant energy depletion. A study conducted by Park et al. (2013) highlighted that energy depletion itself could be a cause of MT depolymerization, which may in turn further promote energy depletion through the inability of the MTs to facilitate movement of mitochondria (Park et al., 2013; Figure 1E). Interestingly, MT pathology was identified prior to alterations to mitochondrial swellings, suggesting it may be a primary event in energy depletion and axon degeneration. This inability to recycle mitochondria may contribute to the abnormal accumulations of these organelles as observed in mutant SOD1 transgenic mice (Sotelo-Silveira et al., 2009). However, aberrant calcium dysregulation as a result of mitochondrial energy depletion, an identified toxic mechanism in ALS, does not seem to directly be a cause of MT dysfunction, suggesting that other, as yet unidentified, mechanisms may be at play (Park et al., 2013).
Excitotoxicity and Calcium Dysregulation Affects Microtubules
Excitotoxicity has also been identified as a primary mechanism in the initiation and maintenance of ALS (Van Damme et al., 2005; Blizzard et al., 2015). Excessive influx and concentrations of intracellular Ca2+, whether from damaged mitochondria (Jaiswal, 2014), glutamatergic over stimulation, lack of glutamate clearance or a loss of Ca2+ buffering capacity, can lead to MN death (Heath and Shaw, 2002). Ca2+ concentrations also have an effect on MT-based transport of mitochondria through inhibition of attachment of kinesins to MTs (Wang and Schwarz, 2009). Indeed, excessive intracellular Ca2+ can lead to aberrant cyclin-dependent kinase 5 (CDK5) activity, which is particularly prominent in the SOD1 mouse model of ALS (Patzke and Tsai, 2002). Moreover, Ca2+ can interact directly with MTs, altering their dynamics (O’Brien et al., 1997; Figure 1E). Interestingly, heightened Ca2+ concentrations are sufficient to cause MAP containing MT preparations to depolymerize, due to an increase in β-tubulin GTP cap hydrolysis. Furthermore, Ca2+ can cause MAP2 loss and MT depolymerization in dendrites treated with NMDA, through calpain proteolysis of MAP2; however, it remains unclear if MT destabilization occurs prior to MAP2 loss (Hoskison et al., 2007). Excitotoxicity has also been shown to have an impact on retrograde transport machinery (Fujiwara and Morimoto, 2012). Indeed, this insult leads to caspase activation and subsequent cleavage and disintegration of the cytoskeleton, reported to be downstream of MT events (King et al., 2013). However, investigation of low dose, long lasting, chronic excitotoxic effects on both MT dynamics and transport are yet to be completed, as recognized ALS insults are chronic or accumulative in their nature. Investigations such as these will identify whether biologically relevant levels of neuronal excitotoxicity alter MT dynamics and transport first, or contrastingly activate the CDK5-p25 and caspase pathways, which lead to cellular pathology.
Oxidative Stress and The Effects on Microtubules
Oxidative stress occurs due to the build-up of oxidative species, which while being a process of general aging of an organism, can cause uncontrollable oxidation of proteins or molecules, leading to cellular dysfunction. Therefore, homeostatic control of reactive oxygen species (ROS) is required for proper cell function. Uncontrollable ROS production and cellular oxidation leads to a number of neurodegenerative diseases (Andersen, 2004). Relevant to ALS, and in addition to Ca2+ toxicity, dysfunctional mitochondria can also drive uncontrollable ROS production (Tahara et al., 2009). While ROS are required for cytoskeletal remodeling and during axonal growth (Munnamalai and Suter, 2009; Wilson and González-Billault, 2015), aberrant ROS and an increased oxidative environment can lead to deleterious impacts on MTs, particularly oxidation of tubulin and selected MAPs (Landino et al., 2004). Although still poorly understood in neurons, increasing ROS in myocytes increase MT depolymerization (Drum et al., 2016). Both α- and β-tubulin contain Cys residues that have the capacity to oxidize (Landino et al., 2007; Wilson and González-Billault, 2015). Oxidative species added to purified tubulin preparations can cause a reduction in polymerization, and increase MT depolymerization, similar to that of increased Ca2+ levels (Landino et al., 2007; Figure 1F). Moreover, it has been established that oxidative stress affects MT integrity; this is evidenced by the presence of methionine sulphaoxides in the β-III tubulin in the brain of AD patients (Boutte et al., 2006). Furthermore, glutathionylation of tubulin, particularly in MNs, occurs during oxidative phases, altering MT dynamics and structure (Carletti et al., 2011). Indeed, the MAPs tau and MAP2 contain Cys residues, which when oxidize, impede MAP function, thus causing MT stability issues (Landino et al., 2004). Others have identified that recapitulating neuronal oxidative stress through the addition of hydrogen peroxide inhibits axonal transport, prior to mitochondrial dysfunction or axonal degeneration (Fang et al., 2012).
Altered Regulation of Microtubule Dynamics has been Associated with ALS
Changes to microtubule dynamics in ALS have also been reported, particularly increases to MT dynamic instability. Fanara et al. (2007) identified that hyperdynamic MTs were present in the SOD1G93A mouse model of ALS, and modulation of MT dynamics can ameliorate disease progression. A subsequent study found that MTs are indeed more dynamic, with an increase in end binding-protein 3 (EB3) +TIP comets on sciatic nerve axonal MTs in the SOD1G93A mouse in comparison to unmutated individuals (Kleele et al., 2014). EB proteins are +TIP MAPs that bind to the growing phase of the labile domain of MTs, aiding in both dynamics and microtubule interactions with other intracellular objects. Increases in +TIP comets signify that the microtubule network is hyperdynamic. It was found that increased MT dynamics consequently slowed axonal transport, occurring presymptomatically (Fanara et al., 2007; Bilsland et al., 2010). Axonal transport dysfunction is then followed by an increase in neuronal pathology and subsequent mortality (Collard et al., 1995; Sasaki and Iwata, 1996; Williamson and Cleveland, 1999; Sasaki et al., 2004). Pharmacological amelioration of MT hyperdynamics not only reverses axonal transport deficits, but also improves clinical symptoms and survival (Fanara et al., 2007). This hints that hyperdynamics can indeed drive transport deficits, followed by cell demise; however, this process is still poorly understood. A possible mechanism through which SOD1 mice develop MT hyperdynamics may be through the interaction of mutant SOD1 and tubulin (Kabuta et al., 2009). Interestingly mutant SOD1/tubulin interactions do not diminish the free tubulin pool; however, it generates a destabilizing effect on MT.
Kabuta et al. (2009) suggest that homology exists between the site of mutant SOD1 binding to tubulin and the binding site of MT destabilizing agents such as colchicine and nocodazole. This may indicate that mutant SOD1 interacts in a similar manner to the MT destabilizing agents, increasing the dynamic instability of MT labile domains, leading to a reactive, hyperdynamic phenotype. A phenomenon less often considered in ALS research is the impact that intracellular aggregates, consisting of proteins such as misfolded SOD1 and neurofilaments (NFs), may have on microtubule dynamics. Indeed, these aberrant structures have the propensity to incorporate many different cytosolic proteins into the aggregate mass, with MT proteins being particularly prone in aggregate localization. This may be an interesting avenue of research as a recent study showed that decreasing the NFs in a pmn mice improves aberrant microtubule dynamics and instability that is associated with this model (Yadav et al., 2016).
An increasing and popular notion is that alterations to transport proteins, tubulin, other MAPs or the dysregulation of MT dynamics can result in aberrant MT structure and function, leading to either developmental disorders or degenerative phenotypes, such as that in ALS (Dubey et al., 2015). Mechanisms driving these alterations in MT dynamics are not well understood, nor are the hypothesized subsequent transport dysfunctions. Identification of whether dysfunction of MT dynamics is a common pathology between familial mutations and sporadic disease is paramount to understanding both disease onset and also its maintenance.
Could Motor Protein Dysfunction Drive ALS Pathology?
