
- Department of Physiology, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
α2-Adrenoceptors lower central sympathetic output and peripheral catecholamine release, and thus may prevent sympathetic hyperactivity and hypertension. α2AR also influence vascular tension. These α2AR are malfunctioning in spontaneously hypertensive rats (SHR). Here I tested if an interaction between α2AR subtypes and the angiotensin AT1 receptor (AT1R) precipitated these disorders. Blood pressure was monitored through a femoral artery catheter and cardiac output by ascending aorta flow in anesthetized rats. Catecholamine concentrations were determined in plasma collected at the end of a 15-min tyramine-infusion. Tyramine stimulates norepinephrine release through the re-uptake transporter, thus preventing re-uptake. Presynaptic control of vesicular release is therefore reflected as differences in overflow to plasma. Previous experiments showed surgical stress to activate some secretion of epinephrine, also subjected to α2AR-auto-inhibition. Normotensive rats (WKY) and SHR were pre-treated with (1) vehicle or α2AR-antagonist (L-659,066), followed by fadolmidine (α2C>B>A + α1AR-agonist), ST-91 (α2non-A-selective agonist), or m-nitrobiphenyline (α2CAR-agonist + α2A+B-antagonist), or (2) AT1R-antagonist losartan, losartan + L-659,066, or losartan + clonidine. In WKY, L-659,066 alone, L-659,066 + agonist or losartan + L-659,066 increased catecholamine overflow to plasma after tyramine and eliminated the norepinephrine-induced rise in total peripheral vascular resistance (TPR). In SHR, L-659,066 + fadolmidine/ST-91/m-nitrobiphenyline and losartan + L-659,066 greatly increased, and losartan + clonidine reduced, catecholamine concentrations, and L-659,066 + ST-91, losartan + L-659,066 and losartan + clonidine eliminated the tyramine-induced rise in TPR. Separately, these drugs had no effect in SHR. In conclusion, peripheral α2CAR-stimulation or AT1R-inhibition restored failing α2AAR-mediated auto-inhibition of norepinephrine and epinephrine release and control of TPR in SHR.
Introduction
Sympathetic hyperactivity is a major force in initiating and sustaining spontaneous hypertension (Guyenet, 2006; Esler, 2011). α2-adrenoceptors (AR) lower sympathetic output from the central nervous system (CNS), and inhibit release of norepinephrine from peripheral sympathetic nerves and catecholamines from the adrenal medulla (Starke, 2001). Their activation is tonic, and they hamper release even in the anesthetized rat without stimulation of norepinephrine release (Berg et al., 2012). They therefore represent the last line of defense against sympathetic hyperactivity, and, if not functioning, plasma norepinephrine levels and blood pressure (BP) will increase, as demonstrated in genetically modified mice (Makaritsis et al., 1999). In the spontaneously hypertensive rat (SHR), deficiencies have been detected in both central and peripheral α2AR-mediated inhibition of release (Remie et al., 1992; Zugck et al., 2003). We have recently demonstrated that during tyramine-stimulated norepinephrine release, α2AR failed to lower norepinephrine and epinephrine release in SHR, and also failed to control vascular tension (Berg and Jensen, 2013). These malfunctions were not detected without activation of norepinephrine release (Berg et al., 2012), indicating that they resulted from the released catecholamine itself, or another agent released by, or co-released with norepinephrine or epinephrine. Surprisingly, these peripheral disorders were repaired by the non-selective agonist clonidine, which reduced catecholamine release, and also, through a central action, normalized the high resting BP, heart rate (HR), and total peripheral vascular resistance (TPR) in SHR (Berg et al., 2012).
The restoring effect of clonidine may result from its central action or from an interaction between presynaptic receptors. α2AR are divided into three subtypes, i.e., α2A, α2B, and α2C. The α2A-and α2C-subtypes mediated the inhibition of central sympathetic output, whereas all three subtypes may reduce norepinephrine release from peripheral sympathetic nerves (Hein et al., 1999; Trendelenburg et al., 2003b) and the adrenal medulla (Brede et al., 2003; Moura et al., 2006). Inhibition of adrenal epinephrine release involved the α2C-subtype in the mouse (Brede et al., 2003; Moura et al., 2006), but the α2A-subtype in rat and man (Lymperopoulos et al., 2007; Berg et al., 2012). It has been shown that on-going α2AR-signaling markedly enhanced the stimulating effect of the angiotensin AT1 receptor (AT1R) – phospholipase C – protein kinase C (PKC) pathway on norepinephrine release in the rat vas deferens (Talaia et al., 2006). Similarly, studies on tissues from genetically modified mice (Trendelenburg et al., 2003a) demonstrated that the enhancing effect of release-stimulating receptors, including the AT1R, depended on active α2AR-signaling. However, the interaction involved the α2CAR-subtype only (Figure 1). Since the renin angiotensin system plays a significant role in hypertension pathology in SHR, I hypothesized that the clonidine-dependent restoration of α2AR inhibition of release in SHR involved stimulation of the α2CAR, thus counter-acting an excessive AT1R-signaling.
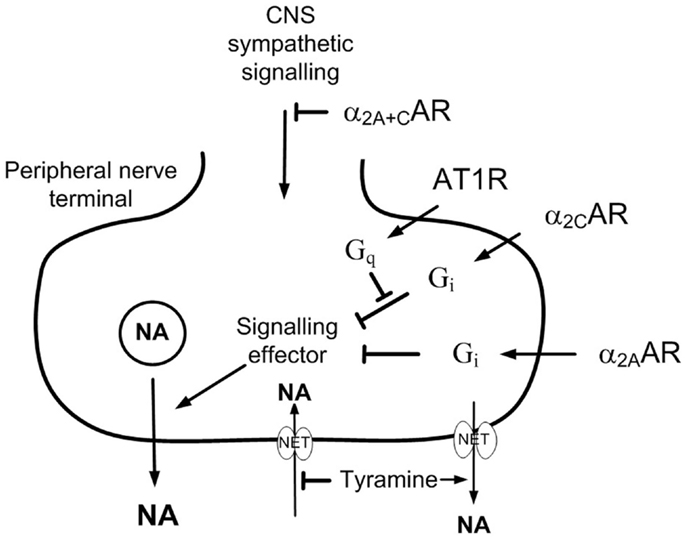
Figure 1. The effect of presynaptic α2CAR and AT1R on norepinephrine release. AT1R-Gq-signaling stimulates norepinephrine release by interfering with the down-stream signaling of Gi (Cox et al., 2000). The AT1R/α2AR interaction involved only the α2C- and not the α2A-subtype (Trendelenburg et al., 2003a). The present results show that α2CAR-stimulation or AT1R-inhibition was required for α2AAR to effectively moderate peripheral norepinephrine release in SHR during tyramine-stimulated norepinephrine release. This malfunction may be due to excessive AT1R-Gq-signaling in this strain, and α2A-signaling was evidently not permitted as long as AT1R-Gq-signaling interfered with the function of the α2CAR. Tyramine stimulates reverse transport of norepinephrine through NET, and therefore also prevents synaptic NET re-uptake, allowing presynaptic control of release to be reflected as differences in overflow and the plasma norepinephrine concentration.
The angiotensin II responsible for a possible AT1R interference in SHR is not likely to origin from the sympathetic nerves themselves. Therefore, to have all components present, a role of the AT1R in the α2AR malfunction in SHR should be tested in vivo, which represents an experimental challenge. Due to synaptic uptake of norepinephrine through the norepinephrine re-uptake transporter (NET), presynaptic modulation of release is not reflected as differences in overflow and the plasma norepinephrine concentration (Berg et al., 2012). However, when NET-mediated re-uptake was blocked by desipramine, α2AR-antagonists greatly increased the plasma concentration of norepinephrine in the resting, anesthetized rat, in which norepinephrine release was not stimulated. Overflow to plasma under resting conditions is low, and inhibition of release by α2AR-agonist had no or little effect on the plasma norepinephrine concentration (Berg et al., 2012). In addition, the α2AR malfunction in SHR was not observed unless norepinephrine release was activated. Peripheral norepinephrine release can be stimulated by tyramine, which activates reverse transport through NET. Most likely by engaging NET in release, thus preventing re-uptake, presynaptic α2AR modulation altered tyramine-induced norepinephrine overflow to plasma, similar to that after desipramine in not-stimulated rats (Berg and Jensen, 2013). Restored α2AR control of release after α2CAR-stimulation or AT1R-antagonist could therefore be tested by the ability of the non-selective α2AR-antagonist L-659,066 to increase tyramine-induced norepinephrine overflow to plasma.
