Carbohydrate-biased control of energy metabolism: the darker side of the selfish brain
- Infotonic Consultancy, Stockholm, Sweden
Introduction
There is evidence that the brain favors consumption of carbohydrates (CHO) rather than fats, this preference resulting in glycolysis-based energy metabolism domination. This metabolic mode, typical for consumers of the “Western diet” (Cordain et al., 2005; Seneff et al., 2011), is characterized by over-generation of reactive oxygen species and advanced glycation products both of which are implicated in many of the neurodegenerative diseases (Tessier, 2010; Vicente Miranda and Outeiro, 2010; Auburger and Kurz, 2011). However, it is not CHO but fat that is often held responsible for metabolic pathologies. This paper, based on analysis of experimental data, offers an opinion that the obesogenic and neurodegenerative effects of dietary fat in the high-fat diets (HFD) cannot be separated from the effects of the CHO compound in them. Since this is not a comprehensive literature review, only essential research results are presented.
It is general knowledge that the glucose homeostasis possesses very limited buffering capacities, while energy homeostasis in its fat-controlling part enjoys practically unlimited energy stores. Logically, a control system with a limited buffer should thoroughly defend the “consumption” part. Indeed, existing experimental data (briefly reviewed here later) show important properties of the CHO intake control that is different from or not shown for the fat intake control:
(1) A mere oral sensation of CHO elicits physiological anticipation response (cephalic phase) that is either inborn or rapidly conditioned.
(2) Oral CHO sensation stimulates reward-specific brain areas.
(3) CHO addiction is essentially similar to typical drug addictions.
These peculiarities can explain the physiologically and metabolically opposite effects of obesogenic HFD versus the ketogenic diet (KD), which is also HFD but lower in CHO.
The Selfish Brain Concept: Two Meanings
There are two ways to look at the CHO-biasing trait of the brain.
(1) The “Selfish Brain” is a term coined by Robert L. DuPont in the title of his book where he wrote: “With respect to aggression, fear, feeding, and sexuality, the brain is selfish. It simply wants what it wants right now” (DuPont, 1997). We must add, “if the environment permits, the brain gets it.” The bad news is, in the long run the body can be harmed as the result.
(2) An elaborate (and rare for modern physiology) systemic concept explaining the fundamental ability of the brain to control priorities of energy allocation has been proposed by Peters and colleagues who also titled their theory the Selfish Brain. They wrote referring to DuPont’s book: “The brain looks after itself first. Such selfishness is reminiscent of an earlier concept in which the brain’s selfishness was addressed with respect to addiction. We chose our title by analogy but applied it in a different context, i.e., the competition for energy resources” (Peters et al., 2004).
These two meaning of the Selfish Brain have important common points if we consider the addiction (highly non-homeostatic) as a result of the “push” principle borrowed from the economic “push–pull” paradigm of supply chains. As early as in 1998, Hill and Peters wrote: “According to the ‘push’ principle, the environment pushes excess amounts of energy into the organism” (Hill and Peters, 1998).
They also share a common mechanism – reward. According to DuPond, “What makes a drug addictive is not that it is ‘psychoactive’ but that it produces specific brain reward. It is not withdrawal that hooks the addict, it is reward” (DuPont, 2008). This reward is hard-wired in the brain, in the loci where both “pull” and “push” systems might be converging, something that is discussed within the Selfish Brain paradigm as the comforting effect of food (Peters et al., 2007), particularly, the CHO-rich foods (Hitze et al., 2010).
Push and Pull Parts of Energy Supply Control System
The role of depots, as determined by a general principle in economic supply chains, is energy buffering in unstable environments (Fischer et al., 2011). Peters and Langemann (2009) analyzed two concepts of environment–organism relationship with opposite views at depots:
(1) An environment offering abundant energy beyond homeostatic need “pushes” it into the body via some evolutionary appropriate gateway. The surplus, naturally, goes into depots. Peters and Langemann, however, remained in doubt about this concept partly due to the fact that this “push” does not work invariably for all animal or human subjects (Martin et al., 2010; Cao et al., 2011).
(2) A somewhat alternative concept, well accepted for the last 50 years, concerns the “pull” character of the open system “organism–environment,” supposedly in accordance with homeostatic needs. In this system, either the size of fat depot (Kennedy, 1953; Woods and Ramsay, 2011) or glucose levels (Mayer, 1953) are being controlled.