Intracellular transport is a major function of neuronal MTs, with alterations in transport associated with a number of neurodegenerative diseases (reviewed in Millecamps and Julien, 2013). Cortical and spinal MNs are thought to be particularly vulnerable to transport dysfunction, due to their axonal length. It is becoming increasingly appreciated that alterations to MT dynamics precede, and thus may aberrantly affect, axonal transport, and that the resultant transport defect can have deleterious effects on neuronal function (Hurd and Saxton, 1996; Fanara et al., 2007; Bilsland et al., 2010; Cartelli et al., 2010; Dubey et al., 2015). The transport MAPs, kinesin and dynein, act as carriers for organelles, proteins and other cellular cargo in a directionally-dependant manner. Kinesin motor proteins transport cell cargo toward the plus end of the MT (anterograde), whereas dynein transport in the minus end direction (retrograde; Maday et al., 2014). Kinesin can have an impact on the stability of MTs, and expression of KIF5, a kinesin motor protein isoform, has been found to be decreased in the spinal cord and sciatic nerves of a mutant SOD1 mouse model (Maximino et al., 2014). This indicates that altered MT-dependent transport may be depleting MNs via the generation of an energy and signaling deficit. This is also substantiated in a study by Tateno et al. (2009), who showed that kinesin-associated protein 3 (KAP3), a kinesin subunit responsible for binding cargo such as choline acetyltransferase (ChAT), was selectively vulnerable to co-aggregation with misfolded SOD1 (Tateno et al., 2009). This phenomenon was also reported to occur in human SOD1 FALS patients, possibly illuminating a further source of MN vulnerability to dysfunctional transport of specific cargos in ALS.
Dynein, in conjunction with its molecular binding partner and activator, dynactin (DCTN1), is also vulnerable, both as a primary driver of ALS and also as a site of convergence of ALS insults (Ligon et al., 2005). Dynein interacts with mutant SOD1 and is located in proteinacious aggregates in SOD1 mice (Ligon et al., 2005; Figure 1B). Mutant SOD1 also interacts directly with the assembled dynein-dynactin complex, occurring prior to disease onset, at a similar age when retrograde dynein-mediated axonal transport dysfunction occurs in SOD1G93A mice (Zhang et al., 2007; Bilsland et al., 2010). The functional consequence of this is yet to be determined, however detrimental impacts of transport dysfunction on the ubiquitin-proteasome system and protein autophagy may create a positive feedback loop, whereby proteins are caught in the incorrect cellular compartment, which compounds aggregation (Goldberg, 2003; Ström et al., 2008; Takalo et al., 2013). Similarly, bidirectional transport of mitochondria is affected presymptomatically, highlighting the cargo and directional specificity of axonal transport dysfunction in this model; however, the exact mechanism is not well understood (Bilsland et al., 2010). Mutations to the p150Glued subunit of DCTN1 are associated with MN degeneration and ALS (Münch et al., 2004, 2005; Levy et al., 2006; Laird et al., 2008). The mutation distorts the folding of the MT-binding domain. An autosomal dominant variant has also been reported to concurrently cause FTD (Münch et al., 2005), highlighting the role of motor proteins in other neurodegenerative diseases.
Expressions of motor protein genes are altered in SALS patients and in mutant SOD1 mouse models. A reduction in dynactin-1 expression is observed, in the absence of alterations to kinesin and dynein expression; down regulation occurs prior to the deposition of NF protein aggregates (Jiang et al., 2007; Rustici et al., 2013). Furthermore, a polymorphism and reduced expression in kinesin-associated protein 3 (KIFAP3) correlates with an extended life span in SALS patients (Landers et al., 2009); however, how this affects transport is unknown. Interestingly, in mutant SOD1 mice, KIFAP3 expression in increased early in the diseases clinical course (Dupuis et al., 2000). Similarly, a number of cytoskeletal genes are altered in the SOD1G93A mouse spinal cord and sciatic nerve, dependant on both age and MN sub compartment evaluated (Maximino et al., 2014). Similar gene expression changes were found in SALS patients, having a decrease in MAP2, MAP1b and tau protein expression, which is also seen in the mutant SOD1G37R mouse model (Farah et al., 2003; Jiang et al., 2007). Motor proteins are also susceptible to alterations in tau levels, as observed in tauopathies, which retard anterograde motor transport (Ebneth et al., 1998). Tau alters the flux at which kinesin and dynein motor complexes bind to the MTs, but not the speed at which they travel the MT tracks (Trinczek et al., 1999), suggesting alterations to tau levels on MTs may impact transport where tau pathology and dysfunction is present. Indeed, multiple upstream effectors such as altered dynamics, MAP dysfunction and protein-protein interactions can produce aberrant axonal transport.
Microtubule Associated Protein Alterations Also Contribute to ALS Pathology
MAPs facilitate MT functions such as cytoskeletal interactions, intracellular signaling and modification of MT dynamics and stability. Indeed, MAPs are thought to “tune” MT dynamics through both direct and indirect interactions (Tortosa et al., 2013; Sayas et al., 2015). Alterations to MAPs are observed in ALS, and are mainly due to the impact of dysregulated signaling and aberrant phosphorylation events. Indeed, MAP dysfunction is a downstream effect of many ALS-related pathological mechanisms. Aberrant hyper-phosphorylation and mutations to the MAP tau are associated with neurodegenerative disorders, such as AD, tauopathies and ALS/FTD (Hanger et al., 2014; Stancu et al., 2014; Huang et al., 2015; Baas et al., 2016). Tau is an axonal specific MAP that localizes to labile domains of MTs (Black et al., 1996). It is found throughout the axonal MT network. Tau alters MT dynamics by increasing the stability of the labile domain, preventing its depolymerization, while promoting assembly (Brandt and Lee, 1993; Panda et al., 1995). Tau influences MT dynamics, as it interacts directly with EB proteins and is required for the localization and density of EB proteins to the plus ends of MTs (Sayas et al., 2015). Another means by which tau mediates MT stability is its role in protecting MTs from the enzymatic severing by katanin (Kempf et al., 1996; Qiang et al., 2006), a process that is thought to be impacted upon in AD and tauopathies, due to the loss of tau-MT localization when tau is phosphorylated (Sudo and Baas, 2011). There are reports of hyper phosphorylated and insoluble tau deposits in cortical neurons of sporadic ALS patients (Strong et al., 2006). Furthermore, mouse models of mutant SOD1 show increased tau phosphorylation (Nguyen et al., 2001); and SOD1 Drosophila models show increased tau toxicity and neuronal degeneration attributed to by tau phosphorylation (Huang et al., 2015). Moreover, mouse models generating hyper activated CDK5, a kinase that causes aberrant tau phosphorylation, show increases in cytoskeletal disintegration and axonal swellings (Ahlijanian et al., 2000). Interestingly, lowering the expression of tau does not improve the phenotype of mutant SOD1 mice, suggesting that while it may play a role in disease progression, it is not necessary for neuronal pathogenesis (Taes et al., 2010). However, identification of tau-mediated alterations to microtubule dynamics in ALS are yet to be completed, and is a necessary avenue of research.
Another MAP that may play an important role in ALS is MAP1b. MAP1b has been shown to play a role in neurite and dendritic spine dynamics, having been identified to be enriched in zones of high MT dynamic instability, such as axonal growth cones and branch points and dendritic spines (Gonzalez-Billault et al., 2001; Tortosa et al., 2011; Villarroel-Campos and Gonzalez-Billault, 2014; Ketschek et al., 2015). Furthermore, MAP1b function can be regulated by phosphorylation at specific sites, which alters its interactions with other cytosolic proteins (Lucas et al., 1998; Villarroel-Campos and Gonzalez-Billault, 2014). MAP1b preferentially binds to, and stabilizes, the labile domain of MTs. MAP1b has recently been implicated in mutant TDP-43 pathology (Feiguin et al., 2009; Alami et al., 2014; Coyne et al., 2014). It was identified that mutated TDP-43 leads to a reduction in local NMJ translation of the MAP1b homolog futsch in a Drosophila model of ALS (Coyne et al., 2014). MAP1b also sequesters the plus TIP binding protein EB3 from the growing plus ends of MTs (Tortosa et al., 2013). MAP1b over expression leads to a loss of EB3 colocalization, and down regulation leads to an increase in EB3 binding to the MT, inducing MT stability defects and aberrant growth (Tortosa et al., 2013). This may not only be in part due to the physical interactions of MAP1b with EB3, but also due to MAP1b’s role in certain signaling cascades, in which end binding proteins and MT dynamics can be affected (Montenegro-Venegas et al., 2010; Villarroel-Campos and Gonzalez-Billault, 2014; Ketschek et al., 2015). Therefore, MAP1b, through multiple mechanisms, affects dynamic pools of MTs due to roles in signaling and by direct physical interactions (Tymanskyj et al., 2012).
