
- 1Divisions of Hypothalamic Research and Endocrinology and Metabolism, Department of Medicine, The University of Texas Southwestern Medical Center, Dallas, TX, USA
- 2Department of Psychiatry, The University of Texas Southwestern Medical Center, Dallas, TX, USA
- 3Laboratory of Neurophysiology of the Multidisciplinary Institute of Cell Biology [Argentine Research Council (CONICET) and Scientific Research Commission, Province of Buenos Aires (CIC-PBA)], La Plata, Buenos Aires, Argentina
Ghrelin is an octanoylated peptide hormone, produced by endocrine cells of the stomach, which acts in the brain to increase food intake and body weight. Our understanding of the mechanisms underlying ghrelin's effects on eating behaviors has been greatly improved by the generation and study of several genetically manipulated mouse models. These models include mice overexpressing ghrelin and also mice with genetic deletion of ghrelin, the ghrelin receptor [the growth hormone secretagogue receptor (GHSR)] or the enzyme that post-translationally modifies ghrelin [ghrelin O-acyltransferase (GOAT)]. In addition, a GHSR-null mouse model in which GHSR transcription is globally blocked but can be cell-specifically reactivated in a Cre recombinase-mediated fashion has been generated. Here, we summarize findings obtained with these genetically manipulated mice, with the aim to highlight the significance of the ghrelin system in the regulation of both homeostatic and hedonic eating, including that occurring in the setting of chronic psychosocial stress.
Introduction
Ghrelin is a 28-amino acid octanoylated peptide hormone secreted predominantly from a distinct population of endocrine “ghrelin” cells located within the gastric oxyntic mucosa, previously referred to as P/D1-type cells in humans and A like-type cells in rodents (Kojima and Kangawa, 2010). Ghrelin is the only known naturally occurring peptide to be modified by Ser3 O-octanoylation, a reaction catalyzed by the enzyme ghrelin O-acyl transferase (GOAT) (Yang et al., 2008; Kojima and Kangawa, 2010). This post-translational modification is essential for the binding of ghrelin to and subsequently activation of its specific receptor, the growth hormone secretagogue receptor type 1a (GHSR). GHSR is a 7 transmembrane protein coupled to the Gq subfamily of heterotrimeric GTP-binding proteins that activates phospholipase C and in turn, induces release of calcium from intracellular stores (Howard et al., 1996; Damian et al., 2012). Stimulation of other signal transduction cascades also have been demonstrated upon activation of GHSR, although much of that data was characterized using transfected cell systems (Cruz and Smith, 2008). GHSR is expressed in several regions of the central nervous system and some peripheral organs (Cruz and Smith, 2008).
Ghrelin's actions are diverse. Ghrelin is a potent growth hormone (GH) secretagogue and orexigenic peptide (Kojima and Kangawa, 2010). Ghrelin also prevents falls in blood glucose upon extreme caloric restriction and minimizes depressive-like behavior upon chronic psychosocial stress (Lutter et al., 2008; Goldstein et al., 2011). Moreover, ghrelin stimulates motor activity in the gastrointestinal tract and accelerates gastric emptying (Peeters, 2003). Although desacyl-ghrelin, the non-octanoylated form, does not bind to GHSR, some studies report that it can modify certain physiological responses, including food intake and glucose homeostasis (Delhanty et al., 2012).
Genetically-modified mouse models have been instrumental in dissecting out the physiological roles of ghrelin, as well as the specific sites of ghrelin action. Genetic modifications in mice can be grouped in two classes: (1) Standard transgenic models, in which a foreign DNA construct is randomly integrated into the mouse genome (commonly used for overexpression models) and (2) Gene-targeted transgenic models, in which a gene disruption or manipulation is introduced in a single gene via homologous recombination between the foreign DNA and the endogenous gene (commonly used for “knockout” and “knock-in” models) (Davey and Maclean, 2006). In this review, we discuss ghrelin's action on eating behaviors as determined using mice with genetic manipulations in the ghrelin signaling system (Tables 1 and 2). We first focus on the role of ghrelin in homeostatic eating and subsequently discuss the role of ghrelin in hedonic eating.
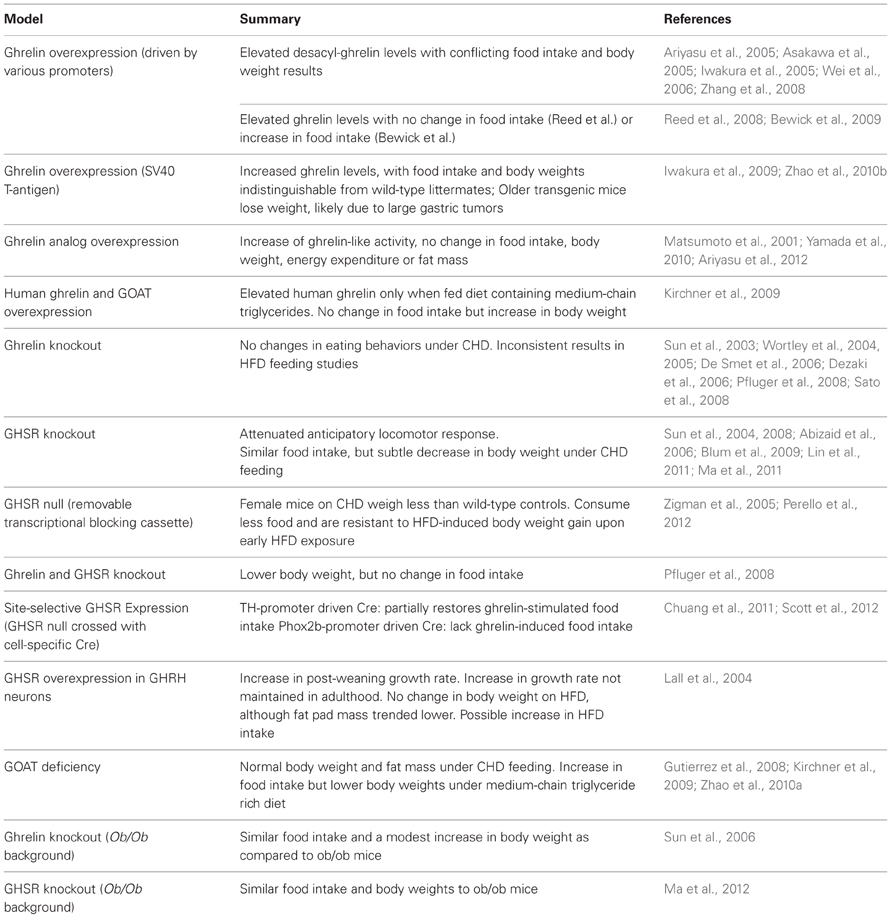
Table 1. Studies of homeostatic eating behaviors in mouse models with genetic alterations in ghrelin-signaling system.

Table 2. Studies of hedonic eating behaviors in mouse models with genetic alterations in ghrelin-signaling system.
Ghrelin and Homeostatic Eating
Homeostatic eating is defined as food intake driven by necessity, due to energy deficiency as perceived by the brain and body. Ghrelin is the only known circulating peptide hormone that stimulates food intake (Cummings, 2006). Ghrelin triggers eating even at times of minimal spontaneous food intake, and its orexigenic actions are rapid, short-lived and potent (Nakazato et al., 2001; Wren et al., 2001; Cummings, 2006). It has been suggested that ghrelin is a physiologic meal initiator, since its levels increase preprandially and decrease post-prandially (Cummings et al., 2001). Ghrelin levels, in individuals under restricted feeding schedules, increase approximately 1 h prior to food presentation. In animals, this preprandial rise in ghrelin occurs simultaneously or possibly precedes the increased locomotion in anticipation of a scheduled meal (Drazen et al., 2006). Indeed, ghrelin administration increases locomotor activity in rats and foraging-like activities in hamsters (Keen-Rhinehart and Bartness, 2005; Jerlhag et al., 2007). Thus, Blum et al. hypothesized that ghrelin is secreted in anticipation of a meal and correlates with anticipatory locomotor activity (Blum et al., 2009).
