 Marie-Amélie Papon1
Marie-Amélie Papon1- 1 Département de Psychiatrie et Neurosciences, Centre Hospitalier de l’Université Laval, Québec, QC, Canada
- 2 Department of Anesthesiology, College of Physicians and Surgeons, Columbia University, New York, NY, USA
Cognitive disorders such as postoperative cognitive dysfunction, confusion, and delirium, are common following anesthesia in the elderly, with symptoms persisting for months or years in some patients. Alzheimer’s disease (AD) patients appear to be particularly at risk of cognitive deterioration following anesthesia, and some studies suggest that exposure to anesthetics may increase the risk of AD. Here, we review the literature linking anesthesia to AD, with a focus on the biochemical consequences of anesthetic exposure on AD pathogenic pathways.
Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder in elderly people; it afflicts an estimated 26.6 million people worldwide, and without a major therapeutic breakthrough, the prevalence of AD is expected to increase to more than 100 million by 2050 (Brookmeyer et al., 2007). This disease is the most common form of dementia and the prevalence rates at ages 65, 75, and 85 years are 0.9, 4.2, and 14.7%, respectively (Brookmeyer et al., 2007).
The two histopathological hallmarks of AD are senile plaques composed of extracellular aggregates of the beta-amyloid peptide (Aβ; Selkoe, 2001) and intraneuronal neurofibrillary tangles (NFTs), composed of abnormally hyperphosphorylated tau protein assembled into paired helical filaments (PHFs; Grundke-Iqbal et al., 1986; Buee et al., 2000). Only a small proportion of AD is due to genetic variants, the large majority of cases (over 99%) are late onset and sporadic in origin (Blennow et al., 2006). The cause of sporadic AD is likely to be multifactorial, with external factors interacting with biological or genetic susceptibility to accelerate the manifestation of the disease. The main risk factor for AD is age, and persons over age 85 have a six-fold increase of getting the disease (Harman, 2002; Bufill et al., 2009).
Exposure to anesthesia might be such an external factor. In 2004, it was estimated that 234.2 million people worldwide have surgery each year, approximately one operation for every 25 people (Weiser et al., 2008) and, as overall life expectancy has increased, an increasing number of elderly patients are undergoing anesthesia. In this article, we review the relationship between anesthesia and AD as well as the impact of anesthesia on the two pathological hallmarks of AD: Aβ accumulation and aberrant tau phosphorylation.
Clinical Studies
The evidence that anesthesia produces a persistent decrement in human cognition in certain susceptible patients is reasonably strong. Postoperative cognitive dysfunction (POCD), confusion, and delirium are common after general anesthesia in the elderly, and symptoms may even persist for months or years in some patients (Bedford, 1955; O’Keeffe and Ni Chonchubhair, 1994; Moller et al., 1998; Ancelin et al., 2001; Ahlgren et al., 2003; Seymour and Severn, 2009; Fodale et al., 2010). Increasing age has been consistently identified as a risk factor for POCD (Moller, 1998; Moller et al., 1998; Monk et al., 2008). In a recent study, 25% of patients older than 65 years developed delirium following surgery (Morimoto et al., 2009); moreover, various studies have demonstrated short-term and long-term POCD following cardiac and non-cardiac surgery (Moller et al., 1998; Newman et al., 2001; Monk et al., 2008; Yoshitani and Ohnishi, 2009). Despite advances in perioperative care, POCD remains a major concern in AD patients as well as the elderly surgical population in general, as this condition has been associated with poor functional recovery, prolonged length of hospital stay, increased hospital costs, decreased patient quality of life, and increased mortality (Francis et al., 1990; Marcantonio et al., 1994; Bergmann et al., 2005; Xie and Tanzi, 2006). Interestingly, it has been suggested that the pathological mechanism(s) underlying POCD mimic AD (Xie and Tanzi, 2006; Fodale et al., 2010).
Indeed, anesthesia might be a risk factor for the development of neurodegenerative disorders such as AD and Parkinson’s disease. Patients with AD are considered to be particularly at risk for some of the cognitive side effects of anesthesia (e.g., delirium, POCD; Rees and Gaines III, 1985; Bone and Rosen, 2000; Fernandez et al., 2003; Xie and Tanzi, 2006); however, there is also now concern that general anesthesia is a risk factor for AD with in vitro and in vivo studies suggesting that anesthetics may promote and intensify the neuropathogenesis of AD (Eckenhoff et al., 2004; Planel et al., 2007; Xie et al., 2007; Fodale et al., 2010). Several investigators have thus examined whether general anesthesia is associated with AD. Some studies find no association (Breteler et al., 1991; Bohnen et al., 1994b; Gasparini et al., 2002; Zuo and Zuo, 2010), while interestingly, others suggest that exposure to anesthetics may increase the risk of AD. For example, in a population 80 years of age or greater, general anesthesia was associated with a greater risk of developing AD (OR: 3.22; 95% CI: 1.03–10.09; p < 0.05; Bufill et al., 2009). Bohnen et al. (1994a) have demonstrated an inverse correlation between the age of AD onset and cumulative exposure to general and spinal anesthesia before the age of 50.
To date, few clinical studies have evaluated AD biomarkers in the cerebrospinal fluid (CSF) before and after surgery and anesthesia. In a study limited to patients with idiopathic normal pressure hydrocephalus, Agren-Wilsson et al. (2007) reported that CSF AD biomarkers such as total tau (T-tau) and tau phosphorylated at Thr181 (P-tau) were significantly elevated following shunt surgery, but Aβ42 did not change. It is well known that the pattern of AD CSF biomarkers is characterized by an increase in T-tau and P-Tau, but a decrease in Aβ42 (Andreasen et al., 2001). However, whether the effects are due to surgery or anesthesia is not clear, as a general increase of CSF spinal proteins after shunt insertion has previously been reported (Poca et al., 2001); moreover, a standardized anesthesia protocol was not utilized in these studies. Similarly, Palotas et al. (2010) have reported gradual cognitive decline, significantly decreased levels of Aβ peptides, and markedly increased tau protein concentrations in the CSF of patients 6 months after coronary artery bypass surgery.