The role of CHO depot was not conferred, perhaps due to its negligible storing capacities. Indeed, the sizes of CHO and fat depots are incomparable. In the survival terms, CHO can provide energy support for less than 2 days (Bilsborough and Crowe, 2003), while a year-long complete water–vitamin fast, with body fat as a sole energy source, has been reported (Stewart and Fleming, 1973).
Non-Homeostatic Effects of Cho Versus Fat
From the teleological standpoint, the strong drive for CHO intake beyond homeostatic needs exists very likely due to limited CHO-storing capacities. For fat with its vast depots, there is less (or none at all) evidence for a drive of similar magnitude. It seems to contradict the overwhelming reports on obesogenic properties of HFD but we shall see later that in reality there is no conflict.
Oral stimulation with both sweet and non-sweet CHO activated brain regions associated with reward – insula/frontal operculum, orbitofrontal cortex, and striatum. These regions were unresponsive to sweet, non-CHO stimulation with saccharin (Jeukendrup and Chambers, 2010). On the other hand, experiments with the no-calorie fat substitute (Olestra) revealed an impaired ability to use sensory cues associated with fat to predict caloric outcomes (Swithers et al., 2011). In humans, the intra-amniotic injection of fat (Lipiodol) reduced fetal drinking, while injection of sodium saccharin stimulated it; infants consumed the same amounts of milk formulas with different fat contents. Oral fat stimulation had no positive or negative mood-related effects, whereas sucrose shifted emotional spectrum toward positive scores (Mattes, 2005). CHO-rich food intake (buffet, KR 0.511:1) relieved neuroglycopenic and mood responses to stress independently from oral or i.v. administration of energy (Hitze et al., 2010).
Besides, HFD often fails in inducing obesity. Consequently, it is not uncommon in diet-induced obesity experiments that obesity-resistant subjects are eliminated from analysis or CHO are added to the diet to encourage overeating. Also, more accurate approximation of biologically adequate environments, e.g., allowing voluntary exercising, prevented HFD-induced obesity through radical modulation of hypothalamic control of fat metabolism (Cao et al., 2011).
To sum it up, fat per se is neither as highly rewarding as CHO nor it is as addictive (Wojnicki et al., 2008; Avena et al., 2009; Pickering et al., 2009; Berthoud et al., 2011). But why, then, it is the dietary fat that is blamed for overconsumption, obesity, and neuro-deteriorating effects?
The Role of Macronutrient Composition
Interestingly, the diet categorization (HFD, low-CHO, KD, etc.) is not fixed and varies from author to author (Figure 1B) although there is a clear-cut tool for labeling a diet according to its metabolic consequences.
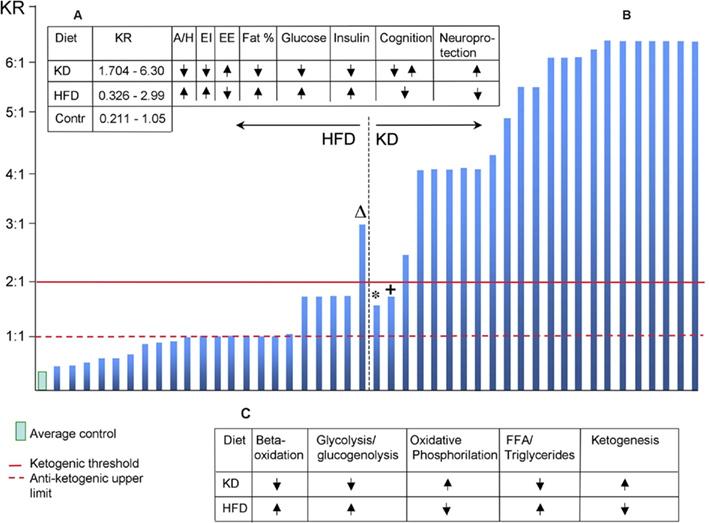
Figure 1. (A) Summary of major physiological effects of HFD and KD. KR, ketogenic ratio; A/H, appetite/hunger; EI, energy intake; EE, energy expenditure. (B) Ketogenic ratio distribution of diets classified by authors as HFD or KD. ΔDiet defined as HFD (Holloway et al., 2011); KR = 2.994:1, resulted in cognitive impairment. The diet’s duration was 5 days or within the adaptation period for KD when it does not exert its neuroprotective properties (Brinkworth et al., 2009). *Diet defined as KD (Johnstone et al., 2008); KR = 1.545:1 and gradually decreasing in the course of 10-week study since five extra grams of CHO was added each week. +Diet defined as KD (Johnstone et al., 2008); KR = 0.704:1 and 0.525:1 in two series. This finding is in agreement with (Puchowicz et al., 2008). (C) Metabolic profiles of HFD and KD (based on Kennedy et al., 2007).
