
- Department of Neurology and Neurobiology of Aging, Kanazawa University Graduate School of Medical Science, Kanazawa, Japan
Cerebral amyloid angiopathy (CAA) of amyloid β-protein (Aβ) type is common in Alzheimer’s disease (AD). Aβ immunotherapies have been reported to induce CAA-related intracerebral hemorrhages (ICH) or vasogenic edema. For the purpose of developing a method to predict CAA-related ICH and other cerebrovascular disorders in AD, the biomarkers, and risk factors are reviewed. The biomarkers include (1) greater occipital uptake on amyloid positron emission tomography imaging and a decrease of cerebrospinal fluid Aβ40 levels as markers suggestive of CAA, and (2) symptomatic lobar ICH, lobar microhemorrhages, focal subarachnoidal hemorrhages/superficial siderosis, cortical microinfarcts, and subacute encephalopathy (caused by CAA-related inflammation or angiitis) as imaging findings of CAA-related ICH and other disorders. The risk factors include (1) old age and AD, (2) CAA-related gene mutations and apolipoprotein E genotype as genetic factors, (3) thrombolytic, anti-coagulation, and anti-platelet therapies, hypertension, and minor head trauma as hemorrhage-inducing factors, and (4) anti-amyloid therapies. Positive findings for one or more biomarkers plus one or more risk factors would be associated with a significant risk of CAA-related ICH and other cerebrovascular disorders. To establish a method to predict future occurrence of CAA-related ICH and other cerebrovascular disorders in AD, prospective studies with a large number of AD patients are necessary, which will allow us to statistically evaluate to what extent each biomarker or risk factor would increase the risk. In addition, further studies with progress of technologies are necessary to more precisely detect CAA and CAA-related cerebrovascular disorders.
Introduction
Cerebral amyloid angiopathy (CAA) is cerebrovascular amyloid deposition, and is classified into several types according to amyloid protein involved; among them, sporadic CAA of amyloid β-protein (Aβ) type is most commonly found in elderly individuals as well as in patients with Alzheimer’s disease (AD; see review Yamada and Naiki, 2012).
Cerebral amyloid angiopathy is pathophysiologically related to both cerebrovascular disorders and dementia as shown in Figure 1 (Yamada and Naiki, 2012). A subset of patients with AD spontaneously develop CAA-related intracerebral hemorrhages (ICH) and other cerebrovascular events, which may affect the clinical course of AD; furthermore, in clinical trials for AD, Aβ immunotherapies induced CAA-related ICH and other vascular complications (Cordonnier and van der Flier, 2011). To develop a method to predict CAA-related ICH and other cerebrovascular events in AD, this article reviews the prevalence, clinical manifestations, biomarkers, and risk factors for CAA and CAA-related cerebrovascular disorders in patients with AD.
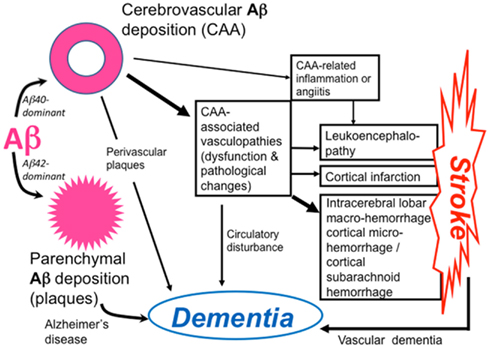
Figure 1. Pathophysiology of cerebral amyloid angiopathy (CAA)-related disorders. Aβ shows parenchymal or vascular deposition, depending on dominance of Aβ42 or Aβ40, respectively, in elderly individuals and patients with Alzheimer’s disease (AD). Cerebrovascular amyloid deposition, CAA, is related to stroke and dementia.
Spectrum of Cerebral Aβ Amyloidosis: CAA with or without AD
High prevalence of CAA in patients with AD as well as of cerebral parenchymal Aβ deposition (senile plaques, SPs) in patients with CAA can be explained by the fact that AD and CAA represent two sides of a single condition, i.e., cerebral Aβ amyloidosis. The length of Aβ of SPs is mainly 42–43 residues (Aβ42), while that of cerebrovascular Aβ is mainly 39–40 residues (Aβ40) (Prelli et al., 1988; Suzuki et al., 1994). Tissue levels of soluble Aβ40 in the brain correlate with severity of CAA (Suzuki et al., 1994). In the process of vascular Aβ deposition, Aβ42 is initially deposited, and later Aβ40 is massively accumulated (Shinkai et al., 1995). It has been indicated in both human and experimental studies that an increase of Aβ40 to Aβ42 ratio significantly shifts Aβ deposition toward the cerebral vasculature, resulting in prominent CAA, as demonstrated in hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D) with a point mutation at codon 693 of the Aβ precursor protein (APP) gene that causes a single amino acid substitution (Glu to Gln) at position 22 of Aβ, and the transgenic mouse model of this disease (Herzig et al., 2004). However, determinants for the pattern of parenchymal or vascular amyloid deposition remain to be elucidated for sporadic cases of AD as well as CAA.
Pathologically, CAA is commonly found in AD, with a prevalence of more than 80% (Yamada et al., 1987; Yamada, 2002). The prevalence and severity of CAA in AD are significantly higher compared with non-AD cases (Yamada, 2002). Actually, some of patients with AD are complicated with CAA-related cerebrovascular disorders as reviewed in the next section. On the other hand, a relatively small subset of patients with symptomatic CAA-related ICH present with dementia/AD at onset of ICH, suggesting vascular-dominant distribution of amyloid deposition in the brain (Cordonnier et al., 2010; Hirohata et al., 2010).