Overall, clinical evidence establishing a link between anesthetic exposure and the risk of AD is inconsistent, with contradictory findings between studies. This is probably due to the lack of prospective, randomized controlled studies that are designed to control for critical variables such as the type of surgery, duration of the procedure, age at exposure, pre-existing co-morbid disease, anesthetic technique, and patient temperature, thus making it extremely difficult to directly compare the results of these published studies. In addition, it is presently difficult to dissociate the effects of anesthesia from those of surgery. However, studies demonstrating postoperative cognitive impairment associated with changes in biomarkers similar to that seen in AD, suggest some unifying pathognomonic factors between the two disorders (Palotas et al., 2010).
Preclinical Studies
Anesthesia and Aβ Oligomerization
Amyloid-beta (Aβ) is a peptide of 39–43 amino acids generated in vivo by specific proteolytic cleavage of amyloid precursor protein (APP), a transmembrane glycoprotein. Aggregated Aβ is the main constituent of amyloid plaques found in the brain of AD patients. It appears that soluble oligomeric Aβ forms, rather than amyloid plaques, contribute to the cellular pathology of the disease and correlate with the severity of cognitive impairment in humans (Lue et al., 1999). Different studies have shown that anesthetics can promote the oligomerization of Aβ peptide, supporting a potential link between anesthesia and the acceleration of Aβ-related toxicity (Eckenhoff et al., 2004; Carnini et al., 2007; Figure 1).
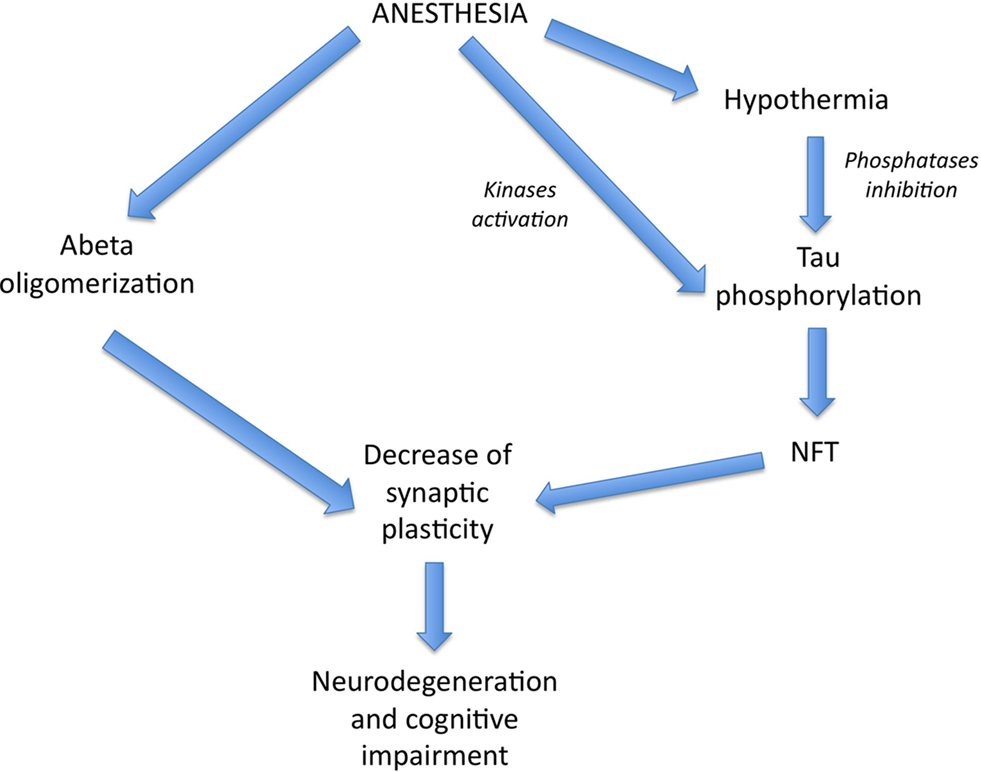
Figure 1. Theoretical pathways by which anesthesia could affect AD pathogenesis. Volatile anesthetics could promote Aβ pathology and NFT formation, both leading to a decrease in synaptic plasticity and neurodegeneration. Interestingly, anesthesia could enhance tau phosphorylation, either directly, through kinases activation, or indirectly, through hypothermia-induced phosphatases inhibition.
Initially,Eckenhoff et al. (2004) demonstrated that inhaled anesthetics (halothane and isoflurane) enhance fibril formation and the cytotoxicity of Aβ. Subsequent studies have shown that these inhaled anesthetics favor the formation of intermediate Aβ oligomers (1–40) and reduce soluble Aβ (1–40) monomer forms (Carnini et al., 2007). Mandal and Pettegrew (2008) have demonstrated by nuclear magnetic resonance (NMR) that halothane and isoflurane, used at high concentrations, interact with a specific region of the Aβ peptide leading to Aβ oligomerization. Another study by Mandal and Fodale (2009) showed a similar effect with clinically relevant concentrations of isoflurane and desflurane. Therefore, it seems that volatile anesthetics can promote Aβ oligomerization.