Epinephrine released in the adrenals is not subjected to re-uptake, and is not stimulated by tyramine. However, the stress induced by the surgical procedure activated some secretion of epinephrine, which was also subjected to α2AR-mediated release-control (Berg et al., 2012; Berg and Jensen, 2013).
Due to the activation of norepinephrine release, tyramine in addition induced a sympathetic cardiovascular response. This response was not influenced by baroreceptor activation, demonstrated by that baroreceptor control of HR was abolished by the pentobarbital-anesthesia (Berg et al., 2012). Moreover, epinephrine secretion is not regulated by the baroreceptor reflex. Thus, by recording BP and cardiac output (CO), the implications of altered catecholamine release and a possible postsynaptic α2AR/AT1R interaction in the control of TPR could be evaluated.
The results will show that the failing α2AR control of norepinephrine and epinephrine release and modulation of the norepinephrine-induced rise in TPR in SHR was restored by stimulation of peripheral α2CAR or inhibition of the AT1R.
Materials and Methods
Experimental Procedure
All experiments were approved by the institutional review committee, and conducted in accordance with the Directive 2010/63/EU of the European Parliament. About 12–14 weeks old, male normotensive rats (Wistar Kyoto, WKY, n = 99, 284 ± 3 g b.w.) and SHR (Okamoto, SHR/NHsd strain, n = 107, 288 ± 2 g b.w.) on 12/12 h light/dark cycles were allowed conventional rat chow diet (0.7% NaCl) and water ad lib until the time of the experiment. The rats were anesthetized with pentobarbital (70–75 mg/kg, i.p.). As previously described (Berg et al., 2010; Berg and Jensen, 2013), mean arterial BP [MBP = (systolic BP − diastolic BP)/3 + diastolic BP] was monitored through a catheter in the femoral artery, flushed with 0.15 ml PBS (0.01 M Na-phosphate, 0.14 M NaCl, pH 7.4) containing 500 IU heparin/ml. CO and HR were recorded by a flow probe on the ascending aorta. TPR (MBP/CO) was calculated. The rats were on a positive-pressure ventilator throughout the experiment, ventilated with air. Previous measurements of blood gas parameters demonstrated adequate ventilation in both strains (Berg, 2002, 2003). Positive-pressure ventilation reduces right atrium ejection, and consequently lowered CO and MBP. This reduction was significant in SHR, but did not appear to influence the stimulated adrenergic responses, as previously discussed (Berg and Jensen, 2013). Body temperature was maintained at 37–38 °C by external heating, guided by a thermo sensor, inserted inguinally into the abdominal cavity.
Experimental Design
Control rats were pre-treated with PBS and infused for 15 min with tyramine to induce NET-mediated norepinephrine release. Since subtype-selective α2AR-agonists, which do not cross the blood-brain barrier, are not available, I used α2AR-agonists with different subtype profiles and different ability to cross the blood-brain barrier. Rats were therefore pre-treated with PBS or the α2AR-antagonist L-659,066, followed 10 min later by α2AR-agonist, i.e., fadolmidine, ST-91, or (R)-(+)-m-nitrobiphenyline oxalate. Rats were also pre-treated with the AT1R-antagonist losartan, alone or followed by L-659,066, clonidine, or ST-91. Drug specificity and dose are given in Table 1. Blood for the measurement of catecholamines was collected from the arterial catheter after the 15-min tyramine-observation period, but without discontinuing the infusion.
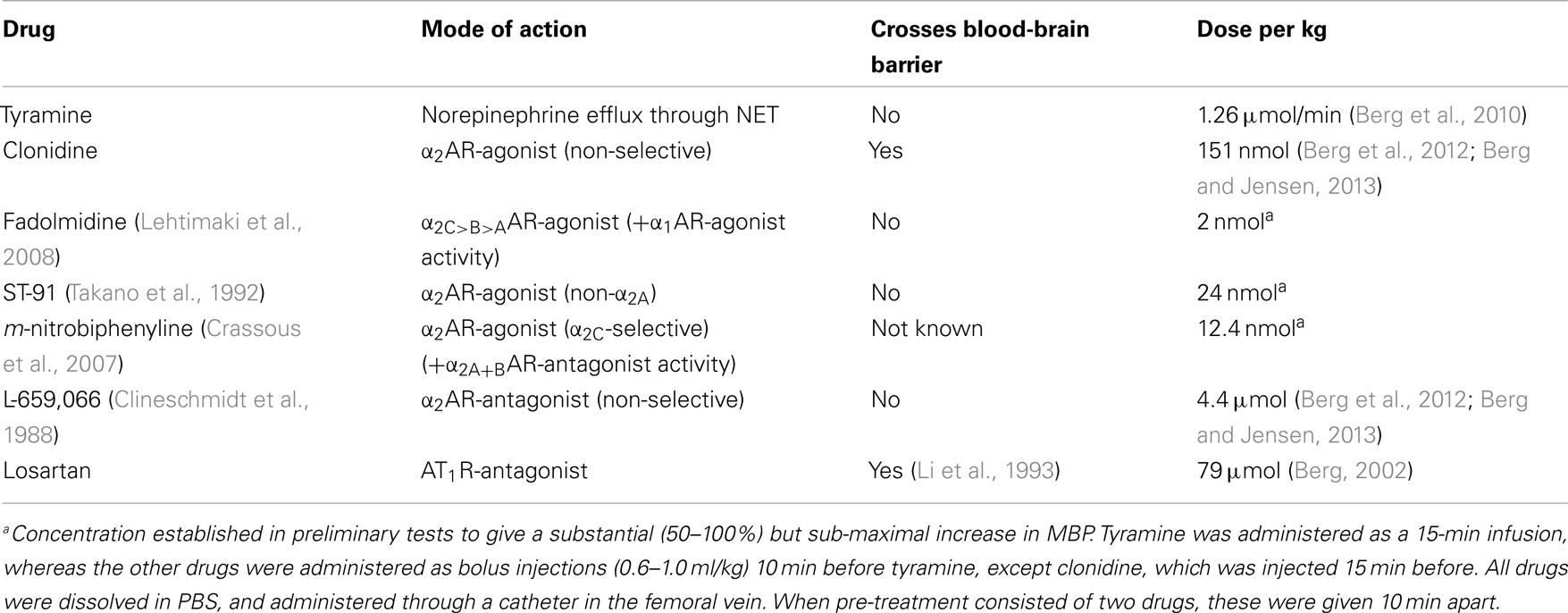
Table 1. Mode of action and dose of the pharmacological agents used.
Measurement of Plasma Catecholamines
About 1.5 ml blood was collected into tubes containing 40 μl 0.2 M glutathione and 0.2 M EGTA (4 °C). Plasma was stored at −80 °C until the norepinephrine and epinephrine concentrations were determined, using 400 μl plasma and the 5000 Reagent kit for HPLC analysis of Catecholamines in plasma from Chromsystems GmbH, Munich, Germany, as described by the manufacturer.
Drugs
Pentobarbital was from the Norwegian National Hospital, Oslo, Norway. L-659,066 was a kind gift from Merck, Sharp, and Dohme Labs, Rahway, NJ, USA, and fadolmidine HCl from Orion Corporation, Espoo, Finland. ST-91 was from TOCRIS bioscience, Bristol, UK; and (R)-(+)-m-nitrobiphenyline oxalate from Santa Cruz Biotechnology, Heidelberg, Germany. The remaining drugs were from Sigma Chemical Co., St. Louis, MO, USA.
