Insights into the metabolic response to traumatic brain injury as revealed by 13C NMR spectroscopy
- 1Department of Radiology, Loma Linda University School of Medicine, Loma Linda, CA, USA
- 2Department of Neurosurgery, David Geffen School of Medicine at University of California Los Angeles, Los Angeles, CA, USA
The present review highlights critical issues related to cerebral metabolism following traumatic brain injury (TBI) and the use of 13C labeled substrates and nuclear magnetic resonance (NMR) spectroscopy to study these changes. First we address some pathophysiologic factors contributing to metabolic dysfunction following TBI. We then examine how 13C NMR spectroscopy strategies have been used to investigate energy metabolism, neurotransmission, the intracellular redox state, and neuroglial compartmentation following injury. 13C NMR spectroscopy studies of brain extracts from animal models of TBI have revealed enhanced glycolytic production of lactate, evidence of pentose phosphate pathway (PPP) activation, and alterations in neuronal and astrocyte oxidative metabolism that are dependent on injury severity. Differential incorporation of label into glutamate and glutamine from 13C labeled glucose or acetate also suggest TBI-induced adaptations to the glutamate-glutamine cycle.
Introduction
A significant body of work has shown that traumatic brain injury (TBI) initiates a cascade of cellular events including potassium efflux (Katayama et al., 1990; Kawamata et al., 1995), Ca++ accumulation (Fineman et al., 1993; Osteen et al., 2001), glutamate release (Katayama et al., 1990; Nilsson et al., 1990; Rose et al., 2002), and increased oxidative stress (Hall et al., 1993; Lewen and Hillered, 1998; Vagnozzi et al., 1999; Tyurin et al., 2000; Marklund et al., 2001) that contribute to reduced ATP production. In addition, TBI results in an immediate increase in cerebral metabolic rates for glucose (CMRglc) (Yoshino et al., 1991; Sutton et al., 1994; Lee et al., 1999; Kelly et al., 2000) that can endure for days in TBI patients (Bergsneider et al., 1997). This increase is thought to represent an increase in glycolysis (hyperglycolysis) in an attempt to meet the cellular energy demand required to restore ionic balance and maintain the neuronal membrane potential (Hovda, 1996). Studies have shown that the duration and severity of regional decreases in ATP are dependent upon TBI severity (Lee et al., 1999; Aoyama et al., 2008; Signoretti et al., 2010), and during the post-injury period where ATP production is reduced, secondary insults or activation of the injured brain can further reduce ATP levels and result in secondary cellular damage (Ip et al., 2003; Zanier et al., 2003; Aoyama et al., 2008). A secondary and enduring reduction of CMRglc (metabolic “depression”) is a common finding in models of experimental TBI (Hovda et al., 1991; Yoshino et al., 1991; Sutton et al., 1994; Jiang et al., 2000; Moore et al., 2000; Prins and Hovda, 2001) and after human TBI (Langfitt et al., 1986; Yamaki et al., 1996; Bergsneider et al., 2000, 2001). Moreover, an increase in energy demand or decreased glucose availability after TBI would potentially compromise neuronal viability and functional outcomes (Vespa et al., 2003, 2007; Parkin et al., 2005; Marcoux et al., 2008).
Potential Factors Contributing to the Metabolic Depression after TBI
Some underlying mechanisms responsible for the hypometabolic response following TBI are the overproduction of reactive oxygen and nitrogen species which can lead to poly(ADP) ribose polymerases (PARP) activation (Laplaca et al., 1999; Clark et al., 2001; Arundine et al., 2004; Mendez et al., 2004; Kauppinen, 2007; Besson, 2009) and related reductions of nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+; Satchell et al., 2003; Clark et al., 2007; Signoretti et al., 2010). Consequently, a diminished supply of reducing equivalents for oxidoreductive reactions involved in glucose metabolism, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and pyruvate dehydrogenase (PDH), could inhibit glycolysis and the entry of pyruvate into the TCA cycle resulting in energy depletion and cell death. Reduced GAPDH activity has also been shown to act as a “molecular switch” resulting in increased flux of glucose into the PPP (Ralser et al., 2007; Grant, 2008). Direct evidence for reduced activity of these enzyme complexes following TBI is an active area of research, and to date, studies have shown PDH nitrosylation (Opii et al., 2007) and alterations in the expression and phosphorylation of PDH E1alpha1 subunit (Sharma et al., 2009; Xing et al., 2009, 2012). Both increased intracellular Ca++ or PARP activity (Lai et al., 2008) following TBI can also lead to an uncoupling of the mitochondrial electron transport chain (Dugan et al., 1995) and mitochondrial permeability transition (Gunter et al., 1994; Schinder et al., 1996; Zamzami et al., 1997), with decreases in state 3 respiratory rates (Xiong et al., 1997, 1998; Verweij et al., 2000) that would contribute to energy loss and cell death. Studies have shown that PARP inhibitors can attenuate NAD+ reductions, decrease neuronal damage, and improve behavioral outcome following experimental TBI (Laplaca et al., 2001; Komjati et al., 2005; Clark et al., 2007; Besson, 2009).
13C Studies of TBI
To more finely resolve the TBI-induced changes in glucose metabolic pathways, a number of studies have employed the use of stable isotopes (13C) of glucose, lactate and acetate to determine the metabolic fate of these fuels and characterize changes in oxidative metabolism and neuroglia metabolic compartmentation during the acute and hypometabolic periods following experimental and clinical TBI (Bartnik et al., 2005, 2007; Dusick et al., 2007; Gallagher et al., 2009; Scafidi et al., 2009; Bartnik-Olson et al., 2010; Clausen et al., 2011). In most studies these isotopes were used in conjunction with ex vivo 13C nuclear magnetic resonance (NMR) spectroscopy, which allows for the simultaneous assessment of multiple metabolic pathways. The primary advantage of this technique arises from its ability to distinguish 13C incorporation into multiple metabolites as well as into the specific carbon positions within the same metabolite, resulting in a detailed analysis of the metabolic “fate” of the 13C label (Bachelard and Badar-Goffer, 1993; Cruz and Cerdan, 1999). The relative 13C enrichment at each carbon position and the ratios between isotopomers of glutamate and glutamine gives additional information regarding enzyme usage, neurotransmitter synthesis, and neuroglia metabolic compartmentation (Badar-Goffer et al., 1990; Shank et al., 1993; Hassel et al., 1995; Aureli et al., 1997). As shown in Figure 1, 13C NMR spectroscopy can be used to measure TBI-induced changes in glycolysis (13C lactate labeling), oxidative metabolism within, and interactions between, the neuron and astrocyte compartments (glutamate and glutamine labeling). By using [1, 2 13C2] glucose as the substrate, injury-induced changes in the activity of the pentose phosphate pathway (PPP) can be assessed using the ratio between the lactate labeled in two carbon positions (doublet) via glycolysis (C2 and C3) and lactate labeled in the singlet C3 carbon position (Cruz et al., 1998; Lee et al., 1998). However, the formation of a lactate C3 singlet also results via the pyruvate recycling pathway (Hassel and Sonnewald, 1995). In contrast, lactate labeling using [1, 2 13C2] acetate is derived exclusively from the pyruvate recycling pathway and comparisons between the lactate C3 singlet/doublet ratio from [1, 2 13C2] glucose and the lactate labeling from [1, 2 13C2] acetate could provide a valid method for more accurately measuring the contribution of the PPP to the post-injury response. The cytosolic NAD+/NADH redox state of the injured tissue can be estimated by the ratio of glutamate labeled in two carbon positions via glycolysis (C3 and C4) to the labeling of glutamate labeled in the C4 position via the recycling of pyruvate from labeled oxaloacetate via malic enzyme using either [1, 2 13C2] glucose or [1, 2 13C2] acetate (ME; Cruz et al., 1998; Cerdan et al., 2006). This more finely-grained approach to monitoring the fate of labeled fuels provides an ideal platform from which to determine how fuel supplementation after injury may prevent on-going metabolic deficits.