Anesthesia and Aβ Production
Certain anesthetics have been demonstrated to also increase Aβ production in cell culture (Xie et al., 2006; Zhang et al., 2008). Xie et al. (2006) have shown that a concentration of 2% isoflurane for 6 h altered processing of APP and increased production of Aβ in H4 human neuroglioma cells. Furthermore, H4-APP cells treated with a combination of 12% desflurane and hypoxia (18% O2) for 6 h also altered APP processing and increased Aβ production (Zhang et al., 2008). The isoflurane-mediated elevation and aggregation of Aβ then induces further caspase-3 activation and apoptosis (Xie et al., 2006). Moreover, isoflurane-induced apoptosis can, in turn, increase β and γ-secretase levels, thus promoting the two sequential cleavages of the APP to Aβ (Tagawa et al., 1991), perpetuating a vicious cycle (Xie et al., 2007).
The impact of anesthetics on Aβ production has also been examined in vivo. Bianchi et al. (2008) have demonstrated in female Tg2576 transgenic mice (harboring the human APP-Swedish mutation thus increasing Aβ levels) that more amyloid plaques were observed following the administration of volatile anesthetics in comparison with control animals. In this study, mice were exposed to halothane (0.8–1%) or isoflurane (0.9–1%) for 120 min per day for 5 days. These investigators observed that halothane enhanced plaque deposition in the Tg2576 mouse model whereas isoflurane did not. On the other hand, isoflurane caused a decrease in cognitive performance in wild-type animals that was not observed with halothane. Another group has observed that repetitive isoflurane anesthesia (4% for 1 min and maintained with 2% for 20–30 min) in Tg2576 mice, twice a week for 3 months, led to increased mortality, less responsiveness, increased apoptosis, and increased Aβ aggregation, changes that were not present in control mice (Perucho et al., 2010). In contrast, we did not detect any anesthesia-induced changes in either endogenous APP or APP C-terminal fragment levels; moreover, neither Aβ 1–40 nor Aβ 1–42 levels were altered following anesthesia (Planel et al., 2007). However, we only studied non-transgenic (C57BL/6J) mice 1 h following the induction of chloral hydrate anesthesia (i.p. 500 mg/kg). Our data are consistent with a report demonstrating that acute (6, 12, 24, and 96 h) or chronic (1, 2, 3, and 4 weeks) anesthesia with thiopental or propofol, injected intraperitoneally at therapeutic (5 and 4 mg/kg respectively) or toxic doses (20 mg/kg), does not change APP protein and mRNA concentrations in the rat brain (Palotas et al., 2005). However, in our study as well as the investigation by Palotas et al. (2005) injected anesthetics were used, which could have different effects than the inhaled anesthetics used in the studies by Bianchi et al. (2008) and Perucho et al. (2010). Therefore, it seems that injected anesthetics have no effect on APP metabolism in wild-type animals, in contrast to inhaled anesthetics in AD models. These results suggest that animals with pre-existing Aβ pathology appear to be more at risk for the deleterious effects of certain inhalational anesthetics.
Anesthesia and Tau
Tau is a microtubule-associated protein (MAP) that is abundant in the CNS and expressed mainly in axons. In the human brain, tau is encoded by a single gene (located on chromosome 17) of 16 exons, generating 6 isoforms of 352–441 amino acids. The amino-terminal region, which is characterized by the presence or absence of one or two aminio acid inserts, has been found to interact with the plasma membrane and is essential for determining axonal diameter (Chen et al., 1992). The carboxy-terminal region is characterized by the presence of 3 or 4 repeats that mediate the microtubule (MT) binding properties of tau and promote MT stabilization and polymerization (Himmler et al., 1989; Butner and Kirschner, 1991; Gustke et al., 1994). These functions are negatively regulated by phosphorylation at multiple sites in, and around, the MT binding domain (Buee et al., 2000; Avila et al., 2004). Tau phosphorylation is regulated by numerous kinases and phosphatases; furthermore, glycogen synthase kinase-3 beta (GSK-3β) and protein phosphatase 2A (PP2A) are considered to be the major tau kinase and phosphatase, respectively, regulating tau hyperphosphorylation in vivo (Planel et al., 2002; Tian and Wang, 2002).
Intracellular aggregates of abnormally hyperphosphorylated tau are present in a group of neurodegenerative diseases called tauopathies that include AD (Buee et al., 2000). Tau hyperphosphorylation can induce its aggregation in vitro (Alonso et al., 2001a; Sato et al., 2002), and is thought to induce NFT formation in the brain of AD patients (Trojanowski and Lee, 1994). Tau pathology has been observed to closely reflect the progression of dementia and memory loss in AD (Wilcock and Esiri, 1982; Arriagada et al., 1992; Guillozet et al., 2003; Bretteville and Planel, 2008).