Statistical Analyses
Results are presented as mean values ± SEM. Changes in the cardiovascular parameters were expressed in % of baseline. Data were averaged every min in all experiments. For the narrow peak-pressor response to ST-91 and m-nitrobiphenyline, data were averaged every 5 s. The cardiovascular response-curves to agonists and tyramine were analyzed using Repeated Measures Analyses of Variance and Covariance, first as over-all tests within each strain, and subsequently for each group separately or between groups. Significant responses and groups differences were subsequently located using one- and two-sample Student’s t-tests, respectively, at specific times. The plasma catecholamine concentrations, the cardiovascular baselines, and the effect of pre-treatment were first analyzed using one-way ANOVA, and group differences were subsequently located by two-sample Student’s t-tests or, in the presence of out-liers, non-parametric Kruskal–Wallis tests. For all analyses, testing proceeded only when significant responses, differences and/or interactions were indicated. The P-value was for all tests and each step adjusted according to Bonferroni, except for the catecholamine data, where P ≤ 0.05 was considered significant.
Results
α2AR- and AT1R-Influence on the Plasma Catecholamine Concentrations
Norepinephrine
Similar to that previously described (Berg and Jensen, 2013), the non-selective α2AR-antagonist L-659,066 increased the tyramine-induced norepinephrine overflow to plasma in WKY (P = 0.015) (Table 2). A similar increase was not seen in SHR, where the plasma norepinephrine concentration was already elevated (P < 0.001, WKY compared to SHR controls). Pre-treatment with α2AR-agonist alone, i.e., fadolmidine (α2C>B>A), ST-91 (α2(non-A)), or m-nitrobiphenyline (α2C) had no effect on overflow in either strain, except for an increase after ST-91 in WKY. After L-659,066 + agonist + tyramine, norepinephrine overflow was not different from that after L-659,066 + tyramine in WKY (P = NS), but was much higher in SHR (P ≤ 0.025–0.004), also when compared to the SHR PBS + tyramine or corresponding PBS + agonist + tyramine groups (P ≤ 0.004).
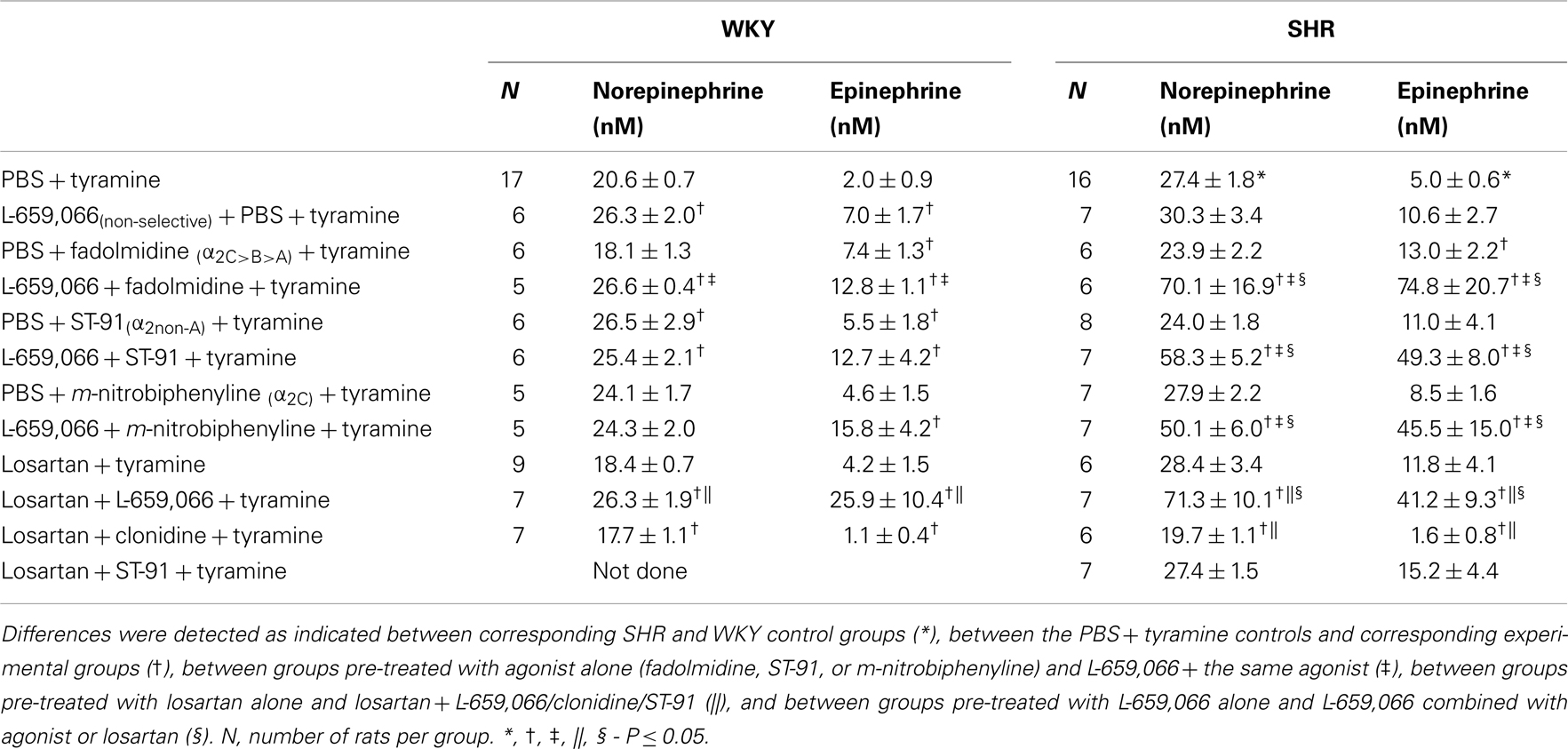
Table 2. The plasma concentration of norepinephrine and epinephrine at the end of the tyramine-infusion period.
Losartan alone had no effect on the tyramine-induced norepinephrine overflow in either strain (P = NS compared to the controls). Losartan also did not influence the augmenting effect of L-659,066 in WKY (P = NS compared to the L-659,066 + tyramine group, and P = 0.001 compared to the WKY PBS + tyramine and losartan + tyramine groups). However, in SHR, losartan allowed L-659,066 to greatly increase norepinephrine overflow (P ≤ 0.005 compared to PBS/L-659,066/losartan + tyramine groups). Pre-treatment with losartan + clonidine reduced the tyramine-induced norepinephrine overflow in SHR (P ≤ 0.048 compared to the PBS/losartan + tyramine groups), and was lower than that in the controls, although not different from that in the losartan + tyramine group, in WKY. Norepinephrine overflow after pre-treatment with losartan + ST-91 was not different from that in the PBS + tyramine or losartan + tyramine groups (tested in SHR only).
Epinephrine
The effect of α2AR-agonists and antagonist on the surgery-activated epinephrine secretion mostly paralleled their effect on the tyramine-induced norepinephrine overflow in both strains. However, pre-treatment with fadolmidine in both strains, and L-659,066 + m-nitrobiphenyline in WKY, increased circulating epinephrine without altering the concentration of norepinephrine.
The Cardiovascular Responses
The α2AR- and AT1R-influence on the cardiovascular baselines
L-659,066 reduced baseline MBP and TPR in both strains (Table 3). All α2AR-agonists induced a transient rise in MBP and TPR (Figure 2, the response to clonidine was similar to that previously published, Berg et al., 2012). Pre-treatment with L-659,066 reduced these TPR-responses, except that of fadolmidine in SHR (Figure 2A), although the MBP-responses were not necessarily reduced. Only fadolmidine subsequently induced an L-659,066-sensitive reduction in MBP and TPR to below baseline in both strains, and also HR in SHR. The agonists had otherwise little effect on baseline HR. Losartan reduced baseline MBP in both strains, HR in WKY, and TPR in SHR (Table 3). Losartan + L-659,066 induced a significant reduction in both HR and TPR in both strains. Losartan increased the MBP-response to ST-91 (Figure 2B) and also the transient rise in CO and MBP in response to clonidine in SHR but had no effect on the HR- or TPR-response to clonidine in either strain (not shown).
