 Mohammad A. Khanfar
Mohammad A. Khanfar Anna Affini
Anna Affini Kiril Lutsenko
Kiril Lutsenko Katarina Nikolic
Katarina Nikolic Stefania Butini
Stefania Butini Holger Stark
Holger Stark- 1Stark Lab, Institut fuer Pharmazeutische and Medizinische Chemie, Heinrich-Heine-Universitaet Duesseldorf, Duesseldorf, Germany
- 2Faculty of Pharmacy, The University of Jordan, Amman, Jordan
- 3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Belgrade, Belgrade, Serbia
- 4Department of Biotechnology, Chemistry, and Pharmacy, European Research Centre for Drug Discovery and Development, University of Siena, Siena, Italy
With the very recent market approval of pitolisant (Wakix®), the interest in clinical applications of novel multifunctional histamine H3 receptor antagonists has clearly increased. Since histamine H3 receptor antagonists in clinical development have been tested for a variety of different indications, the combination of pharmacological properties in one molecule for improved pharmacological effects and reduced unwanted side-effects is rationally based on the increasing knowledge on the complex neurotransmitter regulations. The polypharmacological approaches on histamine H3 receptor antagonists on different G-protein coupled receptors, transporters, enzymes as well as on NO-signaling mechanism are described, supported with some lead structures.
Introduction
The idea of synthesizing multiple targeting compounds arises from the fact that the paradigm “one drug—one target” or “single-target drug” is not sufficiently meeting the need for the treatment of a large number of complex diseases caused by multifunctional pathophysiological processes. Since central nervous system (CNS) disorders are characterized by diverse physiological dysfunctions and deregulations of a complex network of signaling pathways, optimal multipotent drugs should simultaneously and specifically modulate selected groups of biological targets. Polypharmacology is a new scientific area focused on discovery, development, and pharmacological study of Multiple Targeting Designed Ligands (MTDL) able to simultaneously modify the activities of several interacting pharmacological targets (Hopkins, 2008).
This emerging approach suggests that multifactorial CNS diseases such as depression (Millan, 2014), schizophrenia (Ye et al., 2014), Parkinson's disease (PD) and Alzheimer's disease (AD; Youdim and Buccafusco, 2005; Leon et al., 2013) can be treated with higher efficacy, lower toxicity, less drug-drug interactions, and also with unified pharmacokinetic profile if a single drug molecule is able to simultaneously interact with multiple targets (Anighoro et al., 2014; Huang et al., 2015).
Despite the positive effects of MTDL, there are several potential disadvantages, which need to be taken into consideration. In order to identify multiple targeting hits, a more detailed and extensive pharmacological characterization of current drug-target interactions is needed (Peters, 2013). In most previous cases, the need for a polypharmacology to reach a therapeutic effect is discovered retrospectively. After finding a lead compound for a specific group of targets, the optimization of complex structure-activity relationships (SAR) profile is one of the first challenging tasks from a medicinal chemistry point of view. Most importantly, simultaneous targeting of several receptors may lead to a wider and sometimes unpredictable spectrum of biological activities such as side effects. Therefore, a balance between polypharmacological benefits and potential drawbacks brought by promiscuous scaffolds needs to be evaluated at least as carefully as with all other candidates, but based on a more complex behavior (Anighoro et al., 2014). Herein we describe the current implementation of target-oriented polypharmacological approaches with histamine H3 receptor (H3R) ligands based on research findings (Figure 1).
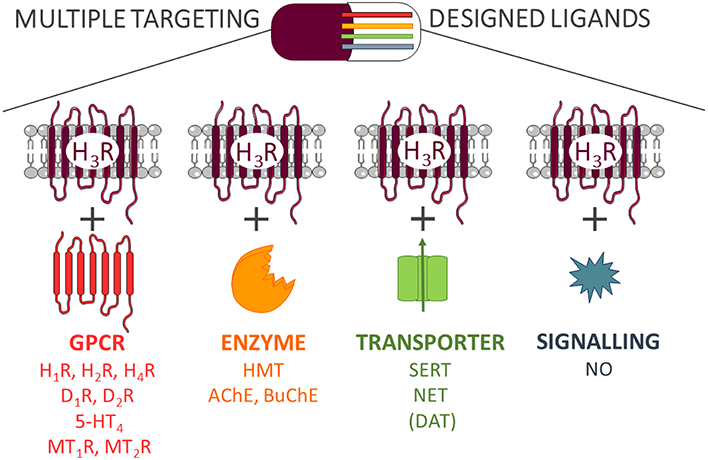
Figure 1. Multi-targeting designed ligands with H3R.
