Environmental enrichment may protect against hippocampal atrophy in the chronic stages of traumatic brain injury
- 1Applied Psychology and Human Development, Ontario Institute for Studies in Education, University of Toronto, Toronto, ON, Canada
- 2Cognitive Neurorehabilitation Sciences Lab, Research Department, Toronto Rehab-University Health Network, Toronto, ON, Canada
- 3fMRI Laboratory, Division of Applied and Interventional Research, Toronto Western Research Institute, University Health Network, Toronto, ON, Canada
- 4Faculty of Medicine, Department of Medical Imaging, University of Toronto, Toronto, ON, Canada
- 5Brain Stimulation and Neuroimaging Laboratory, Monash Alfred Psychiatry Research Centre, Alfred Hospital, Melbourne, VIC, Australia
- 6Graduate Department of Rehabilitation Science, University of Toronto, Toronto, ON, Canada
Objective: To examine the relationship between environmental enrichment (EE) and hippocampal atrophy in the chronic stages of moderate to severe traumatic brain injury (TBI).
Design: Retrospective analysis of prospectively collected data; observational, within-subjects.
Participants: Patients (N = 25) with moderate to severe TBI.
Measures: Primary predictors: (1) An aggregate of self-report rating of EE (comprising hours of cognitive, physical, and social activities) at 5 months post-injury; (2) pre-injury years of education as a proxy for pre-morbid EE (or cognitive reserve). Primary outcome: bilateral hippocampal volume change from 5 to 28 months post-injury.
Results: As predicted, self-reported EE was significantly negatively correlated with bilateral hippocampal atrophy (p < 0.05), with greater EE associated with less atrophy from 5 to 28 months. Contrary to prediction, years of education (a proxy for cognitive reserve) was not significantly associated with atrophy.
Conclusion: Post-injury EE may serve as a buffer against hippocampal atrophy in the chronic stages of moderate-severe TBI. Clinical application of EE should be considered for optimal maintenance of neurological functioning in the chronic stages of moderate-severe TBI.
Introduction
Conventionally, moderate-severe traumatic brain injury (TBI) has been viewed as a non-progressive brain disorder with a predictable trajectory of recovery leading to a stable course thereafter (Lezak et al., 2004). However, growing research findings show progressive gray matter atrophy and loss of white matter integrity in the post-acute and chronic phases of injury (Trivedi et al., 2007; Greenberg et al., 2008; Ng et al., 2008; Farbota et al., 2012; Adnan et al., in press). Further, some individuals with TBI also show progressive cognitive and functional declines in the ensuing months and years following injury (Corkin et al., 1989; Millis et al., 2001; Himanen et al., 2006; Till et al., 2008), supporting the notion that TBI is not a stable condition (Ng et al., 2008; Till et al., 2008).
A brain structure of key importance in TBI is the hippocampus. Memory impairment is one of the most common complaints following TBI, in part due to the acute effects of hippocampal injury, which include excitotoxic and hypoxic insult (Rosenfeld et al., 2012). On top of these acute injuries, post-acute atrophy of the hippocampus has now been demonstrated in several TBI studies (Bigler, 1997; Ng et al., 2008). Tate and Bigler (2000) have suggested that post-acutely, hippocampal cell loss may be the result of transneuronal degeneration secondary to hippocampal deafferentation and/or deefferentation. Indeed, the unique pattern of neuronal projections within the hippocampus (Duvernoy et al., 2005), which is comprised of six architecturally distinct regions linked via unidirectional projections (Amaral and Witter, 1995), has been referred to as an Achilles' heel of sorts, whereby damage to one region of the hippocampus can lead to downstream damage to other regions via loss of activity-dependent survival factors (McCarthy, 2003). Thus, disuse-mediated loss, either secondary to disconnection or to a dearth of behavioral stimulation, may be an important factor in observed hippocampal volume loss in the post-acute stages of injury.
Disuse-mediated loss—or “use it or lose it”—is a concept that has received extensive attention in the older adult literature. It has been argued for decades that greater day-to-day cognitive stimulation is associated with less cognitive decline and delayed onset of dementia. The “negative neuroplasticity” framework of Mahncke et al. (2006), advanced to explain functional losses in normal aging, offers a disuse-mediated framework that can also be applied to atrophy in the chronic stages of TBI (Evans et al., 2008). In their framework, the authors posit that cognitive, perceptual, emotional, physical, psychosocial, and vocational changes associated with aging limit engagement in the busy schedules and complexity of activities of earlier life, leading to a cycle of behavioral inactivation and avoidance—or learned disuse (Taub et al., 2006), and resulting in reduced activation of brain networks (Blake et al., 2006). Although behavioral losses are more sudden in brain injury, moderate-severe TBI patients, too, are commonly unable to engage in the complex activities of work, school and social activities as a result of TBI-induced decrements in cognitive, physical, perceptual and emotional functioning. These patients are especially at risk of reduced behavioral (and thereby neural) stimulation in the sub-acute and chronic stages of injury (after discharge from in-patient rehabilitation facilities), where there is often reduced environmental stimulation and/or reduced supports to foster engagement in the environment (Evans et al., 2008; Frasca et al., 2013).
Related to the above, the concept of environmental enrichment (EE) refers to exposure to and engagement with complex and stimulating environments, and there is extensive evidence that EE can beneficially influence the size, morphology and function of the brain, through synaptic modification and synaptogenesis, the shape and size of dendritic spines (or spine remodeling), axon collateral sprouting, hippocampal (dentate gyrus) neurogenesis and survival of neurons, and brain network connectivity; and such changes have been associated with improvements in functional performance (Rosenzweig and Bennett, 1996; Diamond, 2001; Mohammed et al., 2002; Draganski et al., 2006; Kasai et al., 2010; Kolb et al., 2010; Berlucchi, 2011). Indeed, seminal reviews of the literature conclude that brains that have received increased stimulation, via enhanced mental and physical activity, are better able to mount neuroprotective responses against neurodegenerative processes, traumatic insults and other forms of adult-onset neural dysfunction (Nithianantharajah and Hannan, 2009).