Anesthesia and tau phosphorylation
We previously observed in the brain of non-transgenic mice that short-term (30–60 min) anesthesia induced by chloral hydrate, sodium pentobarbital, or isoflurane resulted in a robust hyperphosphorylation of tau at the pS199 (Ser199), AT8 (Ser202/Thr205), TG3 (Thr231), MC6 (Thr235), pS262 (Ser262), PHF-1 (Ser396/Ser404), and pS422 (Ser422) epitopes (Planel et al., 2007). Some of these phosphoepitopes are associated with early pre-tangle formation (pS422) (Augustinack et al., 2002) as well as later in PHF and NFT formation (AT8, PHF-1; Bramblett et al., 1993; Goedert et al., 1993, 1994). Furthermore, some phosphorylation sites have been linked to specific aspects of tau pathology such as the sequestration of normal tau (e.g., Thr231/Ser235; Alonso et al., 1997; Alonso Adel et al., 2004), the inhibition of tau MT binding (e.g., Ser262; Sengupta et al., 1998), and the promotion of tau aggregation (e.g., Ser396, Ser422; Gong and Iqbal, 2008). As the restoration of normothermia in the mice completely returned phosphorylation to normal levels, tau hyperphosphorylation following these anesthetics was not mediated by the anesthetics per se, but was the consequence of anesthesia-induced hypothermia (Planel et al., 2007). Tau phosphorylation is exquisitely sensitive to temperature, increasing by 80% per degree Celsius drop under 37°C in mice (Planel et al., 2007). We also demonstrated in JNLP3 mice, a mouse model of tauopathy expressing P301L mutant tau (Lewis et al., 2001), that exposure to 1.3% isoflurane for 4 h increased tau phosphorylation at the AT8, CP13(Ser202), pS262, MC6, and PHF-1 epitopes (Planel et al., 2009). Recently, another study confirmed these results in rats and demonstrated that 1.5% isoflurane for 2 h resulted in tau hyperphosphorylation at the Thr205 and Ser396 epitopes in the hippocampus, and that this effect was also due to anesthesia-induced hypothermia (Tan et al., 2010). Other investigators have also confirmed our results of increased tau phosphorylation following anesthesia with sodium pentobarbital (Run et al., 2009), ketamine, or urethane (Holscher et al., 2008). One hour of pentobarbital anesthesia was associated with a decrease in body temperature and induced tau phosphorylation at the Thr181, Thr205, Thr212, and Ser262/Ser356 epitopes to a much larger extent than did anesthesia for 5 min (which did not induce hypothermia; Run et al., 2009). Therefore, it appears that a short exposure to anesthesia induces a slight increase in tau phosphorylation in a hypothermia-independent mechanism contrary to longer periods of anesthesia. Ether anesthesia, which also produces hypothermia (Hemingway, 1948), has also been reported to induce tau hyperphosphorylation in mice at Ser202/Thr205 and Thr231/Ser235, although these investigators did not report the temperature of the animals (Ikeda et al., 2007). Hence, general anesthetics are powerful thermoregulatory depressants leading to tau hyperphosphorylation through hypothermia, suggesting that hypothermia might increase tau pathology (Whittington et al., 2010); however, some anesthetics might have a temperature-independent effect on tau phosphorylation as well (Run et al., 2010; Figure 1).
Mechanisms of tau phosphorylation during anesthesia
We have also demonstrated that anesthesia-induced hypothermia leads to hyperphosphorylation of tau in the mouse brain due to the inhibition of PP2A activity (Planel et al., 2004, 2007). Another recent study has confirmed this result in rats (Tan et al., 2010). Tau hyperphosphorylation during hypothermia is mainly due to the direct inhibition of PP2A, as no single kinase is responsible for the hyperphosphorylation of tau at multiple epitopes observed under these conditions. However, it is important to understand that this statement does not imply that kinases are not involved in tau phosphorylation during hypothermia. On the contrary, in vivo tau hyperphosphorylation can be induced by basal kinase activity, simply by a shift in the phosphorylation equilibrium as a result of phosphatase inhibition (Planel et al., 2007). In other words, inhibiting PP2A is the functional equivalent of activating all the tau kinases together because PP2A dephosphorylates all the known tau epitopes while each kinase is specific for only a set of given sites (Wang et al., 2007). Indeed, even when GSK-3β is inhibited as a consequence to hypothermia, it is still participating to the phosphorylation of tau, as we previously demonstrated with lithium, a GSK-3β inhibitor, in mice rendered hypothermic by starvation (Planel et al., 2001). Thus, hyperphosphorylation during anesthesia-induced hypothermia is due to direct inhibition of PP2A activity, an important finding since PP2A is inhibited in AD and seems to be an important factor in the evolution of AD pathology (Gong et al., 1993, 1995; Vogelsberg-Ragaglia et al., 2001). However, Run et al. (2009) have demonstrated that anesthesia for short periods (pentobarbital for 5 min) induced tau hyperphosphorylation in a hypothermia-independent mechanism. This mechanism is probably due to activation of stress-activated protein kinases, since c-Jun N-terminal kinase (JNK) and mitogen-activated protein kinase (MAPK) were activated during this phase. Therefore, with certain anesthetics, it appears that two mechanisms may underlie the induction of tau hyperphosphorylation: one indirect, linked to hypothermia and due to PP2A inhibition, and the other one more direct, independent of hypothermia, and involving protein kinases activation.
Functional consequences of tau phosphorylation during anesthesia
In vitro studies have demonstrated that hyperphosphorylation of tau can induce MT disruption (Ebneth et al., 1999) as well as tau aggregation (Alonso et al., 2001b; Sato et al., 2002). However, in adult C57BL/6J mice, we have observed that tau hyperphosphorylation by anesthesia-induced hypothermia impaired tau’s ability to bind and polymerize MTs, but did not lead to MT detachment (Planel et al., 2008). MT-bound tau was more resistant to hyperphosphorylation compared with free tau, and tau did not dissociate from MTs in wild-type mice. Surprisingly, hyperphosphorylation of tau in hTau transgenic mice expressing all six isoforms of human tau (Andorfer et al., 2003, 2005), led to the specific dissociation of tau from the MTs in old mice. However, neither normal nor hTau mice, displayed a breakdown of MTs. Of note, in these studies, the mice were examined after only a few hours of anesthesia-induced hypothermia, and it is indeed possible that more prolonged anesthesia-induced hypothermia, even to a mild degree, could lead to more pronounced effects on tau function and MT network integrity. We subsequently confirmed these results in JNPL3 mice, a transgenic mouse strain harboring the human tau P301L mutation (Lewis et al., 2001). Following exposure to 4 h of 1.3% isoflurane twice a week for 2 weeks, we observed, in JNPL3 mice, the detachment of tau from MT without overall collapse of the MT network (Planel et al., 2009). One explanation for the lack of MT disassembly might be that the remaining tau bound to MTs is sufficient to stabilize the cytoskeletal structure. Indeed, tau is known to fulfill its MT-stabilizing function in very minute amounts (Levy et al., 2005).