Histone Deacetylase 6 (HDAC6) deacetylates lysine 40 on α-tubulin, which is the hallmark of MT stability. Alterations to the function and expression of HDAC6 therefore have implications for MT transport (Reed et al., 2006). Indeed, this is found to be the case in SOD1 mouse models of ALS. Interactions between HDAC6 and mutant SOD1 lead to the development of intracellular aggregates containing HDAC6 in vitro and consequently result in the inhibition of HDAC6 deacetylase activity (Gal et al., 2013). This sequestering of HDAC6 reduces its deacetylating action, resulting in greater tubulin acetylation. Furthermore, HDAC6 facilitates the degradation of poly-ubiquinated proteins, such as SOD1 and TDP-43, through the autophagosome (Kawaguchi et al., 2003; Lee et al., 2015). Permanent binding of HDAC6 to mutant SOD1 may account for the loss of deacetylase activity and the increase in SOD1 aggregates due to ineffective action of HDAC6 in protein degradation pathways, effectively providing a double-hit mechanism (Gal et al., 2013). Gal et al. (2013) hypothesized that the described SOD1-HDAC6 model leads to an increase in axonal transport in mutant SOD1 mice; therefore increasing the transport, spread and deposition of misfolded SOD1 aggregates. Deletion of HDAC6 in vivo delays disease progression in SOD1G93A mice, highlighting a possible non-cell autonomous action of HDAC6 that may add to the diseases phenotype, or the gain of function of the SOD1-HDAC6 complex in these mice (Taes et al., 2013). However, the increase in acetylation in mutant SOD1 models is counter-intuitive to the identified increase in microtubule dynamics: acetylation is a marker of stable microtubule domains, highlighting the need for further research (Fanara et al., 2007; Gal et al., 2013; Kleele et al., 2014). Furthermore, HDAC6 has been identified as a substrate of TDP-43; TDP-43 and FUS act in a complex to regulate HDAC6 expression (Fiesel et al., 2010; Kim et al., 2010), indicating that a number of mutations may impact HDAC6 activity, although, these are yet to be explored.
Stable, polyaminated domains of neuronal MTs are associated with MAP6 (originally stable-tubule-only-peptide—STOP), a protein that confers cold stability on MTs. MAP6 also prevents destabilization of MTs by pharmacological means (Slaughter and Black, 2003; Baas et al., 2016). Microtubule dysfunction as a primary driver of neurological pathology has been highlighted by the development of the MAP6 knockout mouse model. Interestingly, MAP6 knockout mice develop a schizophrenic-like phenotype, which can be rescued by pharmacologically increasing MT stability (Andrieux et al., 2006). This is also associated with transport deficits, with evidence suggesting it may be driven by a loss of network stability; pharmacologically increasing MT stability ameliorates this phenotype (Daoust et al., 2014). Of further interest is the identified localization of MAP6 to NF spheroids in the cortex and spinal cord of ALS patients (Letournel et al., 2003). However, the exact impact this has on disease is still not understood. In addition, MAP6 has been found to impact dendritic lysosome transport and trafficking, which can be impaired by the expression of an FTD risk factor with TDP-43 pathology (Kim et al., 2010; Schwenk et al., 2014).
MTs also interact with other cytoskeletal networks, such as the neuronal microfilament actin, and the neuronal specific NFs and are known to influence MT structure and function. Although current understanding of the interactions between these filaments is not fully appreciated, shared signaling cascades (Wittmann and Waterman-Storer, 2005) and shared associated or connecting proteins (Goryunov and Liem, 2016) are potential mechanisms underlying this communication. Cytoskeletal elements may affect one another in a more direct mechanism. This is evidenced by key proteins associated with actin assembly, such as formins, which bind to and regulate MT dynamics (Bartolini et al., 2008). Alternately, the MT TIP protein EB1 also has binding sites on actin, but this binding is mutually exclusive, and therefore competitive exclusion affects the stability neuronal MTs (Alberico et al., 2016). Importantly, NF accumulations are present in several neurodegenerative diseases, including ALS, and reviewed previously (Vickers et al., 2009). It has been suggested that accumulation of NF contributes to axonal degeneration by impeding axonal transport, causing defects in the cells’ ability to maintain the synapse (Collard et al., 1995). Depletion of NFs has been observed to increase lifespan and improve the phenotype of SOD1 mice (Williamson et al., 1998). While aggregation of misfolded proteins such as SOD1 and NF in ALS has been suggested to cause axonal transport dysfunction, the sequestration of toxic, misfolded protein or hyper phosphorylated proteins into subcellular compartments may in turn be neuroprotective (as previously reviewed in Patzke and Tsai, 2002; Takalo et al., 2013). This sequestration potentially reduces toxic oligomer interaction with endogenous proteins required for cellular function, or may act as a “sink” for aberrant phosphorylation; and therefore may extend the life of the cell. Alternately, this compartmentalization may detrimentally cause the sequestration of endogenous proteins that preferentially bind to aggregate proteins, and is still a cause for debate (Tateno et al., 2009).
Collectively, accumulating evidence indicates that MTs and their associated proteins may play an important role in both the initiation and progression of ALS. MT dysfunction may sit on a pathological continuum, whereby MTs can act as the primary driver of MN degeneration, or where other genetic or molecular mechanisms converge to cause MT pathology. Indeed, MT dysfunction in ALS points to aberrant alterations in dynamics, with resultant dysregulated axonal transport driving disease pathogenesis and ultimately cytoskeletal and cellular destruction. Thus these disease processes offer attractive targets for therapeutic intervention.
Targeting Neuronal Microtubules for Therapy: An Approach for ALS
The only available treatment for ALS is the anti-excitotoxic drug, Riluzole. Riluzole acts on the presynaptic neuron to limit the release of glutamate into the synapse therefore reducing the excitotoxic effect of glutamate on the postsynaptic cell (Bensimon et al., 1994). Notwithstanding the reduction in excitotoxicity, treatment strategies involving riluzole have limited effectiveness, and are only able to extend patient life by approximately 3–6 months (Gurney et al., 1998). ALS is a multi-factorial disease, with many cellular components affected; therefore there are a range of available targets for therapy, however many of them have had limited success (Turner and Talbot, 2008). This review has discussed MT involvement in ALS and in particular how dynamics and function are impaired in disease states. Pharmacological manipulation of MTs to improve disease phenotype offers an attractive target in ALS. Indeed, this therapeutic approach has previously been undertaken in multiple neurodegenerative disease models, including ALS.
Fanara et al. (2007), established that hyperdynamic neuronal MTs were present in a SOD1 mouse model of ALS and administered the MT modulating agent noscapine to attenuate this phenotype. Noscapine, which can cross the blood brain barrier (BBB) effectively dampens hyperdynamics, leading to less depolymerization and polymerization events from occurring at the growing plus tip of the MT (Landen et al., 2002, 2004). Noscapine treatment extended lifespan, attenuated MT dynamics and normalized aspects of axonal transport. This in itself gives evidence for hyperdynamics driving transport deficits in these mice. However, more cargo-specific and rate-specific transport assays are required to identify if this is the case. Moreover, this study provides supporting evidence for the use of MT stabilizing agents in the treatment of ALS.
HDAC6 inhibitors and HDAC knockout mice have also been trialed to improve outcomes in ALS models, with the intention of increasing the acetylation of stable MTs to improve stability and axonal transport. The inhibition or removal of HDAC6 was found to improve the phenotype of mutant SOD1 mice (Taes et al., 2013). However, axonal transport was not directly measured. A similar study utilizing HDAC6 inhibition in Charcot-Marie-Tooth (CMT) disease showed an improvement in axonal mitochondrial transport, supporting HDAC6 inhibition as a candidate therapeutic for multiple neurodegenerative diseases (d’Ydewalle et al., 2011).