Thus, the severity of each of vascular and parenchymal amyloid deposition varies. The wide spectrum of cerebral Aβ amyloidosis is shown in Figure 2, including “AD with severe CAA,” “severe CAA without AD,” “AD with no or mild CAA,” and “no AD with no or mild CAA.” Severe CAA with or without AD may be associated with CAA-related disorders. Figure 3 shows the distribution of the severity of CAA in elderly individuals [age (mean ± SD), 85.9 ± 8.0 years] with and without AD from our autopsy series (Yamada, 2002). “No or mild CAA,” that would be clinically free of CAA-related cerebrovascular disorders, is found in 86% of non-AD elderly individuals, while in 66% of AD patients, which would explain that AD patients are associated with higher risk of CAA-related cerebrovascular disorders than non-AD elderly subjects.
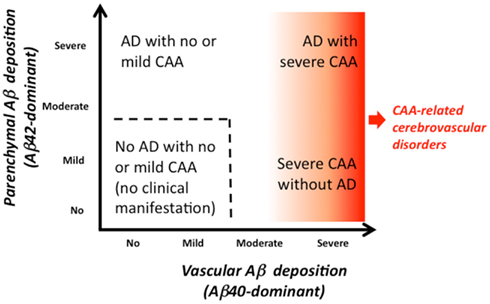
Figure 2. The spectrum of cerebral Aβ amyloidosis.
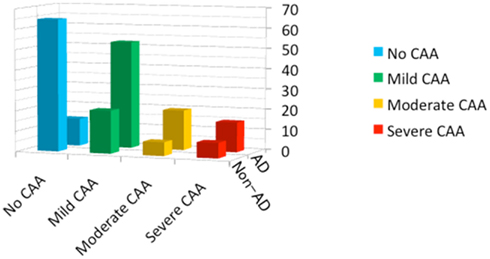
Figure 3. The distribution of the severity of CAA in elderly individuals (n = 201; age, 85.9 ± 8.0 years) with and without Alzheimer’s disease (AD), based on an autopsy series including AD (n = 82; age, 86.1 ± 7.9 years) and non-AD cases (n = 119; age, 85.7 ± 8.0 years). Frequencies are shown in the percentages in each of the AD and non-AD group (The modified data from the previous report; Yamada, 2002).
Patients who initially have the onset of symptomatic CAA-related ICH without AD may increase parenchymal Aβ deposition during the clinical course, leading to development of AD (Hirohata et al., 2010). On the other hand, patients who initially have AD with no or mild CAA may increase vascular Aβ deposition during the clinical course, which might lead to development of CAA-related cerebrovascular disorders (Figure 2). The long time course after onset of AD may allow progression of vascular Aβ deposition, as observed in the kinetic study of CAA in a transgenic mouse model (Robbins et al., 2006).
CAA-Related Intracerebral Hemorrhage and Other Cerebrovascular Disorders in AD
Intracerebral Hemorrhage
Symptomatic ICH
Incidence of symptomatic CAA-related ICH in patients with AD (Figure 4C) has not been elucidated as yet, requiring further prospective studies. Prevalence of ICH (excluding microhemorrhages) among AD cases at autopsy was reported to be 5.6%, including both CAA-related and non-CAA-related ICH, in a large autopsy series (Attems et al., 2008); it is suggested that additional risk is important to develop symptomatic macrohemorrhages, even if AD patients have CAA or CAA-related microhemorrhages in the brain.
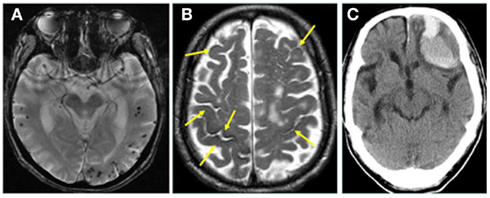
Figure 4. (A) Cerebral microhemorrhages with lobar distribution in a patient with Alzheimer’s disease (AD) on gradient-echo T2* imaging. (B,C) A patient with AD had presented with multiple, old focal subarachnoid hemorrhages (superficial siderosis; arrows) on T2-weighted MRI at the time of the diagnosis of AD (B). The patient later developed a large intracerebral hemorrhage in the left frontal lobe as shown on CT (C), probably related to CAA (by courtesy of Dr. Toshiya Fukui, Showa University).
Microhemorrhages in AD
The frequencies of cerebral microhemorrhages on sensitive magnetic imaging (MRI), such as gradient-echo T2*, were reported to be 16.7–32%, which are higher than those in general population (5–6%) (Hanyu et al., 2003; Cordonnier et al., 2006, 2007; Nakata-Kudo et al., 2006; Pettersen et al., 2008); microhemorrhages in AD are frequently distributed in lobar distribution with more numbers compared with those in control subjects (Figure 4A). A recent study with amyloid positron emission tomography (PET) using 11C-Pittsburgh compound B (PiB) reported high prevalence of lobar microhemorrhages in PiB-positive mild cognitive impairment (MCI) and healthy subjects as well as AD patients, indicating strong association of Aβ deposition with lobar microhemorrhages (Yates et al., 2011). Microhemorrhages were reported to preferentially occur in local regions of concentrated amyloid indicated by PiB retention (Dierksen et al., 2010).
Convexal Subarachnoid Hemorrhage or Superficial Siderosis
Superficial siderosis associated with CAA is detected on MRI in AD patients (Figure 4B) and was suggested to be a marker of CAA in AD (Feldman et al., 2008). It was reported that CAA was a frequent cause of convexal SAH, a subtype of non-aneurysmal subarachnoid bleeding, in patients over the age of 60 (Kumar et al., 2010). Superficial siderosis closely accompanied microhemorrhages in lobar locations, suggesting a close link between superficial siderosis and CAA (Vernooij et al., 2009b). Focal subarachnoid hemorrhages in conjunction with or without lobar microhemorrhages were reported to present with transient repetitive migraine auras or transient ischemic attack mimics (Izenberg et al., 2009).