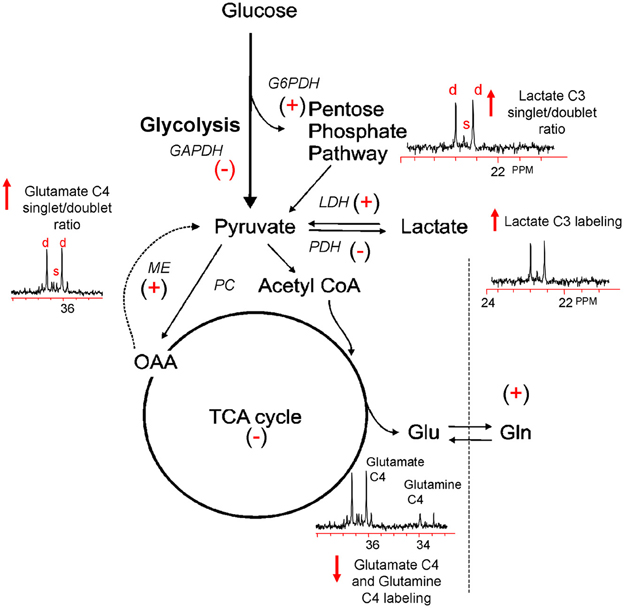
Figure 1. Simplified illustration of the metabolic response to TBI as determined by 13C NMR spectroscopy using [1, 2 13C2] glucose. TBI-induced ion fluxes and neurotransmitter release can increase anaerobic metabolism and initiate an injury cascade including increased oxidative/nitrosative stress, PARP-1 activation, and NAD+ and/or NAPD+ reductions. These in turn are thought to result in the activation (+) or inhibition (−) of enzymes contributing to the metabolic response to TBI (see text). Using [1, 2 13C2] glucose as a precursor, 13C NMR spectroscopy can be used to measure increases (↑) or decreases (↓) in glycolysis, the PPP, oxidative metabolism in the TCA cycle, and the pyruvate recycling system by monitoring the production of [2, 3 13C2] lactate, [2 13C] lactate, and [4, 5 13C2] glutamate and [4 13C] glutamate.
Experimental Models Used in 13C Studies of TBI
To date 13C studies have been conducted using the lateral fluid percussion injury (FPI) or the unilateral controlled cortical impact (CCI) injury models of TBI. The FPI model can produce a diffuse pattern of neuronal and axonal injury, while CCI is often termed a more focal injury model due to the contusion induced (Cernak, 2005) although widespread axonal injury occurs after CCI (Hall et al., 2008). With moderate injury severity the period of hyperglycolysis and the extent of ATP reduction in cortex is reduced in the FPI model compared to CCI (Lee et al., 1999), but in both TBI models a widespread reduction in CMRglc is observed throughout the injured hemisphere within a few hours (Hovda et al., 1991; Yoshino et al., 1991; Sutton et al., 1994; Moore et al., 2000). Although CMRglc recovers to baseline within 10 days after FPI (Yoshino et al., 1991; Moore et al., 2000) substantial reductions of CMRglc are still present by 15 days following CCI injury (Moro et al., 2011).
Glycolysis and PPP Metabolism Following TBI
Numerous experimental studies have reported an acute increase in extracellular lactate levels consistent with hyperglycolysis following TBI (Inao et al., 1988; Kawamata et al., 1995; Chen et al., 2000). In keeping with these observations, studies using ex vivo 13C NMR spectroscopy have reported increased 13C labeling of lactate from 13C glucose in the injured cortex within the first 6 h following a CCI injury in both the adult (Bartnik et al., 2005) and immature rat brain (Scafidi et al., 2009). In the adult brain this increase was seen at 3.5 h after injury which then normalized by 24 h (Bartnik et al., 2005). Increased lactate labeling from 13C-labeled glucose was also detected by gas chromatography/mass spectroscopy (GC-MS) from microdialysis samples of CCI injured cerebral cortex in rats (Clausen et al., 2011) and in blood samples from moderate-severe human TBI patients (Dusick et al., 2007). In adult rats with FPI, lactate 13C labeling was reduced at 24 h post-injury (Bartnik et al., 2007) indicating reduced glucose metabolism via glycolysis in this more diffuse TBI model. It remains to be determined whether this finding is the result of reduced GAPDH activity and whether similar findings are replicated at all developmental stages.
Ex vivo 13C NMR spectroscopy studies of cerebral cortex after CCI or FPI using [1, 2 13C2] glucose also reported a significant increase in lactate labeling via the PPP (Bartnik et al., 2005, 2007). This finding was supported by a clinical study of severe TBI patients, where evidence of increased glucose metabolism via the PPP was detected in blood samples using GC-MS following an infusion of [1, 2 13C2] glucose (Dusick et al., 2007). The PPP functions in producing reducing equivalents of NADPH for biosynthetic reactions (Baquer et al., 1988) and it has been demonstrated that this pathway has an enormous reserve capacity that can be drawn on during periods of oxidative stress to act as a proton donor during the redox cycling of glutathione (Hothersall et al., 1982; Schrader et al., 1993). Peroxynitrite has been shown to activate glucose-6-phosphate dehydrogenase (G6PDH), the enzyme catalyzing the rate limiting step of the oxidative branch of the PPP, resulting in the rapid activation of the PPP and increased NADPH accumulation in astrocytes and neurons (Garcia-Nogales et al., 2003). In addition, in vitro studies of neurons and astrocytes in high glucose environments show increased PPP activity and glutathione levels in astrocytes, which can reduce levels of oxidative stress and protect neurons in mixed cultures (Takahashi et al., 2012). Recently, it was shown that pyruvate generated from metabolism via the PPP can be metabolized in the TCA cycle and contributes to the formation of glutamate in neurons (Brekke et al., 2012). Thus, it is tempting to hypothesize, but remains to be proven that increased PPP activity following TBI reflects a response by injured cells to combat oxidative/nitrosative stress and/or provide additional substrates for oxidative metabolism.
Oxidative Metabolism Following TBI
As previously described, mitochondrial dysfunction is thought to play a key role in the pathophysiology of TBI. Studies using cytochrome C oxidase histochemistry as a measure of oxidative phosphorylation on the mitochondrial membrane, show a diffuse decrease in staining throughout the injured hemisphere of both FPI (Hovda et al., 1991) and CCI (Moro and Sutton, 2010) injured adult rats. Moreover, measurements of mitochondrial respiration rates have shown TBI-induced reductions in mitochondrial state 3 respiratory rates in immature and adult rat experimental models and humans (Xiong et al., 1997; Verweij et al., 2000; Kilbaugh et al., 2011). Neuroprotective strategies targeting mitochondrial dysfunction such as cyclosporin A (or its analog), oxidative/nitrosative species scavengers, or alternative metabolic substrates to glucose have shown reduced cell death, improvements in mitochondrial function, and/or functional outcome (Fukushima et al., 2009; Moro and Sutton, 2010; Mustafa et al., 2010; Kilbaugh et al., 2011; Readnower et al., 2011; Singh et al., 2013). Mitochondrial dysfunction, specifically changes in the oxidative metabolism of metabolic fuels, can be measured using 13C NMR spectroscopy by determining the amount of 13C incorporation into glutamate and glutamine. However, oxidative metabolism in astrocytes and the specific contribution of glutamine to metabolic compartmentation is more accurately measured using acetate, a glial specific substrate (Waniewski and Martin, 1998; Lebon et al., 2002; Deelchand et al., 2009; Shen, 2013) or [2 13C] glucose that preferentially labels glutamine via pyruvate carboxylase (PC).