The mechanism for ghrelin's action on stimulating food intake was initially thought to be through homeostatic hypothalamic circuits originating in the arcuate nucleus (ARC) (Willesen et al., 1999; Nakazato et al., 2001; Kageyama et al., 2010). The expression of GHSR, the only known receptor for ghrelin, has been confirmed within the ARC by many techniques, including in situ hybridization histochemistry, RT-PCR, immunohistochemistry and Western blot analysis (Howard et al., 1996; Guan et al., 1997; Willesen et al., 1999; Zigman et al., 2006). Several studies have demonstrated that ghrelin has the capacity to bind its receptor on arcuate NPY/AgRP neurons, resulting in their activation and ensuing orexigenic behaviors (Willesen et al., 1999; Nakazato et al., 2001; Wren et al., 2001; Kageyama et al., 2010; Schaeffer et al., 2013). Interaction of ghrelin with several other neuronal subtypes and brain sites also has been shown to increase intake of freely-available food. These include, among others, orexin-producing neurons of the lateral hypothalamus (Olszewski et al., 2003; Toshinai et al., 2003). Additionally, some evidence indicates that vagus nerve integrity is required for ghrelin-induced food intake as well (Date et al., 2002; Date, 2012).
Homeostatic Eating in Mice Over Expressing Ghrelin
In order to confirm the effects of ghrelin on homeostatic eating, several groups have attempted to produce transgenic mice with chronically elevated ghrelin. These initial transgenes contained the ghrelin gene driven by various promoters—including those for CAG (Ariyasu et al., 2005; Asakawa et al., 2005), rat insulin II (Iwakura et al., 2005), rat glucagon (Iwakura et al., 2005), CMV (Wei et al., 2006) and FABP4 (Zhang et al., 2008)—resulting in elevated ghrelin gene expression mainly in cell types in which ghrelin gene expression does not usually occur. These mouse models resulted in elevated desacyl-ghrelin rather than elevations of ghrelin, presumably because this overexpression of the ghrelin gene was targeted mostly to tissues lacking the ghrelin-acylating enzyme, GOAT. Indeed, GOAT was not discovered until 2008. Most mouse models overproducing desacyl-ghrelin exhibited similar food intake and body weight as compared to wild-type mice (Iwakura et al., 2005; Wei et al., 2006); nonetheless, some studies found reduced body weight, fat mass, nose-to-anus length and/or food intake in their transgenic mice overexpressing desacyl-ghrelin (Ariyasu et al., 2005; Asakawa et al., 2005; Zhang et al., 2008). These findings may suggest that desacyl-ghrelin has metabolic effects opposite to those of ghrelin.
Two groups successfully produced standard transgenic mice with ghrelin overexpression. Reed et al. generated a transgenic mouse model overexpressing ghrelin in neurons by using a neuron-specific enolase promoter. These mice showed increased ghrelin expression in brain and increased circulating ghrelin (~5-fold), although they did not differ from wild-type controls in body weight, food intake, energy expenditure or fat mass (Reed et al., 2008). Likewise, Bewick et al. generated a transgenic mouse model overexpressing ghrelin, using a bacterial artificial chromosome containing the ghrelin gene and its promoter (Bewick et al., 2009). These mice exhibited increased circulating ghrelin (~1.5-fold) and increased food intake without long-term body weight gain, perhaps due to a paradoxical increase in energy expenditure (Bewick et al., 2009). Bewick et al. proposed that the differences between their model and the Reed et al. mouse model could be due to differential peripheral vs. central nervous system ghrelin concentrations (Bewick et al., 2009). Of note, results regarding the existence, sites of production and physiological significance of endogenous central ghrelin production in non-genetically-manipulated models have been inconsistent (Furness et al., 2011), although ghrelin from the periphery does make its way to the central nervous system.
After GOAT was identified, Kirchner et al. generated a double-transgenic mouse model overexpressing both human ghrelin and MBOAT4 (the membrane-bound O-acyltransferase domain containing 4 or human variant of GOAT) genes in the liver via the human apo-lipoprotein E promoter (Kirchner et al., 2009). These transgenic mice, which exhibited elevated concentrations of human ghrelin only when fed on a diet containing medium-chain triglycerides, showed decreased energy expenditure and increased rate of body weight gain without changes in food intake (Kirchner et al., 2009).
Other unique techniques have been employed to increase ghrelin expression. Yamada and colleagues generated transgenic mice overexpressing a ghrelin analog, which replaced Ser3 with Trp3, under the control of human serum amyloid-P promoter (Yamada et al., 2010). Trp3-ghrelin possesses lower levels of ghrelin-like activity without the need for post-transcriptional modification (Matsumoto et al., 2001). Although mice overexpressing Trp3-ghrelin displayed a ~6-fold increase of ghrelin-like activity, neither body weight, food intake, energy expenditure or fat mass differed from wild-type controls (Ariyasu et al., 2012). Our group generated transgenic mice that express the immortalizing SV40 large T-antigen in ghrelin cells (Zhao et al., 2010b). These mice develop ghrelin-secreting tumors, which result in ~25-fold higher plasma ghrelin concentrations and ~37-fold higher plasma desacyl-ghrelin levels by the time they reach 20 weeks of age (Zhao et al., 2010b). Despite the sustained increase in plasma ghrelin levels, food intake and body weights in these transgenic mice were indistinguishable from those in wild-type mice; older transgenic mice lost weight, presumably due to the development of large gastric tumors (Zhao et al., 2010b). Iwakura et al. also created ghrelin promoter SV40 large T-antigen transgenic mice (Iwakura et al., 2009). These mice showed no difference in body weights as compared to wild-type mice before 12 weeks of age, after which body weight and food intake declined, likely due to tumor growth (Iwakura et al., 2009).
Homeostatic Eating in Mice with Ghrelin Deficiency
To our knowledge, four different ghrelin knockout mouse models have been reported (Sun et al., 2003; Wortley et al., 2004; De Smet et al., 2006; Dezaki et al., 2006). Regardless of the complete absence of ghrelin in circulation in these models, under ad libitum standard rodent chow diet (CHD) feeding, food intake and body weights in these ghrelin knockout mice were indistinguishable from those in wild-type mice. In addition, no differences were observed when other parameters of food intake were measured, including post-fasting hyperphagia (Sun et al., 2003; Wortley et al., 2004; Pfluger et al., 2008), forced dark cycle-induced eating (De Smet et al., 2006) or eating memory (Sato et al., 2008). These findings suggest that genetic ghrelin deficiencies fail to cause significant alterations in homeostatic aspects of eating behaviors.
Furthermore, three of the four ghrelin knockout models have been studied under chronic high fat diet (HFD) feeding, but yielded inconsistent results (Wortley et al., 2005; Dezaki et al., 2006; Sun et al., 2008). Surprisingly, none of the studies exhibited the expected reduction of HFD intake. Wortley et al. did demonstrate that ghrelin deficiency resulted in reduced body weight and fat mass, but these results were not recapitulated in the other mouse ghrelin knockout mouse models. Differences in the age at which the mice were first exposed to HFD diet are thought to account for the inconsistencies in the body weight phenotype among the different ghrelin knockout models. In particular, resistance to the development of diet-induced obesity may manifest in ghrelin knockout mice upon early exposure to HFD, but not when HFD challenge is initiated later in life (Wortley et al., 2004, 2005; Sun et al., 2008).