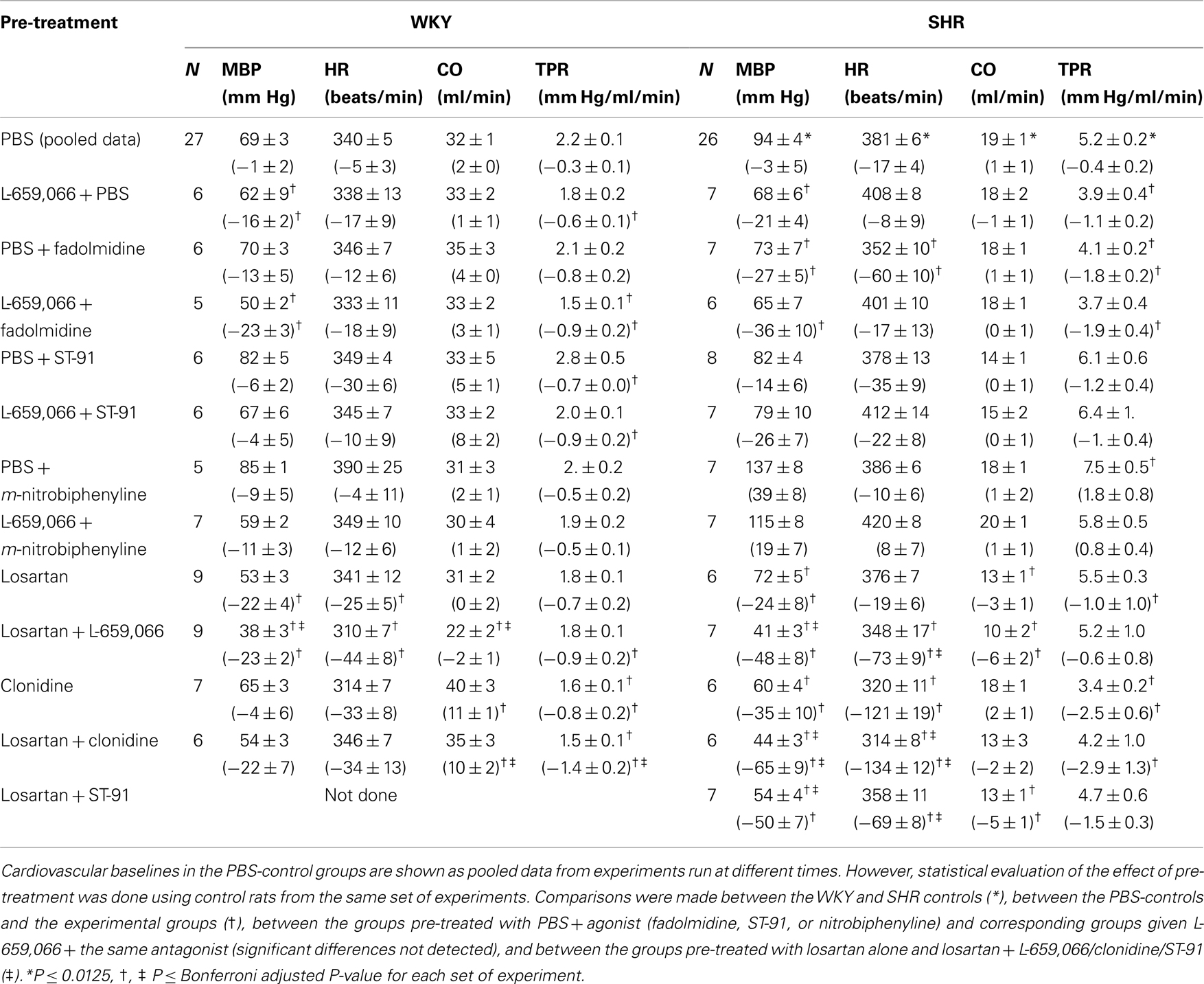
Table 3. Cardiovascular baselines prior to tyramine and, in parenthesis, the response to pre-treatment.
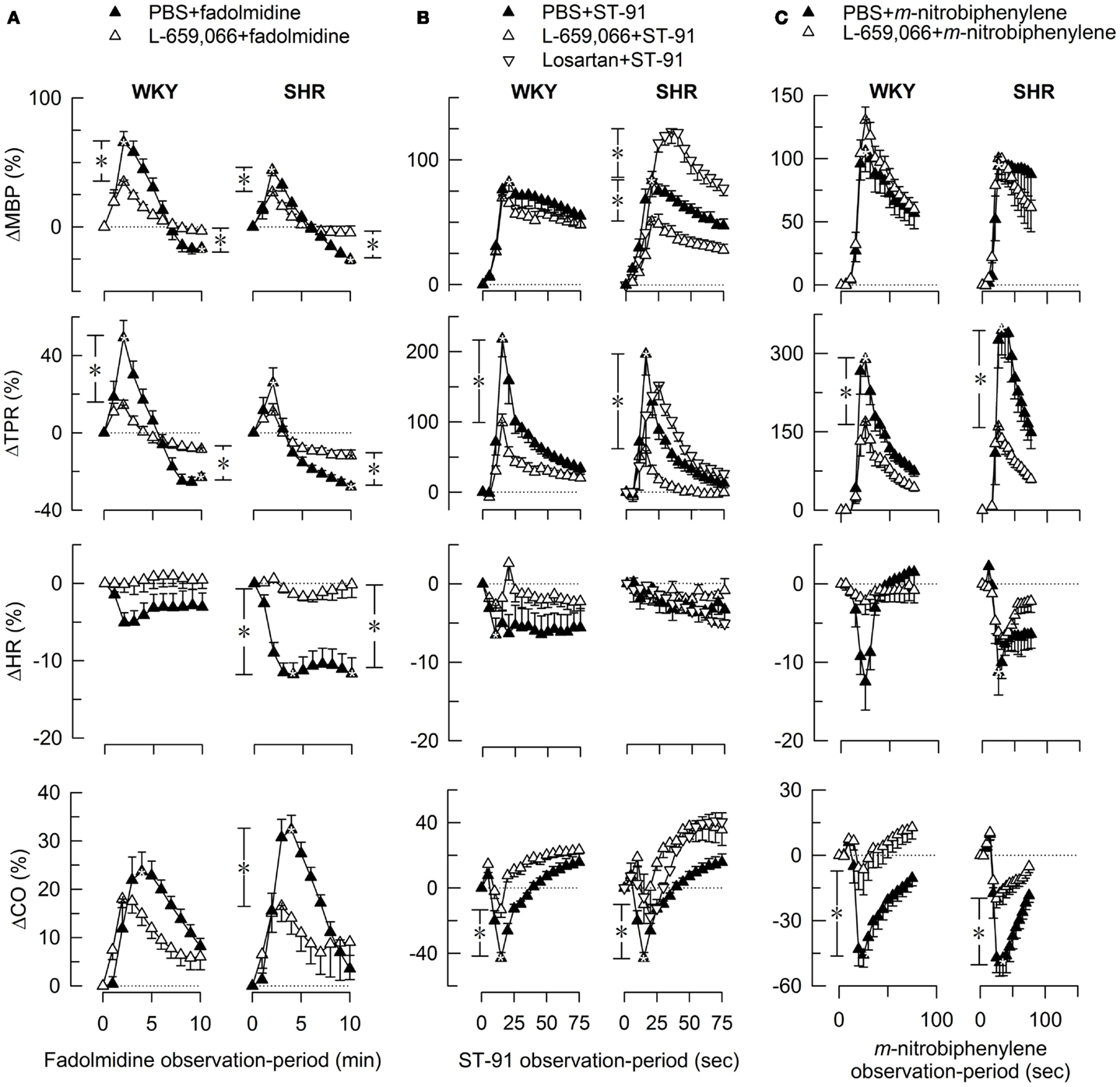
Figure 2. The MBP-, TPR-, HR-, and CO-response to α2AR-agonists. Fadolmidine (α2C>B>AAR) (A), ST-91 [α2(non-A)AR] (B), and m-nitrobiphenyline (α2CAR, with additional α2A+BAR-antagonistic activity) (C) were injected alone or after pre-treatment with the non-selective α2AR-antagonist L-659,066. The response-curves were analyzed using Repeated Measures Analyses of Variance and Covariance (please see Materials and Methods for details). Significant responses (*within symbols) and group differences (*in brackets) were located as indicated at peak response (all agonists) (brackets left of curves) and after 15 min (fadolmidine only) (brackets right of curves). *, *P ≤ 0.025 for (A), and ≤0.05 for (B,C) after curve evaluations.