Direct MT stabilizing agents have been previously used in the medical setting in the treatment of cancers, as the addition of these agents perturb the formation of the mitotic spindle and therefore inhibit cell division (Schiff et al., 1979). The most studied MT targeting agent taxol has, in the last decade, developed a newfound use in modulating MTs in neurodegenerative diseases (Michaelis et al., 2006; Brunden et al., 2011; Das and Miller, 2012; King et al., 2013). At high doses, taxol treated systems develop over-stabilized MTs, preventing cell division in cancer cells; however, this can generate a painful peripheral neuropathy (Reyes-Gibby et al., 2009). At lower doses taxol has been found to limit MT depolymerization and stabilize the MT network in a number of neurodegenerative models (Michaelis et al., 2006; Brunden et al., 2011; Das and Miller, 2012; King et al., 2013). Indeed, treatment of an in vivo tauopathy model with taxol improves MN fast axonal transport, highlighting the therapeutic potential of taxol for disorders effecting MNs (Zhang et al., 2005). The range of taxol concentrations which yield beneficial effects on neurodegeneration are very narrow (Shemesh and Spira, 2011). Coupled with the off target effects identified due to taxol therapy, limited crossover at the BBB and the dramatic alterations to MT stability that it delivers, the benefits of taxol administration in neurodegenerative disease have been brought into question (Baas and Ahmad, 2013; Baas, 2014). In relation to ALS, taxol has been shown to reduce MT disintegration in an in vitro kainic acid excitotoxic model of ALS (King et al., 2013). Excitotoxicity exposure was reported to induce MT instability upstream of caspase-3 activation, which is mitigated with taxol treatment. However, as characterization of MT dynamics has not been completed with regard to excitotoxicity due to kainic acid exposure, it is difficult to assess whether taxol is indeed limiting the hypothesized dynamic instability of MTs, or preventing the breakdown of the MT network. Indeed, inhibition of caspase-3 activation in other excitotoxicity models reports neuroprotection and increased cell survival (Chen et al., 2001). However, it is important to note that Taxol derivatives, which exhibit higher BBB permeability, need to be investigated to determine if this form of MT stabilization is a viable treatment for ALS.
Conclusion
MTs play a fundamental role in normal neuronal functions. MT dysfunction has been implicated in the pathogenesis of a number of neurodegenerative diseases including ALS, however, the precise mechanisms underlying this dysfunction are not fully understood. Currently our understanding of MT involvement in ALS suggests that generation of hyperdynamic MTs, transport dysfunction and alterations to cytoskeletal gene and protein expression may help drive disease pathogenesis. Identification of the chain of events, which lead to the dysfunction of the MT network in ALS, is paramount to our understanding of how MTs are involved in disease initiation and maintenance. Indeed, increasing evidence suggests that MT dysfunction is both a primary driver of pathology, and also a site for pathological convergence from associated familial and molecular ALS mechanisms. Modulation of these events and MT structural integrity is an attractive therapeutic target, with benefits to this approach being shown in other models of disease. Increasing our knowledge of the mechanisms behind MT dysfunction in ALS will hopefully uncover many more targets to manipulate pharmacologically to extend life or cure this debilitating and ultimately fatal disease.
Author Contributions
JaAC (primary author) drafted the majority of the manuscript, along with the initial planning. EJY contributed to the remaining text of the manuscript, initial planning and through literature research of key themes. Various disease mechanisms were investigated by CAB, who also had a primary role in the planning and revision of the manuscript. JyAC has experience with the literature surrounding therapeutic treatment, and also played a primary role in the planning and revision of the manuscript. TCD contributed to the initial planning, literature research and revision of the manuscript. JaAC, EJY, CAB, JyAC and TCD all agreed for the final version to be published and agree to be accountable for all aspects of the work.
Funding
This project was funded through the generous support provided by the National Health and Medical Research Council, the Motor Neurone Disease Research Institute of Australia, the Masonic Centenary Medical Research Foundation of Tasmania, the Dwyer Family, the Cape Hope Foundation and Diagnostic Services Pty Ltd.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlijanian, M. K., Barrezueta, N. X., Williams, R. D., Jakowski, A., Kowsz, K. P., McCarthy, S., et al. (2000). Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Natl. Acad. Sci. U S A 97, 2910–2915. doi: 10.1073/pnas.040577797
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Alberico, E. O., Zhu, Z. C., Wu, Y. F., Gardner, M. K., Kovar, D. R., and Goodson, H. V. (2016). Interactions between the microtubule binding protein EB1 and F-Actin. J. Mol. Biol. 428, 1304–1314. doi: 10.1016/j.jmb.2016.01.032
Al-Chalabi, A., Andersen, P. M., Nilsson, P., Chioza, B., Andersson, J. L., Russ, C., et al. (1999). Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 8, 157–164. doi: 10.1093/hmg/8.2.157
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and Van Den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Alim, M. A., Ma, Q. L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., et al. (2004). Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 6, 435–442; discussion 443–449.
Allen, C., and Borisy, G. G. (1974). Structural polarity and directional growth of microtubules of Chlamydomonas flagella. J. Mol. Biol. 90, 381–402. doi: 10.1016/0022-2836(74)90381-7
Andersen, J. K. (2004). Oxidative stress in neurodegeneration: cause or consequence? Nat. Rev. Neurosci. 10, S18–S25. doi: 10.1038/nrn1434
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Andrieux, A., Salin, P., Schweitzer, A., Bégou, M., Pachoud, B., Brun, P., et al. (2006). Microtubule stabilizer ameliorates synaptic function and behavior in a mouse model for schizophrenia. Biol. Psychiatry 60, 1224–1230. doi: 10.1016/j.biopsych.2006.03.048
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Baas, P. W. (2013). Microtubule stability in the axon: new answers to an old mystery. Neuron 78, 3–5. doi: 10.1016/j.neuron.2013.03.012
Baas, P. W. (2014). Beyond taxol: microtubule-based strategies for promoting nerve regeneration after injury. Neural Regen. Res. 9, 1265–1266. doi: 10.4103/1673-5374.137572
Baas, P. W., and Ahmad, F. J. (2013). Beyond taxol: microtubule-based treatment of disease and injury of the nervous system. Brain 136, 2937–2951. doi: 10.1093/brain/awt153
Baas, P. W., and Black, M. M. (1990). Individual microtubules in the axon consist of domains that differ in both composition and stability. J. Cell Biol. 111, 495–509. doi: 10.1083/jcb.111.2.495
Baas, P. W., and Lin, S. (2011). Hooks and comets: the story of microtubule polarity orientation in the neuron. Dev. Neurobiol. 71, 403–418. doi: 10.1002/dneu.20818
Baas, P. W., Rao, A. N., Matamoros, A. J., and Leo, L. (2016). Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). doi: 10.1002/cm.21286 [Epub ahead of print].
Baird, F. J., and Bennett, C. L. (2013). Microtubule defects and neurodegeneration. J. Genet. Syndr. Gene Ther. 4:203. doi: 10.4172/2157-7412.1000203
Bartolini, F., Moseley, J. B., Schmoranzer, J., Cassimeris, L., Goode, B. L., and Gundersen, G. G. (2008). The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 181, 523–536. doi: 10.1083/jcb.200709029
Belly, A., Moreau-Gachelin, F., Sadoul, R., and Goldberg, Y. (2005). Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 379, 152–157. doi: 10.1016/j.neulet.2004.12.071
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole study group. N. Engl. J. Med. 330, 585–591. doi: 10.1056/NEJM199403033300901
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U S A 107, 20523–20528. doi: 10.1073/pnas.1006869107
Black, M. M., Slaughter, T., Moshiach, S., Obrocka, M., and Fischer, I. (1996). Tau is enriched on dynamic microtubules in the distal region of growing axons. J. Neurosci. 16, 3601–3619.
Blizzard, C. A., Southam, K. A., Dawkins, E., Lewis, K. E., King, A. E., Clark, J. A., et al. (2015). Identifying the primary site of pathogenesis in amyotrophic lateral sclerosis - vulnerability of lower motor neurons to proximal excitotoxicity. Dis. Model Mech. 8, 215–224. doi: 10.1242/dmm.018606
Boutte, A. M., Woltjer, R. L., Zimmerman, L. J., Stamer, S. L., Montine, K. S., Manno, M. V., et al. (2006). Selectively increased oxidative modifications mapped to detergent-insoluble forms of Aβ and β-III tubulin in Alzheimer’s disease. FASEB J. 20, 1473–1483. doi: 10.1096/fj.06-5920com
Brandt, R., and Lee, G. (1993). Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation and bundle formation in vitro. J. Biol. Chem. 268, 3414–3419.