H3R is a member of transmembrane class A of G protein–coupled receptors (GPCR) family (Arrang et al., 1983; Schwartz et al., 1991). It influences several intracellular pathways through its coupling to Gαi/o (Bongers et al., 2007). Analysis of H3R mRNA in rat (Héron et al., 2001) and human (Jin and Panula, 2005) brains showed that H3R is largely expressed on the histaminergic neurons of the CNS (located presynaptically and postsynaptically; Jadhav and Singh, 2013). As auto-receptor, H3R plays an important role in histamine biosynthesis and release and as hetero-receptor in the modulation of different neurotransmitters release (e.g., acetylcholine, noradrenaline, dopamine, GABA, glutamate, and serotonin; Schlicker et al., 1989, 1990). A lower level of H3R is distributed in the peripheral nervous system and is responsible for the regulation of sympathetic effector systems and pain sensation (Héron et al., 2001). Therefore, modulation of the H3R can potentially prevent the activation of the negative feedback mechanism leading to increased neurotransmitter release. Consequently, targeting of H3R with antagonist/inverse agonist may have therapeutic applications in CNS-related disorders, such as depression, schizophrenia, PD, and AD (Esbenshade et al., 2008; Gemkow et al., 2009; Chazot, 2010; Raddatz et al., 2010; Lin et al., 2011; Ghasemi and Tavakoli, 2012) as well as in inflammatory and gastrointestinal diseases (Vuyyuru et al., 1995; Ceras et al., 2012). Recently, several substances have entered late clinical phases for the treatment of several CNS disorders (Sander et al., 2008; Panula et al., 2015).
H3R/H1R
The drug Betahistine (N-methyl-2-(2-pyridyl)ethanamine), indicated for the treatment of vestibular Morbus Menière, can be considered as the first MTDL in this category by working as an agonist at histamine H1 receptor (H1R) and antagonist at H3R (Lian et al., 2014, 2016; Møller et al., 2015). The H3R antagonism leads to inhibition of vestibular neurotransmission, central vasodilatation with potential antipsychotic effects, whereas the H1R agonism have an immune-regulatory effect (Dagli et al., 2008; Zhou et al., 2013).
Currently, the main focus on polypharmacological targeting of H3R/H1R is to develop dual agonist or dual antagonist ligands. Dual acting H3/H1 receptor (H3R/H1R) antagonists were synthesized for the treatment of allergic diseases. These diseases are associated with the degranulation of the mast cell and histamine release which can activate H1R and consequently stimulates phospholipase C that ultimately liberate inositol-1,4,5-trisphosphate and Ca2+; thereby improves mucus secretion and vasodilatation (McLeod et al., 1999; Bakker et al., 2002).
H1R antagonists play a key role in the treatment of allergic rhinitis; however, there are several limitations to their clinical use. The first generation of H1R antagonists (e.g., Diphenhydramine, Chlorpheniramine) show sedative effects whereas second generation H1R blockers (e.g., Loratadine, Mizolastine) have poor penetration to the CNS, thus generating non-sedating antihistaminic activity (Cowart et al., 2004; Stark et al., 2004). However, the second generation H1R blockers are often combined with α-adrenergic agonists to stimulate normal vascular tone and to reduce nasal congestion. Such combination is associated with serious cardiac side effects (QT time prolongation, ventricular arrhythmia).
These findings have encouraged several research groups to consider if other histamine receptor subtypes may contribute to the histamine-induced nasal congestion. Several studies confirmed that H3R may play an important role in histamine-induced nasal congestion because the vasodilatation is caused by activation of H3R in peripheral post-sympathetic ganglionic neurons (Hey et al., 1992). The activation of the H3R hetero-receptors located on neighboring noradrenergic neurons (Berlin et al., 2011) modulates the release of the neurotransmitter noradrenaline in the nasal blood vessel. Therefore, a compound that antagonizes H1R on one hand and inhibits H3R on the other hand may treat allergic diseases without having nasal congestion.