White Matter Lesions
Although white matter lesions or hyperintensities detected by MRI are found in AD, MCI, and healthy elderly subjects as well as CAA, CAA-related perfusion impairments result in progressive damage of white matter and cognitive impairment (Smith et al., 2004; Chen et al., 2006; Yoshita et al., 2006; Viswanathan et al., 2008). Patients with CAA-related ICH showed occipital dominant white matter hyperintensities on MRI, compatible with predilection of CAA pathology for posterior brain regions (Zhu et al., 2012).
Microinfarcts
Cerebral amyloid angiopathy was revealed to be an important risk factor for microinfarcts restricted to the watershed cortical zones in AD, suggesting a role of cerebral hypoperfusion in the pathogenesis of cortical microinfarcts (Suter et al., 2002).
Contribution of CAA to Dementia in AD
Patients with AD and severe CAA may have mixed dementia of AD and CAA-related vascular cognitive impairment, and there may be occasionally “a vascular variant of AD” in which most of Aβ plaques are formed around CAA (Yamada et al., 1997a). CAA had deleterious effects on cognition even after controlling for age and AD pathology, and CAA was associated with white matter abnormalities and cognitive impairment (Greenberg et al., 2004). It was reported that moderate-to-very severe CAA, but not mild-to-moderate CAA, is associated with lower performance in specific cognitive domains, most notably perceptual speed, separately from the effect of AD pathology (Arvanitakis et al., 2011). Multiple microhemorrhages in AD were reported with worse cognitive function and higher abnormalities of cerebrospinal fluid (CSF) markers (Goos et al., 2009).
CAA-Related Inflammation
A subset of patients with CAA present with CAA-associated vascular inflammation or angiitis showing subacute encephalopathy characterized by cognitive symptoms, seizure, headaches, and focal neurological deficits with white matter lesions on MRI (Eng et al., 2004; Oh et al., 2004; Scolding et al., 2005; Kinnecom et al., 2007). In the clinical course all patients with CAA-related inflammation presented with subacute cognitive impairment, but not with AD-like slowly progressive dementia (Kinnecom et al., 2007). Parenchymal Aβ load in CAA-related angiitis was significantly lower than in AD (Scolding et al., 2005).
CAA-Related Cerebrovascular Disorders Induced by Aβ Immunotherapies in AD
Recent clinical as well as experimental studies with Aβ immunotherapies for AD have demonstrated that CAA has important implications for the future of anti-amyloid therapies. Experimental studies with AD model mice indicated that Aβ immunotherapies can induce CAA-related cerebral hemorrhage, although long-term Aβ immunotherapies could decrease CAA (Pfeifer et al., 2002; Wilcock et al., 2004, 2007; Racke et al., 2005; Prada et al., 2007; Schroeter et al., 2008).
In clinical trials with Aβ42 immunization for AD (AN1792, Elan), immunized patients showed significantly higher levels of CAA and cortical microhemorrhages and microvascular lesions compared with unimmunized AD controls, although the longest living had virtually complete absence of both plaques and CAA (Boche et al., 2008). A CAA-related macrohemorrhage was reported in an AD patient in the phase 2a AN1792 trial (Orgogozo et al., 2003). The findings suggest that Aβ immunization results in solubilization of plaque Aβ42, which flows out of the brain parenchyma via the perivascular pathway, causing an increase of CAA and CAA-related hemorrhages. Eventually, this flow also results in Aβ clearance from the cerebral vasculature.
In the AN1792 trial, 6% of patients treated with the AN1792 suffered meningoencephalitis (Orgogozo et al., 2003) and there was perivascular infiltration of lymphocytes around vessels with CAA (Nicoll et al., 2003). In a phase 2 trial of bapineuzumab, a humanized monoclonal anti-Aβ antibody, vasogenic edema occurred in 9.7% of patients, suggesting a vascular permeability change of CAA-laden vessels (Salloway et al., 2009). Vasogenic edema was found only in 2 of 2762 patients with AD at baseline of clinical trials for AD; this indicates that naturally occurring vasogenic edema is a very rare event in untreated patients with AD, though one case of vasogenic edema was presumably related to CAA-related angiitis (Carlson et al., 2011).
Markers for CAA and CAA-Related Cerebrovascular Disorders in AD
Imaging Markers
CT/MRI
Although cranial CT is available for symptomatic CAA-related ICH (macrohemorrhages), MRI is useful to detect small hemorrhagic lesions (lobar microhemorrhages and focal subarachnoid hemorrhages), and ischemic lesions (various white matter lesions and microinfarcts) as discussed above. Sensitive MR imagings, such as gradient-echo T2* imaging and susceptibility-weighted images (SWI), are particularly useful to detect microhemorrhagic lesions (Nandigam et al., 2009). Although subacute, silent cortical, or subcortical infarctions were recognized on diffusion-weighted images (DWI) in patients with CAA (Kimberly et al., 2009), cortical microinfarcts observed in neuropathological studies are often undetected on MRI because of their limited size (Brundel et al., 2012). Introduction of ultra-high strength clinical MR scanners with a higher spatial resolution may make their detection possible.
Amyloid-PET
In amyloid imaging with a PET ligand, PiB, it was reported that PiB binding was moderately increased in most patients with probable CAA-related ICH and that the occipital uptake was greater in CAA-related ICH compared with AD (Johnson et al., 2007; Ly et al., 2010). In an autopsy-confirmed case, PiB–PET could detect CAA as the dominant source of signal (Bacskai et al., 2007). Microhemorrhages are found to be associated with PiB retention (Dierksen et al., 2010; Yates et al., 2011).
Biochemical Markers
Cerebrospinal fluid markers are useful for the diagnosis of AD; the CSF findings in AD are characterized by a decrease of Aβ42 levels, an increase of the ratio of Aβ40 to Aβ42, and an increase of total and phosphorylated tau protein. A significant decrease of CSF Aβ40 as well as Aβ42 was reported in patients with probable CAA, suggesting the trapping of Aβ40 as well as Aβ42 in the cerebral vasculature (Verbeek et al., 2009). Plasma Aβ40 levels were reported to be associated with extent of white matter hyperintensity in AD, MCI, or CAA (Gurol et al., 2006).