Brockington, A., Heath, P. R., Holden, H., Kasher, P., Bender, F. L., Claes, F., et al. (2010). Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis. BMC Genomics 11:203. doi: 10.1186/1471-2164-11-203
Brockington, A., Wharton, S. B., Fernando, M., Gelsthorpe, C. H., Baxter, L., Ince, P. G., et al. (2006). Expression of vascular endothelial growth factor and its receptors in the central nervous system in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 65, 26–36. doi: 10.1097/01.jnen.0000196134.51217.74
Brunden, K. R., Yao, Y., Potuzak, J. S., Ferrer, N. I., Ballatore, C., James, M. J., et al. (2011). The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol. Res. 63, 341–351. doi: 10.1016/j.phrs.2010.12.002
Bunton-Stasyshyn, R. K., Saccon, R. A., Fratta, P., and Fisher, E. M. (2015). SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist 21, 519–529. doi: 10.1177/1073858414561795
Buratti, E., and Baralle, F. E. (2001). Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343. doi: 10.1074/jbc.m104236200
Cairns, N. J., Lee, V. M., and Trojanowski, J. Q. (2004). The cytoskeleton in neurodegenerative diseases. J. Pathol. 204, 438–449. doi: 10.1002/path.1650
Callicott, J. H., Straub, R. E., Pezawas, L., Egan, M. F., Mattay, V. S., Hariri, A. R., et al. (2005). Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. U S A 102, 8627–8632. doi: 10.1073/pnas.0500515102
Cappelletti, G., Surrey, T., and Maci, R. (2005). The parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilising factor. FEBS Lett. 579, 4781–4786. doi: 10.1016/j.febslet.2005.07.058
Carletti, B., Passarelli, C., Sparaco, M., Tozzi, G., Pastore, A., Bertini, E., et al. (2011). Effect of protein glutathionylation on neuronal cytoskeleton: a potential link to neurodegeneration. Neuroscience 192, 285–294. doi: 10.1016/j.neuroscience.2011.05.060
Cartelli, D., Ronchi, C., Maggioni, M. G., Rodighiero, S., Giavini, E., and Cappelletti, G. (2010). Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+-induced neurodegeneration. J. Neurochem. 115, 247–258. doi: 10.1111/j.1471-4159.2010.06924.x
Chandran, J., Ding, J., and Cai, H. (2007). Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 36, 224–231. doi: 10.1007/s12035-007-0034-x
Chen, B. L., Hall, D. H., and Chklovskii, D. B. (2006). Wiring optimization can relate neuronal structure and function. Proc. Natl. Acad. Sci. U S A 103, 4723–4728. doi: 10.1073/pnas.0506806103
Chen, T. A., Yang, F., Cole, G. M., and Chan, S. O. (2001). Inhibition of caspase-3-like activity reduces glutamate induced cell death in adult rat retina. Brain Res. 904, 177–188. doi: 10.1016/s0006-8993(01)02485-4
Collard, J. F., Côté, F., and Julien, J. P. (1995). Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375, 61–64. doi: 10.1038/375061a0
Coyne, A. N., Siddegowda, B. B., Estes, P. S., Johannesmeyer, J., Kovalik, T., Daniel, S. G., et al. (2014). Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 34, 15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014
Daoust, A., Bohic, S., Saoudi, Y., Debacker, C., Gory-Faure, S., Andrieux, A., et al. (2014). Neuronal transport defects of the MAP6 KO mouse–a model of schizophrenia - and alleviation by Epothilone D treatment, as observed using MEMRI. Neuroimage 96, 133–142. doi: 10.1016/j.neuroimage.2014.03.071
Das, V., and Miller, J. H. (2012). Microtubule stabilization by peloruside A and paclitaxel rescues degenerating neurons from okadaic acid-induced tau phosphorylation. Eur. J. Neurosci. 35, 1705–1717. doi: 10.1111/j.1460-9568.2012.08084.x
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K. F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Desai, A., and Mitchison, T. J. (1997). Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 13, 83–117. doi: 10.1146/annurev.cellbio.13.1.83
Devon, R. S., Orban, P. C., Gerrow, K., Barbieri, M. A., Schwab, C., Cao, L. P., et al. (2006). Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. U S A 103, 9595–9600. doi: 10.1073/pnas.0510197103
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Droppelmann, C. A., Campos-Melo, D., Volkening, K., and Strong, M. J. (2014). The emerging role of guanine nucleotide exchange factors in ALS and other neurodegenerative diseases. Front. Cell. Neurosci. 8:282. doi: 10.3389/fncel.2014.00282
Drubin, D. G., and Kirschner, M. W. (1986). Tau protein function in living cells. J. Cell Biol. 103, 2739–2746. doi: 10.1083/jcb.103.6.2739
Drum, B. M., Yuan, C., Li, L., Liu, Q., Wordeman, L., and Santana, L. F. (2016). Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J. Mol. Cell. Cardiol. 93, 32–43. doi: 10.1016/j.yjmcc.2016.02.012
Dubey, J., Ratnakaran, N., and Koushika, S. P. (2015). Neurodegeneration and microtubule dynamics: death by a thousand cuts. Front. Cell. Neurosci. 9:343. doi: 10.3389/fncel.2015.00343
Dupuis, L., de Tapia, M., René, F., Lutz-Bucher, B., Gordon, J. W., Mercken, L., et al. (2000). Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol. Dis. 7, 274–285. doi: 10.1006/nbdi.2000.0292
d’Ydewalle, C., Krishnan, J., Chiheb, D. M., Van Damme, P., Irobi, J., Kozikowski, A. P., et al. (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974. doi: 10.1038/nm.2396
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., and Mandelkow, E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria and endoplasmic reticulum: implications for Alzheimer’s disease. J. Cell Biol. 143, 777–794. doi: 10.1083/jcb.143.3.777
Evans, D. B., Rank, K. B., Bhattacharya, K., Thomsen, D. R., Gurney, M. E., and Sharma, S. K. (2000). Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 275, 24977–24983. doi: 10.1074/jbc.m000808200
Eymard-Pierre, E., Lesca, G., Dollet, S., Santorelli, F. M., Di Capua, M., Bertini, E., et al. (2002). Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 71, 518–527. doi: 10.1086/342359
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21, 3703–3718. doi: 10.1093/hmg/dds205
Fanara, P., Banerjee, J., Hueck, R. V., Harper, M. R., Awada, M., Turner, H., et al. (2007). Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem. 282, 23465–23472. doi: 10.1074/jbc.m703434200
Fang, C., Bourdette, D., and Banker, G. (2012). Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Mol. Neurodegener. 7:29. doi: 10.1186/1750-1326-7-29
Farah, C. A., Nguyen, M. D., Julien, J. P., and Leclerc, N. (2003). Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 84, 77–86. doi: 10.1046/j.1471-4159.2003.01505.x
Feiguin, F., Godena, V. K., Romano, G., D’ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Ferreira, A., and Cáceres, A. (1989). The expression of acetylated microtubules during axonal and dendritic growth in cerebellar macroneurons which develop in vitro. Brain Res. Dev. Brain Res. 49, 205–213. doi: 10.1016/0165-3806(89)90022-9
Fiesel, F. C., Voigt, A., Weber, S. S., Van Den Haute, C., Waldenmaier, A., Gorner, K., et al. (2010). Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209–221. doi: 10.1038/emboj.2009.324
Francis, F., Meyer, G., Fallet-Bianco, C., Moreno, S., Kappeler, C., Socorro, A. C., et al. (2006). Human disorders of cortical development: from past to present. Eur. J. Neurosci. 23, 877–893. doi: 10.1111/j.1460-9568.2006.04649.x
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Fujiwara, T., and Morimoto, K. (2012). Cooperative effect of p150Glued and microtubule stabilization to suppress excitotoxicity-induced axon degeneration. Biochem. Biophys. Res. Commun. 424, 82–88. doi: 10.1016/j.bbrc.2012.06.071
Gal, J., Chen, J., Barnett, K. R., Yang, L., Brumley, E., and Zhu, H. (2013). HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J. Biol. Chem. 288, 15035–15045. doi: 10.1074/jbc.M112.431957
Gillardon, F. (2009). Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability–a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 110, 1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Gonzalez-Billault, C., Avila, J., and Cáceres, A. (2001). Evidence for the role of MAP1B in axon formation. Mol. Biol. Cell 12, 2087–2098. doi: 10.1091/mbc.12.7.2087
Goryunov, D., and Liem, R. K. (2016). Microtubule-actin cross-linking factor 1: domains, interaction partners and tissue-specific functions. Methods Enzymol. 569, 331–353. doi: 10.1016/bs.mie.2015.05.022
Gros-Louis, F., Larivière, R., Gowing, G., Laurent, S., Camu, W., Bouchard, J. P., et al. (2004). A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J. Biol. Chem. 279, 45951–45956. doi: 10.1074/jbc.m408139200
Guerrini, R., Dobyns, W. B., and Barkovich, A. J. (2008). Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 31, 154–162. doi: 10.1016/j.tins.2007.12.004
Gurney, M. E., Fleck, T. J., Himes, C. S., and Hall, E. D. (1998). Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology 50, 62–66. doi: 10.1212/wnl.50.1.62
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Hadano, S., Benn, S. C., Kakuta, S., Otomo, A., Sudo, K., Kunita, R., et al. (2006). Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 15, 233–250. doi: 10.1093/hmg/ddi440
Hadano, S., Hand, C. K., Osuga, H., Yanagisawa, Y., Otomo, A., Devon, R. S., et al. (2001). A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173. doi: 10.1038/ng1001-166
Hanger, D. P., Lau, D. H., Phillips, E. C., Bondulich, M. K., Guo, T., Woodward, B. W., et al. (2014). Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis. 40, S37–S45. doi: 10.3233/JAD-132054
Heath, P. R., and Shaw, P. J. (2002). Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458. doi: 10.1002/mus.10186
Heidemann, S. R., Landers, J. M., and Hamborg, M. A. (1981). Polarity orientation of axonal microtubules. J. Cell Biol. 91, 661–665. doi: 10.1083/jcb.91.3.661
Hersheson, J., Mencacci, N. E., Davis, M., Macdonald, N., Trabzuni, D., Ryten, M., et al. (2013). Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 73, 546–553. doi: 10.1002/ana.23832
Hoskison, M. M., Yanagawa, Y., Obata, K., and Shuttleworth, C. W. (2007). Calcium-dependent NMDA-induced dendritic injury and MAP2 loss in acute hippocampal slices. Neuroscience 145, 66–79. doi: 10.1016/j.neuroscience.2006.11.034
Howes, S. C., Alushin, G. M., Shida, T., Nachury, M. V., and Nogales, E. (2014). Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 25, 257–266. doi: 10.1091/mbc.E13-07-0387
Huang, Y., Wu, Z., and Zhou, B. (2015). hSOD1 promotes tau phosphorylation and toxicity in the Drosophila model. J. Alzheimers Dis. 45, 235–244. doi: 10.3233/JAD-141608
Hurd, D. D., and Saxton, W. M. (1996). Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics 144, 1075–1085.