Homeostatic Eating in Mice with GHSR Deficiency
To our knowledge, three different mouse models with GHSR deficiency have been reported in the literature (Sun et al., 2004; Zigman et al., 2005; Abizaid et al., 2006). Two are traditional GHSR knockout models, in which the GHSR gene has been removed (Sun et al., 2004; Abizaid et al., 2006), and the other is a GHSR-null model in which the GHSR locus has been modified by the insertion of a loxP-flanked transcriptional blocking cassette (Zigman et al., 2005). More specifically, this transcriptional blocking cassette sequence disrupts GHSR gene expression and, as a consequence, the GHSR-null mice (harboring 2 copies of the recombinant GHSR allele) have no detectable functional GHSR expression. Cre recombinase mediates reactivation of GHSR gene expression by removal of the transcriptional blocking cassette sequence.
GHSR-deficient mice show a subtle but significant decrease in body weight. Sun et al. reported that GHSR knockout mice fed on CHD show a lower body weight than wild-type mice from 16 to 24 weeks of age (Sun et al., 2004). GHSR-deficient mice do not differ from wild-type controls in food intake under CHD (Sun et al., 2004; Zigman et al., 2005). Sun et al. found that GHSR knockout mice have a similar susceptibility to diet-induced obesity compared to wild-type mice upon exposure to HFD as adults (Sun et al., 2008); however, GHSR knockout mice manifest reduced age-associated obesity mainly due to reduced adiposity and increased thermogenesis (Lin et al., 2011; Ma et al., 2011). Using a restrictive eating paradigm, Abizaid et al. showed that GHSR knockout mice did not increase their food intake in response to repeated fasts as seen in wild-type mice. This was also true in ghrelin knockout mice (Abizaid et al., 2006).
In the GHSR-null mice from our group, female GHSR-null mice on CHD were significantly lighter than wild-type controls starting at 12 weeks of age (Zigman et al., 2005). Also, in two independent studies, GHSR-null mice were resistant to HFD-induced body weight gain upon early HFD exposure (Zigman et al., 2005; Perello et al., 2012).
As previously mentioned, in animals, a preprandial rise in ghrelin occurs simultaneously or possibly precedes the increased locomotion exhibited in anticipation of a scheduled meal (Drazen et al., 2006). In preliminary studies, Gooley et al. examined food-entrainable circadian rhythms in a very small cohort of female GHSR-null mice (Gooley et al., 2006). Similar to wild-type littermates, the GHSR-null mice continued to show a preprandial rise in locomotor activity and body temperature, suggesting that intact ghrelin signaling was not required for the expression of food-entrainable circadian rhythms that are likely related to finding food (Gooley et al., 2006). Furthermore, Blum et al. also demonstrated increased activity prior to scheduled mealtime in both wild-type and GHSR knockout mice, although in their study, the response in GHSR knockout mice was attenuated (Blum et al., 2009). Others have also similarly reported that GHSR knockout mice under a restricted feeding paradigm demonstrate attenuated anticipatory locomotor responses and reduced expression of the marker of cellular activation c-fos in the mesolimbic dopamine pathway (Blum et al., 2009; Lamont et al., 2012). Using a different paradigm, Verhagen et al. have shown that GHSR knockout mice did not anticipate food when exposed to an activity-based anorexia model, in which mice are given free access to a running wheel and fed once per day for 2 h (Verhagen et al., 2011). Thus, most of these studies suggest that a disruption in the ghrelin signaling results in the lack of food anticipation behavior.
Homeostatic Eating in Mice with Both Ghrelin and GHSR Deficiency
In another study, significant differences in body weight (decreased), energy expenditure (increased) and locomotor activity (increased) were observed when both ghrelin and GHSR were deficient in mice (double knockout mice), whereas neither ghrelin deficiency nor GHSR deficiency independently affected energy balance under CHD feeding conditions (Pfluger et al., 2008). Thus, the authors speculated that additional components of the ghrelin signaling system might be present.
Homeostatic Eating in Mice with Tissue-Specific GHSR Expression
The GHSR-null mouse model with Cre recombinase-dependent reactivation of GHSR has been valuable in establishing the physiological roles of certain of ghrelin's brain targets (Zigman et al., 2005). In one study, we have used GHSR-null mice to assess the role of direct action of ghrelin on the ventral tegmental area (VTA) and other catecholaminergic neurons. We achieved this by crossing GHSR-null mice with mice containing Cre recombinase expression driven by the tyrosine hydroxylase (TH) promoter, resulting in mice expressing GHSR selectively in TH-containing cells normally programmed to express this receptor (Chuang et al., 2011). This selective GHSR expression did not affect body weight, but partially restored ghrelin-stimulated food intake to levels achievable in wild-type littermates (Chuang et al., 2011). In another study, we crossed GHSR-null mice with mice containing expression driven by the paired-like homeobox 2b (Phox2b) promoter, resulting in mice expressing GHSR selectively in specific hindbrain nuclei, including the nucleus of the solitary tract, dorsomotor nucleus of the vagus, area postrema, nucleus ambiguus and facial motor nucleus (Scott et al., 2012). In contrast to mice with catecholaminergic neuron-selective GHSR expression, mice with hindbrain-selective GHSR expression lacked ghrelin-induced acute food intake (Scott et al., 2012). Thus, as opposed to GHSR-expressing catecholaminergic neurons, GHSR-expressing hindbrain neurons are not sufficient to mediate the acute orexigenic effects of ghrelin.
Homeostatic Eating in Mice with GHSR Over Expression in Growth Hormone Releasing Hormone Neurons
Lall et al. generated a mouse model overexpressing GHSR in GH releasing hormone (GHRH) neurons using a rat GHRH cosmid promoter (GHRH-GHSR) (Lall et al., 2004). GHRH-GHSR mice had an increased post-weaning growth rate compared to wild-type littermates. When placed on HFD at 2 months of age, weight gain of GHRH-GHSR mice was similar to that of their wild-type counterparts, although fat pad mass trended lower. Food intake was not measured for individually-housed mice, however groups of GHRH-GHSR mice ate significantly more calories when fed HFD than when fed CHD, while groups of wild-type mice ate similar caloric amounts of HFD and CHD (Lall et al., 2004).
Homeostatic Eating in Mice with Goat Deficiency
Two GOAT knockout mouse lines have been reported (Gutierrez et al., 2008; Zhao et al., 2010a). Using one of these lines, GOAT knockout mice demonstrated higher levels of desacyl-ghrelin compared to their wild-type littermates, and under CHD feeding had normal body weight and fat mass (Kirchner et al., 2009). However, when fed HFD, these GOAT knockout mice displayed significantly lower body weights compared to wild-type animals, despite not having significant changes in body composition. Challenging GOAT knockout mice with HFD rich in medium-chain triglycerides significantly reduced body weight and fat mass compared to similarly-treated wild-type littermates (Kirchner et al., 2009). Such was likely due to increased energy expenditure, since food intake in the GOAT knockout mice was higher (Kirchner et al., 2009). In contrast, the GOAT knockout mice generated by Zhao et al. had similar body weight, body composition and food intake as wild-type littermates, both on CHD and HFD (Zhao et al., 2010a). They also demonstrated similar weight loss curves when subjected to a severe caloric restriction protocol (Zhao et al., 2010a).