Using an adult rat CCI injury model (Bartnik et al., 2005), the amount of 13C label incorporated into the glutamate C2, C3, and C4 isotopomers did not differ from naive, suggesting that oxidative metabolism and the activity of PDH in glutamatergic neurons is maintained in the injured cortex over the first 24 h after injury. In the same study, a significant increase in 13C labeling of the glutamine C3 isotopomer was detected at 3.5 h after injury. Since the specific contribution of glutamine labeling via oxidative metabolism in astrocytes is difficult to ascertain using [1, 2 13C2] labeled glucose, this study could not clarify if the increased labeling of glutamine reflected the de novo synthesis of glutamine or increased glutamate uptake by astrocytes in response to injury. In support of the latter mechanism, increased glutamate metabolism via the astrocytic TCA cycle occurs when extracellular glutamate concentrations are increased (McKenna et al., 1996) and during ischemia (Haberg et al., 1998; Pascual et al., 1998). Also, excitotoxic injury in rats alters glutamate-glutamine cycle enzymes to favor increased glutamine synthesis (Ramonet et al., 2004). In contrast to CCI, adult rats with FPI showed reduced 13C labeling of all glutamate and glutamine isotopomers at 3.5 h post injury, indicating reduced oxidative metabolism in both neurons and astrocytes in the injured cortex (Bartnik et al., 2007). In this model, the 13C labeling of glutamate returned to non-injury levels by 24 h while reductions in glutamine labeling persisted. The divergent pattern of 13C labeling between these two injury models likely represents previously reported differences in the extent and severity of CMRglc changes in the two models (Yoshino et al., 1991; Sutton et al., 1994; Lee et al., 1999; Moore et al., 2000).
In contrast to what is observed in studies using adult models, a 13C NMR spectroscopy study of the injured immature rat brain found increased labeling of glutamate and glutamine C3 and C4 isotopomers at 5.5 and 6 h following CCI injury (Scafidi et al., 2009). Scafidi et al. (2009) proposed that there could be an accumulation of glutamate due to impaired glutamate entry into the mitochondria via reduced activity of the aspartate-glutamate carrier (McKenna et al., 2006; McKenna, 2007), or reduced glutamate oxidation to α-ketoglutarate via decreased activity of α-ketoglutarate dehydrogenase due to oxidative stress (Starkov et al., 2004). The delayed increase in labeling also suggests a delay in the metabolic response in the immature brain and highlight important developmental differences in the response to injury (Scafidi et al., 2009).
Astrocyte Metabolism and Neuroglia Metabolic Compartmentation Following TBI
Astrocytes show pronounced changes in gene expression, cellular hypertrophy and proliferation, in a degree relative to the severity of brain injury. Studies in both experimental and human brain injury have demonstrated the presence of reactive astrocytes (Bourke et al., 1980; Cortez et al., 1989; Castejón, 1998). Reactive astrocytes play dual roles following injury, one that may result in a detrimental increase in glutamate excitotoxicity or inflammation, the other being brain protection or repair (Laird et al., 2008). A transient down regulation of glutamate transporters GLT-1, GLT-1v, and GLAST on astrocytes after experimental (Rao et al., 1998, 2001; Yi and Hazell, 2006) and human TBI (van Landeghem et al., 2006; Beschorner et al., 2007) may well contribute to the injury process. However, ablation of reactive astrocytes following experimental CCI in transgenic mice resulted in greater loss of cortical tissue and inflammation, suggesting an essential protective role for astrocytes after TBI (Myer et al., 2006).
Determining the role that astrocytes play in the metabolic response to TBI is an important research direction. Astrocytes play a pivotal role in meeting the energy requirements of neurons through the glutamate-glutamine cycle that links the exchange of glutamate and glutamine between glutamatergic neurons and astrocytes (Van den Berg et al., 1969). Another proposed mechanism of metabolite trafficking between these cells is the lactate shuttle, where astrocytes preferentially metabolize glucose via glycolysis and transfer lactate to neurons during high metabolic demand (Magistretti and Pellerin, 1999; Bouzier-Sore et al., 2002; Pellerin et al., 2007), although yet to be proven and a topic of ongoing debate (Jolivet et al., 2010; Mangia et al., 2011). The net synthesis of glutamate in neurons also requires a compensatory flux of TCA cycle intermediates, notably glutamine from astrocytes (Schousboe et al., 1997), as neurons lack the capacity to generate TCA cycle intermediates. This net synthesis of TCA cycle intermediates, glutamate and glutamine depends upon the entry of pyruvate, via an anaplerotic pathway, into the TCA cycle. In the brain this is exclusively achieved by PC, an astrocyte specific enzyme (Yu et al., 1983; Shank et al., 1985). Numerous in vitro studies have shown that astrocytes supply TCA cycle substrates to neurons during periods of glucose and/or oxygen deprivation (Hertz, 2003; Bambrick et al., 2004; Peng et al., 2007), suggesting that astrocytes may play an even greater nutritional role for neurons in the injured state. Given the essential role of neuroglia metabolic coupling in normal brain, a greater appreciation of the effect of TBI on metabolic coupling is an important and necessary contribution to understanding the metabolic response to TBI.
The 13C NMR studies detailed in section Glycolysis and PPP Metabolism Following TBI suggest that neuroglia metabolic coupling is altered in two different rat models of TBI. To more clearly define the contribution of this metabolic coupling over the hypometabolic period, a 13C NMR spectroscopy study using [1 13C] glucose, which is consumed in both neuronal and glial compartments, and [1, 2 13C2] acetate, which is metabolized solely within the glial compartment, was undertaken using an adult rat FPI model (Bartnik-Olson et al., 2010). Figure 2 illustrates the metabolic alterations to neuronal and astrocyte metabolism determined using this strategy. Similar to previous findings, decreased 13C labeling of all glutamate isotopomers from the metabolism of glucose was observed early post-injury, but recovered over time, indicating that injury-induced decreases in the oxidative metabolism of glucose in neurons is consistent with the time course of reduced CMRglc following FPI (Yoshino et al., 1991; Moore et al., 2000). Although the 13C labeling of glutamine C4 from glucose in the first turn of the astrocyte TCA cycle was reduced, the labeling of glutamine C2 and C3 remained unchanged, indicating that the metabolism of glucose via PC was unaffected by FPI. In addition, the incorporation of 13C label from acetate into glutamine and glutamate C4 was maintained, indicating that oxidative metabolism in astrocytes and the functional activity of the glutamate-glutamine cycle were preserved during the hypometabolic period following FPI. 13C labeling of glutamine from 13C acetate was also demonstrated following human TBI using microdialysis samples and 13C NMR spectroscopy (Gallagher et al., 2009), although glutamate labeling was seen in only a few patients. It is important to note that acetate enters the astrocyte TCA cycle as acetyl CoA, bypassing any dysfunction in glycolysis or at the level of PDH, which may relate to the ability of an acetate precursor to improve ATP and improve motor performance after CCI (Arun et al., 2010).