Taken together, the above literature (Mohammed et al., 2002; Kolb et al., 2010; Berlucchi, 2011) suggests that environmental influences, such as intensified cognitive stimulation, might buffer against chronic stage atrophy in moderate-severe TBI. The primary objective of the present study, therefore, was to examine the relationship between post-injury EE, and hippocampal atrophy in the chronic stages of injury. To our knowledge, no studies have yet examined this relationship. We hypothesized that greater EE (i.e., frequency of cognitive, physical, and social engagement) measured in the early post-acute stages of injury (i.e., 5 months post-injury) would be associated with less long-term bilateral hippocampal atrophy over the ensuing years (measured from 5 to 28 months post-injury).
The secondary objective concerned pre-injury EE factors, namely exposure to higher levels of education in development and early adulthood, which has been associated with better cognitive recovery following TBI (Kesler et al., 2003). Given evidence of (1) a positive association between education and cognitive and functional recovery following TBI (Green et al., 2008) and stroke (Elkins et al., 2006), (2) a positive association between education and dendritic branching in healthy adult humans (Jacobs et al., 1993), and (3) a positive correlation between years of education and cognitive functioning in normal aging (Corral et al., 2006; Fritsch et al., 2007), it was hypothesized that greater years of education would be associated with less bilateral hippocampal atrophy, measured from 5 to 28 months post-injury.
Methods
Participants
The 25 clinical participants in this study (see Table 1) were part of a larger research study being conducted at the Toronto Rehabilitation Institute, a large publicly-funded inpatient neurorehabilitation hospital. The focus of the larger study was to investigate the natural history and mechanisms of recovery following moderate-severe TBI. Informed consent was obtained from all participants in the study, and procedures for the present study were approved by the Research Ethics Board of the Toronto Rehabilitation Institute and the Office of Research Ethics at the University of Toronto. Participants underwent prospective assessments at approximately 5, 12, and 28 months post-injury. As the current design was a retrospective analysis of data collected in the course of the larger study data were only available at the 5, 12, and 28 month time points.
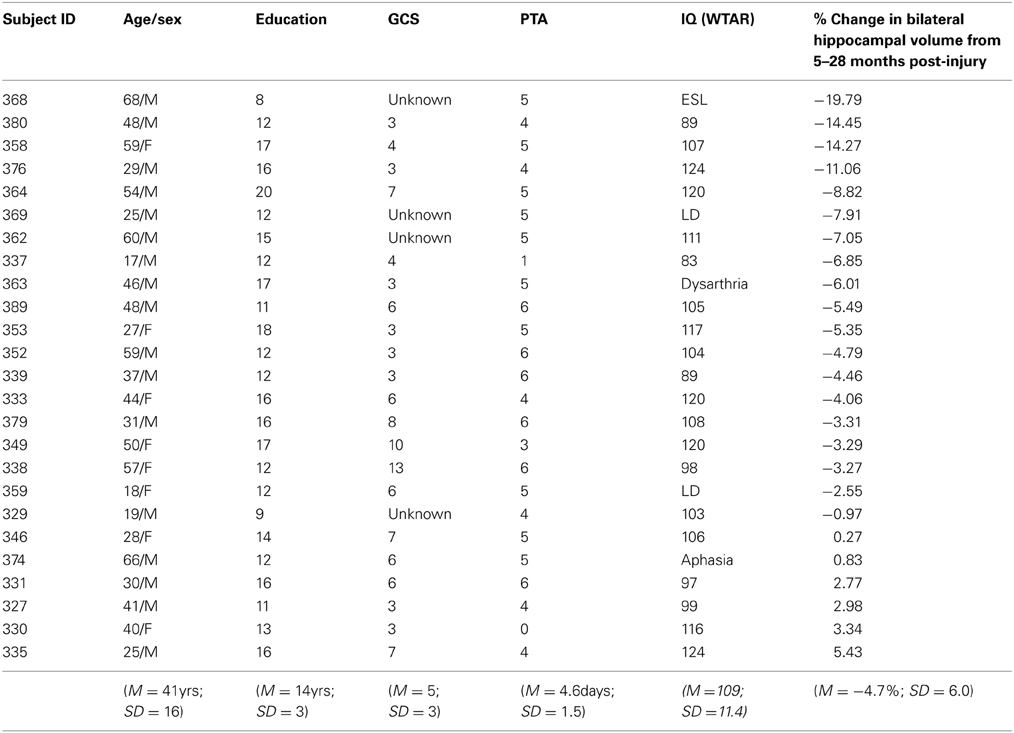
Table 1. Demographic information and hippocampal volume loss.
Inclusion criteria for the larger study comprised: (1) acute care medical diagnosis of TBI; (2) posttraumatic amnesia (PTA) of 1 h or more and/or Glasgow Coma Scale (GCS) score of 12 or less either at emergency or at the scene of the accident and/or positive CT or MRI findings; (3) age between 17 and 80 years old; (4) able to follow simple commands in English based upon Speech Language Pathologist intake assessment; and, (5) competent to provide informed consent for study or availability of a legal decision maker.
Exclusion criteria for the larger study included: (1) orthopaedic injuries affecting both upper extremities; (2) diseases primarily or frequently affecting the central nervous system, including dementia of Alzheimer's type, Parkinson's disease, multiple sclerosis, Huntington's disease, lupus, or stroke; (3) a history of psychotic disorder; (4) non-emergence from posttraumatic amnesia by 6 weeks post-injury, as measured by the Galveston Orientation Amnesia Test (Levin et al., 1979); (5) TBI secondary to another neurological event, such as a fall due to stroke; and, (6) failure on a symptom validity test (Test of Memory Malingering) (Tombaugh, 1996) at any of the assessments. The demographic and injury characteristics of the clinical sample are shown in Table 1. The additional inclusion criteria for the current study were: relevant behavioral and MRI outcome measures at 5 months post-injury plus MRI data at 28 months post-injury.