Homeostatic Eating in Mice with Deficiencies in Ghrelin Signaling and Leptin
Ghrelin and leptin are both regulators of feeding, but have opposite effects on food intake. Unlike ghrelin, leptin increases with positive energy balance and inhibits food intake. Furthermore, leptin- and ghrelin-responsive central targets partially overlap (Zigman and Elmquist, 2003; Perello et al., 2012). To investigate the physiological roles of ghrelin and leptin signaling, and their potential interaction in regulating energy balance, Sun et al. crossed ghrelin knockout mice with leptin deficient (ob/ob) mice to generate mice lacking both ghrelin and leptin (ghrelin knockout ob/ob). Although they predicted that the ghrelin knockout ob/ob mice would have a leaner and relatively hypophagic phenotype, the double knockout mice instead displayed similar food intake and a modest increase in body weight as compared to ob/ob mice (Sun et al., 2006). Ma and colleagues generated mice with GHSR deletion on the ob/ob background (GHSR knockout ob/ob) (Ma et al., 2012). They found that food intake and body weights of GHSR knockout ob/ob mice were similar to those of ob/ob mice, possibly suggesting that the obesity in ob/ob mice is unrelated to unopposed ghrelin action.
Ghrelin and Hedonic Eating
Hedonic eating is defined as food intake motivated primarily by pleasure. Hedonic eating also encompasses behaviors aimed at facilitating access to pleasurable food. Ghrelin is thought to increase the rewarding value of palatable foods. Recent studies show that ghrelin is able to regulate different aspects of hedonic eating. The two-food choice test is a conventional behavioral method for determining preference. In this test, rodents are offered a pair of different types of food simultaneously, and the consumed amount is measured for a defined time. Using this test, it has been shown that ghrelin administration shifts food preference toward diets rich in fat (Shimbara et al., 2004). Ghrelin administration also increases intake of palatable saccharin solution and preference for saccharin-flavored foods in mice (Disse et al., 2010). Similarly, rats treated with a GHSR antagonist consume less peanut butter and Ensure®, but do not change intake of CHD in a free choice protocol (Egecioglu et al., 2010). As will be discussed below, ghrelin administration also affects other models of hedonic eating, including the conditioned place preference (CPP) test and operant responding.
The central distribution of GHSRs within mesolimbic centers supports the likelihood of ghrelin playing a major role in hedonic eating. In particular, GHSR is highly expressed in dopaminergic VTA neurons, which project to the nucleus accumbens (NAc) and strongly drive reward behaviors (Perello and Zigman, 2012). Mesolimbic circuitries also may be indirectly regulated by cholinergic neurons located in the laterodorsal tegmental area (LDTg), which express GHSR and innervate the VTA (Dickson et al., 2010). In addition, we have recently shown that ghrelin action on food reward requires intact orexin signaling, although the neuronal circuits linking orexin neurons and ghrelin action remain unclear (Perello et al., 2010).
Hedonic Eating in Mice with Genetic Deletion of GHSR
The use of mouse models deficient in GHSR have been instrumental in demonstrating an obligatory role for intact ghrelin signaling in various hedonic aspects of eating. Egecioglu et al. showed that GHSR knockout mice have a suppressed intake of the rewarding food in a free choice (CHD/rewarding food) paradigm and a reduced accumbal dopamine release induced by rewarding foods (Egecioglu et al., 2010). We recently subjected GHSR-null mice to a cue-potentiated feeding protocol, in which cue-potentiated feeding was demonstrated by enhanced intake of grain-based pellets selectively upon presentation of a positive conditioned stimulus (Walker et al., 2012). While wild-type mice demonstrated cue-potentiated feeding behavior, GHSR-null mice lacked this behavior. More specifically, GHSR-null mice displayed enhanced food intake in response to both positive and negative conditioned stimuli (Walker et al., 2012). This study supports pharmacologic studies suggesting a key role for intact ghrelin signaling pathways in establishing a specific positive cue-food association (Kanoski et al., 2013).
Altered eating behaviors are observed under stress as well as depression. In order to study the role of ghrelin in stress-associated hedonic eating, our group subjected both GHSR-null and wild-type littermates to a mouse behavioral model of chronic psychosocial stress and major depression, known as chronic social defeat stress (CSDS). Mice were challenged with ten daily bouts of social defeat by aggressive CD1 male mice, a protocol which results in an increase in plasma ghrelin (Lutter et al., 2008). Despite an increase in plasma ghrelin levels, GHSR-null mice failed to increase intake of CHD in response to chronic stress, unlike wild-type controls which increased their food intake. This suggests that activation of GHSR signaling is required for a stress-induced increase in caloric intake. In addition, stressed GHSR-null and wild-type mice were also tested in a food CPP test, which models a more complex, reward-related type of eating behavior. In the food CPP test, animals are conditioned to associate one chamber of the CPP apparatus with CHD and a second, visually and texturally distinct chamber with an equal calorie amount of a more pleasurable food, such as HFD. After conditioning, animals are given free access to both chambers in the absence of the food, and CPP is demonstrated when more time is spent in the chamber previously paired to the food reward. The CSDS-CPP protocol seems to model stress-based comfort food eating. Following exposure to the CSDS protocol, GHSR-null mice did not demonstrate CPP for HFD, whereas wild-type mice did (Chuang et al., 2011).
The above findings in GHSR-null mice confirm pharmacological studies; as administration of ghrelin and other means of achieving increases in plasma ghrelin (caloric restriction) both enable acquisition of food CPP (Perello et al., 2010; Disse et al., 2011), while administration of GHSR antagonists blocks food CPP (Egecioglu et al., 2010; Perello et al., 2010). Likewise, in our studies using mice subjected to a caloric restriction in which mice were limited to 80% of their usual daily food intake, GHSR-null mice did not demonstrate CPP for HFD, whereas wild-type mice did (Perello et al., 2010). Interestingly, when the study was repeated in mice exposed to a more intense calorie restriction protocol, in which mice were limited to ad libitum access to CHD for 4 h per day, no differences between genotypes were detected in the food CPP test. Thus, endogenous increases in GHSR signaling following stress as modeled with the CSDS protocol or under mild food restrictive conditions enable acquisition of CPP for HFD (Perello et al., 2010).
Hedonic Eating in Mice with Tissue-Specific Reactivation of GHSR
We also assessed food CPP performance following CSDS or as induced by administered ghrelin in mice with catecholaminergic selective GHSR expression, using the mice described above in section Homeostatic Eating in Mice with Tissue Specific GHSR Expression (Chuang et al., 2011). Such selective GHSR expression in TH-expressing cells was sufficient to mediate the ability of administered ghrelin or chronic stress to induce CPP for HFD.
Hedonic Eating in Mice with Goat Deficiency
Recently, it was determined that GOAT-mediated acylation of ghrelin is a critical modulator of food reward. Following a 24-h food deprivation period, GOAT-deficient mice displayed an attenuated motivation for food in an operant responding model (Davis et al., 2012). This supports several pharmacologic studies in which ghrelin administration increased operant lever pressing for sucrose, peanut butter-flavored sucrose or HFD pellets in rodents, and in which GHSR antagonist reduced operant responding for sucrose solution (Perello et al., 2010; Finger et al., 2012; Landgren et al., 2012; Skibicka et al., 2012). GOAT knockout mice also showed a decreased hedonic feeding response as examined in a “dessert effect” protocol, wherein the intake of a palatable HFD “dessert” was assessed in calorically-sated mice (Davis et al., 2012). Furthermore, GOAT knockout mice displayed atypical transcriptional changes in orexin 1 receptor gene expression in the NAc, potentially implying the involvement of orexin signaling in these effects of ghrelin (Davis et al., 2012). Taken together, these results suggest that the motivation to obtain food is regulated by endogenous GOAT activity.