A century ago, Woodyatt wrote: “antiketogenesis is an effect due to certain products which occur in the oxidation of glucose, an interaction between these products on the one hand and one or more of the acetone bodies on the other” (Woodyatt, 1910). Shaffer (1921) calculated the number of “ketogenic” molecules versus molecules of glucose and concluded that the maximal ratio compatible with the oxidation of the “ketogenic” molecules becomes possible when their ratio is at least 1:1. Later, Woodyatt (1921) suggested the following equation for calculating KD composition:
KR = (0.46 pg + 0.90 fg):(1.0 cg + 0.58 pg + 0.1 fg)
Where KR is “ketogenic ratio,” g is grams, P is protein, F is fat, and C is CHO.
Wilder and Winter (1922) defined the threshold of ketogenesis explaining it from the standpoint of condition where either ketone bodies or glucose can be oxidized. They arrived, together with Shaffer and Woodyatt, at the conclusion that KR for induction of ketogenesis should be 2:1 or higher.
This is a very important point, not only methodologically, but also ideologically. The KR invariably indicates whether the CHO proportion is low enough for allowing the fat-mobilizing pathway and ketogenesis, or high enough for blocking it and supporting glycolysis instead. The latter option opens the energy “push” opportunity through CHO intake gateway with the consequences discussed above. On the other hand, ketogenesis introduces a fuel alternative to glucose, which can be crucial in metabolic pathologies.
In the clinical KD, KR is usually 4:1; in the experimental KD, it’s often 6:1 or higher. KR of HFD is more often than not undeclared, making it difficult to compare effects of different HFD – unless diet composition is known and calculation of KR is possible.
Ketogenesis-to-Antiketogenesis Ratios: The Physiological Effects
Using the Woodyartt’s equation, I calculated KR of 45 experimental works (2005–2011), wherever there was enough information regarding macronutrient composition. It is evident (Figure 1B) that in the HFD, KR is almost uniformly below the threshold of ketogenesis indicating a too-high proportion of CHO. It is interesting that even below the ketogenic threshold, the lower CHO proportion was, the higher neuroprotective effects were reported, e.g., against hypoxia (Puchowicz et al., 2007, 2008).
Among the most frequently reported consequences of HFD are features typical for metabolic syndrome – increased hunger/appetite, insulin resistance, elevated body fat deposition, and glucose intolerance along with decreased neuronal resistance to damaging conditions. A set of completely opposite effects is well documented for KD – decreased hunger/appetite, decreased levels of glucose and insulin in the blood, lower body fat deposition, increased neuronal resistance to damaging conditions (Figure 1A). The metabolic state caused by KD (Figure 1C) was called “unique” (Kennedy et al., 2007) and it closely resembles effects of calorie restriction (Domouzoglou and Maratos-Flier, 2011).
The Ketogenic Ratio and the “Push” Component of Energy Metabolism
The environment in Western-type societies can be characterized as “pushing” the energy into our organisms via activation of reward and addiction circuits of our selfish brains. In the standard experimental “Western Diet” (5TJN) with KR close to 1:1, CHO proportion is high enough to continuously maintain glycolysis, overconsumption, and the subsequent chain of events resulting in metabolic disturbances detrimental for the brain (Langdon et al., 2011).
Interestingly, this view of the environmental “push” is in line with a socioeconomic explanation of the modern obesity epidemic. The NHANES surveys of 1971–2006 (Austin et al., 2011) revealed that in the USA population, the trend toward increased CHO intake and decreased fat intake (KR shift from 0.716 to 0.620) resulted in the increase of obesity incidence. In a recent article entitled “Using Marketing Muscle to Sell Fat: The Rise of Obesity in the Modern Economy,” J. Zimmerman wrote: “In this paradigm, overeating results from more extensive advertising, new product development, increased portion sizes, and other tactics of food marketers that have caused shifts in the underlying demand for total food calories” (Zimmerman, 2011).
On the other hand, the diets with KR of 2:1 or higher are repeatedly described as metabolically beneficial, non-addictive, hunger–reducing, and neuroprotective (Figure 1A). In this, they mimic many of the effects of calorie restriction (Kennedy et al., 2007; Domouzoglou and Maratos-Flier, 2011) and are seen as evolutionary and genetically adequate but “no longer adaptive under current circumstances” (Stranahan and Mattson, 2011).