Risk Factors for CAA and CAA-Related Cerebrovascular Disorders in AD
In addition to old age and AD, both genetic and non-genetic risk factors have been reported for CAA and CAA-related cerebrovascular disorders.
Genetic Risk Factors
Besides hereditary cases of AD/CAA-associated with mutations of the AβPP or presenilin genes (see review Yamada and Naiki, 2012), the ApoE gene (ApoE) has been reported to be risk for sporadic CAA as well as AD; the ε4 allele for CAA itself, and the ε2 allele for CAA-related ICH (Greenberg et al., 1995; Premkumar et al., 1996; Nicoll et al., 1997). Carriers of the ApoE ε2 or ε4 allele, particularly, of the ApoE ε2/ε4 genotype were associated with early recurrence of lobar ICH in patients who survived a lobar ICH (O’Donnell et al., 2000). The ApoE ε4 allele constituted a risk factor for capillary CAA (CAA-Type 1) (Thal et al., 2002) and CAA-related inflammation (Kinnecom et al., 2007). The presence of CAA in head injured cases was significantly associated with the ApoE ε4 allele, suggesting an interaction between gene and environment in development of CAA (Leclercq et al., 2005). Another CAA-related gene reported by more than one research group is transforming growth factor (TGF)-β1 (TGF-β1) (Hamaguchi et al., 2005; Peila et al., 2007).
In addition, CAA was reported to be associated with other gene polymorphisms, including the presenilin 1 (PS1), α1-antichymotrypsin (ACT), neprilysin, low-density lipoprotein-receptor related protein (LRP-1), and angiotensin-converting enzyme (ACE) genes (Yamada et al., 1997b, 1998, 2003; Yamada, 2002, 2004; Christoforidis et al., 2005; Thal et al., 2010; Domingues-Montanari et al., 2011). A recent study reported that a CR1 gene polymorphism, reported to increase risk for AD, was also associated with risk of CAA–ICH as well as CAA (Biffi et al., 2012).
Non-Genetic Risk Factors
Hypertension
Lowering of blood pressure reduced risk of CAA-related ICH, suggesting that high blood pressure could be a factor inducing ICH in patients with CAA (Arima et al., 2010).
Thrombolytic, anti-coagulation, and anti-platelet therapies
There is increasing evidence that CAA could be a risk factor for ICH in thrombolytic therapies for acute myocardial infarction, pulmonary embolism, or ischemic stroke, and for ICH in warfarin therapies (Rosand et al., 2000; McCarron and Nicoll, 2004). The use of anti-platelet drugs such as aspirin was related to the presence of microbleeds, and to strictly lobar microhemorrhages suggestive of CAA (Vernooij et al., 2009a). In a systematic review of clinical trials with aspirin in AD, ICH was found in 3.2% of the aspirin group, while, in 0% of the control group, suggesting that aspirin use in AD might pose an increased risk of ICH; a possible explanation for an increased risk of ICH could be CAA in AD (Thoonsen et al., 2010).
Anti-amyloid therapies
As discussed above, clinical trials with Aβ immunotherapies for AD could induce CAA-related ICH and microvascular lesions (Orgogozo et al., 2003; Boche et al., 2008), inflammation around CAA (Nicoll et al., 2003), and vasogenic edema related probably to vascular permeability changes of CAA-laden vessels (Salloway et al., 2009) These findings in the Aβ immunotherapies (Nicoll et al., 2003; Orgogozo et al., 2003; Boche et al., 2008; Salloway et al., 2009) suggest that, generally, anti-amyloid therapies with the removal effects of parenchymal Aβ may be complicated with CAA-related cerebrovascular disorders, because solubilized parenchymal Aβ would flow via the perivascular pathway and may deposit on vascular walls resulting in an increase of CAA and CAA-related cerebrovascular disorders; in addition, in the case of Aβ immunotherapies, inflammatory, or permeability changes may be also induced in vessels with CAA by immune reactions with vascular Aβ.
Predicting CAA-Related Intracerebral Hemorrhage and Other Cerebrovascular Disorders in AD
Biomarker findings and risk factors for CAA and CAA-related ICH and other cerebrovascular disorders are summarized in Table 1. In the table, CAA-related white matter hyperintensities are not included as a biomarker, because they are common findings in elderly subjects and AD patients with small vessel diseases including both CAA and non-CAA. As for CAA-related ICH, the Boston criteria were proposed for the diagnosis (Knudsen et al., 2001). When a given person presents with one or more biomarkers plus one or more risk factors, we need to be careful about future development of CAA-related ICH and other cerebrovascular disorders, and should avoid additional risk factors if they are avoidable (Table 1).

Table 1. Biomarker findings and risk factors for cerebral amyloid angiopathy (CAA) and CAA-related intracerebral hemorrhages (ICH) and other cerebrovascular disorders.
Alzheimer’s disease is a definite risk factor for CAA. When a patient with AD presents with positive findings for one or more biomarkers with additional risk factors such as anti-amyloid therapies, the patient would be associated with higher risk of CAA-related ICH and other cerebrovascular disorders compared with AD patients with no positive biomarkers or additional risk factors. However, the reported data for AD were based on cross-sectional or retrospective studies; prospective studies of CAA-related ICH and other cerebrovascular disorders with a large number of AD patients are necessary to establish a method to predict them in AD. Such studies will allow us to statistically evaluate to what extent each biomarker or risk factor would increase the risk of future development of CAA-related ICH and other cerebrovascular disorders in AD patients.