The α2AR- and AT1R-influence on the cardiovascular response to tyramine
As previously documented (Berg et al., 2010; Berg and Jensen, 2013), tyramine induced an immediate, but transient rise in TPR (Figure 3) and a sustained increase in MBP, HR, and CO. The present results focused on the effect of pre-treatment on the TPR-response to tyramine, and the concomitant changes in MBP, HR, and CO (all expressed in % of baselines) are therefore shortly described but not shown.
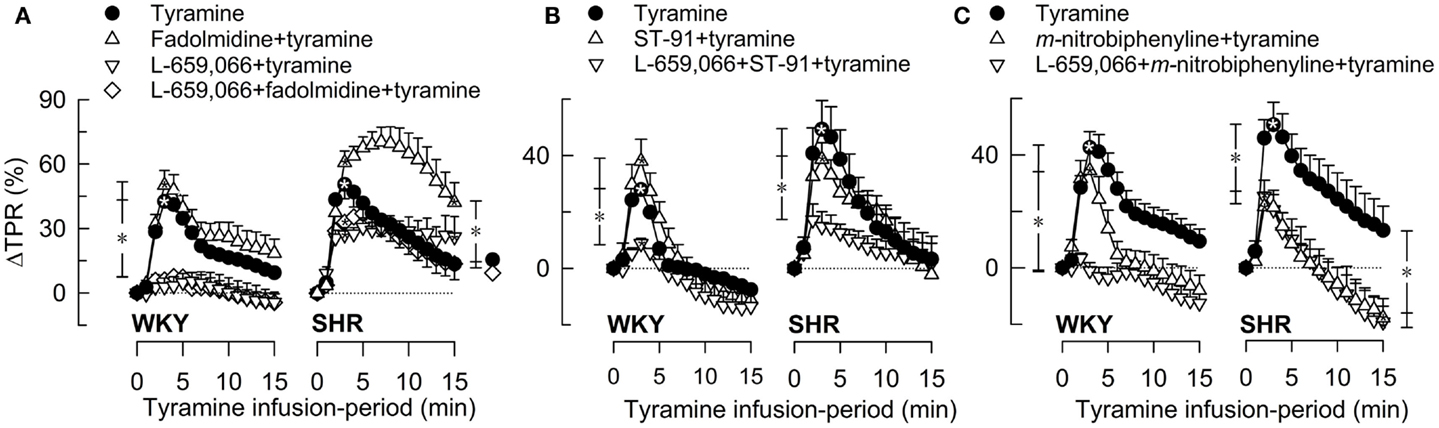
Figure 3. The TPR-response to tyramine-induced norepinephrine release after pre-treatment with α2(non-A)AR-selective agonist, alone or combined with L-659,066. The peripherally restricted α2C>B>AAR-agonist fadolmidine (A), the peripherally restricted α2(non-A)AR-selective agonist ST-91 (B), and the α2C-selective agonist m-nitrobiphenyline with additional α2A+BAR-antagonistic activity (C) were injected alone or after pre-treatment with the peripherally restricted α2AR-antagonist L-659,066. Baselines prior to tyramine are shown in Table 3. Significant responses (*within symbol) and differences between the control and experimental groups were located at peak response (*brackets left of curves) and at 15 min (*brackets right of curves). *, *P ≤ 0.025 after curve evaluations.
Pre-treatment with α2AR-agonist alone (Figures 3A–C), i.e., fadolmidine, ST-91, or m-nitrobiphenyline, had no effect on the TPR-response to tyramine in WKY (P = NS). In SHR, the TPR-response to tyramine was increased after fadolmidine (P = 0.023 at 15 min), not influenced by ST-91, and decreased after m-nitrobiphenyline (P = 0.003 at 3 min). L-659,066 alone (Figure 3A) virtually eliminated the TPR-response in WKY (P ≤ 0.008), with no additional effect when combined with agonist (Figures 3A–C). In SHR, L-659,066 alone did not change the tyramine-induced rise in TPR, but abolished the response when combined with ST-91 (Figures 3A,B). The response to tyramine in L-659,066 + fadolmidine-pre-treated SHR was less than that after fadolmidine alone, although not different from that in the controls (Figure 3A). Moreover, ΔTPR was not further reduced after L-659,066 + m-nitrobiphenyline compared to that after m-nitrobiphenyline alone in SHR (Figure 3C).
A reduced MBP-response to tyramine after L-659,066, alone or combined with agonist (fadolmidine, ST-91, or m-nitrobiphenyline), was observed in WKY, but only after L-659,066 + agonist in SHR. m-Nitrobiphenyline alone reduced ΔMBP in both strains. The agonists had little effect on the tyramine-induced tachycardia, except fadolmidine which increased ΔHR in SHR. A lower tyramine-induced rise in CO was observed after fadolmidine and ST-91 in WKY, after fadolmidine in SHR, and in all groups given L-659,066 as part of the pre-treatment.
Losartan alone had no effect on the TPR-peak response to tyramine in either strain, but induced a vasodilatory TPR-response at the end of the tyramine-infusion in WKY (Figure 4). Like L-659,066 alone (Figure 3A), losartan + L-659,066 eliminated the TPR-peak response to tyramine in WKY (Figure 4), and in addition caused a fall in TPR to below baseline. Losartan + clonidine, like clonidine alone, had no effect on the TPR-response to tyramine in WKY (Figure 4). In SHR, losartan + L-659,066 and losartan + clonidine, unlike losartan, L-659,066 or clonidine alone, eliminated the TPR-response to tyramine. The TPR-peak response was reduced also after pre-treatment with losartan + ST-91 (tested in SHR only, Figure 4). Losartan did not alter the MBP-response to tyramine, but increased the CO-response in both strains. This increase was eliminated when losartan was combined with L-659,066, and in WKY also with clonidine. The tyramine-induced tachycardia was increased in SHR after losartan + clonidine, similar to that seen after clonidine alone.
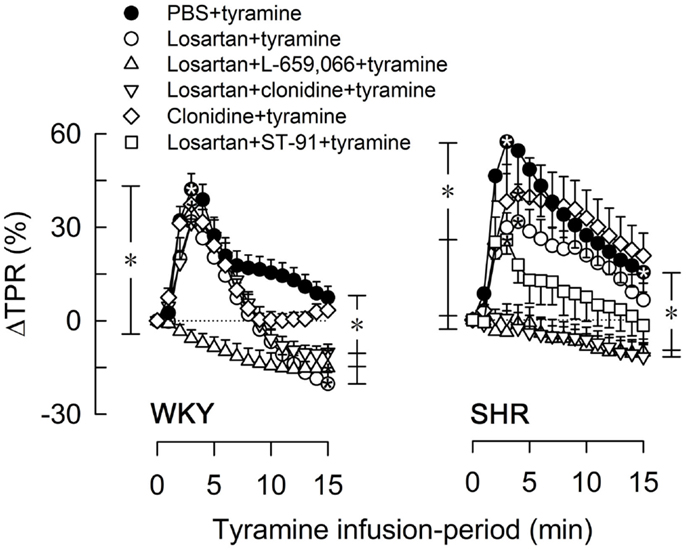
Figure 4. The TPR-response to tyramine after pre-treatment with the AT1R-antagonist losartan, alone or combined with L-659,066, clonidine, or ST-91. The effect of losartan + ST-91 was tested in SHR only. Baselines prior to tyramine are shown in Table 3. Significant responses (*within symbol) and group differences were detected at peak response (*brackets left of curves) and at 15 min (*brackets right of curves) as indicated. *, *P ≤ 0.025 after curve evaluations.
Discussion
The main finding in the present study was that the failing α2AAR inhibition of peripheral norepinephrine and epinephrine release in SHR during tyramine-stimulated norepinephrine release was restored by stimulation of the α2CAR or inhibition of the AT1R. α2CAR-stimulation and AT1R-inhibition also restored the failing postsynaptic α2AR control of vascular tension in SHR.