Based on first and second generations of H3Rantagonists, imidazole and non-imidazole H3R/H1R ligands were designed. Several imidazole-derivatives taking Chlorphenamine 1 (hH1R Ki = 2 nM) as an additional pharmacophore for the introduction of H1R antagonist activity show dual H3R/H1R inhibitory affinity (Wieland et al., 1999). Limited variations of the linker in both sides of the aliphatic amino moiety provided compounds with good H3R binding affinity. Like all aminergic GPCR, H1R, and H3R contain an aspartate residue in the transmembrane domain III, that is involved in electrostatic interaction with protonated amino functionality (Wieland et al., 1999). Therefore, replacement of the basic amino linker by a neutral linker such as amide or urea, resulted in activity loss on the H1R. However, incorporating a tertiary amine led to the synthesis of the most potent dual inhibitor in that series (compound 2, Figure 2) that displays affinities at low nanomolar concentration range for both H1R and H3R.
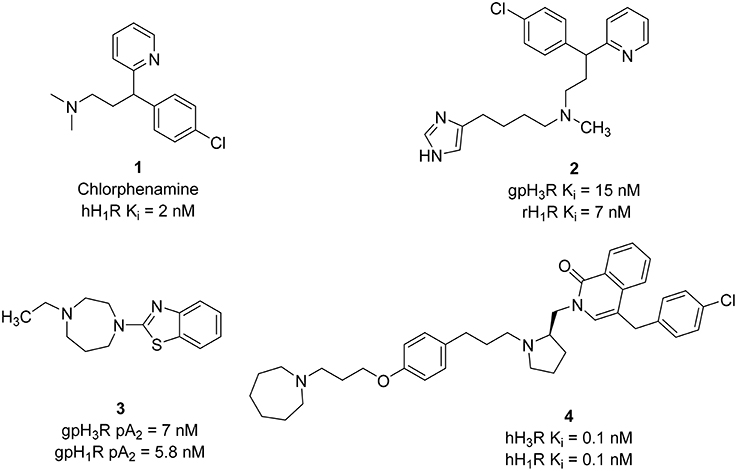
Figure 2. Structures and biological activities of selected H3R/H1R ligands.
Further structural optimization was conducted by replacing the imidazole ring with different heterocycles in order to avoid potential interactions with CYP450 enzymes. In one of the trials, the non-imidazole heterocycles were combined with a benzothiazole structure (Walczyński et al., 1999). In vitro results of this series from guinea pig ileum system showed increasing H3R antagonist potency in the presence of an alkyl-substituted azepane (compound 3, Figure 2). However, this compound showed weak H1R antagonist activity, with pA2 value of 5.77. A similar approach was applied in designing H3R/H1R dual inhibitors by combining nitrogen-containing heterocycles, with a benzylphthalazinone (GSK-1004723), compound 4 (Figure 2), or a quinoline structure (GSK-835726) (Slack et al., 2011; Daley-Yates et al., 2012), and WO-094643 (Norman, 2011). Compounds 4 and GSK-835726 were potent H3R/H1R antagonists in vitro and in vivo systems. Compound 3 has a major advantage associated with its long duration of action (t1/2 of 1.2–1.5 h, Table 1) which allows once a day intranasal dosing for the treatment of allergic rhinitis. GSK–1004723 completed phase II of clinical trials for the treatment of allergic rhinitis.

Table 1. Selected pharmacokinetic data of preclinical candidates (Ly et al., 2008; Slack et al., 2011; Daley-Yates et al., 2012).
H3R/H2R
Limited efforts have been conducted so far for the designing of dual H3R/H2R ligands. However, guanidine-based histamine H2R ligands demonstrate additional H3R antagonist potencies. Recently, Buschauer et al. investigated dimeric carbamoylguanidine derivatives for the synthesis of potent H2R agonists (Kagermeier et al., 2015). Compounds containing two imidazole moieties, display selectivity for H3R and H2R in radioligand competition binding studies, whereas compound 5 (Figure 3) shows high H2R affinity with simultaneous high H3R inhibitory affinity. Since the brain penetration of these compounds is quite low, they can mostly be used on cells and isolated tissues.
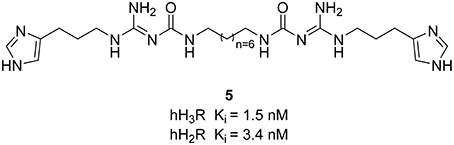
Figure 3. Structures and biological activities of selected H3R/H2R ligands.