Pathological consequences of tau phosphorylation during anesthesia
After detaching from MTs, tau can aggregate, and it has been suggested that aggregated tau is the toxic species that causes cognitive decline in AD (Bretteville and Planel, 2008). Tau hyperphosphorylation is thought to induce the formation of insoluble aggregates in vivo (Trojanowski and Lee, 1994). JNPL3 mice readily develop such aggregates in the brain stem and constitute a good model to study whether hyperphosphorylation accelerates tau aggregation in vivo. After 4 h of 1.3% isoflurane without temperature control, there was no change in the level of sarkosyl-insoluble tau in male and female mice. Although insoluble tau levels did not increase significantly immediately following a single exposure to isoflurane, they were elevated a week later in female mice only. Male mice are known to develop pathology at a slower rate than females due to reduced overall tau levels. Interestingly, multiple exposures to isoflurane (4 h to 1.3% isoflurane twice a week for 2 weeks without temperature control) were accompanied by elevated tau aggregation in the brain stem of the male mice 1 week after the last exposure (Planel et al., 2009). These results confirm that exposure to anesthesia-induced hypothermia can enhance tau pathology in JNPL3 mice, and suggest that repeated exposure is necessary to increase the pathology in male mice, in which tau aggregates are slower to form relative to female mice.
We thus evaluated whether increases in aggregated tau following anesthesia affects the degenerative phenotype in JNPL3 mice. Although we observed the accumulation of aggregated tau 1 week after a single exposure to isoflurane in female mice, we did not observe a worsening of the degenerative phenotype as evaluated by a battery of motor function tests (rope hanging, tail hanging, and righting reflex). A similar result was observed following repeated exposure to isoflurane. One explanation could be that the levels of aggregated tau that accumulated in neurons were not enough to induce motor deficits. Indeed, our results show that insoluble tau levels correlate with the degree of motor impairment in JNPL3 mice, but that anesthesia-induced accumulation of aggregated tau did not reach the threshold necessary for motor impairment (Planel et al., 2009).
Conclusion
Overall, epidemiological evidence establishing a link between anesthetic exposure and the risk of AD remains controversial, with some studies finding an increased risk after anesthesia-exposure, and others finding no risk. Moreover, there is currently no direct evidence that anesthesia has a negative influence on the clinical course and outcome of patients with AD. On the other hand, clinical studies examining AD biomarkers postoperatively, and preclinical studies exploring the impact of anesthetics on Aβ and tau, converge to indicate that anesthetics could affect AD pathogenesis, either directly or indirectly.
The therapeutic objective of general anesthesia is to produce analgesia, amnesia, hypnosis, and muscle relaxation to facilitate surgery. As such, general anesthetics provide immense benefits that likely outweigh the possible deleterious outcomes discussed above. Nevertheless, these potential toxic effects should be identified and their mechanisms investigated in order to mitigate any detrimental effects of anesthetic exposure, by the use of the safest drugs and techniques in patients with or at risk for AD (Baranov et al., 2009). Therefore, further clinical studies in humans examining the influence of anesthesia (both direct and indirect) on the subsequent development and progression of AD are warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agren-Wilsson, A., Lekman, A., Sjoberg, W., Rosengren, L., Blennow, K., Bergenheim, A. T., and Malm, J. (2007). CSF biomarkers in the evaluation of idiopathic normal pressure hydrocephalus. Acta Neurol. Scand. 116, 333–339.
Ahlgren, E., Lundqvist, A., Nordlund, A., Aren, C., and Rutberg, H. (2003). Neurocognitive impairment and driving performance after coronary artery bypass surgery. Eur. J. Cardiothorac. Surg. 23, 334–340.
Alonso, A., Zaidi, T., Novak, M., Grundke-Iqbal, I., and Iqbal, K. (2001a). Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928.
Alonso, A. D., Zaidi, T., Novak, M., Barra, H. S., Grundke-Iqbal, I., and Iqbal, K. (2001b). Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J. Biol. Chem. 276, 37967–37973.
Alonso, A. D., Grundke-Iqbal, I., Barra, H. S., and Iqbal, K. (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303.
Alonso Adel, C., Mederlyova, A., Novak, M., Grundke-Iqbal, I., and Iqbal, K. (2004). Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 279, 34873–34881.
Ancelin, M. L., de Roquefeuil, G., Ledesert, B., Bonnel, F., Cheminal, J. C., and Ritchie, K. (2001). Exposure to anaesthetic agents, cognitive functioning and depressive symptomatology in the elderly. Br. J. Psychiatry 178, 360–366.
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454.
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K. L., Barde, Y. A., Duff, K., and Davies, P. (2003). Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590.
Andreasen, N., Minthon, L., Davidsson, P., Vanmechelen, E., Vanderstichele, H., Winblad, B., and Blennow, K. (2001). Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch. Neurol. 58, 373–379.
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639.
Augustinack, J. C., Schneider, A., Mandelkow, E. M., and Hyman, B. T. (2002). Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35.
Avila, J., Perez, M., Lim, F., Gomez-Ramos, A., Hernandez, F., and Lucas, J. J. (2004). Tau in neurodegenerative diseases: tau phosphorylation and assembly. Neurotox. Res. 6, 477–482.