As previously described (Berg and Jensen, 2013), α2AR-mediated auto-inhibition of peripheral catecholamine release was demonstrated in tyramine-stimulated WKY by an increased norepinephrine overflow to plasma after pre-treatment with the non-selective α2AR-antagonist L-659,066. This increase was eliminated after addition of the non-selective α2AR-agonist clonidine (Berg and Jensen, 2013), but not, as demonstrated by the present experiment, by agonists with less or no α2AAR reactivity, such as fadolmidine, ST-91, or m-nitrobiphenyline. Clonidine reduced the tyramine-induced norepinephrine overflow in SHR, and this reduction was fully reversed by L-659,066 (Berg and Jensen, 2013), and, again, a similar decrease was not seen after fadolmidine, ST-91, or m-nitrobiphenyline. Both tyramine and L-659,066 are peripherally restricted, i.e., do not pass the blood-brain barrier (Oldendorf, 1971; Clineschmidt et al., 1988). Inhibition of tyramine-stimulated norepinephrine overflow therefore involved in both strains peripherally located α2AR, predominantly of the α2A-subtype, in agreement with that previously observed by others (Starke, 2001; Brede et al., 2004).
Epinephrine is secreted directly into blood and not subjected to local re-uptake, and release is therefore not stimulated by tyramine (Berg and Jensen, 2013). However, the stress induced by the surgical procedure activated some secretion of epinephrine from the adrenals (Berg et al., 2012). Clonidine precipitated an L-659,066-sensitive reduction in this secretion in both strains (Berg et al., 2012; Berg and Jensen, 2013), whereas fadolmidine, ST-91, or m-nitrobiphenyline did not. It therefore appeared that the α2AAR inhibited also the secretion of epinephrine, in agreement with previous studies on the rat adrenal gland (Lymperopoulos et al., 2007). This differed from that in the mouse, where the α2C-subtype inhibited epinephrine secretion (Brede et al., 2003; Moura et al., 2006).
Although clonidine reduced tyramine-induced norepinephrine overflow to plasma in SHR, the antagonist L-659,066 failed to increase overflow in this strain (Berg and Jensen, 2013). This malfunction depended on the tyramine-stimulated release of norepinephrine, since L-659,066, and also the α2AR-antagonist yohimbine, clearly increased norepinephrine overflow in SHR not stimulated with tyramine but where NET-re-up-take was blocked by desipramine (Berg et al., 2012). However, norepinephrine overflow was greatly increased in tyramine-stimulated SHR when L-659,066 was combined with the α2CAR-reactive agonist fadolmidine, which has a 35 and 10 times higher affinity for the α2C- and α2BAR than the rat α2A-subtype, respectively (Lehtimaki et al., 2008). Overflow was also greatly increased when L-659,066 was combined with the non-A-selective ST-91 (Takano et al., 1992), or the α2CAR-selective m-nitrobiphenyline, which in addition has an α2A+BAR-antagonistic effect (Crassous et al., 2007). Since fadolmidine and ST-91 do not cross the blood-brain barrier (Clineschmidt et al., 1988; Lehtimaki et al., 2008), stimulation of peripheral α2CAR appeared to re-establish α2A-auto-inhibition in SHR (Figure 1).
Augmented tyramine-induced norepinephrine overflow was also observed in SHR but not in WKY after pre-treatment with losartan + L-659,066, whereas losartan alone had no effect. G-protein Gq-signaling agents, including angiotensin II through the AT1R, have been shown in isolated mouse atria to stimulate norepinephrine release by interfering with down-stream signaling of the inhibitory α2AR-Gi pathway (Figure 1) (Cox et al., 2000; Trendelenburg et al., 2003a). The AT1R interaction involved only the α2C- and not the α2A-subtype (Cox et al., 2000; Trendelenburg et al., 2003a). α2CAR-agonist may therefore restore α2A-auto-inhibition by counter-acting the AT1R-Gq-interference, and losartan by eliminating the AT1R-interference. Thus, as could be expected, ST-91 did not alter the tyramine-induced norepinephrine overflow after losartan in SHR. The present results were therefore compatible with studies showing that the reduced afferent renal nerve signaling observed in response to efferent renal sympathetic nerve activation was increased in SHR by the α2AR-antagonist rauwolscine, and further potentiated when rauwolscine was combined with losartan, whereas losartan alone had no effect (Kopp et al., 2011).
However, the experimental approach is indirect and performed in the whole animal, and other explanations should therefore also be considered. For instance, α2CAR-stimulation will hamper renal renin release (Michel and Rump, 1996), and, through that, may lower AT1R-activation and stimulation of release. However, if this was the mechanism responsible, one might have expected losartan alone to lower the release of norepinephrine, which it did not. Unlike vesicular release, NET-mediated release has been considered not to be regulated by presynaptic receptors (Starke, 2001). However, recent studies show that NET may indeed be influenced by presynaptic control, as demonstrated by the hampering effect of muscarinic receptor activation on the NET transport rate (Parker et al., 2010), a response which in other cells is mediated through a PKC-dependent pathway (Apparsundaram et al., 1998). However, PKC did not seem to influence tyramine-induced transport through NET, since preliminary studies showed that the PKC-inhibitor staurosporine, like losartan alone, did alter norepinephrine overflow (plasma norepinephrine concentration = 19.8 ± 2.3 and 27.1 ± 2.3 nM in WKY and SHR, respectively, five rats/group, P = NS compared to the controls, Berg, unpublished observations). α2AR-agonists have also been shown to bind to NET and to competitively inhibit re-uptake of a norepinephrine analog (Park et al., 2013). This response was not prevented by α2AR-antagonist, and was therefore likely to result from their structural similarity to norepinephrine and not from α2AR-signaling. Agonist inhibition of NET did not seem to alter the tyramine-induced reversed transport of norepinephrine through NET, since none of the present agonists lowered tyramine-induced overflow, and the reduction observed in SHR after clonidine was abolished by L-659,066 (Berg and Jensen, 2013).
The secretion of epinephrine mostly followed the same pattern as that of norepinephrine overflow, indicating that α2AAR failed to inhibit also epinephrine secretion in SHR, and that this malfunction could be restored by α2CAR-stimulation or AT1R-inhibition.
The tyramine-stimulated norepinephrine overflow after L-659,066 + agonist and losartan + L-659,066 was about two times greater, and that of epinephrine 10 times greater, than that in the control or L-659,066-only groups in SHR, but not higher than that after pre-treatment with L-659,066 alone in WKY, i.e., 28% higher than in the controls. L-659,066 and yohimbine greatly increased the plasma concentration of norepinephrine and epinephrine also in desipramine-treated, non-stimulated SHR (Berg et al., 2012). These observations suggested an up-regulation of peripheral, presynaptic α2AAR in SHR, in order to down-regulate the elevated sympathetic tone and/or to compensate for the failing α2AR-auto-inhibition in this strain.
L-659,066 reduced baseline MBP and TPR in both strains, but abolished the tyramine-induced rise in TPR in WKY only. Also the Gi-inhibitor pertussis toxin eliminated the TPR-response to tyramine in this strain alone (Berg et al., 2009). The abolished TPR-response was most likely due to that L-659,066 inhibited postsynaptic, VSMC α2AR-Gi-signaling, thereby allowing VSMC βAR-adenylyl cyclase-mediated dilatation to oppose the norepinephrine-induced, α1AR-mediated vasoconstriction. Also this α2AR-function failed in SHR. The malfunction appeared to be precipitated by the stimulated release of norepinephrine, since a strain-related difference was not seen in the moderating effect of L-659,066 on the TPR-response to exogenous α1AR-agonist (Berg et al., 2012). Like the failing control of catecholamine release, also this disorder was repaired by AT1R-inhibition or α2CAR-stimulation, since losartan + L-659,066 and L-659,066 + ST-91 eliminated the TPR-response to tyramine. This may be due to the high norepinephrine and/or epinephrine release in these SHR groups, which, in the presence of the α2AR-antagonist inhibiting VSMC α2AR, may be sufficient to re-establish a βAR-mediated counter-action of the norepinephrine-induced α1AR-mediated vasoconstriction. This conclusion is in agreement with our previous study showing that neuronally activated, β1AR-mediated vasodilatation counter-acted the TPR-response to tyramine in WKY only, whereas β2+3AR activated by epinephrine from the adrenals opposed the late half of the TPR-response in SHR (Berg et al., 2010). The TPR-response to tyramine in SHR was also eliminated after losartan + clonidine and reduced after losartan + ST-91, in spite of a normal plasma norepinephrine concentration. It is therefore possible that also the failing β1AR contribution to TPR-control in SHR resulted from VSMC AT1R-activation.