H3R/H4R
Similarly, dual targeting is also often applied on histamine H3R and H4 receptors (H4R). Because of the relative high H4R homology with H3R (37% in entire sequence, 68% within transmembrane domains) many potent histamine H3R ligands containing imidazole moieties (6–8; Figure 4) show off-target affinity at H4R (Neumann et al., 2013). The human H4R is the last receptor subtype that has been identified in the histamine receptor family (Corrêa and Fernandes, 2015). The H4R is mainly located on cells of hematopoietic origin and, therefore, may be a promising target for the treatment of inflammatory diseases like allergic rhinitis, asthma, and pruritus (Thurmond et al., 2008). The expression of H4R in the CNS is a controversial topic because immunostaining methods are critically discussed and inconsistent mRNA screening results were obtained (Panula et al., 2015). Dual H3R/H4R ligands could be promising targets for pain and cancer since it is likely that these two targets contribute to the development of pain sensation and itching as well as cell-proliferation-associated effects (Medina and Rivera, 2010). However, further investigation is required to fully understand and evaluate their functions for therapeutic applications.
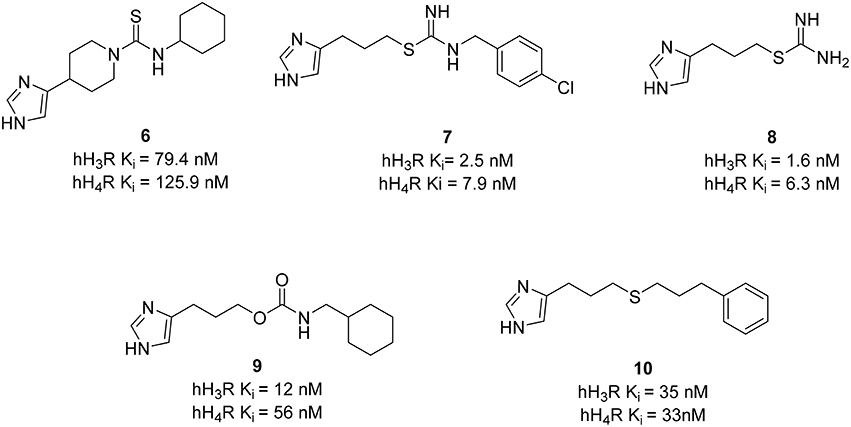
Figure 4. Structures and biological activities of selected H3R/H4R ligands.
Clobenpropit (7), a potent reference H3R antagonist, was identified as a template for dual H3R/H4R ligands. Variations in substituents of the phenyl moiety as well as in the length of the alkyl chain between the central core isothiourea and the lipophilic aromatic residue were performed (Lim et al., 2009). Elongation of the spacer and introduction of bulky groups in the east part of these molecules such as diphenyl residue led to moderate affinity for both H3R and H4R. Nevertheless, most of these compounds showed moderate to high affinity at both H3R and H4R in a similar concentration range [human H3 receptor (hH3R) Ki=2.5–79.4, hH4R Ki= 1.6–158.4]. Compounds with a halogen substituent at the 4-position of the benzyl moiety showed the best binding affinities at both receptors. Further structural modifications were performed to expand the SAR on imidazole-containing histamine receptor ligands. Changing the polarity of the central core isothiourea by introducing different moieties such as amide, carbamate, urea, ester, ketone, and ethers was exploited (Kottke et al., 2011). Amide derivatives were unsuccessful because they had poor affinity at the hH3R. In contrast, all the other moieties bound to both receptors in a comparable concentration range, showing that these central cores of the alkyl imidazole can be used as a lead structure for dual acting H3R/H4R ligands. Among the carbamate series, the presence of a cycloalkyl moiety in the east part is important to have Ki values for both receptors below 200 nM. Cyclohexylmethyl derivative 9 (Figure 4) is the most potent H3R/H4R antagonist in that series (Wicek et al., 2011). It must be stressed that the affinity is not the only criteria for the MTDL selection. Some compounds may have similar affinities, but different efficacies. In this respect, replacing the carbamate function with a thioether group led to the synthesis of a potent dual H3R antagonist and H4R partial agonist 10 (Figure 4). These compounds are potent dual H3R/H4R ligands that can be optimized for further pre-clinical trials; however, no further work has been reported. Therefore, efficacy and not only affinity data has to be considered for the pharmacological profile evaluation of new drugs.