There were 30 individuals from the larger study with all relevant outcome measures at 5 months post-injury who had reached or surpassed the 28 month post-injury time-point at the time of data analysis. Of those, 25 underwent the 28-month post-injury MRI and were, therefore, eligible for inclusion in the present study, indicating a retention rate of 83%.
Of the sample included in the present study, none showed evidence of severe anxiety or depression, and less than 30% reported experiencing mild or moderate depression or anxiety during the first year post-injury based on the Beck Depression Inventory (Beck, 1987) and Beck Anxiety Inventory (Beck, 1990).
Materials
Lifestyle activities questionnaire
Assessment of post-injury EE was based on participants' self-reported frequency of engagement in a variety of activities involving cognitive, physical, and social demands. Given the absence of reliable or valid published measures of EE in humans at the time of data analysis, EE activities were listed on a self-report questionnaire designed for the purpose of the present study, entitled the Lifestyle Activities Questionnaire (LAQ). Cognitive, physical, and social activities on the LAQ were obtained from a theoretically-derived and empirically-tested inventory developed by Salthouse et al. (2002), which was constructed by specifying 22 common activities that a sample of 1200 adults, ranging from the age of 18–97 years, rated in terms of their cognitive demand (where 1 = low demand, corresponding to sleeping, and 5 = high demand, corresponding to working on a tax form). An activity added to the LAQ that was not on Salthouse's inventory was the frequency of engagement in sports or physical activity at the gym. Participants were asked to rate the frequency with which they engaged in each of the listed activities on an ordinal scale ranging from 1 to 5.
The scale comprised descriptors that ranged from (1) didn't do at all to (5) several hours, every day of the week. For statistical analysis, each number of the scale was weighted based on estimated associated hours. “Didn't do at all and less than once a week” was assigned a weight of zero; “once or twice a week” was assigned a weight of 1 (i.e., for 1 h/week); “several times a week” was assigned a weight of 3 (3 h/week); “an hour or so most days” was assigned a weight of 7; and “several hours a day” was assigned a weight of 20 (20 h/week). Transformation of ordinal scores into weighted scale scores corresponding to estimated hours/week provided a meaningful and more ecological marker of EE. However, for patient responses, the scale was kept as ordinal (i.e., 1–5), because self-reported ratings were deemed by clinical experts consulted to be more easily understood by patients and less likely to result in missing or inaccurate data. We suggest that this approach enabled us to provide richer data than previous methods of aggregating EE activities, in which individuals were classified as “active” if they endorsed doing even one mentally-challenging activity for 1 h per week or more, for example (Bosma et al., 2002; Richards et al., 2003).
Items were sub-classified into cognitive, social or physical activity types to allow for a comparison of the respective influences of each on hippocampal volume loss. For all items, consensus was reached by two trained clinicians regarding assignment to a cognitive, social or physical aggregate. Nineteen items were classified as cognitive activities, 8 items were classified as social activities, and 2 items were classified as physical activities. For each subject, the weighted score for each item type (i.e., social, cognitive, physical) was summed to create a sub-aggregate score for each component of EE; the grand sum was the total score for the larger EE aggregate.
MRI acquisition protocol
MRI scans were acquired on a General Electric (GE) Signa-Echospeed 1.5 Tesla HD scanner (SIGNA EXCITE, GE Healthcare, Milwaukee WI), using an eight channel head coil. Sequences included sagittal T1 (TR/TE = 300/13 ms), slice thickness = 5 mm, space 2.5 mm, matrix 256 × 128 axial gradient recalled echo TR/TE = 450/20, flip angle = 20°, slice thickness = 3 mm no gap, matrix 256 × 192 axial fluid-attenuated-inversion-recovery TR/TE = 9000/45 ms, TI (inversion time) = 2200 ms, slice thickness = 5 mm no gap, matrix 256 × 192 axial fast spin echo proton density (PD)/T2 TR/TE 5500/30, 90 ms, slice thickness = 3 mm no gap, matrix 256 × 192. All above mentioned sequences were obtained with a 22 cm field of view. The high-resolution isotropic T1 weighted, three-dimensional IR prepped radio-frequency spoiled-gradient recalled-echo images TI/TR/TE = 12/300/5, TI, FA = 20, slice thickness = 1 mm no gap, matrix = 256 × 256 were acquired in the axial plane utilizing a 25 cm field of view. The entire scanning session lasted ~55 min.
Image processing and analysis
The MR images were transferred to a workstation for image processing. The scans were received in the Digital Imaging and Communications in Medicine file format and were subsequently converted into (Medical Imaging Network Common Data Form) file format that was created at McConnell Brain Imaging Centre of the Montreal Neurological Institute. Following this procedure, the files were anonymized.
A number of image processing steps were performed in order to make the MRI data usable for image analysis. First, an intensity non-uniformity correction was performed, followed by linear registration of the images into stereotaxic coordinates based on the Talairach atlas. The linear registration to Talairach coordinates was accomplished through 3D cross-correlation between a given volume and an average MR brain image previously converted into the Talairach coordinate system allowing for direct anatomical comparisons between subjects. Finally, a second non-uniformity correction was performed after the registration, which helped to remove any residual non-uniformity artifacts.
The hippocampi were manually outlined using Analyze 7.0 (Brain Imaging Resource, Mayo Clinic, MN) by an experienced tracer (JM) from coronally orientated MR images in the anterior-posterior direction. Calculations of volumes were computed automatically by multiplying the number of voxels traced in each slice, by their depth (i.e., slice thickness). As described by Watson et al. (1992, 1997), the anterior tip of the hippocampus until the slice before the opening of the crux of the fornix was measured as the hippocampal head and body and included the subiculum, CA1-(4) areas, and dentate gyrus. The hippocampus tail was measured from the slice immediately posterior to that which represented the last slice according to the Watson protocol (Watson et al., 1992, 1997) (see Maller et al., 2007 for a more detailed description of this procedure).