Discussion
In this review, we have summarized the findings of ghrelin action on eating behavior as determined or confirmed using different mouse models with genetic manipulations of the ghrelin signaling system. Of note, the various transgenic mouse models more consistently demonstrated alterations in hedonic eating, while changes to homeostatic eating and body weight changes were more variable and subtle.
An important consideration when evaluating the eating-related phenotypes of the various transgenic models relates to differences between manipulations of ghrelin vs. GHSR vs. GOAT. For example, variable degrees of altered homeostatic eating, hedonic eating and body weight have been observed in ghrelin-deficient, GHSR-deficient, and GOAT-deficient models. Although the exact reasons for these differences are unclear, they could result from differences inherent to each of those molecules. For instance, GHSR possesses strong ligand-independent constitutive activity when studied in vitro (Holst et al., 2004; Holst and Schwartz, 2004), and thus phenotypic changes in GHSR-null mice could result from the combined loss of ghrelin binding and of baseline GHSR “tone.” Additionally, it has been proposed that ghrelin has other receptors besides GHSR, which could explain why simultaneous deletion of both ghrelin and GHSR result in a more severe phenotype than deletion of either alone (Holst et al., 2004; Pfluger et al., 2008). Furthermore, GOAT may have other substrates besides ghrelin, and thus its loss could directly affect more than just ghrelin bioactivity.
Another concern when evaluating these mouse models, as with any transgenic mouse models, is the possibility of functional redundancy or developmental compensation by another related system, which may mask the true effect of manipulation of the ghrelin system. While this has not yet been demonstrated specifically for the ghrelin system, it has been demonstrated for AgRP neurons that are thought to be crucial for ghrelin's effects on eating. Importantly, unlike the lack of significant body weight or food intake phenotypes in mice with genetic AgRP ablation achieved since inception or in neonates, rapid starvation ensues when AgRP expression is deleted in adult mice (Qian et al., 2002; Gropp et al., 2005; Luquet et al., 2005). Such a time-dependent effect of genetic AgRP deletion may be influenced by a time-sensitive capacity for the development of compensatory neurocircuits (Bouret et al., 2004; Luquet et al., 2005). Indeed, leptin's neurotrophic effects on hypothalamic development are restricted to a critical neonatal period (Bouret et al., 2004). Similar time-sensitive neurotrophic actions have been predicted for ghrelin (Steculorum and Bouret, 2011).
Further considerations when evaluating the effects of genetic engineering—in particular, those changes induced in standard transgenic models in which the transgene is randomly inserted into the genome—include the potential for the site of integration to affect tissue specificity, levels of transgene expression as well as expression of other genes present in the nearby genomic vicinity. For all these reasons, the use of the multiple mouse models generated using different genetic strategies and comparison of these mice to mice treated pharmacologically with agents targeting the ghrelin system is important for best determining the importance of the ghrelin system in eating behavior
It should also be noted that these various genetically-engineered mouse models targeting the ghrelin system additionally have been used to assess the importance of ghrelin action in domains other than those related to eating and body weight. For instance, GHSR-null mice have been used to demonstrate a key role for ghrelin action in minimizing depressive-like behavior following chronic stress (Lutter et al., 2008). Also, ghrelin-overexpressing, ghrelin-knockout, GHSR-knockout, GHSR-null and GOAT-knockout mice all demonstrate alterations in glucose homeostasis, with the most extreme phenotype—namely marked hypoglycemia and near death—appearing upon severe caloric restriction of mice with genetic deletion of either GOAT or ghrelin (Zhao et al., 2010a; Li et al., 2012).
These studies predict eating- and body weight-related phenotypes for people if they were to carry a genetic mutation engendering either ghrelin and/or GOAT overexpression or ghrelin, GHSR or GOAT deletion. Indeed, the extreme hyperphagia, early-onset obesity and debilitating food seeking behaviors associated with Prader-Willi Syndrome, in which ghrelin levels are found to elevated and abnormally regulated, represent much more extreme phenotypes than the mouse models of ghrelin over expression would predict (Feigerlova et al., 2008; Hinton et al., 2010). As of yet, no definitive examples of ghrelin, GHSR or GOAT deletion have been identified in people. However, other types of mutations—in the ghrelin gene and the GHSR gene—have been identified. One, the Leu72Met ghrelin polymorphism has been associated with binge eating disorder in small cohorts (Monteleone et al., 2007). Several GHSR polymorphisms have been found in individuals with short stature, which makes sense given ghrelin's ability to potently stimulate GH secretion. Using in vitro methods, these mutations result in loss of endogenous constitutive activity of GHSR, altered binding of ghrelin, decreased ghrelin-stimulated signal transduction, and/or decrease cell surface expression of GHSR (Wang et al., 2004; Pantel et al., 2006; Inoue et al., 2011). Interestingly, carriers of some of these GHSR mutations demonstrate an incomplete penetrance of overweight and obesity, although the relationship of the mutation and the body weight phenotypes have not been confirmed (Wang et al., 2004).
Given the crucial insight regarding ghrelin action on feeding and other domains provided by these mouse models, we would encourage further investigations using not only the currently available mouse models, but also novel genetically modified mouse models. For instance, a GHSR-conditional knockout mouse in which GHSR is eliminated exclusively from a single cell type or a single brain region by using the Cre-loxP technology is an exciting alternative. Additionally, the Cre-loxP system could be used to investigate the role of GHSR at specific stages of development by breeding GHSR floxed mouse lines with transgenic mice bearing a tamoxifen-dependent Cre recombinase expressed under the control of specific promoters, thus allowing the manipulation of GHSR to be controlled in a spatiotemporal-specific manner. In addition, mouse models containing some of the above-described ghrelin and GHSR polymorphisms described in individuals with binge eating disorder and families with short stature ± overweight/obesity would help clarify and causative role for the mutations in those conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Agency of Scientific and Technological Promotion of Argentina (PICT2010-1954 and PICT2011-2142) to Mario Perelló and by the NIH (T32DK007307-33 to Aki Uchida, R03TW008925 to Mario Perelló and Jeffrey M. Zigman, and R01DA024680 and R01MH085298 to Jeffrey M. Zigman).