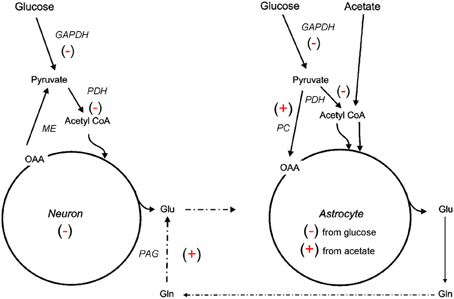
Figure 2. Simplified illustration of changes in neuroglial metabolic coupling following FPI as determined using 13C labeled glucose and acetate. Findings show reduced (−) glucose metabolism in both neuron and astrocyte metabolic compartments, possibly due to reduced activity of GAPDH and/or PDH. The capacity for oxidative metabolism was retained (+) in the astrocyte compartment as 13C labeling was detected in glutamine isotopomers resulting from acetate metabolism via the TCA cycle and PC. Labeling of glutamate from 13C acetate indicates continued activity (+) of the glutamate-glutamine cycle and phosphate activated glutaminase (PAG). These findings could be interpreted to mean that astrocytes have a supportive metabolic role to neurons following TBI.
Limitations and Future Directions
The studies reviewed above highlight alterations to a number of key metabolic processes during the period of metabolic depression following experimental TBI. Although these studies are valuable in their contributions linking the period of metabolic depression to qualitative changes in a number of metabolic processes, they are limited by their descriptive nature. In vivo metabolic reactions are dynamic and future studies making use of mathematical models to extract quantitative flux rates would vastly improve our understanding of TBI-induced changes in neuroglia compartmentation and neurotransmission. Moreover, clinical (human) studies using dynamic 13C NMR spectroscopy is a logical next step in advancing our understanding brain function after TBI.
One goal of future animal and clinical 13C studies should be to understand the cellular basis of metabolic alterations following TBI. It is important to establish how individual cell types respond to TBI. For example, studies employing compartment specific labels (singly or in combination) could delineate a cell-type specific preference for a metabolic fuel that would preferentially enhance outcome. In addition, future studies of metabolic flux during the acute period of hyperglycolysis could provide direct evidence of the metabolic forces (increased neurotransmission and/or energetics) driving this need. Moreover, comparisons between findings from the acute period of hyperglycolysis and the period of metabolic depression could establish key time points and potential targets for metabolic intervention.
Conclusion
TBI induces multiple primary and secondary injury mechanisms that can impact the supply of fuels and/or alter the functions of metabolic enzymes and proteins which can lead to deficits in energy availability. 13C NMR spectroscopy can be utilized to probe multiple aspects of the metabolic response to TBI, including changes in glycolysis, PPP activity, oxidative metabolism, and neuroglial metabolic compartmentation. As illustrated in the materials reviewed above, numerous experimental treatments that improve cerebral metabolism, reduce neuronal injury, and improve functional outcomes after TBI are currently being investigated, and future studies using 13C NMR spectroscopy to evaluate the metabolic responses to such treatments should provide valuable insights into the mechanisms of actions.
Author Contributions
Dr's. Bartnik-Olson and Sutton prepared the initial draft of this manuscript, Dr's. Harris and Shijo contributed additions and edits to the final versions of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the UCLA Brain Injury Research Center and P01NS058489 from the National Institute of Neurological Disorders and Stroke (NINDS). The content is the sole responsibility of the authors and does not necessarily represent official views of the NINDS or the National Institutes of Health.
Abbreviations
ATP, adenosine triphosphate; Ca++, calcium; CCI, controlled cortical impact; CMRglc, cerebral metabolic rate of glucose; FPI, fluid percussion injury; G6PDH, glyceraldehyde-6-phosphate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GC-MS, gas chromatography-mass spectrometry; GLAST, glutamate-aspartate transporter; GLT-1, glutamate transporter 1; GLT-1v, glutamate transporter 1 variant; NAD+, nicotinamide adenine dinucleotide phosphate; NADP+, nicotinamide adenine dinucleotide phosphate; NMR, nuclear magnetic resonance spectroscopy; PARP, poly(ADP) ribose polymerases; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PPP, pentose phosphate pathway; TCA, tricarboxylic acid cycle; TBI, traumatic brain injury.
References
Aoyama, N., Lee, S. M., Moro, N., Hovda, D. A., and Sutton, R. L. (2008). Duration of ATP reduction affects extent of CA1 cell death in rat models of fluid percussion injury combined with secondary ischemia. Brain Res. 1230, 310–319. doi: 10.1016/j.brainres.2008.07.006
Arun, P., Ariyannur, P. S., Moffett, J. R., Xing, G., Hamilton, K., Grunberg, N. E., et al. (2010). Metabolic acetate therapy for the treatment of traumatic brain injury. J. Neurotrauma 27, 293–298. doi: 10.1089/neu.2009.0994
Arundine, M., Aarts, M., Lau, A., and Tymianski, M. (2004). Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J. Neurosci. 24, 8106–8123. doi: 10.1523/JNEUROSCI.1362-04.2004
Aureli, T., Di Cocco, M. E., Calvani, M., and Conti, F. (1997). The entry of [1-13C] glucose into biochemical pathways reveals a complex compartmentation and metabolite trafficking between glia and neurons: a study by 13C-NMR spectroscopy. Brain Res. 765, 218–227. doi: 10.1016/S0006-8993(97)00514-3
Bachelard, H. S., and Badar-Goffer, R. S. (1993). NMR spectroscopy in neurochemistry. J. Neurochem. 61, L412–L429. doi: 10.1111/j.1471-4159.1993.tb02141.x
Badar-Goffer, R. S., Bachelard, H. S., and Morris, P. G. (1990). Cerebral metabolism of acetate and glucose studied by 13C-n.m.r. spectroscopy. A technique for investigating metabolic compartmentation in the brain. Biochem. J. 266, 133–139.
Bambrick, L., Kristian, T., and Fiskum, G. (2004). Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem. Res. 29, 601–608. doi: 10.1023/B:NERE.0000014830.06376.e6
Baquer, N. Z., Hothersall, J. S., and McLean, P. (1988). Function and regulation of the pentose phosphate pathway in brain. Curr. Top. Cell. Regul. 29, 265–289.
Bartnik, B. L., Lee, S. M., Hovda, D. A., and Sutton, R. L. (2007). The fate of glucose during the period of decreased metabolism after fluid percussion injury: a 13C NMR study. J. Neurotrauma 24, 1079–1092. doi: 10.1089/neu.2006.0210
Bartnik, B. L., Sutton, R. L., Fukushima, M., Harris, N. G., Hovda, D. A., and Lee, S. M. (2005). Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J. Neurotrauma 22, 1052–1065. doi: 10.1089/neu.2005.22.1052
Bartnik-Olson, B. L., Oyoyo, U., Hovda, D., and Sutton, R. L. (2010). Astrocyte oxidative metabolism and metabolite trafficking after fluid percussion brain injury in adult rats. J. Neurotrauma 27, 2191–2202. doi: 10.1089/neu.2010.1508
Bergsneider, M., Hovda, D. A., Lee, S. M., Kelly, D. F., McArthur, D. L., Vespa, P. M., et al. (2000). Dissociation of cerebral glucose metabolism and level of consciousness during the period of metabolic depression following human traumatic brain injury. J. Neurotrauma 17, 389–401. doi: 10.1089/neu.2000.17.389
Bergsneider, M., Hovda, D. A., McArthur, D. L., Etchepare, M., Huang, S.-C., Sehati, N., et al. (2001). Metabolic recovery following human traumatic brain injury based on FDG-PET: time course and relationship to neurological disability. J. Head Trauma Rehabil. 16, 135–148. doi: 10.1097/00001199-200104000-00004
Bergsneider, M., Hovda, D. A., Shalmon, E., Kelly, D. F., Vespa, P. M., Martin, N. A., et al. (1997). Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg. 86, 241–251. doi: 10.3171/jns.1997.86.2.0241
Beschorner, R., Dietz, K., Schauer, N., Mittelbronn, M., Schluesener, H. J., Trautmann, K., et al. (2007). Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol. Histopathol. 22, 515–526.