Future Directions
In addition to the prospective studies with AD patients, further studies with progress of technologies are necessary to more precisely detect CAA and CAA-related cerebrovascular disorders. Currently, sensitive MRI, such as gradient-echo T2* and SWI, is widely used to detect hemorrhages with a characteristic distribution of CAA for the clinical diagnosis of CAA-related hemorrhages. However, CAA may not present with hemorrhagic lesions, but with other clinical features, such as dementia. Imaging techniques such as PiB–PET have been used to directly image cerebral amyloid deposits. However, such PET tracers are unable to distinguish Aβ from other amyloid proteins, or to distinguish CAA from amyloid plaques, though the greater uptake in the occipital lobe may be suggestive of CAA (Bacskai et al., 2007; Johnson et al., 2007; Ly et al., 2010). More CAA-specific imaging is required. It was reported that clinical field-strength MRI could directly visualize amyloid plaques, as demonstrated in animal models (Ronald et al., 2009). Postmortem detection of Aβ deposition in CAA as well as AD with 7.0 T MRI was reported (van Rooden et al., 2009). Further progress in high resolution MRI may enable direct visualization of CAA. Regarding amyloid-PET, resolution of amyloid deposits on PET imaging would not be enough to distinguish CAA from amyloid plaques, requiring a new tracer, or technique to identify vascular amyloid.
As for biochemical markers, we need to establish the values of plasma as well as CSF markers for CAA. Blood and brain/CSF levels of Aβ influence each other through transport across the blood–brain barrier, from brain to blood and from blood to brain, using LRP-1 and receptor for advanced glycation end products (RAGE), respectively (Bell and Zlokovic, 2009). The integrity of blood vessels is disrupted in CAA (Bell and Zlokovic, 2009). Changes of plasma as well as CSF levels of Aβ40 and Aβ42 need to be investigated in a longitudinal study of patients with CAA to further clarify their value.
Finally, it is required to establish methods for prevention of CAA and CAA-related cerebrovascular disorders. Some hemorrhage-inducing factors are controllable or avoidable, including hypertension and use of thrombolytic/anti-coagulation/anti-platelet therapies (Rosand et al., 2000; McCarron and Nicoll, 2004; Trouillas and von Kummer, 2006; Vernooij et al., 2009a; Arima et al., 2010). We need future strategies for protection of vessel walls against amyloid-related vascular damage such as toxicity of Aβ oligomers, Aβ-induced oxidative stress, and inflammation.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported in part by a Grant from the Amyloidosis Research Committee from the Ministry of Health, Labour and Welfare, Japan, and by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan. The author is grateful to Drs. Toshiya Fukui and Miharu Samuraki for providing imaging data, and to Ms. Etsuko Tsujiguchi for her excellent secretarial work.
References
Arima, H., Tzourio, C., Anderson, C., Woodward, M., Bousser, M. G., MacMahon, S., Neal, B., and Chalmers, J. (2010). Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the progress trial. Stroke 41, 394–396.
Arvanitakis, Z., Leurgans, S. E., Wang, Z., Wilson, R. S., Bennett, D. A., and Schneider, J. A. (2011). Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327.
Attems, J., Lauda, F., and Jellinger, K. A. (2008). Unexpectedly low prevalence of intracerebral hemorrhages in sporadic cerebral amyloid angiopathy: an autopsy study. J. Neurol. 255, 70–76.
Bacskai, B. J., Frosch, M. P., Freeman, S. H., Raymond, S. B., Augustinack, J. C., Johnson, K. A., Irizarry, M. C., Klunk, W. E., Mathis, C. A., Dekosky, S. T., Greenberg, S. M., Hyman, B. T., and Growdon, J. H. (2007). Molecular imaging with Pittsburgh compound B confirmed at autopsy: a case report. Arch. Neurol. 64, 431–434.
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113.
Biffi, A., Shulman, J. M., Jagiella, J. M., Cortellini, L., Ayres, A. M., Schwab, K., Brown, D. L., Silliman, S. L., Selim, M., Worrall, B. B., Meschia, J. F., Slowik, A., De Jager, P. L., Greenberg, S. M., Schneider, J. A., Bennett, D. A., and Rosand, J. (2012). Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology 78, 334–341.
Boche, D., Zotova, E., Weller, R. O., Love, S., Neal, J. W., Pickering, R. M., Wilkinson, D., Holmes, C., and Nicoll, J. A. (2008). Consequence of Aβ immunization on the vasculature of human Alzheimer’s disease brain. Brain 131, 3299–3310.
Brundel, M., de Bresser, J., van Dillen, J. J., Kappelle, L. J., and Biessels, G. J. (2012). Cerebral microinfarcts: a systematic review of neuropathological studies. J. Cereb. Blood Flow Metab. 32, 425–436.
Carlson, C., Estergard, W., Oh, J., Suhy, J., Jack, C. R., Siemers, E., and Barakos, J. (2011). Prevalence of asymptomatic vasogenic edema in pretreatment Alzheimer’s disease study cohorts from phase 3 trials of semagacestat and solanezumab. Alzheimers Dement. 7, 396–401.
Chen, Y. W., Gurol, M. E., Rosand, J., Viswanathan, A., Rakich, S. M., Groover, T. R., Greenberg, S. M., and Smith, E. E. (2006). Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 67, 83–87.
Christoforidis, M., Schober, R., and Krohn, K. (2005). Genetic-morphologic association study: association between the low density lipoprotein-receptor related protein (LRP) and cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 31, 11–19.
Cordonnier, C., Al-Shahi Salman, R., and Wardlaw, J. (2007). Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain 130, 1988–2003.
Cordonnier, C., Leys, D., Dumont, F., Deramecourt, V., Bordet, R., Pasquier, F., and Hénon, H. (2010). What are the causes of pre-existing dementia in patients with intracerebral haemorrhages? Brain 133, 3281–3289.
Cordonnier, C., and van der Flier, W. M. (2011). Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain 134, 335–344.
Cordonnier, C., van der Flier, W. M., Sluimer, J. D., Leys, D., Barkhof, F., and Scheltens, P. (2006). Prevalence and severity of microbleeds in a memory clinic setting. Neurology 66, 1356–1360.