Baranov, D., Bickler, P. E., Crosby, G. J., Culley, D. J., Eckenhoff, M. F., Eckenhoff, R. G., Hogan, K. J., Jevtovic-Todorovic, V., Palotas, A., Perouansky, M., Planel, E., Silverstein, J. H., Wei, H., Whittington, R. A., Xie, Z., and Zuo, Z. (2009). Consensus statement: first international workshop on anesthetics and Alzheimer’s disease. Anesth. Analg. 108, 1627–1630.
Bergmann, M. A., Murphy, K. M., Kiely, D. K., Jones, R. N., and Marcantonio, E. R. (2005). A model for management of delirious postacute care patients. J. Am. Geriatr. Soc. 53, 1817–1825.
Bianchi, S. L., Tran, T., Liu, C., Lin, S., Li, Y., Keller, J. M., Eckenhoff, R. G., and Eckenhoff, M. F. (2008). Brain and behavior changes in 12-month-old Tg2576 and nontransgenic mice exposed to anesthetics. Neurobiol. Aging 29, 1002–1010.
Bohnen, N., Warner, M. A., Kokmen, E., and Kurland, L. T. (1994a). Early and midlife exposure to anesthesia and age of onset of Alzheimer’s disease. Int. J. Neurosci. 77, 181–185.
Bohnen, N. I., Warner, M. A., Kokmen, E., Beard, C. M., and Kurland, L. T. (1994b). Alzheimer’s disease and cumulative exposure to anesthesia: a case–control study. J. Am. Geriatr. Soc. 42, 198–201.
Bramblett, G. T., Goedert, M., Jakes, R., Merrick, S. E., Trojanowski, J. Q., and Lee, V. M. (1993). Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10, 1089–1099.
Breteler, M. M., van Duijn, C. M., Chandra, V., Fratiglioni, L., Graves, A. B., Heyman, A., Jorm, A. F., Kokmen, E., Kondo, K., Mortimer, J. A., Rocca, W. A., Shalat, S. L., Soininen, H., and Hofman, A. (1991). Medical history and the risk of Alzheimer’s disease: a collaborative re-analysis of case–control studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 20(Suppl. 2), S36–S42.
Bretteville, A., and Planel, E. (2008). Tau aggregates: toxic, inert, or protective species? J. Alzheimers Dis. 14, 431–436.
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191.
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130.
Bufill, E., Bartes, A., Moral, A., Casadevall, T., Codinachs, M., Zapater, E., Carles Rovira, J., Roura, P., Oliva, R., and Blesa, R. (2009). Genetic and environmental factors that may influence in the senile form of Alzheimer’s disease: nested case control studies. Neurologia 24, 108–112.
Butner, K. A., and Kirschner, M. W. (1991). Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 115, 717–730.
Carnini, A., Lear, J. D., and Eckenhoff, R. G. (2007). Inhaled anesthetic modulation of amyloid beta(1-40) assembly and growth. Curr. Alzheimer Res. 4, 233–241.
Chen, J., Kanai, Y., Cowan, N. J., and Hirokawa, N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677.
Ebneth, A., Drewes, G., Mandelkow, E. M., and Mandelkow, E. (1999). Phosphorylation of MAP2c and MAP4 by MARK kinases leads to the destabilization of microtubules in cells. Cell Motil. Cytoskeleton 44, 209–224.
Eckenhoff, R. G., Johansson, J. S., Wei, H., Carnini, A., Kang, B., Wei, W., Pidikiti, R., Keller, J. M., and Eckenhoff, M. F. (2004). Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology 101, 703–709.
Fernandez, C. R., Fields, A., Richards, T., and Kaye, A. D. (2003). Anesthetic considerations in patients with Alzheimer’s disease. J. Clin. Anesth. 15, 52–58.
Fodale, V., Santamaria, L. B., Schifilliti, D., and Mandal, P. K. (2010). Anaesthetics and postoperative cognitive dysfunction: a pathological mechanism mimicking Alzheimer’s disease. Anaesthesia 65, 388–395.
Francis, J., Martin, D., and Kapoor, W. N. (1990). A prospective study of delirium in hospitalized elderly. JAMA 263, 1097–1101.
Gasparini, M., Vanacore, N., Schiaffini, C., Brusa, L., Panella, M., Talarico, G., Bruno, G., Meco, G., and Lenzi, G. L. (2002). A case–control study on Alzheimer’s disease and exposure to anesthesia. Neurol. Sci. 23, 11–14.
Goedert, M., Jakes, R., Crowther, R. A., Cohen, P., Vanmechelen, E., Vandermeeren, M., and Cras, P. (1994). Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem. J. 301(Pt 3), 871–877.
Goedert, M., Jakes, R., Crowther, R. A., Six, J., Lubke, U., Vandermeeren, M., Cras, P., Trojanowski, J. Q., and Lee, V. M. (1993). The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. U.S.A. 90, 5066–5070.
Gong, C. X., and Iqbal, K. (2008). Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 15, 2321–2328.
Gong, C. X., Shaikh, S., Wang, J. Z., Zaidi, T., Grundke-Iqbal, I., and Iqbal, K. (1995). Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J. Neurochem. 65, 732–738.
Gong, C. X., Singh, T. J., Grundke-Iqbal, I., and Iqbal, K. (1993). Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 61, 921–927.
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Guillozet, A. L., Weintraub, S., Mash, D. C., and Mesulam, M. M. (2003). Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729–736.
Gustke, N., Trinczek, B., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (1994). Domains of tau protein and interactions with microtubules. Biochemistry 33, 9511–9522.
Harman, D. (2002). Alzheimer’s disease: role of aging in pathogenesis. Ann. N. Y. Acad. Sci. 959, 384–395; discussion 463–385.
Hemingway, A. (1948). Rate of recovery of temperature-regulating responses after ether anesthesia. Am. J. Physiol. 152, 663–670.