Besson, V. C. (2009). Drug targets for traumatic brain injury from poly(ADP-ribose)polymerase pathway modulation. Br. J. Pharmacol. 157, 695–704. doi: 10.1111/j.1476-5381.2009.00229.x
Bourke, R. S., Kimelberg, H. K., Nelson, L. R., Barron, K. D., Auen, E. L., Popp, A. J., et al. (1980). Biology of glial swelling in experimental brain edema. Adv. Neurol. 28, 99–109.
Bouzier-Sore, A. K., Merle, M., Magistretti, P. J., and Pellerin, L. (2002). Feeding active neurons: (re)emergence of a nursing role for astrocytes. J. Physiol. Paris 96, 273–282. doi: 10.1016/S0928-4257(02)00016-5
Brekke, E. M., Walls, A. B., Schousboe, A., Waagepetersen, H. S., and Sonnewald, U. (2012). Quantitative importance of the pentose phosphate pathway determined by incorporation of (13)C from [2-(13)C]- and [3-(13)C]glucose into TCA cycle intermediates and neurotransmitter amino acids in functionally intact neurons. J. Cereb. Blood Flow Metab. 32, 1788–1799. doi: 10.1038/jcbfm.2012.85
Castejón, O. J. (1998). Morphological astrocytic changes in complicated human brain trauma. A light and electron microscopic study. Brain Inj. 12, 409–427. doi: 10.1080/026990598122539
Cerdan, S., Rodrigues, T. B., Sierra, A., Benito, M., Fonseca, L. L., Fonseca, C. P., et al. (2006). The redox switch/redox coupling hypothesis. Neurochem. Int. 48, 523–530. doi: 10.1016/j.neuint.2005.12.036
Chen, T., Qian, Y. Z., Di, X., Rice, A., Zhu, J. P., and Bullock, R. (2000). Lactate/glucose dynamics after rat fluid percussion brain injury. J. Neurotrauma 17, 135–142. doi: 10.1089/neu.2000.17.135
Clark, R. S., Vagni, V. A., Nathaniel, P. D., Jenkins, L. W., Dixon, C. E., and Szabo, C. (2007). Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+ depletion and improves water maze performance after traumatic brain injury in mice. J. Neurotrauma 24, 1399–1405. doi: 10.1089/neu.2007.0305
Clark, R. S. B., Chen, M., Kochanek, P. M., Watkins, S. C., Jin, K. L., Draviam, R., et al. (2001). Detection of single- and double-strand DNA breaks after traumatic brain injury in rats: comparison of in situ labeling techniques using DNA polymerase I, the Klenow fragment of DNA polymerase I, and terminal deoxynucleotidyl transferase. J. Neurotrauma 18, 675–689. doi: 10.1089/089771501750357627
Clausen, F., Hillered, L., and Gustafsson, J. (2011). Cerebral glucose metabolism after traumatic brain injury in the rat studied by 13C-glucose and microdialysis. Acta Neurochir.(Wien.) 153, 653–658. doi: 10.1007/s00701-010-0871-7
Cortez, S. C., McIntosh, T. K., and Noble, L. J. (1989). Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 482, 271–282. doi: 10.1016/0006-8993(89)91190-6
Cruz, F., Scott, S. R., Barroso, I., Santisteban, P., and Cerdan, S. (1998). Ontogeny and cellular localization of the pyruvate recycling system in rat brain. J. Neurochem. 70, 2613–2619. doi: 10.1046/j.1471-4159.1998.70062613.x
Cruz, F., and Cerdan, S. (1999). Quantitative 13C NMR studies of metabolic compartmentation in the adult mammalian brain. NMR Biomed. 12, 451–462. doi: 10.1002/(SICI)1099-1492(199911)12:7<451::AID-NBM571>3.3.CO;2-5
Deelchand, D. K., Shestov, A. A., Koski, D. M., Uğurbil, K., and Henry, P. G. (2009). Acetate transport and utilization in the rat brain. J. Neurochem. 109, 46–54. doi: 10.1111/j.1471-4159.2009.05895.x
Dugan, L. L., Sensi, S. L., Canzoniero, L. M., Handran, S. D., Rothman, S. M., Lin, T. S., et al. (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J. Neurosci. 15, 6377–6388.
Dusick, J. R., Glenn, T. C., Lee, W. N., Vespa, P. M., Kelly, D. F., Lee, S. M., et al. (2007). Increased pentose phosphate pathway flux after clinical traumatic brain injury: a [1,2-(13)C(2)]glucose labeling study in humans. J. Cereb. Blood Flow Metab. 27, 1593–1602. doi: 10.1038/sj.jcbfm.9600458
Fineman, I., Hovda, D. A., Smith, M., Yoshino, A., and Becker, D. P. (1993). Concussive brain injury is associated with a prolonged accumulation of calcium: a 45 Ca autoradiographic study. Brain Res. 624, 94–102. doi: 10.1016/0006-8993(93)90064-T
Fukushima, M., Lee, S. M., Moro, N., Hovda, D. A., and Sutton, R. L. (2009). Metabolic and histologic effects of sodium pyruvate treatment in the rat after cortical contusion injury. J. Neurotrauma 26, 1095–1110. doi: 10.1089/neu.2008.0771
Gallagher, C. N., Carpenter, K. L., Grice, P., Howe, D. J., Mason, A., Timofeev, I., et al. (2009). The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 132, 2839–2849. doi: 10.1093/brain/awp202
Garcia-Nogales, P., Almeida, A., and Bolanos, J. P. (2003). Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 278, 864–874. doi: 10.1074/jbc.M206835200
Grant, C. M. (2008). Metabolic reconfiguration is a regulated response to oxidative stress. J. Biol. 7, 1. doi: 10.1186/jbiol63
Gunter, T. E., Gunter, K. K., Sheu, S. S., and Gavin, C. E. (1994). Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol. 267, C313–C339.