In agreement with studies on genetically modified mice, where the initial clonidine-induced vasoconstriction was due to activation of VSMC α2BAR (Link et al., 1996), the present agonists, and as previously described also clonidine (Berg et al., 2012), induced a transient rise in TPR, which was reduced or eliminated by L-659,066, except that of fadolmidine in SHR. The L-659,066-sensitive fraction of this vasoconstriction may be mediated through the α2BAR on VSMC, although the present experiments could not exclude a role of the α2AAR. However, the L-659,066-sensitive fraction of the response to m-nitrobiphenyline was likely to be mediated through VSMC α2CAR, since this α2C-selective agonist also acted as an α2A+BAR-antagonist (Crassous et al., 2007). Although VSMC α2CAR did not contribute to BP control in genetically modified mice (MacDonald et al., 1997), stimulated α2CAR-mediated vasoconstriction has been demonstrated in veins and arterioles (Chotani et al., 2004; Görnemann et al., 2007). The L-659,066-insensitive part of the agonist-induced vasoconstriction was likely to be mediated through α1AR, since at least fadolmidine contained α1AR-agonistic activity (Lehtimaki et al., 2008). The latter component may also explain why fadolmidine increased the TPR-response to tyramine in SHR. This increase was absent after additional pre-treatment with L-659,066, possibly due to that L-659,066, by inhibiting the VSMC α2AR-Gi pathway, allowed norepinephrine-stimulated, βAR-mediated vasodilatation, in that manner opposing the tyramine-induced, α1AR-mediated vasoconstriction. Fadolmidine was the only agonist which induced a late L-659,066-sensitive fall in MBP, TPR, and HR in SHR, possibly due to its α2AAR-component, which may lower catecholamine release prior to tyramine-stimulation and/or stimulate endothelial, vasodilatory α2AAR (Shafaroudi et al., 2005). The TPR-response to tyramine was reduced by m-nitrobiphenyline. This reduction was not further influenced by additional pre-treatment with L-659,066, and was therefore likely to result from the α2A+BAR-antagonistic effect of this agonist. The TPR-response was therefore more sensitive to the promiscuity of the α2AR-agonists than the α2AR-mediated control of catecholamine release.
Conclusion
Peripheral α2AR represent the last line of defense against adrenergic hyperactivity. The α2A-subtype played a dominating role in limiting peripheral catecholamine release in WKY, but failed to do so in SHR. This malfunction was restored after α2CAR-stimulation or AT1R-inhibition, suggesting that an AT1R-Gq/α2CAR-Gi-interaction disturbed normal α2AAR-mediated control of catecholamine release in SHR. This α2CAR-AT1R-interaction may be responsible for the elevated plasma norepinephrine concentrations observed in SHR, and contribute to the sympathetic hyperactivity and hypertension in this strain. A loss-of-function α2CAR deletion polymorphism has been shown to be more frequent in African–Americans and connected to a greater HR- and BP-response in the cold-pressor-test (Kurnik et al., 2008). An augmented sympathetic response to this stress-test is linked to increased cardiovascular morbidity (Matthews et al., 2004), and heart failure patients with the same α2CAR polymorphism had a worsened prognosis and increased risk of heart failure (Small et al., 2002, 2003). Estrogen stimulated the expression of α2CAR in human dermal arteriole VSMC (Eid et al., 2007), and may from the present results provide a mechanism whereby estrogen protects against hypertension. A failing α2AAR auto-inhibition of catecholamine release due to an AT1R-α2CAR interaction may therefore be highly relevant for development of hypertension, the major risk factor for cardiovascular events.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by The Norwegian Council on Cardiovascular Diseases and Anders Jahre’s Fond.
References
Apparsundaram, S., Galli, A., Defelice, L. J., Hartzell, H. C., and Blakely, R. D. (1998). Acute regulation of norepinephrine transport: I. protein kinase C-linked muscarinic receptors influence transport capacity and transporter density in SK-N-SH cells. J. Pharmacol. Exp. Ther. 287, 733–743.
Berg, T. (2002). Analysis of the pressor response to the K+ channel inhibitor 4-aminopyridine. Eur. J. Pharmacol. 452, 325–337. doi:10.1016/S0014-2999(02)02306-3
Berg, T. (2003). The vascular response to the K+ channel inhibitor 4-aminopyridine in hypertensive rats. Eur. J. Pharmacol. 466, 301–310. doi:10.1016/S0014-2999(03)01555-3
Berg, T., Degerman, E., and Tasken, K. (2009). Increased cAMP signaling can ameliorate the hypertensive condition in spontaneously hypertensive rats. J. Vasc. Res. 46, 25–35. doi:10.1159/000135662
Berg, T., and Jensen, J. (2013). Tyramine reveals failing alpha2-adrenoceptor control of catecholamine release and total peripheral vascular resistance in hypertensive rats. Front. Neurol. 4:19. doi:10.3389/fneur.2013.00019
Berg, T., Piercey, B. W., and Jensen, J. (2010). Role of beta1-3-adrenoceptors in blood pressure control at rest and during tyramine-induced norepinephrine release in spontaneously hypertensive rats. Hypertension 55, 1224–1230. doi:10.1161/HYPERTENSIONAHA.109.149286
Berg, T., Walaas, S. I., Roberg, B. A., Huynh, T. T., and Jensen, J. (2012). Plasma norepinephrine in hypertensive rats reflects alpha(2)-adrenoceptor release control only when re-uptake is inhibited. Front. Neurol. 3:160. doi:10.3389/fneur.2012.00160
Brede, M., Nagy, G., Philipp, M., Sorensen, J. B., Lohse, M. J., and Hein, L. (2003). Differential control of adrenal and sympathetic catecholamine release by alpha 2-adrenoceptor subtypes. Mol. Endocrinol. 17, 1640–1646. doi:10.1210/me.2003-0035
Brede, M., Philipp, M., Knaus, A., Muthig, V., and Hein, L. (2004). Alpha2-adrenergic receptor subtypes – novel functions uncovered in gene-targeted mouse models. Biol. Cell 96, 343–348. doi:10.1111/j.1768-322X.2004.tb01424.x
Chotani, M. A., Mitra, S., Su, B. Y., Flavahan, S., Eid, A. H., Clark, K. R., et al. (2004). Regulation of alpha(2)-adrenoceptors in human vascular smooth muscle cells. Am. J. Physiol. 286, H59–H67.
Clineschmidt, B. V., Pettibone, D. J., Lotti, V. J., Hucker, H. B., Sweeney, B. M., Reiss, D. R., et al. (1988). A peripherally acting alpha-2 adrenoceptor antagonist: L-659,066. J. Pharmacol. Exp. Ther. 245, 32–40.
Cox, S. L., Schelb, V., Trendelenburg, A. U., and Starke, K. (2000). Enhancement of noradrenaline release by angiotensin II and bradykinin in mouse atria: evidence for cross-talk between G(q/11) protein- and G(i/o) protein-coupled receptors. Br. J. Pharmacol. 129, 1095–1102. doi:10.1038/sj.bjp.0703167
Crassous, P. A., Cardinaletti, C., Carrieri, A., Bruni, B., Di, V. M., Gentili, F., et al. (2007). Alpha2-adrenoreceptors profile modulation. 3.1 (R)-(+)-m-nitrobiphenyline, a new efficient and alpha2C-subtype selective agonist. J. Med. Chem. 50, 3964–3968. doi:10.1021/jm061487a
Eid, A. H., Maiti, K., Mitra, S., Chotani, M. A., Flavahan, S., Bailey, S. R., et al. (2007). Estrogen increases smooth muscle expression of alpha2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am. J. Physiol. 293, H1955–H1961.