Himmler, A., Drechsel, D., Kirschner, M. W., and Martin, D. W. Jr. (1989). Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 9, 1381–1388.
Holscher, C., van Aalten, L., and Sutherland, C. (2008). Anaesthesia generates neuronal insulin resistance by inducing hypothermia. BMC Neurosci. 9, 100. doi: 10.1186/1471-2202-9-100
Ikeda, Y., Ishiguro, K., and Fujita, S. C. (2007). Ether stress-induced Alzheimer-like tau phosphorylation in the normal mouse brain. FEBS Lett. 581, 891–897.
Levy, S. F., Leboeuf, A. C., Massie, M. R., Jordan, M. A., Wilson, L., and Feinstein, S. C. (2005). Three- and four-repeat tau regulate the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners. Implications for neurodegeneration. J. Biol. Chem. 280, 13520–13528.
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., Yen, S. H., Sahara, N., Skipper, L., Yager, D., Eckman, C., Hardy, J., Hutton, M., and McGowan, E. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491.
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E., and Rogers, J. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862.
Mandal, P. K., and Fodale, V. (2009). Isoflurane and desflurane at clinically relevant concentrations induce amyloid beta-peptide oligomerization: an NMR study. Biochem. Biophys. Res. Commun. 379, 716–720.
Mandal, P. K., and Pettegrew, J. W. (2008). Abeta peptide interactions with isoflurane, propofol, thiopental and combined thiopental with halothane: a NMR study. Biochim. Biophys. Acta 1778, 2633–2639.
Marcantonio, E. R., Goldman, L., Mangione, C. M., Ludwig, L. E., Muraca, B., Haslauer, C. M., Donaldson, M. C., Whittemore, A. D., Sugarbaker, D. J., Poss, R., Haas, S., Cook, E. F., Orav, E. J., and Lee, T. H. (1994). A clinical prediction rule for delirium after elective noncardiac surgery. JAMA 271, 134–139.
Moller, J. T. (1998). Postoperative cognitive decline: the extent of the problem. Eur. J. Anesthesiol. 15, 765–767.
Moller, J. T., Cluitmans, P., Rasmussen, L. S., Houx, P., Rasmussen, H., Canet, J., Rabbitt, P., Jolles, J., Larsen, K., Hanning, C. D., Langeron, O., Johnson, T., Lauven, P. M., Kristensen, P. A., Biedler, A., van Beem, H., Fraidakis, O., Silverstein, J. H., Beneken, J. E., and Gravenstein, J. S. (1998). Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet 351, 857–861.
Monk, T. G., Weldon, B. C., Garvan, C. W., Dede, D. E., van der Aa, M. T., Heilman, K. M., and Gravenstein, J. S. (2008). Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology 108, 18–30.
Morimoto, Y., Yoshimura, M., Utada, K., Setoyama, K., Matsumoto, M., and Sakabe, T. (2009). Prediction of postoperative delirium after abdominal surgery in the elderly. J. Anesth. 23, 51–56.
Newman, M. F., Grocott, H. P., Mathew, J. P., White, W. D., Landolfo, K., Reves, J. G., Laskowitz, D. T., Mark, D. B., and Blumenthal, J. A. (2001). Report of the substudy assessing the impact of neurocognitive function on quality of life 5 years after cardiac surgery. Stroke 32, 2874–2881.
O’Keeffe, S. T., and Ni Chonchubhair, A. (1994). Postoperative delirium in the elderly. Br. J. Anaesth. 73, 673–687.
Palotas, A., Reis, H. J., Bogats, G., Babik, B., Racsmany, M., Engvau, L., Kecskemeti, E., Juhasz, A., Vieira, L. B., Teixeira, A. L., Mukhamedyarov, M. A., Rizvanov, A. A., Yalvac, M. E., Guimarees, M. M., Ferreira, C. N., Zefirov, A. L., Kiyasov, A. P., Wang, L., Janka, Z., and Kalman, J. (2010). Coronary artery bypass surgery provokes Alzheimer’s disease-like changes in the cerebrospinal fluid. J. Alzheimers Dis. doi: 10.3233/JAD-2010-100702. [Epub ahead of print].
Palotas, M., Palotas, A., Bjelik, A., Pakaski, M., Hugyecz, M., Janka, Z., and Kalman, J. (2005). Effect of general anesthetics on amyloid precursor protein and mRNA levels in the rat brain. Neurochem. Res. 30, 1021–1026.
Perucho, J., Rubio, I., Casarejos, M. J., Gomez, A., Rodriguez-Navarro, J. A., Solano, R. M., De Yebenes, J. G., and Mena, M. A. (2010). Anesthesia with isoflurane increases amyloid pathology in mice models of Alzheimer’s disease. J. Alzheimers Dis. 19, 1245–1257.
Planel, E., Bretteville, A., Liu, L., Virag, L., Du, A. L., Yu, W. H., Dickson, D. W., Whittington, R. A., and Duff, K. E. (2009). Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB J. 23, 2595–2604.
Planel, E., Krishnamurthy, P., Miyasaka, T., Liu, L., Herman, M., Kumar, A., Bretteville, A., Figueroa, H. Y., Yu, W. H., Whittington, R. A., Davies, P., Takashima, A., Nixon, R. A., and Duff, K. E. (2008). Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J. Neurosci. 28, 12798–12807.
Planel, E., Miyasaka, T., Launey, T., Chui, D. H., Tanemura, K., Sato, S., Murayama, O., Ishiguro, K., Tatebayashi, Y., and Takashima, A. (2004). Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J. Neurosci. 24, 2401–2411.
Planel, E., Richter, K. E., Nolan, C. E., Finley, J. E., Liu, L., Wen, Y., Krishnamurthy, P., Herman, M., Wang, L., Schachter, J. B., Nelson, R. B., Lau, L. F., and Duff, K. E. (2007). Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J. Neurosci. 27, 3090–3097.