Haberg, A., Qu, H., Haraldseth, O., Unsgard, G., and Sonnewald, U. (1998). In vivo injection of [1-13C]glucose and [1,2-13C]acetate combined with ex vivo 13C nuclear magnetic resonance spectroscopy: a novel approach to the study of middle cerebral artery occlusion in the rat. J. Cereb. Blood Flow Metab. 18, 1223–1232. doi: 10.1097/00004647-199811000-00008
Hall, E. D., Andrus, P. K., and Yonkers, P. A. (1993). Brain hydroxyl radical generation in acute experimental head injury. J. Neurochem. 60, 588–594. doi: 10.1111/j.1471-4159.1993.tb03189.x
Hall, E. D., Bryant, Y. D., Cho, W., and Sullivan, P. G. (2008). Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de olmos silver and fluorojade staining methods. J. Neurotrauma 25, 235–247. doi: 10.1089/neu.2007.0383
Hassel, B., Sonnewald, U., and Fonnum, F. (1995). Glial-neuronal interactions as studied by cerebral metabolism of [2-13C]acetate and [1-13C]glucose: an ex vivo 13C NMR spectroscopic study. J. Neurochem. 64, 2773–2782. doi: 10.1046/j.1471-4159.1995.64062773.x
Hassel, B., and Sonnewald, U. (1995). Glial formation of pyruvate and lactate from TCA cycle intermediates: implications for the inactivation of transmitter amino acids. J. Neurochem. 65, 2227–2234. doi: 10.1046/j.1471-4159.1995.65052227.x
Hertz, L. (2003). Astrocytic amino acid metabolism under control conditions and during oxygen and/or glucose deprivation. Neurochem. Res. 28, 243–258. doi: 10.1023/A:1022377100379
Hothersall, J. S., Greenbaum, A. L., and McLean, P. (1982). The functional significance of the pentose phosphate pathway in synaptosomes: protection against peroxidative damage by catecholamines and oxidants. J. Neurochem. 39, 1325–1332. doi: 10.1111/j.1471-4159.1982.tb12574.x
Hovda, D. A. (1996). “Metabolic dysfunction,” in Neurotrauma, eds R. K.Narayan, J. E. Wilberger, and J. T. Povlishock (New York, NY: McGraw-Hill, Inc.), 1459–1478.
Hovda, D. A., Yoshino, A., Kawamata, T., Katayama, Y., and Becker, D. P. (1991). Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 567, 1–10. doi: 10.1016/0006-8993(91)91429-5
Inao, S., Marmarou, A., Clarke, G. D., Andersen, B. J., Fatouros, P. P., and Young, H. F. (1988). Production and clearance of lactate from brain tissue, cerebrospinal fluid, and serum following experimental brain injury. J. Neurosurg. 69, 736–744. doi: 10.3171/jns.1988.69.5.0736
Ip, E. Y., Zanier, E. R., Moore, A. H., Lee, S. M., and Hovda, D. A. (2003). Metabolic, neurochemical, and histologic responses to vibrissa motor cortex stimulation after traumatic brain injury. J. Cereb. Blood Flow Metab. 23, 900–910. doi: 10.1097/01.WCB.0000076702.71231.F2
Jiang, X. B., Ohno, K., Qian, L., Tominaga, B., Kuroiwa, T., Nariai, T., et al. (2000). Changes in local cerebral blood flow, glucose utilization, and mitochondrial function following traumatic brain injury in rats. Neurol. Med. Chir. (Tokyo) 40, 16–28. doi: 10.2176/nmc.40.16
Jolivet, R., Allaman, I., Pellerin, L., Magistretti, P. J., and Weber, B. (2010). Comment on recent modeling studies of astrocyte-neuron metabolic interactions. J. Cereb. Blood Flow Metab. 30, 1982–1986. doi: 10.1038/jcbfm.2010.132
Katayama, Y., Becker, D. P., Tamura, T., and Hovda, D. A. (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900. doi: 10.3171/jns.1990.73.6.0889
Kauppinen, T. M. (2007). Multiple roles for poly(ADP-ribose)polymerase-1 in neurological disease. Neurochem. Int. 50, 954–958. doi: 10.1016/j.neuint.2006.11.010
Kawamata, T., Katayama, Y., Hovda, D. A., Yoshino, A., and Becker, D. P. (1995). Lactate accumulation following concussive brain injury: the role of ionic fluxs induced by excitatory amino acids. Brain Res. 674, 196–204. doi: 10.1016/0006-8993(94)01444-M
Kelly, D. F., Kozlowski, D. A., Haddad, E., Echiverri, A., Hovda, D. A., and Lee, S. M. (2000). Ethanol reduces metabolic uncoupling following experimental head injury. J. Neurotrauma 17, 261–272. doi: 10.1089/neu.2000.17.261
Kilbaugh, T. J., Bhandare, S., Lorom, D. H., Saraswati, M., Robertson, C. L., and Margulies, S. S. (2011). Cyclosporin a preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J. Neurotrauma 28, 763–774. doi: 10.1089/neu.2010.1635
Komjati, K., Besson, V. C., and Szabo, C. (2005). Poly (adp-ribose) polymerase inhibitors as potential therapeutic agents in stroke and neurotrauma. Curr. Drug Targets CNS Neurol. Disord. 4, 179–194. doi: 10.2174/1568007053544138
Lai, Y., Chen, Y., Watkins, S. C., Nathaniel, P. D., Guo, F., Kochanek, P. M., et al. (2008). Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 104, 1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x
Laird, M. D., Vender, J. R., and Dhandapani, K. M. (2008). Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals 16, 154–164. doi: 10.1159/000111560
Langfitt, T. W., Obrist, W. D., Alavi, A., Grossman, R. I., Zimmerman, R., Jaggi, J., et al. (1986). Computerized tomography, magnetic resonance imaging, and positron emission tomography in the study of brain trauma. Preliminary observations. J. Neurosurg. 64, 760–767. doi: 10.3171/jns.1986.64.5.0760
Laplaca, M. C., Raghupathi, R., Verma, A., Pieper, A. A., Saatman, K. E., Snyder, S. H., et al. (1999). Temporal patterns of poly(ADP-ribose) polymerase activation in the cortex following experimental brain injury in the rat. J. Neurochem. 73, 205–213. doi: 10.1046/j.1471-4159.1999.0730205.x
Laplaca, M. C., Zhang, J., Raghupathi, R., Li, J. H., Smith, F., Bareyre, F. M., et al. (2001). Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J. Neurotrauma 18, 369–376. doi: 10.1089/089771501750170912
Lebon, V., Petersen, K. F., Cline, G. W., Shen, J., Mason, G. F., Dufour, S., et al. (2002). Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J. Neurosci. 22, 1523–1531.
Lee, S. M., Wong, D. A., Samii, A., and Hovda, D. A. (1999). Evidence for energy failure following irreversible traumatic brain injury. Ann. N.Y. Acad. Sci. 893, 337–340. doi: 10.1111/j.1749-6632.1999.tb07849.x
Lee, W. N., Boros, L. G., Puigjaner, J., Bassilian, S., Lim, S., and Cascante, M. (1998). Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. 274, E843–E851.