H3R and Non-Histaminergic GPCRs
In addition to combined properties with other histamine receptor subtypes, other aminergic GPCRs have also been addressed with polypharmacological targeting of H3R. Dopamine is an important neurotransmitter in the human brain. It affects almost all mental functions, such as movement control, motivation, emotion, learning, and memory. Dysregulations of dopamine neurotransmitter system of the CNS may cause schizophrenia and related mood disorders (Schlicker et al., 1993; Witkin and Nelson, 2004). Neuroleptics used for the treatment of schizophrenia usually inhibit dopamine D2-like receptors and other aminergic receptors, such as serotonin 5-HT2A receptor, dopamine D1 receptor (D1R) receptors, and other serotonin receptor subtypes (Remington, 2003). The most important side effects of these neuroleptics are extrapyramidal side effects and weight gain problems (Vuyyuru et al., 1995; Deng et al., 2010; Lian et al., 2016). These side effects are related to their antagonistic properties at the dopamine D2-like and H1R, respectively (Kroeze et al., 2003; Von Coburg et al., 2009). Additionally, schizophrenic patients usually showed a significantly high level of N-methylhistamine in cerebral cerebrospinal fluid (Ligneau et al., 2007). There are several studies showing an interaction between histamine H3R and dopamine D2 receptors (D2R) as well as H3R and D1R as oligomeric hetero-receptors (Humbert-Claude et al., 2007; Ferrada et al., 2008). Furthermore, H3R inverse agonists/antagonists showed a reduction of undesirable side effects like weight gain, somnolence, and cognitive impairment in several rodent models of schizophrenia while displaying a significant inhibitory activity (Ligneau et al., 2007). Combining the known H3R antagonists pharmacophore 4-(3-piperidinopropoxy)phenyl with known neuroleptics may provide novel multi-acting antipsychotic drugs with an improved pharmacological profile and reduced side effects by decreasing H1R affinity and introducing H3R activity while maintaining D2R/D3R affinity (Humbert-Claude et al., 2007; Von Coburg et al., 2009). For this approach 4-(3-piperidinopropoxy)phenyl was linked to several known neuroleptics. Resulting compounds showed high H3R affinity with Ki values between 4.90 nM and 42 pM while simultaneously reduced the H1R affinity by a factor of 10–600 as off-target and maintained the D2-like receptor subtypes affinity (Figure 5; Deng et al., 2010). Compound 11 (Figure 5) with a good overall profile and high H3R affinity was synthesized by merging 4-(3-piperidinopropoxy)phenyl fragment with amitriptyline 12 (Figure 5). This compound was selected for an early in vivo screening for central H3R antagonist potency on male Swiss mice. To determine the in vivo potency, an increase in N-methylhistamine level in the brain 90 min after the oral application of the compound was measured (Von Coburg et al., 2009). Unfortunately, this compound seems to be inactive (ED50 > 10 mg/kg p.o.) with unclear reasons mostly for absorption, distribution, or metabolism.
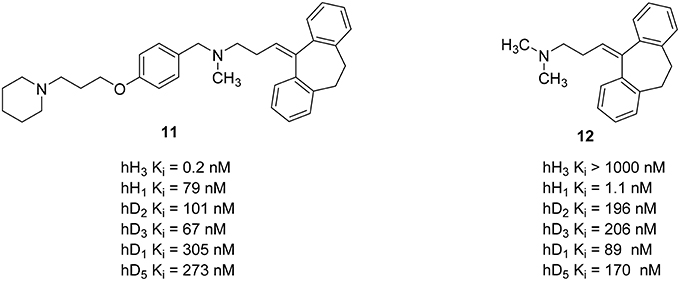
Figure 5. Structures and biological activities of selected H3R/D2R ligands.