Dierksen, G. A., Skehan, M. E., Khan, M. A., Jeng, J., Nandigam, R. N., Becker, J. A., Kumar, A., Neal, K. L., Betensky, R. A., Frosch, M. P., Rosand, J., Johnson, K. A., Viswanathan, A., Salat, D. H., and Greenberg, S. M. (2010). Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann. Neurol. 68, 545–548.
Domingues-Montanari, S., Hernandez-Guillamon, M., Fernandez-Cadenas, I., Mendioroz, M., Boada, M., Munuera, J., Rovira, A., Maisterra, O., Pares, M., Gutierrez, M., Alvarez-Sabin, J., Chacon, P., Delgado, P., and Montaner, J. (2011). ACE variants and risk of intracerebral hemorrhage recurrence in amyloid angiopathy. Neurobiol. Aging 32, 9.
Eng, J. A., Frosch, M. P., Choi, K., Rebeck, G. W., and Greenberg, S. M. (2004). Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann. Neurol. 55, 250–256.
Feldman, H. H., Maia, L. F., Mackenzie, I. R., Forster, B. B., Martzke, J., and Woolfenden, A. (2008). Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke 39, 2894–2897.
Goos, J. D., Kester, M. I., Barkhof, F., Klein, M., Blankenstein, M. A., Scheltens, P., and van der Flier, W. M. (2009). Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke 40, 3455–3460.
Greenberg, S. M., Gurol, M. E., Rosand, J., and Smith, E. E. (2004). Amyloid angiopathy-related vascular cognitive impairment. Stroke 35, 2616–2619.
Greenberg, S. M., Rebeck, G. W., Vonsattel, J. P., Gomez-Isla, T., and Hyman, B. T. (1995). Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol. 38, 254–259.
Gurol, M. E., Irizarry, M. C., Smith, E. E., Raju, S., Diaz-Arrastia, R., Bottiglieri, T., Rosand, J., Growdon, J. H., and Greenberg, S. M. (2006). Plasma β-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 66, 23–29.
Hamaguchi, T., Okino, S., Sodeyama, N., Itoh, Y., Takahashi, A., Otomo, E., Matsushita, M., Mizusawa, H., and Yamada, M. (2005). Association of a polymorphism of the transforming growth factor-beta1 gene with cerebral amyloid angiopathy. J. Neurol. Neurosurg. Psychiatr. 76, 696–699.
Hanyu, H., Tanaka, Y., Shimizu, S., Takasaki, M., and Abe, K. (2003). Cerebral microbleeds in Alzheimer’s disease. J. Neurol. 250, 1496–1497.
Herzig, M. C., Winkler, D. T., Burgermeister, P., Pfeifer, M., Kohler, E., Schmidt, S. D., Danner, S., Abramowski, D., Sturchler-Pierrat, C., Burki, K., van Duinen, S. G., Maat-Schieman, M. L., Staufenbiel, M., Mathews, P. M., and Jucker, M. (2004). Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 7, 954–960.
Hirohata, M., Yoshita, M., Ishida, C., Ikeda, S. I., Tamaoka, A., Kuzuhara, S., Shoji, M., Ando, Y., Tokuda, T., and Yamada, M. (2010). Clinical features of non-hypertensive lobar intracerebral hemorrhage related to cerebral amyloid angiopathy. Eur. J. Neurol. 17, 823–829.
Izenberg, A., Aviv, R. I., Demaerschalk, B. M., Dodick, D. W., Hopyan, J., Black, S. E., and Gladstone, D. J. (2009). Crescendo transient aura attacks: a transient ischemic attack mimic caused by focal subarachnoid hemorrhage. Stroke 40, 3725–3729.
Johnson, K. A., Gregas, M., Becker, J. A., Kinnecom, C., Salat, D. H., Moran, E. K., Smith, E. E., Rosand, J., Rentz, D. M., Klunk, W. E., Mathis, C. A., Price, J. C., Dekosky, S. T., Fischman, A. J., and Greenberg, S. M. (2007). Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 62, 229–234.
Kimberly, W. T., Gilson, A., Rost, N. S., Rosand, J., Viswanathan, A., Smith, E. E., and Greenberg, S. M. (2009). Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 72, 1230–1235.
Kinnecom, C., Lev, M. H., Wendell, L., Smith, E. E., Rosand, J., Frosch, M. P., and Greenberg, S. M. (2007). Course of cerebral amyloid angiopathy-related inflammation. Neurology 68, 1411–1416.
Knudsen, K. A., Rosand, J., Karluk, D., and Greenberg, S. M. (2001). Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56, 537–539.
Kumar, S., Goddeau, R. P. Jr., Selim, M. H., Thomas, A., Schlaug, G., Alhazzani, A., Searls, D. E., and Caplan, L. R. (2010). Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology 74, 893–899.
Leclercq, P. D., Murray, L. S., Smith, C., Graham, D. I., Nicoll, J. A., and Gentleman, S. M. (2005). Cerebral amyloid angiopathy in traumatic brain injury: association with apolipoprotein E genotype. J. Neurol. Neurosurg. Psychiatr. 76, 229–233.
Ly, J. V., Donnan, G. A., Villemagne, V. L., Zavala, J. A., Ma, H., O’Keefe, G., Gong, S. J., Gunawan, R. M., Saunder, T., Ackerman, U., Tochon-Danguy, H., Churilov, L., Phan, T. G., and Rowe, C. C. (2010). 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology 74, 487–493.
McCarron, M. O., and Nicoll, J. A. (2004). Cerebral amyloid angiopathy and thrombolysis-related intracerebral haemorrhage. Lancet Neurol. 3, 484–492.
Nakata-Kudo, Y., Mizuno, T., Yamada, K., Shiga, K., Yoshikawa, K., Mori, S., Nishimura, T., Nakajima, K., and Nakagawa, M. (2006). Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr. Cogn. Disord. 22, 8–14.