Ikeda, K., Zhapparova, O., Brodsky, I., Semenova, I., Tirnauer, J. S., Zaliapin, I., et al. (2011). CK1 activates minus-end-directed transport of membrane organelles along microtubules. Mol. Biol. Cell 22, 1321–1329. doi: 10.1091/mbc.e10-09-0741
Ikenaka, K., Katsuno, M., Kawai, K., Ishigaki, S., Tanaka, F., and Sobue, G. (2012). Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 13, 1225–1238. doi: 10.3390/ijms13011225
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Jaiswal, M. K. (2014). Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol. Cell. Ther. 2:26. doi: 10.1186/2052-8426-2-26
Janke, C. (2014). The tubulin code: molecular components, readout mechanisms and functions. J. Cell Biol. 206, 461–472. doi: 10.1083/jcb.201406055
Janke, C., and Kneussel, M. (2010). Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33, 362–372. doi: 10.1016/j.tins.2010.05.001
Jiang, Y. M., Yamamoto, M., Kobayashi, Y., Yoshihara, T., Liang, Y., Terao, S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. doi: 10.1002/ana.20379
Jiang, Y. M., Yamamoto, M., Tanaka, F., Ishigaki, S., Katsuno, M., Adachi, H., et al. (2007). Gene expressions specifically detected in motor neurons (dynactin 1, early growth response 3, acetyl-CoA transporter, death receptor 5 and cyclin C) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66, 617–627. doi: 10.1097/nen.0b013e318093ece3
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kabuta, T., Kinugawa, A., Tsuchiya, Y., Kabuta, C., Setsuie, R., Tateno, M., et al. (2009). Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem. Biophys. Res. Commun. 387, 121–126. doi: 10.1016/j.bbrc.2009.06.138
Kanai, Y., Dohmae, N., and Hirokawa, N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525. doi: 10.1016/j.neuron.2004.07.022
Kawaguchi, Y., Kovacs, J. J., Mclaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738. doi: 10.1016/s0092-8674(03)00939-5
Kempf, M., Clement, A., Faissner, A., Lee, G., and Brandt, R. (1996). Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J. Neurosci. 16, 5583–5592.
Ketschek, A., Jones, S., Spillane, M., Korobova, F., Svitkina, T., and Gallo, G. (2015). Nerve growth factor promotes reorganization of the axonal microtubule array at sites of axon collateral branching. Dev. Neurobiol. 75, 1441–1461. doi: 10.1002/dneu.22294
Kieran, D., Hafezparast, M., Bohnert, S., Dick, J. R., Martin, J., Schiavo, G., et al. (2005). A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 169, 561–567. doi: 10.1083/jcb.200501085
Kim, S. H., Shanware, N. P., Bowler, M. J., and Tibbetts, R. S. (2010). Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 285, 34097–34105. doi: 10.1074/jbc.M110.154831
King, A. E., Southam, K. A., Dittmann, J., and Vickers, J. C. (2013). Excitotoxin-induced caspase-3 activation and microtubule disintegration in axons is inhibited by taxol. Acta Neuropathol. Commun. 1:59. doi: 10.1186/2051-5960-1-59
Kleele, T., Marinkovic, P., Williams, P. R., Stern, S., Weigand, E. E., Engerer, P., et al. (2014). An assay to image neuronal microtubule dynamics in mice. Nat. Commun. 5:4827. doi: 10.1038/ncomms5827
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi: 10.1038/nn.3230
Lai, C., Xie, C., McCormack, S. G., Chiang, H. C., Michalak, M. K., Lin, X., et al. (2006). Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J. Neurosci. 26, 11798–11806. doi: 10.1523/jneurosci.2084-06.2006
Laird, F. M., Farah, M. H., Ackerley, S., Hoke, A., Maragakis, N., Rothstein, J. D., et al. (2008). Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 28, 1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008
Landen, J. W., Hau, V., Wang, M., Davis, T., Ciliax, B., Wainer, B. H., et al. (2004). Noscapine crosses the blood-brain barrier and inhibits glioblastoma growth. Clin. Cancer Res. 10, 5187–5201. doi: 10.1158/1078-0432.ccr-04-0360
Landen, J. W., Lang, R., McMahon, S. J., Rusan, N. M., Yvon, A. M., Adams, A. W., et al. (2002). Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 62, 4109–4114.
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., van den Berg, L. H., Van Es, M. A., et al. (2009). Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 106, 9004–9009. doi: 10.1073/pnas.0812937106
Landino, L. M., Koumas, M. T., Mason, C. E., and Alston, J. A. (2007). Modification of tubulin cysteines by nitric oxide and nitroxyl donors alters tubulin polymerization activity. Chem. Res. Toxicol. 20, 1693–1700. doi: 10.1021/tx7001492
Landino, L. M., Robinson, S. H., Skreslet, T. E., and Cabral, D. M. (2004). Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 323, 112–117. doi: 10.1016/j.bbrc.2004.08.065
Lee, J. Y., Kawaguchi, Y., Li, M., Kapur, M., Choi, S. J., Kim, H. J., et al. (2015). Uncoupling of protein aggregation and neurodegeneration in a mouse amyotrophic lateral sclerosis model. Neurodegener. Dis. 15, 339–349. doi: 10.1159/000437208
Letournel, F., Bocquet, A., Dubas, F., Barthelaix, A., and Eyer, J. (2003). Stable tubule only polypeptides (STOP) proteins co-aggregate with spheroid neurofilaments in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 62, 1211–1219. doi: 10.1093/jnen/62.12.1211
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Levy, J. R., Sumner, C. J., Caviston, J. P., Tokito, M. K., Ranganathan, S., Ligon, L. A., et al. (2006). A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 172, 733–745. doi: 10.1083/jcb.200511068
Ligon, L. A., Lamonte, B. H., Wallace, K. E., Weber, N., Kalb, R. G., and Holzbaur, E. L. (2005). Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 16, 533–536. doi: 10.1097/00001756-200504250-00002
Lim, S. S., Sammak, P. J., and Borisy, G. G. (1989). Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J. Cell Biol. 109, 253–263. doi: 10.1083/jcb.109.1.253
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, J. S., Schubert, C. R., Fu, X., Fourniol, F. J., Jaiswal, J. K., Houdusse, A., et al. (2012). Molecular basis for specific regulation of neuronal kinesin-3 motors by doublecortin family proteins. Mol. Cell 47, 707–721. doi: 10.1016/j.molcel.2012.06.025
Lopez-Fanarraga, M., Carranza, G., Bellido, J., Kortazar, D., Villegas, J. C., and Zabala, J. C. (2007). Tubulin cofactor B plays a role in the neuronal growth cone. J. Neurochem. 100, 1680–1687. doi: 10.1111/j.1471-4159.2006.04328.x
Lucas, F. R., Goold, R. G., Gordon-Weeks, P. R., and Salinas, P. C. (1998). Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J. Cell Sci. 111, 1351–1361.