Planel, E., Sun, X., and Takashima, A. (2002). Role of GSK-3 beta in Alzheimer’s disease pathology. Drug Dev. Res. 56, 491–510.
Planel, E., Yasutake, K., Fujita, S. C., and Ishiguro, K. (2001). Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase five inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J. Biol. Chem. 276, 34298–34306.
Poca, M. A., Mataro, M., Sahuquillo, J., Catalan, R., Ibanez, J., and Galard, R. (2001). Shunt related changes in somatostatin, neuropeptide Y, and corticotropin releasing factor concentrations in patients with normal pressure hydrocephalus. J. Neurol. Neurosurg. Psychiatr. 70, 298–304.
Rees, D. I., and Gaines, G. Y. III. (1985). Anesthetic considerations for patients with Alzheimer’s disease. Tex. Med. 81, 45–48.
Run, X., Liang, Z., and Gong, C. X. (2010). Anesthetics and tau protein: animal model studies. J. Alzheimers Dis. 22(Suppl. 3), 49–55.
Run, X., Liang, Z., Zhang, L., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2009). Anesthesia induces phosphorylation of tau. J. Alzheimers Dis. 16, 619–626.
Sato, S., Tatebayashi, Y., Akagi, T., Chui, D. H., Murayama, M., Miyasaka, T., Planel, E., Tanemura, K., Sun, X., Hashikawa, T., Yoshioka, K., Ishiguro, K., and Takashima, A. (2002). Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 277, 42060–42065.
Selkoe, D. J. (2001). Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 3, 75–80.
Sengupta, A., Kabat, J., Novak, M., Wu, Q., Grundke-Iqbal, I., and Iqbal, K. (1998). Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 357, 299–309.
Seymour, D. G., and Severn, A. M. (2009). Cognitive dysfunction after surgery and anaesthesia: what can we tell the grandparents? Age Ageing 38, 147–150.
Tagawa, K., Kunishita, T., Maruyama, K., Yoshikawa, K., Kominami, E., Tsuchiya, T., Suzuki, K., Tabira, T., Sugita, H., and Ishiura, S. (1991). Alzheimer’s disease amyloid beta-clipping enzyme (APP secretase): identification, purification, and characterization of the enzyme. Biochem. Biophys. Res. Commun. 177, 377–387.
Tan, W., Cao, X., Wang, J., Lv, H., Wu, B., and Ma, H. (2010). Tau hyperphosphorylation is associated with memory impairment after exposure to 1.5% isoflurane without temperature maintenance in rats. Eur. J. Anaesthesiol. 27, 835–841.
Tian, Q., and Wang, J. (2002). Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neurosignals 11, 262–269.
Trojanowski, J. Q., and Lee, V. M. (1994). Paired helical filament tau in Alzheimer’s disease. The kinase connection. Am. J. Pathol. 144, 449–453.
Vogelsberg-Ragaglia, V., Schuck, T., Trojanowski, J. Q., and Lee, V. M. (2001). PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 168, 402–412.
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (2007). Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68.
Weiser, T. G., Regenbogen, S. E., Thompson, K. D., Haynes, A. B., Lipsitz, S. R., Berry, W. R., and Gawande, A. A. (2008). An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 372, 139–144.
Whittington, R. A., Papon, M. A., Chouinard, F., and Planel, E. (2010). Hypothermia and Alzheimer’s disease neuropathogenic pathways. Curr. Alzheimer Res. 7, 717–725.
Wilcock, G. K., and Esiri, M. M. (1982). Plaques, tangles and dementia. A quantitative study. J. Neurol. Sci. 56, 343–356.
Xie, Z., Dong, Y., Maeda, U., Alfille, P., Culley, D. J., Crosby, G., and Tanzi, R. E. (2006). The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology 104, 988–994.
Xie, Z., Dong, Y., Maeda, U., Moir, R. D., Xia, W., Culley, D. J., Crosby, G., and Tanzi, R. E. (2007). The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J. Neurosci. 27, 1247–1254.
Xie, Z., and Tanzi, R. E. (2006). Alzheimer’s disease and post-operative cognitive dysfunction. Exp. Gerontol. 41, 346–359.
Yoshitani, K., and Ohnishi, Y. (2009). Cognitive dysfunction after cardiac surgery. Masui 58, 308–314.
Zhang, B., Dong, Y., Zhang, G., Moir, R. D., Xia, W., Yue, Y., Tian, M., Culley, D. J., Crosby, G., Tanzi, R. E., and Xie, Z. (2008). The inhalation anesthetic desflurane induces caspase activation and increases amyloid beta-protein levels under hypoxic conditions. J. Biol. Chem. 283, 11866–11875.
Keywords: anesthesia, Alzheimer’s disease, amyloid, tau phosphorylation, hypothermia
Citation: Papon M-A, Whittington RA, El-Khoury NB and Planel E (2011) Alzheimer’s disease and anesthesia. Front. Neurosci. 4:272. doi: 10.3389/fnins.2010.00272
Received: 22 November 2010;
Accepted: 24 December 2010;
Published online: 31 January 2011.
Edited by:
David J. Loane, University of Maryland School of Medicine, USAReviewed by:
Jesus Avila, Centro de Biología Molecular Severo Ochoa CSIC-UAM, SpainCheng-Xin Gong, The City University of New York, USA
Copyright: © 2011 Papon, Whittington, El-Khoury and Planel. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Emmanuel Planel, Centre Hospitalier de l’Université Laval, RC-9800, 2705 Boulevard Laurier, Québec, QC, Canada G1V 4G2. e-mail: emmanuel@planel.org