Nandigam, R. N., Viswanathan, A., Delgado, P., Skehan, M. E., Smith, E. E., Rosand, J., Greenberg, S. M., and Dickerson, B. C. (2009). MR imaging detection of cerebral microbleeds: effect of susceptibility-weighted imaging, section thickness, and field strength. AJNR Am. J. Neuroradiol. 30, 338–343.
Nicoll, J. A., Burnett, C., Love, S., Graham, D. I., Dewar, D., Ironside, J. W., Stewart, J., and Vinters, H. V. (1997). High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann. Neurol. 41, 716–721.
Nicoll, J. A., Wilkinson, D., Holmes, C., Steart, P., Markham, H., and Weller, R. O. (2003). Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat. Med. 9, 448–452.
O’Donnell, H. C., Rosand, J., Knudsen, K. A., Furie, K. L., Segal, A. Z., Chiu, R. I., Ikeda, D., and Greenberg, S. M. (2000). Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N. Engl. J. Med. 342, 240–245.
Oh, U., Gupta, R., Krakauer, J. W., Khandji, A. G., Chin, S. S., and Elkind, M. S. (2004). Reversible leukoencephalopathy associated with cerebral amyloid angiopathy. Neurology 62, 494–497.
Orgogozo, J. M., Gilman, S., Dartigues, J. F., Laurent, B., Puel, M., Kirby, L. C., Jouanny, P., Dubois, B., Eisner, L., Flitman, S., Michel, B. F., Boada, M., Frank, A., and Hock, C. (2003). Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology 61, 46–54.
Peila, R., Yucesoy, B., White, L. R., Johnson, V., Kashon, M. L., Wu, K., Petrovitch, H., Luster, M., and Launer, L. J. (2007). A TGF-beta1 polymorphism association with dementia and neuropathologies: the HAAS. Neurobiol. Aging 28, 1367–1373.
Pettersen, J. A., Sathiyamoorthy, G., Gao, F. Q., Szilagyi, G., Nadkarni, N. K., St George-Hyslop, P., Rogaeva, E., and Black, S. E. (2008). Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch. Neurol. 65, 790–795.
Pfeifer, M., Boncristiano, S., Bondolfi, L., Stalder, A., Deller, T., Staufenbiel, M., Mathews, P. M., and Jucker, M. (2002). Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science 298, 1379.
Prada, C. M., Garcia-Alloza, M., Betensky, R. A., Zhang-Nunes, S. X., Greenberg, S. M., Bacskai, B. J., and Frosch, M. P. (2007). Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J. Neurosci. 27, 1973–1980.
Prelli, F., Castano, E., Glenner, G. G., and Frangione, B. (1988). Differences between vascular and plaque core amyloid in Alzheimer’s disease. J. Neurochem. 51, 648–651.
Premkumar, D. R., Cohen, D. L., Hedera, P., Friedland, R. P., and Kalaria, R. N. (1996). Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am. J. Pathol. 148, 2083–2095.
Racke, M. M., Boone, L. I., Hepburn, D. L., Parsadainian, M., Bryan, M. T., Ness, D. K., Piroozi, K. S., Jordan, W. H., Brown, D. D., Hoffman, W. P., Holtzman, D. M., Bales, K. R., Gitter, B. D., May, P. C., Paul, S. M., and DeMattos, R. B. (2005). Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J. Neurosci. 25, 629–636.
Robbins, E. M., Betensky, R. A., Domnitz, S. B., Purcell, S. M., Garcia-Alloza, M., Greenberg, C., Rebeck, G. W., Hyman, B. T., Greenberg, S. M., Frosch, M. P., and Bacskai, B. J. (2006). Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J. Neurosci. 26, 365–371.
Ronald, J. A., Chen, Y., Bernas, L., Kitzler, H. H., Rogers, K. A., Hegele, R. A., and Rutt, B. K. (2009). Clinical field-strength MRI of amyloid plaques induced by low-level cholesterol feeding in rabbits. Brain 132, 1346–1354.
Rosand, J., Hylek, E. M., O’Donnell, H. C., and Greenberg, S. M. (2000). Warfarin-associated hemorrhage and cerebral amyloid angiopathy: a genetic and pathologic study. Neurology 55, 947–951.
Salloway, S., Sperling, R., Gilman, S., Fox, N. C., Blennow, K., Raskind, M., Sabbagh, M., Honig, L. S., Doody, R., van Dyck, C. H., Mulnard, R., Barakos, J., Gregg, K. M., Liu, E., Lieberburg, I., Schenk, D., Black, R., and Grundman, M. (2009). A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070.
Schroeter, S., Khan, K., Barbour, R., Doan, M., Chen, M., Guido, T., Gill, D., Basi, G., Schenk, D., Seubert, P., and Games, D. (2008). Immunotherapy reduces vascular amyloid-β in PDAPP mice. J. Neurosci. 28, 6787–6793.
Scolding, N. J., Joseph, F., Kirby, P. A., Mazanti, I., Gray, F., Mikol, J., Ellison, D., Hilton, D. A., Williams, T. L., MacKenzie, J. M., Xuereb, J. H., and Love, S. (2005). Aβ-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128, 500–515.
Shinkai, Y., Yoshimura, M., Ito, Y., Odaka, A., Suzuki, N., Yanagisawa, K., and Ihara, Y. (1995). Amyloid β-proteins 1-40 and 1-42(43) in the soluble fraction of extra- and intracranial blood vessels. Ann. Neurol. 38, 421–428.
Smith, E. E., Gurol, M. E., Eng, J. A., Engel, C. R., Nguyen, T. N., Rosand, J., and Greenberg, S. M. (2004). White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology 63, 1606–1612.
Suter, O. C., Sunthorn, T., Kraftsik, R., Straubel, J., Darekar, P., Khalili, K., and Miklossy, J. (2002). Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 33, 1986–1992.
Suzuki, N., Iwatsubo, T., Odaka, A., Ishibashi, Y., Kitada, C., and Ihara, Y. (1994). High tissue content of soluble beta 1-40 is linked to cerebral amyloid angiopathy. Am. J. Pathol. 145, 452–460.