All raw hippocampal volumes were expressed in mm3. For the purpose of the present study, hippocampal volume change between 5 months (T1) post-injury and 28 months (T2) post-injury was measured using the following formula:
Design and Procedures
The study employed a retrospective, within subjects, longitudinal design. For clinical participants, LAQs were collected and initial MRI scans were acquired at a mean of 5.3 months (SD = 1.2; range = 4.3–10.3 months) post-injury; follow-up MRI scans were acquired at 28.48 months (SD = 5.5; range = 24–42 months) post-injury.
The primary dependent measure for the study was bilateral hippocampal volume. The primary independent measures were (1) EE as measured by the total score of the LAQ at 5 months post-injury and (2) years of education prior to the injury. We also examined the relative contributions of the cognitive, social and physical EE sub-aggregate scores.
The primary control variables were injury severity (measured by the Glasgow Coma Scale, duration of PTA, and duration of acute care length of stay), estimated premorbid intelligence (based on the mean score on the Wechsler Test of Adult Reading administered [Wechsler, 2001] at 12 and 28 months post-injury), socioeconomic status, and age at injury.
Results
Using Pearson product moment correlation, primary control variables were correlated with hippocampal atrophy and with the measure of overall EE. Each variable was correlated separately due to limited power secondary to sample size. Any variables with significant correlation were included in subsequent analyses. No significant correlations were observed between any of the primary control variables and EE or hippocampal atrophy, with the exception of age, which correlated significantly with the EE aggregate (r = −0.45, p < 0.05, N = 25) whereby increasing age was associated with less overall EE. As well, there was a trend for higher age at injury to be associated with greater bilateral hippocampal atrophy (r = 0.39, p = 0.06, N = 25). These findings, combined with previous research findings of greater deleterious effects of injury on aging brains (Popa-Wagner et al., 2007; Onyszchuk et al., 2008; Petcu et al., 2008), necessitated controlling for age at injury in the present study analyses.
Partial correlation was used to test the primary hypothesis of the study, namely the relationship between the EE aggregate and bilateral hippocampal atrophy, while controlling for age at injury. A significant negative correlation was observed (r = −0.42, p < 0.05, df = 21) whereby greater general activity level at 5 months post-injury was associated with less bilateral hippocampal atrophy from 5 to 28 months post-injury.
Partial correlation between years of education and hippocampal atrophy, controlling for age at injury was used to examine the secondary hypothesis. Contrary to prediction, no significant relationship was observed (r = −0.05, p = 0.82, df = 22).
Lastly, we also explored preliminarily the respective contribution to hippocampal atrophy of the cognitive, social and physical sub-aggregates. Here, a hierarchical linear regression was conducted, which revealed significant negative correlations between bilateral hippocampal atrophy and cognitive activity (r = −0.53, p < 0.01), and between bilateral hippocampal atrophy and social activity (r = −0.47, p < 0.05) but no significant correlation between bilateral hippocampal atrophy and physical activity.
Discussion
Consistent with our prediction, there was a significant negative association between EE (i.e., engagement in cognitive, physical, and social activities) at 5 months post-injury and bilateral hippocampal atrophy from 5 to 28 months post-injury. Although the observational design of the present study precludes conclusions regarding causality, our findings are consistent with previous associations found between EE and enhanced hippocampal structure, and they are potentially explained by increased production, survival and integration of newly generated dentate gyrus neurons (Kempermann et al., 2002; Olson et al., 2006; Tanti et al., 2013); findings in animals—where EE has been correlated with neurogenesis in TBI and moreover correlated with better recovery (Kovesdi et al., 2011; Matter et al., 2011)—bolster such an interpretation. At a behavioral level, the current findings are consistent with use-it-or-lose-it frameworks, and support the extrapolation of the Mahncke et al. (2006) framework—where reduced schedules of activities and avoidance of cognitively-challenging activities were purported to lead to under-stimulation of critical neural networks and loss of associated functions—to chronic TBI (Evans et al., 2008). The findings are also consistent with Robertson and Murre's (1999) computational model of brain plasticity and guided recovery of function, whereby damaged, but potentially viable circuits may be repaired via cognitive activity, cognitive arousal, and Hebbian learning mechanisms. As well, these findings converge with findings from the cognitive-training literature, which shows evidence that engagement in cognitively-demanding tasks is associated with increased cerebral volume in brain areas that are functionally-related to the demands of the task (Draganski et al., 2006).
Unexpectedly, the results of the present study showed no significant relationship between hippocampal atrophy and pre-injury cognitive reserve, as measured by level of education. This finding was surprising given the extensive evidence of a positive association between level of education and cognitive functioning in normal aging (Corral et al., 2006; Fritsch et al., 2007), stroke recovery (Elkins et al., 2006), and TBI recovery (Kesler et al., 2003). Therefore, the study findings do not offer evidence that this proxy for pre-injury EE protects against the structural progression of subacute hippocampal atrophy following TBI. One interpretation of this null finding is that a fixed degree of neural reserve at the time of brain injury does not confer neuroprotection against progressive pathology (e.g., disconnection, [Tate and Bigler, 2000]; neuroinflammation [Bigler, 2013; Johnson et al., 2013]) as would be logically predicted by conventional theories of brain reserve (Satz, 1993; Stern, 2002) and by negative associations found between premorbid cognitive reserve and progression of pathology in Alzheimers disease (Stern, 2006), multiple sclerosis (Amato et al., 2013) and stroke (Willis and Hakim, 2013). In particular, our findings suggest that in order for EE to positively modulate brain disorders via neuroprotective and/or compensatory mechanisms, EE exposure must occur after the disorder has commenced, and during the period of progression of neuropathology (Nithianantharajah and Hannan, 2009). Such an explanation is consistent with EE influences on neurogenesis, as discussed above (Kovesdi et al., 2011; Matter et al., 2011). As neurogenesis is an active process, it may be that temporally congruent factors, namely current EE, but not past education, play a more significant role (Lee et al., 2009; Surget et al., 2011). Unfortunately, our retrospective analysis did not allow us to quantitate the dentate gyrus, the site of neurogenesis.