References
Abizaid, A., Liu, Z. W., Andrews, Z. B., Shanabrough, M., Borok, E., Elsworth, J. D., et al. (2006). Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116, 3229–3239. doi: 10.1172/JCI29867
Ariyasu, H., Takaya, K., Iwakura, H., Hosoda, H., Akamizu, T., Arai, Y., et al. (2005). Transgenic mice overexpressing des-acyl ghrelin show small phenotype. Endocrinology 146, 355–364. doi: 10.1210/en.2004-0629
Ariyasu, H., Yamada, G., Iwakura, H., Akamizu, T., Kangawa, K., and Nakao, K. (2012). Transgenic mice overexpressing ghrelin or ghrelin analog. Meth. Enzymol. 514, 371–377. doi: 10.1016/B978-0-12-381272-8.00023-4
Asakawa, A., Inui, A., Fujimiya, M., Sakamaki, R., Shinfuku, N., Ueta, Y., et al. (2005). Stomach regulates energy balance via acylated ghrelin and desacyl ghrelin. Gut 54, 18–24. doi: 10.1136/gut.2004.038737
Bewick, G. A., Kent, A., Campbell, D., Patterson, M., Ghatei, M. A., Bloom, S. R., et al. (2009). Mice with hyperghrelinemia are hyperphagic and glucose intolerant and have reduced leptin sensitivity. Diabetes 58, 840–846. doi: 10.2337/db08-1428
Blum, I. D., Patterson, Z., Khazall, R., Lamont, E. W., Sleeman, M. W., Horvath, T. L., et al. (2009). Reduced anticipatory locomotor responses to scheduled meals in ghrelin receptor deficient mice. Neuroscience 164, 351–359. doi: 10.1016/j.neuroscience.2009.08.009
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004). Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110. doi: 10.1126/science.1095004
Chuang, J. C., Perello, M., Sakata, I., Osborne-Lawrence, S., Savitt, J. M., Lutter, M., et al. (2011). Ghrelin mediates stress-induced food-reward behavior in mice. J. Clin. Invest. 121, 2684–2692. doi: 10.1172/JCI57660
Cruz, C. R., and Smith, R. G. (2008). The growth hormone secretagogue receptor. Vitam. Horm. 77, 47–88. doi: 10.1016/S0083-6729(06)77004-2
Cummings, D. E. (2006). Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol. Behav. 89, 71–84. doi: 10.1016/j.physbeh.2006.05.022
Cummings, D. E., Purnell, J. Q., Frayo, R. S., Schmidova, K., Wisse, B. E., and Weigle, D. S. (2001). A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719. doi: 10.2337/diabetes.50.8.1714
Damian, M., Marie, J., Leyris, J. P., Fehrentz, J. A., Verdie, P., Martinez, J., et al. (2012). High constitutive activity is an intrinsic feature of Ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J. Biol. Chem. 287, 3630–3641. doi: 10.1074/jbc.M111.288324
Date, Y. (2012). Ghrelin and the vagus nerve. Methods Enzymol. 514, 261–269. doi: 10.1016/B978-0-12-381272-8.00016-7
Date, Y., Murakami, N., Toshinai, K., Matsukura, S., Niijima, A., Matsuo, H., et al. (2002). The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123, 1120–1128. doi: 10.1053/gast.2002.35954
Davey, R. A., and Maclean, H. E. (2006). Current and future approaches using genetically modified mice in endocrine research. Am. J. Physiol. Endocrinol. Metab. 291, E429–E438. doi: 10.1152/ajpendo.00124.2006
Davis, J. F., Perello, M., Choi, D. L., Magrisso, I. J., Kirchner, H., Pfluger, P. T., et al. (2012). GOAT induced ghrelin acylation regulates hedonic feeding. Horm. Behav. 62, 598–604. doi: 10.1016/j.yhbeh.2012.08.009
Delhanty, P. J., Neggers, S. J., and Van Der Lely, A. J. (2012). Mechanisms in endocrinology: ghrelin: the differences between acyl- and des-acyl ghrelin. Eur. J. Endocrinol. 167, 601–608. doi: 10.1530/EJE-12-0456
De Smet, B., Depoortere, I., Moechars, D., Swennen, Q., Moreaux, B., Cryns, K., et al. (2006). Energy homeostasis and gastric emptying in ghrelin knockout mice. J. Pharmacol. Exp. Ther. 316, 431–439. doi: 10.1124/jpet.105.091504
Dezaki, K., Sone, H., Koizumi, M., Nakata, M., Kakei, M., Nagai, H., et al. (2006). Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes 55, 3486–3493. doi: 10.2337/db06-0878
Dickson, S. L., Hrabovszky, E., Hansson, C., Jerlhag, E., Alvarez-Crespo, M., Skibicka, K. P., et al. (2010). Blockade of central nicotine acetylcholine receptor signaling attenuate ghrelin-induced food intake in rodents. Neuroscience 171, 1180–1186. doi: 10.1016/j.neuroscience.2010.10.005
Disse, E., Bussier, A. L., Deblon, N., Pfluger, P. T., Tschop, M. H., Laville, M., et al. (2011). Systemic ghrelin and reward: effect of cholinergic blockade. Physiol. Behav. 102, 481–484. doi: 10.1016/j.physbeh.2010.12.006
Disse, E., Bussier, A. L., Veyrat-Durebex, C., Deblon, N., Pfluger, P. T., Tschop, M. H., et al. (2010). Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol. Behav. 101, 277–281. doi: 10.1016/j.physbeh.2010.05.017
Drazen, D. L., Vahl, T. P., D'Alessio, D. A., Seeley, R. J., and Woods, S. C. (2006). Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 147, 23–30. doi: 10.1210/en.2005-0973
Egecioglu, E., Jerlhag, E., Salome, N., Skibicka, K. P., Haage, D., Bohlooly, Y. M., et al. (2010). Ghrelin increases intake of rewarding food in rodents. Addict Biol. 15, 304–311. doi: 10.1111/j.1369-1600.2010.00216.x
Feigerlova, E., Diene, G., Conte-Auriol, F., Molinas, C., Gennero, I., Salles, J. P., et al. (2008). Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 93, 2800–2805. doi: 10.1210/jc.2007-2138
Finger, B. C., Dinan, T. G., and Cryan, J. F. (2012). Diet-induced obesity blunts the behavioural effects of ghrelin: studies in a mouse-progressive ratio task. Psychopharmacology (Berl.) 220, 173–181. doi: 10.1007/s00213-011-2468-0
Furness, J. B., Hunne, B., Matsuda, N., Yin, L., Russo, D., Kato, I., et al. (2011). Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience 193, 1–9. doi: 10.1016/j.neuroscience.2011.07.063
Goldstein, J. L., Zhao, T. J., Li, R. L., Sherbet, D. P., Liang, G., and Brown, M. S. (2011). Surviving starvation: essential role of the ghrelin-growth hormone axis. Cold Spring Harb. Symp. Quant. Biol. 76, 121–127. doi: 10.1101/sqb.2011.76.010447
Gooley, J. J., Schomer, A., and Saper, C. B. (2006). The dorsomedial hypothalamic nucleus is critical for the expression of food- entrainable circadian rhythms. Nat. Neurosci. 9, 398–407. doi: 10.1038/nn1651
Gropp, E., Shanabrough, M., Borok, E., Xu, A. W., Janoschek, R., Buch, T., et al. (2005). Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 8, 1289–1291. doi: 10.1038/nn1548
Guan, X. M., Yu, H., Palyha, O. C., McKee, K. K., Feighner, S. D., Sirinathsinghji, D. J., et al. (1997). Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 48, 23–29. doi: 10.1016/S0169-328X(97)00071-5
Gutierrez, J. A., Solenberg, P. J., Perkins, D. R., Willency, J. A., Knierman, M. D., Jin, Z., et al. (2008). Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl. Acad. Sci. U.S.A. 105, 6320–6325. doi: 10.1073/pnas.0800708105
Hinton, E. C., Isles, A. R., Williams, N. M., and Parkinson, J. A. (2010). Excessive appetitive arousal in Prader-Willi syndrome. Appetite 54, 225–228. doi: 10.1016/j.appet.2009.12.002
Holst, B., Holliday, N. D., Bach, A., Elling, C. E., Cox, H. M., and Schwartz, T. W. (2004). Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 279, 53806–53817. doi: 10.1074/jbc.M407676200
Holst, B., and Schwartz, T. W. (2004). Constitutive ghrelin receptor activity as a signaling set-point in appetite regulation. Trends Pharmacol. Sci. 25, 113–117. doi: 10.1016/j.tips.2004.01.010
Howard, A. D., Feighner, S. D., Cully, D. F., Arena, J. P., Liberator, P. A., Rosenblum, C. I., et al. (1996). A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273, 974–977. doi: 10.1126/science.273.5277.974
Inoue, H., Kangawa, N., Kinouchi, A., Sakamoto, Y., Kimura, C., Horikawa, R., et al. (2011). Identification and functional analysis of novel human growth hormone secretagogue receptor (GHSR) gene mutations in Japanese subjects with short stature. J. Clin. Endocrinol. Metab. 96, E373–E378. doi: 10.1210/jc.2010-1570
Iwakura, H., Ariyasu, H., Li, Y., Kanamoto, N., Bando, M., Yamada, G., et al. (2009). A mouse model of ghrelinoma exhibited activated growth hormone-insulin-like growth factor I axis and glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 297, E802–E811. doi: 10.1152/ajpendo.00205.2009
Iwakura, H., Hosoda, K., Son, C., Fujikura, J., Tomita, T., Noguchi, M., et al. (2005). Analysis of rat insulin II promoter-ghrelin transgenic mice and rat glucagon promoter-ghrelin transgenic mice. J. Biol. Chem. 280, 15247–15256. doi: 10.1074/jbc.M411358200
Jerlhag, E., Egecioglu, E., Dickson, S. L., Douhan, A., Svensson, L., and Engel, J. A. (2007). Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict. Biol. 12, 6–16. doi: 10.1111/j.1369-1600.2006.00041.x
Kageyama, H., Takenoya, F., Shiba, K., and Shioda, S. (2010). Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides 44, 133–138. doi: 10.1016/j.npep.2009.11.010
Kanoski, S. E., Fortin, S. M., Ricks, K. M., and Grill, H. J. (2013). Ghrelin signaling in the ventral hippocampus stimulates learned and motivational aspects of feeding via PI3K-Akt signaling. Biol. Psychiatry 73, 915–923. doi: 10.1016/j.biopsych.2012.07.002
Keen-Rhinehart, E., and Bartness, T. J. (2005). Peripheral ghrelin injections stimulate food intake, foraging, and food hoarding in Siberian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R716–R722. doi: 10.1152/ajpregu.00705.2004
Kirchner, H., Gutierrez, J. A., Solenberg, P. J., Pfluger, P. T., Czyzyk, T. A., Willency, J. A., et al. (2009). GOAT links dietary lipids with the endocrine control of energy balance. Nat. Med. 15, 741–745. doi: 10.1038/nm.1997
Kojima, M., and Kangawa, K. (2010). Ghrelin: from gene to physiological function. Results Probl. Cell Differ. 50, 185–205.