Thal, D. R., Ghebremedhin, E., Rub, U., Yamaguchi, H., Del Tredici, K., and Braak, H. (2002). Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293.
Thal, D. R., Papassotiropoulos, A., Saido, T. C., Griffin, W. S., Mrak, R. E., Kolsch, H., Tredici, K. D., Attems, J., and Ghebremedhin, E. (2010). Capillary cerebral amyloid angiopathy identifies a distinct APOE epsilon4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 120, 169–183.
Thoonsen, H., Richard, E., Bentham, P., Gray, R., van Geloven, N., De Haan, R. J., Van Gool, W. A., and Nederkoorn, P. J. (2010). Aspirin in Alzheimer’s disease: increased risk of intracerebral hemorrhage: cause for concern? Stroke 41, 2690–2692.
Trouillas, P., and von Kummer, R. (2006). Classification and pathogenesis of cerebral hemorrhages after thrombolysis in ischemic stroke. Stroke 37, 556–561.
van Rooden, S., Maat-Schieman, M. L., Nabuurs, R. J., van der Weerd, L., van Duijn, S., van Duinen, S. G., Natte, R., van Buchem, M. A., and van der Grond, J. (2009). Cerebral amyloidosis: postmortem detection with human 7.0-T MR imaging system. Radiology 253, 788–796.
Verbeek, M. M., Kremer, B. P., Rikkert, M. O., Van Domburg, P. H., Skehan, M. E., and Greenberg, S. M. (2009). Cerebrospinal fluid amyloid β(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 66, 245–249.
Vernooij, M. W., Haag, M. D., van der Lugt, A., Hofman, A., Krestin, G. P., Stricker, B. H., and Breteler, M. M. (2009a). Use of antithrombotic drugs and the presence of cerebral microbleeds: the Rotterdam Scan Study. Arch. Neurol. 66, 714–720.
Vernooij, M. W., Ikram, M. A., Hofman, A., Krestin, G. P., Breteler, M. M., and van der Lugt, A. (2009b). Superficial siderosis in the general population. Neurology 73, 202–205.
Viswanathan, A., Patel, P., Rahman, R., Nandigam, R. N., Kinnecom, C., Bracoud, L., Rosand, J., Chabriat, H., Greenberg, S. M., and Smith, E. E. (2008). Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 39, 1988–1992.
Wilcock, D. M., Jantzen, P. T., Li, Q., Morgan, D., and Gordon, M. N. (2007). Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience 144, 950–960.
Wilcock, D. M., Rojiani, A., Rosenthal, A., Subbarao, S., Freeman, M. J., Gordon, M. N., and Morgan, D. (2004). Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 1, 24.
Yamada, M. (2002). Risk factors for cerebral amyloid angiopathy in the elderly. Ann. N. Y. Acad. Sci. 977, 37–44.
Yamada, M., Itoh, Y., Suematsu, N., Otomo, E., and Matsushita, M. (1997a). Vascular variant of Alzheimer’s disease characterized by severe-plaque-like β protein angiopathy. Dement. Geriatr. Cogn. Disord. 8, 163–168.
Yamada, M., Sodeyama, N., Itoh, Y., Suematsu, N., Otomo, E., Matsushita, M., and Mizusawa, H. (1997b). Association of presenilin-1 polymorphism with cerebral amyloid angiopathy in the elderly. Stroke 28, 2219–2221.
Yamada, M., and Naiki, H. (2012). Cerebral amyloid angiopathy. Prog. Mol. Biol. Transl. Sci. 107, 41–78.
Yamada, M., Sodeyama, N., Itoh, Y., Suematsu, N., Otomo, E., Matsushita, M., and Mizusawa, H. (1998). Association of α1-antichymotrypsin polymorphism with cerebral amyloid angiopathy. Ann. Neurol. 44, 129–131.
Yamada, M., Sodeyama, N., Itoh, Y., Takahashi, A., Otomo, E., Matsushita, M., and Mizusawa, H. (2003). Association of neprilysin polymorphism with cerebral amyloid angiopathy. J. Neurol. Neurosurg. Psychiatr. 74, 749–751.
Yamada, M., Tsukagoshi, H., Otomo, E., and Hayakawa, M. (1987). Cerebral amyloid angiopathy in the aged. J. Neurol. 234, 371–376.
Yates, P. A., Sirisriro, R., Villemagne, V. L., Farquharson, S., Masters, C. L., Rowe, C. C., and Group, A. R. (2011). Cerebral microhemorrhage and brain β-amyloid in aging and Alzheimer disease. Neurology 77, 48–54.
Yoshita, M., Fletcher, E., Harvey, D., Ortega, M., Martinez, O., Mungas, D. M., Reed, B. R., and DeCarli, C. S. (2006). Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology 67, 2192–2198.
Keywords: cerebral amyloid angiopathy, Alzheimer’s disease, intracerebral hemorrhage, cerebrovascular disorders, dementia, amyloid β-protein, biomarker, risk factor
Citation: Yamada M (2012) Predicting cerebral amyloid angiopathy-related intracerebral hemorrhages and other cerebrovascular disorders in Alzheimer’s disease. Front. Neur. 3:64. doi: 10.3389/fneur.2012.00064
Received: 24 February 2012; Paper pending published: 14 March 2012;
Accepted: 05 April 2012; Published online: 25 April 2012.
Edited by:
Steven M. Greenberg, Harvard, USAReviewed by:
Hidetoshi Kasuya, Tokyo Women’s Medical University, JapanWengui Yu, UT Southwestern Medical Center at Dallas, USA
Copyright: © 2012 Yamada. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Masahito Yamada, Department of Neurology and Neurobiology of Aging, Kanazawa University Graduate School of Medical Science, Kanazawa 920-8640, Japan. e-mail: m-yamada@med.kanazawa-u.ac.jp