EE in humans is viewed conventionally as being comprised of three primary elements: cognitive, physical, and social stimulation (Scarmeas and Stern, 2003; Studenski et al., 2006). Of the three conventional elements of EE, our preliminary findings indicated that cognitive activity accounted for the most outcome variance in bilateral hippocampal atrophy. The question whether it is cognitive, social or physical enrichment that confers the greatest neuroplastic advantage is under active debate; our findings are consistent with those comparison studies in animals that found greater survival and integration of new hippocampal neurons after cognitive stimulation than exercise (Kempermann et al., 2002, 2010; Olson et al., 2006; Curlik and Shors, 2011; Shors et al., 2012; Tanti et al., 2013).
A limitation of the present study was the small sample size and relatively small (albeit typical) representation of female subjects, which limits the generalizability of the study findings and precluded examination of gender as a control variable.
It is important to note that in our preliminary and exploratory analysis of the relative contributions of the sub-aggregates (cognitive vs. social vs. physical stimulation), the greater number of cognitive items on the LAQ may have conferred greater analytic stability to the cognitive outcome measure, with the very small number of physical items (2) providing limited stability. Therefore, further research into the relationship between cognitive vs. other types of stimulation and atrophy in the chronic stages of TBI is needed.
Many studies of EE in humans are observational and correlational in design, including the present study. Future EE research would benefit from inclusion of elements that would permit conclusions regarding causality and directionality of relationships, as offered by Salthouse et al. (2002). They suggested that random assignment to experimental and control groups would minimize influences of relevant pre-existing individual differences, such as initial level of cognitive ability and amount of education. Further, rigorous control of enrichment groups, in terms of type and amount of EE, is needed as well as long-term objective monitoring of the amount and frequency of EE activities. Though, given that objective monitoring of lifestyle activities is often unfeasible, experience sampling might offer greater accuracy than retrospective self-report (Csikszentmihalyi and Larson, 1987). Further general challenges in the measurement and manipulation of EE in humans pertain to inter-individual differences in motivation, interest and engagement: what is cognitively challenging, stimulating, and enjoyable to one may be overly difficult and thus stressful for another, or excessively easy and thus insufficiently stimulating to confer neural benefits to another. These limitations affect the strength of conclusions in our own study and warrant future experimental studies with the controls described above.
With regard to models of rehabilitation, the present study arguably provides preliminary empirical support for the contextualized model of cognitive rehabilitation proposed by Ylvisaker et al. (2002), in which therapeutic activities embedded in the patient's real life outside of the clinical setting may lead to greater recovery following brain injury. Cognitive rehabilitation researchers have recommended that rehabilitation strategies should be embedded in real-life contexts in order to maximize far transfer of learned skills and to impact real-life functioning (Murre and Robertson, 1999; Mateer and Sira, 2006). Consistent with these recommendations and with the Ylvisaker et al. (2002) contextualized model of rehabilitation, the majority of activities examined in the present study were embedded in real-life contexts, thus increasing the practical utility of the present study findings. Future prospective research should compare augmentation or intensification of those activities that are a part of patients' day to day life, with EE conferred by more conventional interventions such as computerized cognitively stimulating activities. Longitudinal measures of EE are also needed. Here, we inferred that EE at 5 months post-injury is a proxy for ongoing EE.
Regarding future research avenues, preliminary findings indicate that the default mode network (DMN; Raichle et al., 2001) in humans is implicated in TBI, with functional connectivity decrements within the DMN predicting sustained attention deficits (Bonnelle et al., 2011; Sandrone and Bacigaluppi, 2012). Future studies could investigate the benefits of EE factors purported to impact connectivity within the DMN, such as meditation practice (Taylor et al., 2013), an area of burgeoning interest in the literature on recovery from TBI.
Conclusions
The present study is the first to examine EE factors and their relationship with hippocampal atrophy in the chronic stages of moderate-severe TBI. EE that followed TBI and was temporally proximal to injury had a negative association with hippocampal atrophy, while years of education, a proxy for pre-injury EE (or cognitive reserve) was not significantly associated. Validation of the present findings through replication with larger samples of TBI patients is needed, ideally in a study in which the type and duration of EE is experimentally manipulated. EE is intimately associated with modification of existing synapses, synaptogenesis as well as neurogenesis. Its role in neurorehabilitation appears critical. The findings of this correlational study can be used to generate testable hypotheses regarding the directional impact of EE, as well as the active ingredients of EE for buffering against cerebral atrophy, and its functional consequences. Such questions have significant implications for the development of effective rehabilitation methods for people suffering the long-term consequences of moderate-severe TBI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported by funding from the Ontario Neurotrauma foundation, Canadian Institutes for Health Research, Canada Research Chair and Physicians' Services Incorporated foundation.
References
Adnan, A., Crawley, A., Mikulis, D., Moscovitch, M., Colella, B., and Green, R. E. A. (in press). Moderate-severe traumatic brain injury causes delayed loss of white matter integrity: evidence of fornix deterioration in the chronic stage of injury. Brain Injury.
Amaral, D. G., and Witter, M. P. (1995). “Hippocampal formation,” in The Rat Nervous System, ed G. Paxinos (New York, NY: Academic Press), 443–493.