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi: 10.1016/j.neuron.2014.10.019
Marangi, G., and Traynor, B. J. (2015). Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 1607, 75–93. doi: 10.1016/j.brainres.2014.10.009
Matsuyama, S. S., and Jarvik, L. F. (1989). Hypothesis: microtubules, a key to Alzheimer disease. Proc. Natl. Acad. Sci. U S A 86, 8152–8156. doi: 10.1073/pnas.86.20.8152
Maximino, J. R., de Oliveira, G. P., Alves, C. J., and Chadi, G. (2014). Deregulated expression of cytoskeleton related genes in the spinal cord and sciatic nerve of presymptomatic SOD1G93A amyotrophic lateral sclerosis mouse model. Front. Cell. Neurosci. 8:148. doi: 10.3389/fncel.2014.00148
Mcmurray, C. T. (2000). Neurodegeneration: diseases of the cytoskeleton? Cell Death Differ. 7, 861–865. doi: 10.1038/sj.cdd.4400764
Mezzapesa, D. M., D’errico, E., Tortelli, R., Distaso, E., Cortese, R., Tursi, M., et al. (2013). Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS One 8:e80748. doi: 10.1371/journal.pone.0080748
Michaelis, M. L., Georg, G., Telikepalli, H., Mcintosh, M., and Rajewski, R. A. (2006). Ongoing in vivo studies with cytoskeletal drugs in tau transgenic mice. Curr. Alzheimer Res. 3, 215–219. doi: 10.2174/156720506777632880
Millecamps, S., and Julien, J. P. (2013). Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. doi: 10.1038/nrn3380
Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. doi: 10.1038/312237a0
Montenegro-Venegas, C., Tortosa, E., Rosso, S., Peretti, D., Bollati, F., Bisbal, M., et al. (2010). MAP1B regulates axonal development by modulating Rho-GTPase Rac1 activity. Mol. Biol. Cell 21, 3518–3528. doi: 10.1091/mbc.e09-08-0709
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morris, J. A., Kandpal, G., Ma, L., and Austin, C. P. (2003). DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum. Mol. Genet. 12, 1591–1608. doi: 10.1093/hmg/ddg162
Münch, C., Rosenbohm, A., Sperfeld, A. D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.wnl.0000134608.83927.b1
Munnamalai, V., and Suter, D. M. (2009). Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J. Neurochem. 108, 644–661. doi: 10.1111/j.1471-4159.2008.05787.x
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, M. D., Larivière, R. C., and Julien, J. P. (2001). Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30, 135–147. doi: 10.1016/s0896-6273(01)00268-9
Nogales, E., and Wang, H. W. (2006). Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr. Opin. Struct. Biol. 16, 221–229. doi: 10.1016/j.sbi.2006.03.005
O’Brien, E. T., Salmon, E. D., and Erickson, H. P. (1997). How calcium causes microtubule depolymerization. Cell Motil. Cytoskeleton 36, 125–135. doi: 10.10.1002/(SICI)1097-0169(1997)36:2<125::AID-CM3>3.0.CO;2-8
Oosthuyse, B., Moons, L., Storkebaum, E., Beck, H., Nuyens, D., Brusselmans, K., et al. (2001). Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28, 131–138. doi: 10.1038/88842
Ou, S. H., Wu, F., Harrich, D., Garcia-Martinez, L. F., and Gaynor, R. B. (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596.
Panda, D., Goode, B. L., Feinstein, S. C., and Wilson, L. (1995). Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 34, 11117–11127. doi: 10.1021/bi00035a017
Panzeri, C., De Palma, C., Martinuzzi, A., Daga, A., De Polo, G., Bresolin, N., et al. (2006). The first ALS2 missense mutation associated with JPLS reveals new aspects of alsin biological function. Brain 129, 1710–1719. doi: 10.1093/brain/awl104
Park, J. Y., Jang, S. Y., Shin, Y. K., Koh, H., Suh, D. J., Shinji, T., et al. (2013). Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience 238, 258–269. doi: 10.1016/j.neuroscience.2013.02.033
Patzke, H., and Tsai, L. H. (2002). Cdk5 sinks into ALS. Trends Neurosci. 25, 8–10. doi: 10.1016/s0166-2236(00)02000-2
Peters, O. M., Ghasemi, M., and Brown, R. H. Jr. (2015). Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 125, 1767–1779. doi: 10.1172/JCI71601
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Puls, I., Jonnakuty, C., Lamonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Qiang, L., Yu, W., Andreadis, A., Luo, M., and Baas, P. W. (2006). Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 26, 3120–3129. doi: 10.1523/jneurosci.5392-05.2006
Reed, N. A., Cai, D., Blasius, T. L., Jih, G. T., Meyhofer, E., Gaertig, J., et al. (2006). Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172. doi: 10.1016/j.cub.2006.09.014
Ren, Y., Liu, W., Jiang, H., Jiang, Q., and Feng, J. (2005). Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 280, 34105–34112. doi: 10.1074/jbc.m503483200
Ren, Y., Zhao, J., and Feng, J. (2003). Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 23, 3316–3324.
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Reyes-Gibby, C. C., Morrow, P. K., Buzdar, A., and Shete, S. (2009). Chemotherapy-induced peripheral neuropathy as a predictor of neuropathic pain in breast cancer patients previously treated with paclitaxel. J. Pain 10, 1146–1150. doi: 10.1016/j.jpain.2009.04.006
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rosen, D. R. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364:362. doi: 10.1038/364362c0
Rustici, G., Kolesnikov, N., Brandizi, M., Burdett, T., Dylag, M., Emam, I., et al. (2013). ArrayExpress update–trends in database growth and links to data analysis tools. Nucleic Acids Res. 41, D987–D990. doi: 10.1093/nar/gks1174
Sakakibara, A., Ando, R., Sapir, T., and Tanaka, T. (2013). Microtubule dynamics in neuronal morphogenesis. Open Biol. 3:130061. doi: 10.1098/rsob.130061
Sasaki, S., and Iwata, M. (1996). Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology 47, 535–540. doi: 10.1212/wnl.47.2.535
Sasaki, S., Warita, H., Abe, K., and Iwata, M. (2004). Slow component of axonal transport is impaired in the proximal axon of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 107, 452–460. doi: 10.1007/s00401-004-0838-y
Sayas, C. L., Tortosa, E., Bollati, F., Ramirez-Rios, S., Arnal, I., and Avila, J. (2015). Tau regulates the localization and function of end-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem. 133, 653–667. doi: 10.1111/jnc.13091
Schiff, P. B., Fant, J., and Horwitz, S. B. (1979). Promotion of microtubule assembly in vitro by taxol. Nature 277, 665–667. doi: 10.1038/277665a0
Schwenk, B. M., Lang, C. M., Hogl, S., Tahirovic, S., Orozco, D., Rentzsch, K., et al. (2014). The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 33, 450–467. doi: 10.1002/embj.201385857
Shemesh, O. A., and Spira, M. E. (2011). Rescue of neurons from undergoing hallmark tau-induced Alzheimer’s disease cell pathologies by the antimitotic drug paclitaxel. Neurobiol. Dis. 43, 163–175. doi: 10.1016/j.nbd.2011.03.008
Slaughter, T., and Black, M. M. (2003). STOP (stable-tubule-only-polypeptide) is preferentially associated with the stable domain of axonal microtubules. J. Neurocytol. 32, 399–413. doi: 10.1023/b:neur.0000011334.70648.87
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Sotelo-Silveira, J. R., Lepanto, P., Elizondo, V., Horjales, S., Palacios, F., Martinez-Palma, L., et al. (2009). Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid. Redox Signal. 11, 1535–1545. doi: 10.1089/ARs.2009.2614
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stancu, I. C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Ström, A. L., Gal, J., Shi, P., Kasarskis, E. J., Hayward, L. J., and Zhu, H. (2008). Retrograde axonal transport and motor neuron disease. J. Neurochem. 106, 495–505. doi: 10.1111/j.1471-4159.2008.05393.x
Strong, M. J., Yang, W., Strong, W. L., Leystra-Lantz, C., Jaffe, H., and Pant, H. C. (2006). Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology 66, 1770–1771. doi: 10.1212/01.wnl.0000218161.15834.db
Sudo, H., and Baas, P. W. (2011). Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum. Mol. Genet. 20, 763–778. doi: 10.1093/hmg/ddq521
Taes, I., Goris, A., Lemmens, R., Van Es, M. A., Van Den Berg, L. H., Chio, A., et al. (2010). Tau levels do not influence human ALS or motor neuron degeneration in the SOD1G93A mouse. Neurology 74, 1687–1693. doi: 10.1212/WNL.0b013e3181e042f7
Taes, I., Timmers, M., Hersmus, N., Bento-Abreu, A., Van Den Bosch, L., Van Damme, P., et al. (2013). HDAC6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 22, 1783–1790. doi: 10.1093/hmg/ddt028
Tahara, E. B., Navarete, F. D., and Kowaltowski, A. J. (2009). Tissue-, substrate- and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 46, 1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008
Takalo, M., Salminen, A., Soininen, H., Hiltunen, M., and Haapasalo, A. (2013). Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2, 1–14.