Lall, S., Balthasar, N., Carmignac, D., Magoulas, C., Sesay, A., Houston, P., et al. (2004). Physiological studies of transgenic mice overexpressing growth hormone (GH) secretagogue receptor 1A in GH-releasing hormone neurons. Endocrinology 145, 1602–1611. doi: 10.1210/en.2003-1509
Lamont, E. W., Patterson, Z., Rodrigues, T., Vallejos, O., Blum, I. D., and Abizaid, A. (2012). Ghrelin-deficient mice have fewer orexin cells and reduced cFOS expression in the mesolimbic dopamine pathway under a restricted feeding paradigm. Neuroscience 218, 12–19. doi: 10.1016/j.neuroscience.2012.05.046
Landgren, S., Simms, J. A., Hyytia, P., Engel, J. A., Bartlett, S. E., and Jerlhag, E. (2012). Ghrelin receptor (GHS-R1A) antagonism suppresses both operant alcohol self-administration and high alcohol consumption in rats. Addict. Biol. 17, 86–94. doi: 10.1111/j.1369-1600.2010.00280.x
Li, R. L., Sherbet, D. P., Elsbernd, B. L., Goldstein, J. L., Brown, M. S., and Zhao, T. J. (2012). Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. J. Biol. Chem. 287, 17942–17950. doi: 10.1074/jbc.M112.358051
Lin, L., Saha, P. K., Ma, X., Henshaw, I. O., Shao, L., Chang, B. H., et al. (2011). Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell 10, 996–1010. doi: 10.1111/j.1474-9726.2011.00740.x
Luquet, S., Perez, F. A., Hnasko, T. S., and Palmiter, R. D. (2005). NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310, 683–685. doi: 10.1126/science.1115524
Lutter, M., Sakata, I., Osborne-Lawrence, S., Rovinsky, S. A., Anderson, J. G., Jung, S., et al. (2008). The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 11, 752–753. doi: 10.1038/nn.2139
Ma, X., Lin, L., Qin, G., Lu, X., Fiorotto, M., Dixit, V. D., et al. (2011). Ablations of ghrelin and ghrelin receptor exhibit differential metabolic phenotypes and thermogenic capacity during aging. PLoS ONE 6:e16391. doi: 10.1371/journal.pone.0016391
Ma, X., Lin, Y., Lin, L., Qin, G., Pereira, F. A., Haymond, M. W., et al. (2012). Ablation of ghrelin receptor in leptin-deficient ob/ob mice has paradoxical effects on glucose homeostasis when compared with ablation of ghrelin in ob/ob mice. Am. J. Physiol. Endocrinol. Metab. 303, E422–E431. doi: 10.1152/ajpendo.00576.2011
Matsumoto, M., Hosoda, H., Kitajima, Y., Morozumi, N., Minamitake, Y., Tanaka, S., et al. (2001). Structure-activity relationship of ghrelin: pharmacological study of ghrelin peptides. Biochem. Biophys. Res. Commun. 287, 142–146. doi: 10.1006/bbrc.2001.5553
Monteleone, P., Tortorella, A., Castaldo, E., Di Filippo, C., and Maj, M. (2007). The Leu72Met polymorphism of the ghrelin gene is significantly associated with binge eating disorder. Psychiatr. Genet. 17, 13–16. doi: 10.1097/YPG.0b013e328010e2c3
Nakazato, M., Murakami, N., Date, Y., Kojima, M., Matsuo, H., Kangawa, K., et al. (2001). A role for ghrelin in the central regulation of feeding. Nature 409, 194–198. doi: 10.1038/35051587
Olszewski, P. K., Li, D., Grace, M. K., Billington, C. J., Kotz, C. M., and Levine, A. S. (2003). Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides 24, 597–602. doi: 10.1016/S0196-9781(03)00105-0
Pantel, J., Legendre, M., Cabrol, S., Hilal, L., Hajaji, Y., Morisset, S., et al. (2006). Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J. Clin. Invest. 116, 760–768. doi: 10.1172/JCI25303
Peeters, T. L. (2003). Central and peripheral mechanisms by which ghrelin regulates gut motility. J. Physiol. Pharmacol. 54 (Suppl. 4), 95–103.