Amato, M. P., Razzolini, L., Goretti, B., Stromillo, M. L., Rossi, F., Giorgio, A., et al. (2013). Cognitive reserve and cortical atrophy in multiple sclerosis: a longitudinal study. Neurology 80, 1728–1733. doi: 10.1212/WNL.0b013e3182918c6f
Berlucchi, G. (2011). Brain plasticity and cognitive neurorehabilitation. Neuropsychol. Rehabil. 21, 560–578. doi: 10.1080/09602011.2011.573255
Bigler, E. D. (2013). Neuroinflammatin and the dynamic lesion. Brain 136, 9–11. doi: 10.1093/brain/aws342
Bigler, E. D., Blatter, D. D., Anderson, C. V., Johnson, S. C., Gale, S. D., Hopkins, R. O., et al. (1997). Hippocampal volume in normal aging and traumatic brain injury. AJNR Am. J. Neuroradiol. 18, 11–23.
Blake, D. T., Heiser, M. A., Caywood, M., and Merzenich, M. M. (2006). Experience-dependent adult cortical plasticity requires cognitive association between sensation and reward. Neuron 52, 371–381. doi: 10.1016/j.neuron.2006.08.009
Bonnelle, V., Leech, R., Kinnunen, K. M., Ham, T. E., Beckmann, C. F., De Boissezon, X., et al. (2011). Default mode network connectivity predicts sustained attention deficits after traumatic brain injury. J. Neurosci. 31, 13442–13451. doi: 10.1523/JNEUROSCI.1163-11.2011
Bosma, H., Van Boxtel, M. P., Ponds, R. W., Jelicic, M., Houx, P., Metsemakers, J., et al. (2002). Engaged lifestyle and cognitive function in middle and old-aged, non-demented persons: a reciprocal association? Z. Gerontol. Geriatr. 35, 575–581. doi: 10.1007/s00391-002-0080-y
Corkin, S., Rosen, T. J., Sullivan, E. V., and Clegg, R. A. (1989). Penetrating head injury in young adulthood exacerbates cognitive decline in later years. J. Neurosci. 9, 3876–3883.
Corral, M., Rodriguez, M., Amenedo, E., Sanchez, J. L., and Diaz, F. (2006). Cognitive reserve, age, and neuropsychological performance in healthy participants. Dev. Neuropsychol. 29, 479–491. doi: 10.1207/s15326942dn2903_6
Csikszentmihalyi, M., and Larson, R. (1987). Validity and reliability of the experience-sampling method. J. Nerv. Ment. Dis. 175, 526–536. doi: 10.1097/00005053-198709000-00004
Curlik, D. M., and Shors, T. J. (2011). Learning increases the survival of newborn neurons provided that learning is difficult to achieve and successful. J. Cogn. Neurosci. 23, 2159–2170. doi: 10.1162/jocn.2010.21597
Draganski, B., Gaser, C., Kempermann, G., Kuhn, H. G., Winkler, J., Buchel, C., et al. (2006). Temporal and spatial dynamics of brain structure changes during extensive learning. J. Neurosci. 26, 6314–6317. doi: 10.1523/JNEUROSCI.4628-05.2006
Duvernoy, H. M., Cattin, F., and Naidich, T. (2005). Human Hippocampus: Functional Anatomy, Vascularization and Serial Sections with MRI. Heidelberg: Springer-Verlag.
Elkins, J. S., Longstreth, W. T. Jr., Manolio, T. A., Newman, A. B., Bhadelia, R. A., et al. (2006). Education and the cognitive decline associated with MRI-defined brain infarct. Neurology 67, 435–440. doi: 10.1212/01.wnl.0000228246.89109.98
Evans, J. J., Bateman, A., Turner, G., and Green, R. E. A. (2008). Understanding brain injury resources and evidence base. Neuropsychol. Rehabil. Res. Dig. 18, 372–384. doi: 10.1080/09602010801909153
Farbota, K. D., Bendlin, B. B., Alexander, A. L., Rowley, H. A., Dempsey, R. J., and Johnson, S. C. (2012). Longitudinal diffusion tensor imaging and neuropsychological correlates in traumatic brain injury patients. Front. Hum. Neurosci. 6:160. doi: 10.3389/fnhum.2012.00160
Frasca, D., Tomaszczyk, J., McFadyen, B. J., and Green, R. E. A. (2013). Traumatic brain injury and post-acute decline: what role does environmental enrichment play? A scoping review. Front. Hum. Neurosci. 7:31. doi: 10.3389/fnhum.2013.00031
Fritsch, T., McClendon, M. J., Smyth, K. A., Lerner, A. J., Friedland, R. P., and Larsen, J. D. (2007). Cognitive functioning in healthy aging: the role of reserve and lifestyle factors early in life. Gerontologist 47, 307–322. doi: 10.1093/geront/47.3.307
Green, R. E. A., Colella, B., Herbert, D. A., Bayley, M., Kang, H. S., Till, C., et al. (2008). Prediction of return to productivity after severe traumatic brain injury: investigations of optimal neuropsychological tests and timing of assessment. Arch. Phys. Med. Rehabil. 89, S51–S60.
Greenberg, G., Mikulis, D. J., Ng, K., Desouza, D., and Green, R. E. (2008). Use of diffusion tensor imaging to examine subacute white matter injury progression in moderate to severe traumatic brain injury. Arch. Phys. Med. Rehabil. 89, 45–50. doi: 10.1016/j.apmr.2008.09.552
Himanen, L., Portin, R., Isoniemi, H., Helenius, H., Kurki, T., and Tenovuo, O. (2006). Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology 66 187–192. doi: 10.1212/01.wnl.0000194264.60150.d3
Jacobs, B., Schall, M., and Scheibel, A. B. (1993). A quantitative dendritic analysis of Wernicke's area in humans. II. Gender, hemispheric, and environmental factors. J. Comp. Neurol. 327, 97–111. doi: 10.1002/cne.903270108
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136(Pt 1), 28–42. doi: 10.1093/brain/aws322
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33 121–129.