Tateno, M., Kato, S., Sakurai, T., Nukina, N., Takahashi, R., and Araki, T. (2009). Mutant SOD1 impairs axonal transport of choline acetyltransferase and acetylcholine release by sequestering KAP3. Hum. Mol. Genet. 18, 942–955. doi: 10.1093/hmg/ddn422
Tischfield, M. A., Cederquist, G. Y., Gupta, M. L. Jr., and Engle, E. C. (2011). Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 21, 286–294. doi: 10.1016/j.gde.2011.01.003
Tischfield, M. A., and Engle, E. C. (2010). Distinct α- and β-tubulin isotypes are required for the positioning, differentiation and survival of neurons: new support for the ‘multi-tubulin’ hypothesis. Biosci. Rep. 30, 319–330. doi: 10.1042/BSR20100025
Todd, T. W., and Petrucelli, L. (2016). Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J. Neurochem. 138, 145–162. doi: 10.1111/jnc.13623
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
Tortosa, E., Galjart, N., Avila, J., and Sayas, C. L. (2013). MAP1B regulates microtubule dynamics by sequestering EB1/3 in the cytosol of developing neuronal cells. EMBO J. 32, 1293–1306. doi: 10.1038/emboj.2013.76
Tortosa, E., Montenegro-Venegas, C., Benoist, M., Härtel, S., González-Billault, C., Esteban, J. A., et al. (2011). Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J. Biol. Chem. 286, 40638–40648. doi: 10.1074/jbc.M111.271320
Trinczek, B., Ebneth, A., Mandelkow, E. M., and Mandelkow, E. (1999). Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 112, 2355–2367.
Tudor, E. L., Perkinton, M. S., Schmidt, A., Ackerley, S., Brownlees, J., Jacobsen, N. J., et al. (2005). ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J. Biol. Chem. 280, 34735–34740. doi: 10.1074/jbc.m506216200
Turner, M. R., and Swash, M. (2015). The expanding syndrome of amyotrophic lateral sclerosis: a clinical and molecular odyssey. J. Neurol. Neurosurg. Psychiatry 86, 667–673. doi: 10.1136/jnnp-2014-308946
Turner, B. J., and Talbot, K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. doi: 10.1016/j.pneurobio.2008.01.001
Tymanskyj, S. R., Scales, T. M., and Gordon-Weeks, P. R. (2012). MAP1B enhances microtubule assembly rates and axon extension rates in developing neurons. Mol. Cell. Neurosci. 49, 110–119. doi: 10.1016/j.mcn.2011.10.003
Vadlamudi, R. K., Barnes, C. J., Rayala, S., Li, F., Balasenthil, S., Marcus, S., et al. (2005). p21-activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin cofactor B. Mol. Cell. Biol. 25, 3726–3736. doi: 10.1128/mcb.25.9.3726-3736.2005
Van Beuningen, S. F., Will, L., Harterink, M., Chazeau, A., Van Battum, E. Y., Frias, C. P., et al. (2015). TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel Microtubule arrays. Neuron 88, 1208–1226. doi: 10.1016/j.neuron.2015.11.012
Van Damme, P., Dewil, M., Robberecht, W., and van Den Bosch, L. (2005). Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener. Dis. 2, 147–159. doi: 10.1159/000089620
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vickers, J. C., King, A. E., Woodhouse, A., Kirkcaldie, M. T., Staal, J. A., Mccormack, G. H., et al. (2009). Axonopathy and cytoskeletal disruption in degenerative diseases of the central nervous system. Brain Res. Bull. 80, 217–223. doi: 10.1016/j.brainresbull.2009.08.004
Villarroel-Campos, D., and Gonzalez-Billault, C. (2014). The MAP1B case: an old MAP that is new again. Dev. Neurobiol. 74, 953–971. doi: 10.1002/dneu.22178
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Warita, H., Itoyama, Y., and Abe, K. (1999). Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 819, 120–131. doi: 10.1016/s0006-8993(98)01351-1
Williamson, T. L., Bruijn, L. I., Zhu, Q., Anderson, K. L., Anderson, S. D., Julien, J. P., et al. (1998). Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc. Natl. Acad. Sci. U S A 95, 9631–9636. doi: 10.1073/pnas.95.16.9631
Williamson, T. L., and Cleveland, D. W. (1999). Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. doi: 10.1038/4553
Wilson, C., and González-Billault, C. (2015). Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front. Cell. Neurosci. 9:381. doi: 10.3389/fncel.2015.00381
Wittmann, T., and Waterman-Storer, C. M. (2005). Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J. Cell Biol. 169, 929–939. doi: 10.1083/jcb.200412114
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Yadav, P., Selvaraj, B. T., Bender, F. L., Behringer, M., Moradi, M., Sivadasan, R., et al. (2016). Neurofilament depletion improves microtubule dynamics via modulation of Stat3/stathmin signaling. Acta Neuropathol. 132, 93–110. doi: 10.1007/s00401-016-1564-y
Yamanaka, K., Miller, T. M., McAlonis-Downes, M., Chun, S. J., and Cleveland, D. W. (2006). Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann. Neurol. 60, 95–104. doi: 10.1002/ana.20888
Yang, F., Jiang, Q., Zhao, J., Ren, Y., Sutton, M. D., and Feng, J. (2005). Parkin stabilizes microtubules through strong binding mediated by three independent domains. J. Biol. Chem. 280, 17154–17162. doi: 10.1074/jbc.m500843200
Yau, K. W., Schätzle, P., Tortosa, E., Pagès, S., Holtmaat, A., Kapitein, L. C., et al. (2016). Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J. Neurosci. 36, 1071–1085. doi: 10.1523/JNEUROSCI.2430-15.2016
Zhang, B., Maiti, A., Shively, S., Lakhani, F., Mcdonald-Jones, G., Bruce, J., et al. (2005). Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. U S A 102, 227–231. doi: 10.1073/pnas.0406361102
Zhang, F., Ström, A. L., Fukada, K., Lee, S., Hayward, L. J., and Zhu, H. (2007). Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 282, 16691–16699. doi: 10.1074/jbc.m609743200
Keywords: amyotrophic lateral sclerosis, microtubules, dynamics, axon transport
Citation: Clark JA, Yeaman EJ, Blizzard CA, Chuckowree JA and Dickson TC (2016) A Case for Microtubule Vulnerability in Amyotrophic Lateral Sclerosis: Altered Dynamics During Disease. Front. Cell. Neurosci. 10:204. doi: 10.3389/fncel.2016.00204
Received: 23 June 2016; Accepted: 15 August 2016;
Published: 13 September 2016.
Edited by:
Eleonora Palma, Sapienza University of Rome, ItalyReviewed by:
Daniel Kaganovich, Hebrew University of Jerusalem, IsraelJavier Fernández-Ruiz, Complutense University of Madrid, Spain
Copyright © 2016 Clark, Yeaman, Blizzard, Chuckowree and Dickson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracey C. Dickson, tracey.dickson@utas.edu.au