Perello, M., Sakata, I., Birnbaum, S., Chuang, J. C., Osborne-Lawrence, S., Rovinsky, S. A., et al. (2010). Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry 67, 880–886. doi: 10.1016/j.biopsych.2009.10.030
Perello, M., Scott, M. M., Sakata, I., Lee, C. E., Chuang, J. C., Osborne-Lawrence, S., et al. (2012). Functional implications of limited leptin receptor and ghrelin receptor coexpression in the brain. J. Comp. Neurol. 520, 281–294. doi: 10.1002/cne.22690
Perello, M., and Zigman, J. M. (2012). The role of ghrelin in reward-based eating. Biol. Psychiatry 72, 347–353. doi: 10.1016/j.biopsych.2012.02.016
Pfluger, P. T., Kirchner, H., Gunnel, S., Schrott, B., Perez-Tilve, D., Fu, S., et al. (2008). Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G610–G618. doi: 10.1152/ajpgi.00321.2007
Qian, S., Chen, H., Weingarth, D., Trumbauer, M. E., Novi, D. E., Guan, X., et al. (2002). Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol. Cell. Biol. 22, 5027–5035. doi: 10.1128/MCB.22.14.5027-5035.2002
Reed, J. A., Benoit, S. C., Pfluger, P. T., Tschop, M. H., D'Alessio, D. A., and Seeley, R. J. (2008). Mice with chronically increased circulating ghrelin develop age-related glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 294, E752–E760. doi: 10.1152/ajpendo.00463.2007
Sato, T., Kurokawa, M., Nakashima, Y., Ida, T., Takahashi, T., Fukue, Y., et al. (2008). Ghrelin deficiency does not influence feeding performance. Regul. Pept. 145, 7–11. doi: 10.1016/j.regpep.2007.09.010
Schaeffer, M., Langlet, F., Lafont, C., Molino, F., Hodson, D. J., Roux, T., et al. (2013). Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 1512–1517. doi: 10.1073/pnas.1212137110
Scott, M. M., Perello, M., Chuang, J. C., Sakata, I., Gautron, L., Lee, C. E., et al. (2012). Hindbrain ghrelin receptor signaling is sufficient to maintain fasting glucose. PLoS ONE 7:e44089. doi: 10.1371/journal.pone.0044089
Shimbara, T., Mondal, M. S., Kawagoe, T., Toshinai, K., Koda, S., Yamaguchi, H., et al. (2004). Central administration of ghrelin preferentially enhances fat ingestion. Neurosci. Lett. 369, 75–79. doi: 10.1016/j.neulet.2004.07.060
Skibicka, K. P., Hansson, C., Egecioglu, E., and Dickson, S. L. (2012). Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict. Biol. 17, 95–107. doi: 10.1111/j.1369-1600.2010.00294.x
Steculorum, S. M., and Bouret, S. G. (2011). Developmental effects of ghrelin. Peptides 32, 2362–2366. doi: 10.1016/j.peptides.2011.06.021
Sun, Y., Ahmed, S., and Smith, R. G. (2003). Deletion of ghrelin impairs neither growth nor appetite. Mol. Cell. Biol. 23, 7973–7981. doi: 10.1128/MCB.23.22.7973-7981.2003
Sun, Y., Asnicar, M., Saha, P. K., Chan, L., and Smith, R. G. (2006). Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell. Metab. 3, 379–386. doi: 10.1016/j.cmet.2006.04.004
Sun, Y., Butte, N. F., Garcia, J. M., and Smith, R. G. (2008). Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 149, 843–850. doi: 10.1210/en.2007-0271
Sun, Y., Wang, P., Zheng, H., and Smith, R. G. (2004). Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. U.S.A. 101, 4679–4684. doi: 10.1073/pnas.0305930101
Toshinai, K., Date, Y., Murakami, N., Shimada, M., Mondal, M. S., Shimbara, T., et al. (2003). Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology 144, 1506–1512. doi: 10.1210/en.2002-220788
Verhagen, L. A., Egecioglu, E., Luijendijk, M. C., Hillebrand, J. J., Adan, R. A., and Dickson, S. L. (2011). Acute and chronic suppression of the central ghrelin signaling system reveals a role in food anticipatory activity. Eur. Neuropsychopharmacol. 21, 384–392. doi: 10.1016/j.euroneuro.2010.06.005
Walker, A. K., Ibia, I. E., and Zigman, J. M. (2012). Disruption of cue-potentiated feeding in mice with blocked ghrelin signaling. Physiol. Behav. 108, 34–43. doi: 10.1016/j.physbeh.2012.10.003
Wang, H. J., Geller, F., Dempfle, A., Schauble, N., Friedel, S., Lichtner, P., et al. (2004). Ghrelin receptor gene: Identification of several sequence variants in extremely obese children and adolescents, healthy normal-weight and underweight students, and children with short normal stature. J. Clin. Endocrinol. Metabol. 89, 157–162. doi: 10.1210/jc.2003-031395
Wei, W., Qi, X., Reed, J., Ceci, J., Wang, H. Q., Wang, G., et al. (2006). Effect of chronic hyperghrelinemia on ingestive action of ghrelin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R803–R808. doi: 10.1152/ajpregu.00331.2005
Willesen, M. G., Kristensen, P., and Romer, J. (1999). Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70, 306–316. doi: 10.1159/000054491
Wortley, K. E., Anderson, K. D., Garcia, K., Murray, J. D., Malinova, L., Liu, R., et al. (2004). Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc. Natl. Acad. Sci. U.S.A. 101, 8227–8232. doi: 10.1073/pnas.0402763101
Wortley, K. E., Del Rincon, J. P., Murray, J. D., Garcia, K., Iida, K., Thorner, M. O., et al. (2005). Absence of ghrelin protects against early-onset obesity. J. Clin. Invest. 115, 3573-3578. doi: 10.1172/JCI26003
Wren, A. M., Seal, L. J., Cohen, M. A., Brynes, A. E., Frost, G. S., Murphy, K. G., et al. (2001). Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992. doi: 10.1210/jc.86.12.5992
Yamada, G., Ariyasu, H., Iwakura, H., Hosoda, H., Akamizu, T., Nakao, K., et al. (2010). Generation of transgenic mice overexpressing a ghrelin analog. Endocrinology 151, 5935–5940. doi: 10.1210/en.2010-0635
Yang, J., Brown, M. S., Liang, G., Grishin, N. V., and Goldstein, J. L. (2008). Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396. doi: 10.1016/j.cell.2008.01.017
Zhang, W., Chai, B., Li, J. Y., Wang, H., and Mulholland, M. W. (2008). Effect of des-acyl ghrelin on adiposity and glucose metabolism. Endocrinology 149, 4710–4716. doi: 10.1210/en.2008-0263
Zhao, T. J., Liang, G., Li, R. L., Xie, X., Sleeman, M. W., Murphy, A. J., et al. (2010a). Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. U.S.A. 107, 7467–7472. doi: 10.1073/pnas.1002271107
Zhao, T. J., Sakata, I., Li, R. L., Liang, G., Richardson, J. A., Brown, M. S., et al. (2010b). Ghrelin secretion stimulated by {beta}1-adrenergic receptors in cultured ghrelinoma cells and in fasted mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15868–15873. doi: 10.1073/pnas.1011116107
Zigman, J. M., and Elmquist, J. K. (2003). Minireview: from anorexia to obesity–the yin and yang of body weight control. Endocrinology 144, 3749–3756. doi: 10.1210/en.2003-0241
Zigman, J. M., Jones, J. E., Lee, C. E., Saper, C. B., and Elmquist, J. K. (2006). Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494, 528–548. doi: 10.1002/cne.20823
Keywords: hedonic eating, homeostatic eating, GOAT, GHSR
Citation: Uchida A, Zigman JM and Perelló M (2013) Ghrelin and eating behavior: evidence and insights from genetically-modified mouse models. Front. Neurosci. 7:121. doi: 10.3389/fnins.2013.00121
Received: 29 April 2013; Paper pending published: 15 May 2013;
Accepted: 25 June 2013; Published online: 16 July 2013.
Edited by:
Alfonso Abizaid, Carleton University, CanadaReviewed by:
Julie A. Chowen, Hospital Infantil Universitario Niño Jesús, SpainStephanie Borgland, University of British Columbia, Canada
Copyright © 2013 Uchida, Zigman and Perelló. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Jeffrey M. Zigman, Department of Medicine, The University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, MC 9077, Dallas, TX 75390-9077, USA e-mail: jeffrey.zigman@utsouthwestern.edu
Mario Perelló, Laboratory of Neurophysiology of the Multidisciplinary Institute of Cell Biology [Argentine Research Council (CONICET) and Scientific Research Commission, Province of Buenos Aires (CIC-PBA)], Calle 526 S/N entre 10 y 11-PO Box 403, La Plata, Buenos Aires 1900, Argentina e-mail: marioperello@yahoo.com
†These authors have contributed equally to this work.