Esler, M. (2011). The sympathetic nervous system through the ages: from Thomas Willis to resistant hypertension. Exp. Physiol. 96, 611–622. doi:10.1113/expphysiol.2011.052332
Görnemann, T., von, W. H., Kleuser, B., Villalon, C. M., Centurion, D., Jahnichen, S., et al. (2007). Characterization of the postjunctional alpha 2C-adrenoceptor mediating vasoconstriction to UK14304 in porcine pulmonary veins. Br. J. Pharmacol. 151, 186–194. doi:10.1038/sj.bjp.0707221
Guyenet, P. G. (2006). The sympathetic control of blood pressure. Nat. Rev. Neurosci. 7, 335–346. doi:10.1038/nrn1902
Hein, L., Altman, J. D., and Kobilka, B. K. (1999). Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 402, 181–184. doi:10.1038/46040
Kopp, U. C., Cicha, M. Z., and Smith, L. A. (2011). Impaired interaction between efferent and afferent renal nerve activity in SHR involves increased activation of alpha2-adrenoceptors. Hypertension 57, 640–647. doi:10.1161/HYPERTENSIONAHA.110.166595
Kurnik, D., Friedman, E. A., Muszkat, M., Sofowora, G. G., Xie, H. G., Dupont, W. D., et al. (2008). Genetic variants in the alpha2C-adrenoceptor and G-protein contribute to ethnic differences in cardiovascular stress responses. Pharmacogenet. Genomics 18, 743–750. doi:10.1097/FPC.0b013e3282fee5a1
Lehtimaki, J., Leino, T., Koivisto, A., Viitamaa, T., Lehtimaki, T., Haapalinna, A., et al. (2008). In vitro and in vivo profiling of fadolmidine, a novel potent alpha(2)-adrenoceptor agonist with local mode of action. Eur. J. Pharmacol. 599, 65–71. doi:10.1016/j.ejphar.2008.10.003
Li, Z., Bains, J. S., and Ferguson, A. V. (1993). Functional evidence that the angiotensin antagonist losartan crosses the blood-brain barrier in the rat. Brain Res. Bull. 30, 33–39. doi:10.1016/0361-9230(93)90036-B
Link, R. E., Desai, K., Hein, L., Stevens, M. E., Chruscinski, A., Bernstein, D., et al. (1996). Cardiovascular regulation in mice lacking alpha2-adrenergic receptor subtypes b and c. Science 273, 803–805. doi:10.1126/science.273.5276.803
Lymperopoulos, A., Rengo, G., Funakoshi, H., Eckhart, A. D., and Koch, W. J. (2007). Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 13, 315–323. doi:10.1038/nm1553
MacDonald, E., Kobilka, B. K., and Scheinin, M. (1997). Gene targeting – homing in on alpha 2-adrenoceptor-subtype function. Trends Pharmacol. Sci. 18, 211–219. doi:10.1016/S0165-6147(97)90625-8
Makaritsis, K. P., Johns, C., Gavras, I., Altman, J. D., Handy, D. E., Bresnahan, M. R., et al. (1999). Sympathoinhibitory function of the alpha(2A)-adrenergic receptor subtype. Hypertension 34, 403–407. doi:10.1161/01.HYP.34.3.403
Matthews, K. A., Katholi, C. R., McCreath, H., Whooley, M. A., Williams, D. R., Zhu, S., et al. (2004). Blood pressure reactivity to psychological stress predicts hypertension in the CARDIA study. Circulation 110, 74–78. doi:10.1161/01.CIR.0000133415.37578.E4
Michel, M. C., and Rump, L. C. (1996). alpha-Adrenergic regulation of human renal function. Fundam. Clin. Pharmacol. 10, 493–503. doi:10.1111/j.1472-8206.1996.tb00606.x
Moura, E., Afonso, J., Hein, L., and Vieira-Coelho, M. A. (2006). Alpha2-adrenoceptor subtypes involved in the regulation of catecholamine release from the adrenal medulla of mice. Br. J. Pharmacol. 149, 1049–1058. doi:10.1038/sj.bjp.0706950
Oldendorf, W. H. (1971). Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am. J. Physiol. 221, 1629–1639.
Park, J. W., Chung, H. W., Lee, E. J., Jung, K. H., Paik, J. Y., and Lee, K. H. (2013). alpha2-Adrenergic agonists including xylazine and dexmedetomidine inhibit norepinephrine transporter function in SK-N-SH cells. Neurosci. Lett. 541, 184–189. doi:10.1016/j.neulet.2013.02.022
Parker, L. K., Shanks, J. A., Kennard, J. A., and Brain, K. L. (2010). Dynamic monitoring of NET activity in mature murine sympathetic terminals using a fluorescent substrate. Br. J. Pharmacol. 159, 797–807. doi:10.1111/j.1476-5381.2009.00574.x
Remie, R., Van Rossum, J. X., Coppes, R. P., and Zaagsma, J. (1992). Dysfunctional presynaptic alpha 2-adrenoceptors expose facilitatory beta 2-adrenoceptors in the vasculature of spontaneously hypertensive rats. Eur. J. Pharmacol. 211, 257–261. doi:10.1016/0014-2999(92)90537-E
Shafaroudi, M. M., McBride, M., Deighan, C., Wokoma, A., MacMillan, J., Daly, C. J., et al. (2005). Two “knockout” mouse models demonstrate that aortic vasodilatation is mediated via alpha2a-adrenoceptors located on the endothelium. J. Pharmacol. Exp. Ther. 314, 804–810. doi:10.1124/jpet.105.085944
Small, K. M., McGraw, D. W., and Liggett, S. B. (2003). Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu. Rev. Pharmacol. Toxicol. 43, 381–411. doi:10.1146/annurev.pharmtox.43.100901.135823
Small, K. M., Wagoner, L. E., Levin, A. M., Kardia, S. L., and Liggett, S. B. (2002). Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N. Engl. J. Med. 347, 1135–1142. doi:10.1056/NEJMoa020803
Starke, K. (2001). Presynaptic autoreceptors in the third decade: focus on alpha2-adrenoceptors. J. Neurochem. 78, 685–693. doi:10.1046/j.1471-4159.2001.00484.x
Takano, Y., Takano, M., and Yaksh, T. L. (1992). The effect of intrathecally administered imiloxan and WB4101: possible role of alpha 2-adrenoceptor subtypes in the spinal cord. Eur. J. Pharmacol. 219, 465–468. doi:10.1016/0014-2999(92)90490-U
Talaia, C., Queiroz, G., Pinheiro, H., Moura, D., and Goncalves, J. (2006). Involvement of G-protein betagamma subunits on the influence of inhibitory alpha2-autoreceptors on the angiotensin AT1-receptor modulation of noradrenaline release in the rat vas deferens. Neurochem. Int. 49, 698–707. doi:10.1016/j.neuint.2006.07.002
Trendelenburg, A. U., Meyer, A., Klebroff, W., Guimaraes, S., and Starke, K. (2003a). Crosstalk between presynaptic angiotensin receptors, bradykinin receptors and alpha 2-autoreceptors in sympathetic neurons: a study in alpha 2-adrenoceptor-deficient mice. Br. J. Pharmacol. 138, 1389–1402. doi:10.1038/sj.bjp.0705223
Trendelenburg, A. U., Philipp, M., Meyer, A., Klebroff, W., Hein, L., and Starke, K. (2003b). All three alpha2-adrenoceptor types serve as autoreceptors in postganglionic sympathetic neurons. Naunyn Schmiedebergs Arch. Pharmacol. 368, 504–512. doi:10.1007/s00210-003-0829-x
Keywords: α2-adrenoceptors, angiotensin AT1 receptor, sympathetic nervous system, norepinephrine, epinephrine, release-control, spontaneously hypertensive rats, total peripheral vascular resistance
Citation: Berg T (2013) Angiotensin AT1 – α2C-adrenoceptor interaction disturbs α2A-auto-inhibition of catecholamine release in hypertensive rats. Front. Neurol. 4:70. doi: 10.3389/fneur.2013.00070
Received: 19 March 2013; Accepted: 26 May 2013;
Published online: 10 June 2013.
Edited by:
James Alexander Brock, University of Melbourne, AustraliaReviewed by:
Miyako Takaki, Nara Medical University, JapanKeith L. Brain, University of Birmingham, UK
Copyright: © 2013 Berg. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Torill Berg, Department of Physiology, Institute of Basic Medical Sciences, University of Oslo, P.O. Box 1103, Blindern, 0317 Oslo, Norway e-mail: torill.berg@medisin.uio.no