Conclusion
Homeostatic control of energy metabolism aims to maintaining energy balance and as soon as it is achieved, energy intake might be expected to decrease or stop. However, this is possible only in deterministic environments. In variable environments, energy storage becomes advantageous and approximately equal parts of energy are allocated for maintenance, reproduction, and depots (Fischer et al., 2011). Energy intake beyond rigid homeostatic regulation relies on behaviors with hedonic, rewarding, and addictive nuances more characteristic for CHO than for fat. Their traits notwithstanding, these behaviors are highly evolutionary significant: “Although at first glance, hijacking of the homeostatic regulatory mechanisms by its hedonic counterpart may seem conflicting, it should be borne in mind that during evolution, humans have lived in an environment where food availability was restricted and uncertain (e.g., hunter–gatherers) and the biological system has been ‘hard-wired’ to maximize energy stores” (Pandit et al., 2011).
To maximize energy stores, energy intake relies on CHO-driven behaviors to allow the environmental “push.” Therefore: (1) dietary fat is not to blame for the diet-induced obesity; it is CHO that is not limited enough in HFD; (2) KR may be an element of common language in experiments with different methodological approaches.
References
Auburger, G., and Kurz, A. (2011). The role of glyoxalases for sugar stress and aging, with relevance for dyskinesia, anxiety, dementia and Parkinson’s disease. Aging (Albany NY) 3, 5–9.
Austin, G. L., Ogden, L. G., and Hill, J. O. (2011). Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am. J. Clin. Nutr. 93, 836–843.
Avena, N. M., Rada, P., and Hoebel, B. G. (2009). Sugar and fat bingeing have notable differences in addictive-like behavior. J. Nutr. 139, 623–628.
Berthoud, H. R., Lenard, N. R., and Shin, A. C. (2011). Food reward, hyperphagia, and obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1266–R1277.
Bilsborough, S. A., and Crowe, T. C. (2003). Low-carbohydrate diets: what are the potential short- and long-term health implications? Asia Pac. J. Clin. Nutr. 12, 396–404.
Brinkworth, G. D., Noakes, M., Buckley, J. D., Keogh, J. B., and Clifton, P. M. (2009). Long-term effects of a very-low-carbohydrate weight loss diet compared with an isocaloric low-fat diet after 12 mo. Am. J. Clin. Nutr. 90, 23–32.
Cao, L., Choi, E. Y., Liu, X., Martin, A., Wang, C., Xu, X., and During, M. J. (2011). White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 14, 324–338.
Cordain, L., Eaton, S. B., Sebastian, A., Mann, N., Lindeberg, S., Watkins, B. A., O’keefe, J. H., and Brand-Miller, J. (2005). Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 81, 341–354.
Domouzoglou, E., and Maratos-Flier, E. (2011). Fibroblast growth factor 21 is a metabolic regulator that plays a role in the adaptation to ketosis. Am. J. Clin. Nutr. 93(Suppl.), 901S–905S.
Fischer, B., Dieckmann, U., and Taborsky, B. (2011). When to store energy in a stochastic environment. Evolution 65, 1221–1232.
Hill, J. O., and Peters, J. C. (1998). Environmental contributions to the obesity epidemic. Science 280, 1371–1374.
Hitze, B., Hubold, C., Van Dyken, R., Schlichting, K., Lehnert, H., Entringer, S., and Peters, A. (2010). How the selfish brain organizes its supply and demand. Front. Neuroenergetics 2:7. doi: 10.3389/fnene.2010.00007
Holloway, C. J., Cochlin, L. E., Emmanuel, Y., Murray, A., Codreanu, I., Edwards, L. M., Szmigielski, C., Tyler, D. J., Knight, N. S., Saxby, B. K., Lambert, B., Thompson, C., Neubauer, S., and Clarke, K. (2011). A high-fat diet impairs cardiac high-energy phosphate metabolism and cognitive function in healthy human subjects. Am. J. Clin. Nutr. 93, 748–755.
Jeukendrup, A. E., and Chambers, E. S. (2010). Oral carbohydrate sensing and exercise performance. Curr. Opin. Clin. Nutr. Metab. Care 13, 447–451.
Johnstone, A. M., Horgan, G. W., Murison, S. D., Bremner, D. M., and Lobley, G. E. (2008). Effects of a high-protein ketogenic diet on hunger, appetite, and weight loss in obese men feeding ad libitum. Am. J. Clin. Nutr. 87, 44–55.
Kennedy, A. R., Pissios, P., Otu, H., Roberson, R., Xue, B., Asakura, K., Furukawa, N., Marino, F. E., Liu, F. F., Kahn, B. B., Libermann, T. A., and Maratos-Flier, E. (2007). A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 292, E1724–E1739.