Lewen, A., and Hillered, L. (1998). Involvement of reactive oxygen species in membrane phospholipid breakdown and energy perturbation after traumatic brain injury in the rat. J. Neurotrauma 15, 521–530. doi: 10.1089/neu.1998.15.521
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 354, 1155–1163. doi: 10.1098/rstb.1999.0471
Mangia, S., DiNuzzo, M., Giove, F., Carruthers, A., Simpson, I. A., and Vannucci, S. J. (2011). Response to ‘comment on recent modeling studies of astrocyte-neuron metabolic interactions’: much ado about nothing. J. Cereb. Blood Flow Metab. 31, 1346–1353. doi: 10.1038/jcbfm.2011.29
Marcoux, J., McArthur, D. A., Miller, C., Glenn, T. C., Villablanca, P., Martin, N. A., et al. (2008). Persistent metabolic crisis as measured by elevated cerebral microdialysis lactate-pyruvate ratio predicts chronic frontal lobe brain atrophy after traumatic brain injury. Crit. Care Med. 36, 2871–2877. doi: 10.1097/CCM.0b013e318186a4a0
Marklund, N., Clausen, F., Ander, T. L., and Hillered, L. (2001). Monitoring of reactive oxygen species production after traumatic brain injury in rats with microdialysis and the 4-hydroxybenzoic acid trapping method. J. Neurotrauma 18, 1217–1227. doi: 10.1089/089771501317095250
McKenna, M. C. (2007). The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J. Neurosci. Res. 85, 3347–3358. doi: 10.1002/jnr.21444
McKenna, M. C., Sonnewald, U., Huang, X., Stevenson, J., and Zielke, H. R. (1996). Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 66, 386–393. doi: 10.1046/j.1471-4159.1996.66010386.x
McKenna, M. C., Waagepetersen, H. S., Schousboe, A., and Sonnewald, U. (2006). Neuronal and Astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents current evidence and pharmacological tools. Biochem. Pharmacol. 71, 399–407. doi: 10.1016/j.bcp.2005.10.011
Mendez, D. R., Cherian, L., Moore, N., Arora, T., Liu, P. K., and Roberstson, C. S. (2004). Oxidative DNA lesions in a rodent model of traumatic brain injury. J. Trauma 56, 1235–1240. doi: 10.1097/01.TA.0000130759.62286.0E
Moore, A. H., Osteen, C. L., Chatziioannou, A. F., Hovda, D. A., and Cherry, S. R. (2000). Quantitative assessment of longitudinal metabolic changes in vivo after traumatic brain injury in the adult rat using FDG-microPET. J. Cereb. Blood Flow Metab. 20, 1492–1501. doi: 10.1097/00004647-200010000-00011
Moro, N., Ghavim, S., Hovda, D. A., and Sutton, R. L. (2011). Delayed sodium pyruvate treatment improves working memory following experimental traumatic brain injury. Neurosci. Lett. 491, 158–162. doi: 10.1016/j.neulet.2011.01.029
Moro, N., and Sutton, R. L. (2010). Beneficial effects of sodium or ethyl pyruvate after traumatic brain injury in the rat. Exp. Neurol. 225, 391–401. doi: 10.1016/j.expneurol.2010.07.013
Mustafa, A. G., Singh, I. N., Wang, J., Carrico, K. M., and Hall, E. D. (2010). Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J. Neurochem. 114, 271–280. doi: 10.1111/j.1471-4159.2010.06749.x
Myer, D. J., Gurkoff, G. G., Lee, S. M., Hovda, D. A., and Sofroniew, M. V. (2006). Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772. doi: 10.1093/brain/awl165
Nilsson, P., Hillered, L., Ponten, U., and Ungerstedt, U. (1990). Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J. Cereb. Blood Flow Metab. 10, 631–637. doi: 10.1038/jcbfm.1990.115
Opii, W. O., Nukala, V. N., Sultana, R., Pandya, J. D., Day, K. M., Merchant, M. L., et al. (2007). Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma 24, 772–789. doi: 10.1089/neu.2006.0229
Osteen, C. L., Moore, A. H., Prins, M. L., and Hovda, D. A. (2001). Age-dependency of 45 calcium accumulation following lateral fluid percussion: acute and delayed patterns. J. Neurotrauma 18, 141–162. doi: 10.1089/08977150150502587
Parkin, M., Hopwood, S., Jones, D. A., Hashemi, P., Landolt, H., Fabricius, M., et al. (2005). Dynamic changes in brain glucose and lactate in pericontusional areas of the human cerebral cortex, monitored with rapid sampling on-line microdialysis: relationship with depolarisation-like events. J. Cereb. Blood Flow Metab. 25, 402–413. doi: 10.1038/sj.jcbfm.9600051
Pascual, J. M., Carceller, F., Roda, J. M., and Cerdan, S. (1998). Glutamate, glutamine, and GABA as substrates for the neuronal and glial compartments after focal cerebral ischemia in rats. Stroke 29, 1048–1056. doi: 10.1161/01.STR.29.5.1048
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., et al. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262. doi: 10.1002/glia.20528
Peng, L., Gu, L., Zhang, H., Huang, X., Hertz, E., and Hertz, L. (2007). Glutamine as an energy substrate in cultured neurons during glucose deprivation. J. Neurosci. Res. 85, 3480–3486. doi: 10.1002/jnr.21262
Prins, M. L., and Hovda, D. A. (2001). Mapping cerebral glucose metabolism during spatial learning: interactions of development and traumatic brain injury. J. Neurotrauma 18, 31–46. doi: 10.1089/089771501750055758
Ralser, M., Wamelink, M. M., Kowald, A., Gerisch, B., Heeren, G., Struys, E. A., et al. (2007). Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 6, 10. doi: 10.1186/jbiol61
Ramonet, D., Rodriguez, M. J., Fredriksson, K., Bernal, F., and Mahy, N. (2004). In vivo neuroprotective adaptation of the glutamate/glutamine cycle to neuronal death. Hippocampus 14, 586–594. doi: 10.1002/hipo.10188
Rao, V. L., Baskaya, M. K., Dogan, A., Rothstein, J. D., and Dempsey, R. J. (1998). Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J. Neurochem. 70, 2020–2027. doi: 10.1046/j.1471-4159.1998.70052020.x
Rao, V. L., Dogan, A., Bowen, K. K., Todd, K. G., and Dempsey, R. J. (2001). Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur. J. Neurosci. 13, 119–128. doi: 10.1046/j.1460-9568.2001.01367.x
Readnower, R. D., Pandya, J. D., McEwen, M. L., Pauly, J. R., Springer, J. E., and Sullivan, P. G. (2011). Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J. Neurotrauma 28, 1845–1853. doi: 10.1089/neu.2011.1755
Rose, M. E., Huerbin, M. B., Melick, J., Marion, D. W., Palmer, A. M., Schiding, J. K., et al. (2002). Regulation of interstitial excitatory amino acid concentrations after cortical contusion injury. Brain Res. 935, 40–46. doi: 10.1016/S0006-8993(02)02445-9
Satchell, M. A., Zhang, X., Kochanek, P. M., Dixon, C. E., Jenkins, L. W., Melick, J., et al. (2003). A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-13-3gamma. J. Neurochem. 85, 697–708. doi: 10.1046/j.1471-4159.2003.01707.x
Scafidi, S., O'Brien, J., Hopkins, I., Robertson, C., Fiskum, G., and McKenna, M. (2009). Delayed cerebral oxidative glucose metabolism after traumatic brain injury in young rats. J. Neurochem. 109(Suppl. 1), 189–197. doi: 10.1111/j.1471-4159.2009.05896.x
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133.