Kempermann, G., Fabel, K., Ehninger, D., Babu, H., Leal-Galicia, P., Garthe, A., et al. (2010). Why and how physical activity promotes experience-induced brain plasticity. Front. Neurosci. 4:189. doi: 10.3389/fnins.2010.00189
Kempermann, G., Gast, D., and Gage, F. H. (2002). Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann. Neurol. 52, 135–143. doi: 10.1002/ana.10262
Kesler, S. R., Adams, H. F., Blasey, C. M., and Bigler, E. D. (2003). Premorbid intellectual functioning, education, and brain size in traumatic brain injury: an investigation of the cognitive reserve hypothesis. Appl. Neuropsychol. 10, 153–162. doi: 10.1207/S15324826AN1003_04
Kolb, B., Teskey, G. C., and Gibb, R. (2010). Factors influencing cerebral plasticity in the normal and injured brain. Front. Hum. Neurosci. 4:204. doi: 10.3389/fnhum.2010.00204
Kovesdi, E., Gyorgy, A. B., Kwon, S. C., Wingo, D. L., Kamnaksh, A., Long, J. B., et al. (2011). The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front. Neurosci. 5:42. doi: 10.3389/fnins.2011.00042
Lee, T., Jarome, T., Li, S. J., Kim, J. J., and Helmstetter, F. J. (2009). Chronic stress selectively reduces hippocampal volume in rats: a longitudinal magnetic resonance imaging study. Neuroreport 20, 1554–1558. doi: 10.1097/WNR.0b013e328332bb09
Levin, H. S., O'donnell, M. A., and Grossman, R. G. (1979). The Galveston orientation and amnesia test: a practical scale to assess cognition after head injury. J. Nerv. Ment. Dis. 167, 675–684. doi: 10.1097/00005053-197911000-00004
Lezak, M. D., Howieson, D. B., and Loring, D. W. (2004). Neuropsychological Assessment. New York, NY: Oxford University Press.
Mahncke, H. W., Bronstone, A., and Merzenich, M. M. (2006). Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog. Brain Res. 157, 81–109.
Maller, J. J., Daskalakis, Z. J., and Fitzgerald, P. B. (2007). Hippocampal volumetrics in depression: the importance of the posterior tail. Hippocampus 17, 1023–1027.
Mateer, C. A., and Sira, C. S. (2006). Cognitive and emotional consequences of TBI: intervention strategies for vocational rehabilitation. Neurorehabilitation 21, 315–326.
Matter, A. M., Folweiler, K. A., Curatolo, L. M., and Kline, A. E. (2011). Temporal effects of environmental enrichment–mediated functional improvement after experimental traumatic brain injury in rats. Neurorehabil. Neural Repair 25, 558–564. doi: 10.1177/1545968310397206
McCarthy, M. M. (2003). Stretching the truth. Why hippocampal neurons are so vulnerable following traumatic brain injury. Exp. Neurol. 184, 40–43. doi: 10.1016/j.expneurol.2003.08.020
Millis, S. R., Rosenthal, M., Novack, T. A., Sherer, M., Nick, T. G., Kreutzer, J. S., et al. (2001). Long-term neuropsychological outcome after traumatic brain injury. J. Head Trauma Rehabil. 16, 343–355. doi: 10.1097/00001199-200108000-00005
Mohammed, A. H., Zhu, S. W., Darmopil, S., Hjerling-Leffler, J., Ernfors, P., Winblad, B., et al. (2002). Environmental enrichment and the brain. Prog. Brain Res. 138, 109–133. doi: 10.1016/S0079-6123(02)38074-9
Murre, R., and Robertson, I. (1999). Rehabilitation of brain damage: brain plasticity and principles of guided recovery. Psychol. Bull. 125, 544–575. doi: 10.1037/0033-2909.125.5.544
Ng, K., Mikulis, D. J., Glazer, J., Kabani, N., Till, C., Greenberg, G., et al. (2008). Magnetic resonance imaging evidence of progression of subacute brain atrophy in moderate to severe traumatic brain injury. Arch. Phys. Med. Rehabil. 89, S35–S44. doi: 10.1016/j.apmr.2008.07.006
Nithianantharajah, J., and Hannan, A. J. (2009). The neurobiology of brain and cognitive reserve: mental and physical activity as modulators of brain disorders. Prog. Neurobiol. 89, 369–382. doi: 10.1016/j.pneurobio.2009.10.001
Olson, A. K., Eadie, B. D., Ernst, C., and Christie, B. R. (2006). Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus 16, 250–260. doi: 10.1002/hipo.20157
Onyszchuk, G., He, Y. Y., Berman, N. E., and Brooks, W. M. (2008). Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. J. Neurotrauma 25, 153–171. doi: 10.1089/neu.2007.0430
Petcu, E. B., Sfredel, V., Platt, D., Herndon, J. G., Kessler, C., and Popa-Wagner, A. (2008). Cellular and molecular events underlying the dysregulated response of the aged brain to stroke: a mini-review. Gerontology 54, 6–17. doi: 10.1159/000112845
Popa-Wagner, A., Carmichael, S. T., Kokaia, Z., Kessler, C., and Walker, L. C. (2007). The response of the aged brain to stroke: too much, too soon? Curr. Neurovasc. Res. 4, 216–227.
Raichle, M. E., MacLeod, A. M., Snyder, A. Z., Powers, W. J., Gusnard, D. A., and Shulman, G. L. (2001). A default mode of brain function. Proc. Natl. Acad. Sci. U.S.A. 98, 676–682. doi: 10.1073/pnas.98.2.676
Richards, M., Hardy, R., and Wadsworth, M. E. (2003). Does active leisure protect cognition? Evidence from a national birth cohort. Soc. Sci. Med. 56, 785–792.