Kennedy, G. (1953). The role of depot fat in the hypothalamic control of food intake in the rat. Proc. R. Soc. Lond. B Biol. Sci. 140, 578–596.
Langdon, K. D., Clarke, J., and Corbett, D. (2011). Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience 182, 82–87.
Martin, B., Ji, S., Maudsley, S., and Mattson, M. P. (2010). “Control” laboratory rodents are metabolically morbid: why it matters. Proc. Natl. Acad. Sci. U.S.A. 107, 6127–6133.
Mayer, J. (1953). Genetic, traumatic and environmental factors in the etiology of obesity. Physiol. Rev. 33, 472–508
Pandit, R., De Jong, J. W., Vanderschuren, L. J., and Adan, R. A. (2011). Neurobiology of overeating and obesity: the role of melanocortins and beyond. Eur. J. Pharmacol. 660, 28–42.
Peters, A., and Langemann, D. (2009). Build-ups in the supply chain of the brain: on the neuroenergetic cause of obesity and type 2 diabetes mellitus. Front. Neuroenergetics 1:2. doi: 10.3389/neuro.14.002.2009
Peters, A., Pellerin, L., Dallman, M. F., Oltmanns, K. M., Schweiger, U., Born, J., and Fehm, H. L. (2007). Causes of obesity: looking beyond the hypothalamus. Prog. Neurobiol. 81, 61–88.
Peters, A., Schweiger, U., Pellerin, L., Hubold, C., Oltmanns, K. M., Conrad, M., Schultes, B., Born, J., and Fehm, H. L. (2004). The selfish brain: competition for energy resources. Neurosci. Biobehav. Rev. 28, 143–180.
Pickering, C., Alsio, J., Hulting, A. L., and Schioth, H. B. (2009). Withdrawal from free-choice high-fat high-sugar diet induces craving only in obesity-prone animals. Psychopharmacology (Berl.) 204, 431–443.
Puchowicz, M. A., Xu, K., Sun, X., Ivy, A., Emancipator, D., and Lamanna, J. C. (2007). Diet-induced ketosis increases capillary density without altered blood flow in rat brain. Am. J. Physiol. Endocrinol. Metab. 292, E1607–E1615.
Puchowicz, M. A., Zechel, J. L., Valerio, J., Emancipator, D. S., Xu, K., Pundik, S., Lamanna, J. C., and Lust, W. D. (2008). Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J. Cereb. Blood Flow Metab. 28, 1907–1916.
Seneff, S., Wainwright, G., and Mascitelli, L. (2011). Nutrition and Alzheimer’s disease: the detrimental role of a high carbohydrate diet. Eur. J. Intern. Med. 22, 134–140.
Stewart, W. K., and Fleming, L. W. (1973). Features of a successful therapeutic fast of 382 days’ duration. Postgrad. Med. J. 49, 203–209.
Stranahan, A. M., and Mattson, M. P. (2011). Bidirectional metabolic regulation of neurocognitive function. Neurobiol. Learn. Mem. 96, 507–516.
Swithers, S. E., Ogden, S. B., and Davidson, T. L. (2011). Fat substitutes promote weight gain in rats consuming high-fat diets. Behav. Neurosci. 125, 512–518.
Tessier, F. J. (2010). The Maillard reaction in the human body. The main discoveries and factors that affect glycation. Pathol. Biol. (Paris) 58, 214–219.
Vicente Miranda, H., and Outeiro, T. F. (2010). The sour side of neurodegenerative disorders: the effects of protein glycation. J. Pathol. 221, 13–25.
Wojnicki, F. H., Charny, G., and Corwin, R. L. (2008). Binge-type behavior in rats consuming trans-fat-free shortening. Physiol. Behav. 94, 627–629.
Woods, S. C., and Ramsay, D. S. (2011). Food intake, metabolism and homeostasis. Physiol. Behav. 104, 4–7.
Woodyatt, R. (1910). The action of glycol aldehyd and glycerin aldehyd in diabetes mellitus and the nature of antiketogenesis. J. Am. Med. Assoc. 1, 2109.
Citation: Zilberter T (2011) Carbohydrate-biased control of energy metabolism: the darker side of the selfish brain. Front. Neuroenerg. 3:8. doi: 10.3389/fnene.2011.00008
Received: 10 October 2011; Accepted: 02 December 2011;
Published online: 20 December 2011.
Copyright: © 2011 Zilberter. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: zilberter@gmail.com