Schrader, M. C., Eskey, C. J., Simplaceanu, V., and Ho, C. (1993). A carbon-13 nuclear magnetic resonance investigation of the metabolic fluxes associated with glucose metabolism in human erythrocytes. Biochim. Biophys. Acta 1182, 162–178. doi: 10.1016/0925-4439(93)90138-Q
Schousboe, A., Westergaard, N., Waagepetersen, H. S., Larsson, O. M., Bakken, I. J., and Sonnewald, U. (1997). Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 21, 99–105. doi: 10.1002/(SICI)1098-1136(199709)21:1<99::AID-GLIA11>3.0.CO;2-W
Shank, R. P., Leo, G. C., and Zilkha, E. (1993). Cerebral metabolic compartmentation as revealed by nuclear magnetic resonance analysis of D- [1-13C] glucose metabolism. J. Neurochem. 61, 315–323. doi: 10.1111/j.1471-4159.1993.tb03570.x
Shank, R. P., Bennett, G. S., Freytag, S. O., and Campbell, G. L. (1985). Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 329, 364–367. doi: 10.1016/0006-8993(85)90552-9
Sharma, P., Benford, B., Li, Z. Z., and Ling, G. S. (2009). Role of pyruvate dehydrogenase complex in traumatic brain injury and measurement of pyruvate dehydrogenase enzyme by dipstick test. J. Emerg.Trauma Shock 2, 67–72. doi: 10.4103/0974-2700.50739
Shen, J. (2013). Modeling the glutamate-glutamine neurotransmitter cycle. Front. Neuroenergetics 5:1. doi: 10.3389/fnene.2013.00001
Signoretti, S., Vagnozzi, R., Tavazzi, B., and Lazzarino, G. (2010). Biochemical and neurochemical sequelae following mild traumatic brain injury: summary of experimental data and clinical implications. Neurosurg. Focus 29, E1. doi: 10.3171/2010.9.FOCUS10183
Singh, I. N., Gilmer, L. K., Miller, D. M., Cebak, J. E., Wang, J. A., and Hall, E. D. (2013). Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J. Cereb. Blood Flow Metab. 33, 593–599. doi: 10.1038/jcbfm.2012.211
Starkov, A. A., Fiskum, G., Chinopoulos, C., Lorenzo, B. J., Browne, S. E., Patel, M. S., et al. (2004). Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004
Sutton, R. L., Hovda, D. A., Adelson, P. D., Benzel, E. C., and Becker, D. P. (1994). Metabolic changes following cortical contusion: relationships to edema and morphological changes. Acta Neurochir. Suppl.(Wien.) 60, 446–448.
Takahashi, S., Izawa, Y., and Suzuki, N. (2012). Astroglial pentose phosphate pathway rates in response to high-glucose environments. ASN Neuro. 4, 71–88. doi: 10.1042/AN20120002
Tyurin, V. A., Tyurina, Y. Y., Borisenko, G. G., Sokolova, T. V., Ritov, V. B., Quinn, P. J., et al. (2000). Oxidative stress following traumatic brain injury in rats: quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 75, 2178–2189. doi: 10.1046/j.1471-4159.2000.0752178.x
Vagnozzi, R., Marmarou, A., Tavazzi, B., Signoretti, S., Di Pierro, D., del Bolgia, F., et al. (1999). Changes of cerebral energy metabolism and lipid peroxidation in rats leading to mitochondrial dysfunction after diffuse brain injury. J. Neurotrauma 16, 903–913. doi: 10.1089/neu.1999.16.903
Van den Berg, C. J., Krzalic, L. J., Mela, P., and Waelsch, H. (1969). Compartmentation of glutamate metabolism in brain. Evidence for the existence of two different tricarboxylic acid cycles in brain. Biochem. J. 113, 281–290.
van Landeghem, F. K. H., Weiss, T., Oehmichen, M., and von Deimling, A. (2006). Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J. Neurotrauma 23, 1518–1528. doi: 10.1089/neu.2006.23.1518
Verweij, B. H., Muizelaar, J. P., Vinas, F. C., Peterson, P. L., Xiong, Y., and Lee, C. P. (2000). Impaired cerebral mitochondrial function after traumatic brain injury in humans. J. Neurosurg. 93, 815–820. doi: 10.3171/jns.2000.93.5.0815
Vespa, P. M., McArthur, D., O'Phelan, K., Glenn, T., Etchepare, M., Kelly, D., et al. (2003). Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J. Cereb. Blood Flow Metab. 23, 865–877. doi: 10.1097/01.WCB.0000076701.45782.EF
Vespa, P. M., Miller, C., McArthur, D., Eliseo, M., Etchepare, M., Hirt, D., et al. (2007). Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit. Care Med. 35, 2830–2836. doi: 10.1097/01.CCM.0000295667.66853.BC
Waniewski, R. A., and Martin, D. L. (1998). Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci. 18, 5225–5233.
Xing, G., Ren, M., O'Neill, J. T., Sharma, P., Verma, A., and Watson, W. D. (2012). Pyruvate dehydrogenase phosphatase1 mRNA expression is divergently and dynamically regulated between rat cerebral cortex, hippocampus and thalamus after traumatic brain injury: a potential biomarker of TBI-induced hyper- and hypo-glycaemia and neuronal vulnerability. Neurosci. Lett. 525, 140–145. doi: 10.1016/j.neulet.2012.07.055
Xing, G., Ren, M., Watson, W. A., O'Neil, J. T., and Verma, A. (2009). Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: a mechanism of dysregulated glucose metabolism. Neurosci. Lett. 454, 38–42. doi: 10.1016/j.neulet.2009.01.047
Xiong, Y., Gu, Q., Peterson, P. L., Muizelaar, J. P., and Lee, C. P. (1997). Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 14, 23–34. doi: 10.1089/neu.1997.14.23
Xiong, Y., Peterson, P. L., Verweij, B. H., Vinas, F. C., Muizelaar, J. P., and Lee, C. P. (1998). Mitochondrial dysfunction after experimental traumatic brain injury: combined efficacy of SNX-111 and U-101033E. J. Neurotrauma 15, 531–544. doi: 10.1089/neu.1998.15.531
Yamaki, T., Yoshino, E., Fujimoto, M., Ohmori, Y., Imahori, Y., and Ueda, S. (1996). Chronological positron emission tomographic study of severe diffuse brain injury in the chronic stage. J. Trauma 40, 50–56. doi: 10.1097/00005373-199601000-00010
Yi, J. H., and Hazell, A. S. (2006). Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 48, 394–403. doi: 10.1016/j.neuint.2005.12.001
Yoshino, A., Hovda, D. A., Kawamata, T., Katayama, Y., and Becker, D. P. (1991). Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 561, 106–119. doi: 10.1016/0006-8993(91)90755-K
Yu, A. C., Drejer, J., Hertz, L., and Schousboe, A. (1983). Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 41, 1484–1487. doi: 10.1111/j.1471-4159.1983.tb00849.x
Zamzami, N., Hirsch, T., Dallaporta, B., Petit, P. X., and Kroemer, G. (1997). Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J. Bioenerg. Biomembr. 29, 185–193. doi: 10.1023/A:1022694131572
Keywords: acetate, glucose, glutamate-glutamine cycle, magnetic resonance spectroscopy, neuroglial compartmentation, oxidative metabolism, pentose phosphate pathway
Citation: Bartnik-Olson BL, Harris NG, Shijo K and Sutton RL (2013) Insights into the metabolic response to traumatic brain injury as revealed by 13C NMR spectroscopy. Front. Neuroenergetics 5:8. doi: 10.3389/fnene.2013.00008
Received: 31 July 2013; Accepted: 12 September 2013;
Published online: 04 October 2013.
Edited by:
Sebastian Cerdan, Instituto de Investigaciones Biomedicas Alberto Sols, SpainReviewed by:
Sebastian Cerdan, Instituto de Investigaciones Biomedicas Alberto Sols, SpainSusanna Scafidi, Johns Hopkins University School of Medicine, USA
Copyright © 2013 Bartnik-Olson, Harris, Shijo and Sutton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brenda L. Bartnik-Olson, Department of Radiology, Loma Linda University Medical Center, 11234 Anderson Street, Room B623, Loma Linda, CA 92354, USA e-mail: bbartnik@llu.edu
†Present address: Katsunori Shijo, Department of Neurological Surgery, Nihon University School of Medicine, Tokyo, Japan