Using pharmacophore-based virtual screening, Lepailleur et al. identified an interesting additional target activity while analyzing the screening hits (Lepailleur et al., 2014). A series of tricyclic derivatives have high serotonin 5-HT4 receptor (5-HT4R) affinity. There is a connection between different serotonin receptor subtypes, especially on 5-HT1AR, 5-HT4R, and 5-HT6R and emerging AD therapies (Sabbagh, 2009; Mangialasche et al., 2010; Herrmann et al., 2011) and other degenerative disorders connected to an impaired cholinergic function (Esbenshade et al., 2008; Sander et al., 2008; Gemkow et al., 2009). 5-HT4R provide symptomatic alleviation of cognitive impairments and neuroprotection by reducing amyloid-β (βA) generation and toxicity (Lezoualc'h, 2007). 5-HT4R activation improves cognitive processes such as learning and memory (Lelong et al., 2001, 2003; Levallet et al., 2009; Hotte et al., 2012). Combined with the beneficial effects of H3R on neurodegenerative diseases, dual targeting of H3R and 5-HT4R would therapeutically be useful. One of the identified hits, compound 13 (Figure 6) showed high affinities with Ki values of 41.6 nM at H3R and 208 nM at 5-HT4R and significant selectivity over 5-HT1AR and 5-HT6R. Compound 13 was able to reverse the scopolamine-induced cognitive impairment partially at 1 mg/kg and completely at 3 mg/kg in a spatial working memory experiment (Klinkenberg and Blokland, 2011). Scopolamine is a nonselective muscarinic antagonist, which partially blocks the cholinergic neurotransmission and is used to examine the cognitive enhancing effects of potential compounds (Snyder et al., 2005; Fredrickson et al., 2008). These results reveal the potential of combined H3R antagonist/5-HT4R agonist profiles in one multi-targeting compound to modify symptomatic effects in Alzheimer's disease.
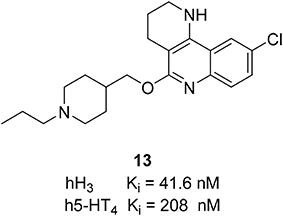
Figure 6. Structure and biological activity of the selected H3R/5-HT4 ligand.
Recently, different combinations between melatonin and another neuroprotection agent, e.g., curcurmin derivatives, have shown that melatonin may have a therapeutic potential in the treatment of cognitive disorders and neurodegenerative pathologies like AD (Chojnacki et al., 2014). Different H3R antagonists also showed neuroprotective actions (Brioni et al., 2011). Therefore, the synthesis of ligands able to bind at both H3R and melatonin receptors could be useful for the treatment of the diseases mentioned above. Pala et al. have synthesized compounds that can interact simultaneously with the H3R and melatonin T1 receptor (MT1R) and melatonin T2 receptor (MT2R; Pala et al., 2014). Melatonin is a methoxyindole-derived hormone secreted mainly by the pineal gland. The activation of MT1R and MT2R is not only important for the regulation of cardiac rhythms, but also for having antioxidant and neuroprotective effects (Srinivasan et al., 2006). For the synthesis of this melatonergic/histaminergic ligands the classical pharmacophore showed for potent H3R antagonists such as Ciproxifan and its analogs, was combined with an anilinoethylamide to have comparable binding affinity with the indol-3-ylrthylamide moiety of the melatonin (Figure 7). The length of the alkyl chain influences more the binding affinities at hMT1R and hMT2R than that at hH3R. Compounds with a short spacer such as a propyl or ethyl chain did not show affinity toward both MT1R and MT2R. One good dual acting ligand was obtained by elongating the alkyl chain between the imidazole ring and the melatonin moiety with a pentyl linker. The introduction of a six methylene unit improved the Ki values for both hMT1R and hMT2R. The elongation of the spacer can store the imidazole in a more peripheral region of the melatonin receptors. In that region, negative interactions with positively charged amino groups are weakened. Therefore, compounds (14, 15; Figure 7) able to bind to both melatonin and histamine H3R with affinity in the micromolar concentration range were designed. The optimization of these ligands can be the next step for discovering new multiple targeting compounds that belong to the new melatonin-histamine combination.
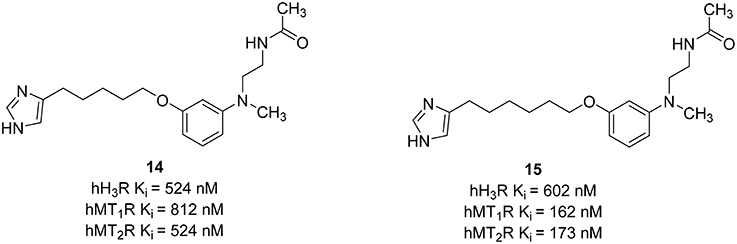
Figure 7. Structures and biological activities of selected H3R/melatonin receptor ligands.
H3R and Transporters
Selective serotonin reuptake inhibitors (SSRI) have been the drugs of choice to treat depression. However, the efficacy of these drugs is noticeable only after weeks of treatment and do not improve cognitive functions of depressive patients, which prompt many physicians to co-prescribe stimulants with SSRI to provide subjective relief. H3R antagonists produce wakefulness in animals without releasing dopamine or producing behavioral activation. Such activation has been avoided due to the risk of allowing patients to act on their suicidal ideation (Menza et al., 2000; Stahl, 2001). Combined H3R/SERT inhibition would provide symptomatic relief for the fatigue during the first weeks of treatment and afford immediate relief from some of the symptoms of depression with possible concurrent cognitive enhancement (Schlicker et al., 1998; Barbier et al., 2007; Nikolic et al., 2014).