Robertson, I. H., and Murre, J. M. (1999). Rehabilitation of brain damage: brain plasticity and principles of guided recovery. Psychol. Bull. 125, 544–575. doi: 10.1037/0033-2909.125.5.544
Rosenzweig, M. R., and Bennett, E. L. (1996). Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav. Brain Res. 78, 57–65. doi: 10.1016/0166-4328(95)00216-2
Rosenfeld, J. V., Maas, A. I., Bragge, P., Morganti-Kossmann, M. C., Manley, G. T., and Gruen, R. L. (2012). Early management of severe traumatic brain injury. Lancet 380, 1088–1098. doi: 10.1016/S0140-6736(12)60864-2
Salthouse, T. A., Berish, D. E., and Miles, J. D. (2002). The role of cognitive stimulation on the relations between age and cognitive functioning. Psychol. Aging 17, 548–557. doi: 10.1037/0882-7974.17.4.548
Sandrone, S., and Bacigaluppi, M. (2012). Learning from default mode network: the predictive value of resting state in traumatic brain injury. J. Neurosci. 32, 1915–1917. doi: 10.1523/JNEUROSCI.5637-11.2012
Satz, P. (1993). Brain reserve capacity on symptom onset after brain injury: a formulation and review of evidence for threshold theory. Neuropsychology 7, 273–295. doi: 10.1037/0894-4105.7.3.273
Scarmeas, N., and Stern, Y. (2003). Cognitive reserve and lifestyle. J. Clin. Exp. Neuropsychol. 25, 625–633. doi: 10.1076/jcen.25.5.625.14576
Shors, T. J., Anderson, M. L., Curlik, D. M., and Nokia, M. S. (2012). Use it or lose it: How neurogenesis keeps the brain fit for learning. Behav. Brain Res. 227, 450–458. doi: 10.1016/j.bbr.2011.04.023
Stern, Y. (2002). What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 8, 448–460. doi: 10.1017/S1355617702813248
Stern, Y. (2006). Cognitive reserve and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 20, 112–117.
Studenski, S., Carlson, M. C., Fillit, H., Greenough, W. T., Kramer, A., and Rebok, G. W. (2006). “From bedside to bench: does mental and physical activity promote cognitive vitality in late life?” in Proceeding of the Conference on Science Aging Knowledge Environment 2006 (Philadelphia, PA), pe21.
Surget, A., Tanti, A., Leonardo, E. D., Laugeray, A., Rainer, Q., Touma, C., et al. (2011). Antidepressants recruit new neurons to improve stress response regulation. Mol. Psychiatry 16, 1177–1188. doi: 10.1038/mp.2011.48
Tanti, A., Westphal, W. P., Girault, V., Brizard, B., Devers, S., Leguisquet, A. M., et al. (2013). Region-dependent and stage-specific effects of stress, environmental enrichment and antidepressant treatment on hippocampal neurogenesis. Hippocampus 23, 797–811. doi: 10.1002/hipo.22134
Tate, and Bigler, E. D. (2000). Fornix and hippocampal atrophy in traumatic brain injury. Learn. Mem. 7, 442–446. doi: 10.1101/lm.33000
Taub, E., Uswatte, G., Mark, V. W., and Morris, D. M. (2006). The learned nonuse phenomenon: implications for rehabilitation. Eura. Medicophys. 42, 241–256.
Taylor, V. A., Daneault, V., Grant, J., Scavone, G., Breton, E., Roffe-Vidal, S., et al. (2013). Impact of meditation training on the default mode network during a restful state. Soc. Cogn. Affect. Neurosci. 8, 4–14. doi: 10.1093/scan/nsr087
Till, C., Colella, B., Verwegen, J., and Green, R. E. (2008). Postrecovery cognitive decline in adults with traumatic brain injury. Arch. Phys. Med. Rehabil. 89(12 Suppl.), S25–S34. doi: 10.1016/j.apmr.2008.07.004
Trivedi, M. A., Ward, M. A., Hess, T. M., Gale, S. D., Dempsey, R. J., Rowley, H. A., et al. (2007). Longitudinal changes in global brain volume between 79 and 409 days after traumatic brain injury: relationship with duration of coma. J. Neurotrauma 24, 766–771. doi: 10.1089/neu.2006.0205
Watson, C., Andermann, F., Gloor, P., Jones-Gotman, M., Peters, T., Evans, A., et al. (1992). Anatomic basis of amygdaloid and hippocampal volume measurement by magnetic resonance imaging. Neurology 42, 1743–1750. doi: 10.1212/WNL.42.9.1743
Watson, C., Cendes, F., Fuerst, D., Dubeau, F., Williamson, B., Evans, A., et al. (1997). Specificity of volumetric magnetic resonance imaging in detecting hippocampal sclerosis. Arch. Neurol. 54, 67–73. doi: 10.1001/archneur.1997.00550130049015
Wechsler, D. (2001). Wechsler Test of Adult Reading (WTAR). San Antonio, TX: The Psychological Corporation.
Willis, K. J., and Hakim, A. M. (2013). Stroke prevention and cognitive reserve: emerging approaches to modifying risk and delaying onset of dementia. Front. Neurol. 4:13. doi: 10.3389/fneur.2013.00013
Keywords: traumatic brain injury, environmental enrichment, subacute atrophy, adult, moderate to severe
Citation: Miller LS, Colella B, Mikulis D, Maller J and Green REA (2013) Environmental enrichment may protect against hippocampal atrophy in the chronic stages of traumatic brain injury. Front. Hum. Neurosci. 7:506. doi: 10.3389/fnhum.2013.00506
Received: 18 December 2012; Accepted: 07 August 2013;
Published online: 24 September 2013.
Edited by:
Alvaro Pascual-Leone, Beth Israel Deaconess Medical Center/Harvard Medical School, USAReviewed by:
Bogdan Draganski, University Lausanne, SwitzerlandStefano Sandrone, Institute of Neuroinformatics, Switzerland
Copyright © 2013 Miller, Colella, Mikulis, Maller and Green. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robin E. A. Green, Cognitive Neurorehabilitation Sciences Lab, Research Department, Toronto Rehab-University Health Network, 550 University Ave., Toronto, ON M5A 2G2, Canada e-mail: robin.green@uhn.ca