Until now, most of the medicinal chemistry effort to develop new dual H3R/SERT inhibitors was conducted by Johnson & Johnson Pharmaceutical Research and Development group. Their effort was started with the identification of lead compounds with desirable SERT affinity, which could then be used as a template to introduce H3R antagonist activity. Two SSRI templates were designated, the first based on fluoxetine, which is the third most prescribed antidepressant drug (16, Figure 8; Wong et al., 1995), and the second based on the hexahydropyrroloisoquinoline scaffold represented by JNJ-7925476 (17, Figure 8), identified by high-throughput screening (Aluisio et al., 2008). Four templates of potent and selective H3R antagonists were considered to develop dual H3R/SERT inhibitors evaluated pre-clinically (18–21, Figure 8; Letavic et al., 2006). Starting from fluoxetine template, the tertiary benzyl amines of 18–21 were replaced with the fluoxetine template, so that the known SSRI would serve as both, the lipophilic core and one of the basic amines. Several H3 amine side moieties were initially 3- or 4-substituted on both phenylene rings of fluoxetine (rings A and B). All the regioisomers had high affinity for the hH3R, but the 3-piperidinyl-propyloxy derivative provided the highest affinity for both the rat serotonin transporter (rSERT) and human serotonin transporter (hSERT) (e.g., compound 22, Figure 8; Stocking et al., 2007). The 4-(trifluoromethyl) substituted phenoxy (B) ring derivatives have no discrepancy between rSERT and hSERT, however, a decrease in affinity for hSERT over rSERT was observed for the unsubstituted derivatives. Electron donating substituents on B ring is associated with 5 to 30-fold decrease in hSERT affinity, however, electron withdrawing substituents displayed a good correlation between rSERT and hSERT (Stocking et al., 2007).
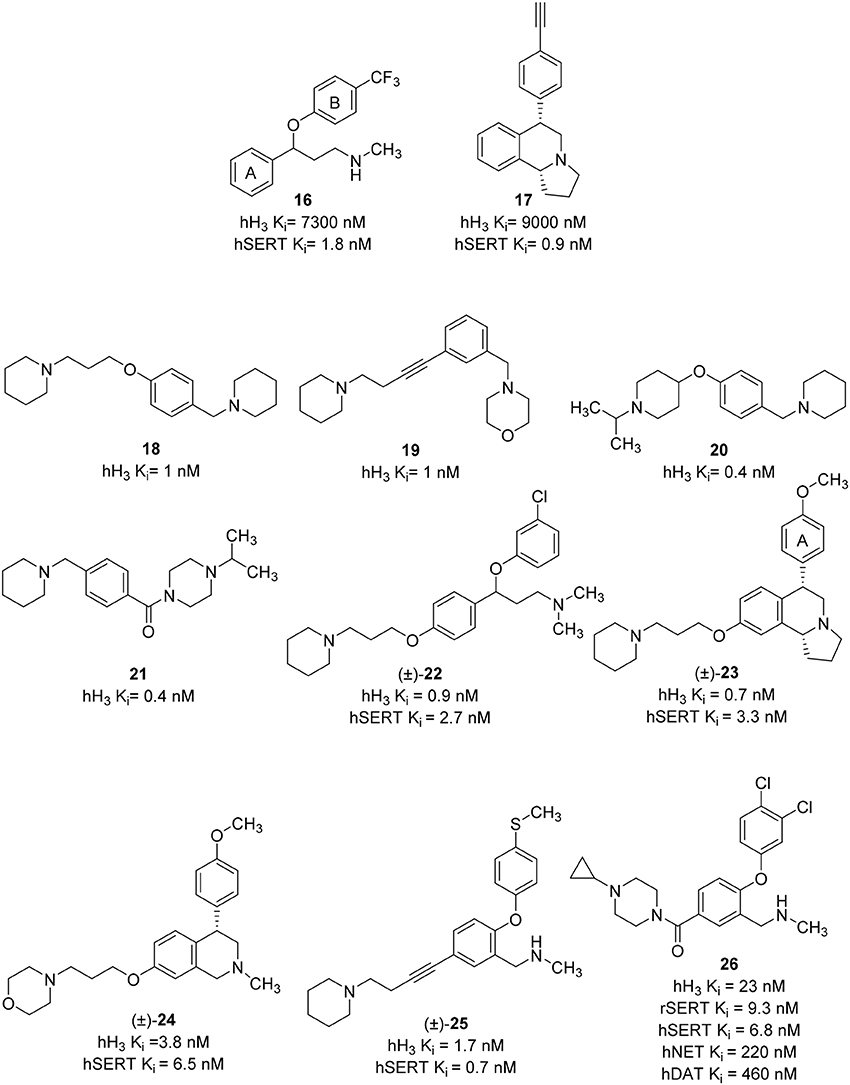
Figure 8. Structures and biological activities of selected H3R/SERT ligands.
The same approach was applied for designing of hexahydropyrroloisoquinolines-derived dual H3R antagonists and SERT inhibitors. The overlap of the H3R antagonist 17 and SERT inhibitor 16 was pictured as exemplified in compound 23 (Figure 8). This approach generated a series of high affinity H3R antagonists with the SERT affinity dependent on aryl ring (A) substitution. Nevertheless, unlike the fluoxetine scaffold, most simple substitutions on the aryl ring (A) of the hexahydropyrroloisoquinoline scaffold provided similar rSERT and hSERT affinity (Keith et al., 2007c). On the other hand, the hydroxyl and the heterocyclic derivatives displayed a slightly higher affinity for rSERT than hSERT. Two high affinity compounds, the 4-methoxy derivative and the 3-pyridyl derivatives demonstrated good in vivo activities in serotonin potentiated head twitch model for SERT inhibition and blockade of imetit-induced drinking model for the H3R inhibition. However, this series showed unsatisfactory pharmacokinetics with low oral bioavailability, long t1/2 and a slow onset of action. In addition, these structures still retained affinity for the dopamine transporter (DAT; Keith et al., 2007c). Consequently, simpler templates from hexahydropyrroloisoquinoline were attempted, initially, by removal of the fused pyrrolidine ring and one chiral center to obtain the tetrahydroisoquinolines (Letavic et al., 2007a). Structural optimization of tetrahydroisoquinolines derivatives was conducted using a large number of amines in order to improve the binding affinity at H3R, varying the physical properties of the resulting compounds and maintaining SERT affinity (Keith et al., 2007b). Several modifications were attempted on the pendant piperidine ring; morpholine and substituted piperidines usually resulted in high affinity compounds. Replacing the piperidine with piperazine afforded compounds that have variable affinity for the hH3R, depending greatly on the basicity of the terminal nitrogen. For example, small alkyl substituents on the piperazine provided compounds with high affinity for the H3R, but decreasing the basicity of the terminal nitrogen by addition of bulky groups lowered the affinity for the H3R. Among the large number of derivatives that were synthesized, compound 24 (Figure 8), which was afforded by removal of the pyrrolidine ring of 23 together with the replacement of the piperidine ring with a morpholine, has improved rat pharmacokinetics and improved pharmacodynamics with a head twitch response (Keith et al., 2007a).
Further simplification was conducted by removing one carbon on the tetrahydroisoquinoline, which deleted the last remaining stereocenter to provide the benzyl amine derivatives (e.g., 25, Figure 8). The benzylic carbon of tetrahydroisoquinolines was replaced with an oxygen in order to improve overall physical properties (Letavic et al., 2007b). The 3-piperidinyl-propyloxy derivatives were not used in this series; instead, they used the alkyne and amide side chains corresponding to the known H3R antagonists 19 and 21. The later modification was important to avoid any potential metabolic problems associated with 1,4-hydroxyquinone. The SAR of alkynes was generally similar to that of the tetrahydroisoqinolines and most of the compounds have high affinity toward H3R and SERT. Selected compounds had good brain penetration in rat with brain levels of above 1 μM when dosed at 10 mg/kg p.o. (Letavic et al., 2007b). The benzamides benzyl amine derivatives were very potent with good selectivity over the norepinephrine transporter (NET) and DAT. One of the compounds, 26 (Figure 8), was extensively profiled in vivo and was found to have good rat pharmacokinetic and pharmacodynamics properties (Table 1; Ly et al., 2008). Although not yet tested on humans, inhibition of the H3R makes it an attractive combination with SERT blockade in order to create a novel antidepressant treatment.
The serotonin/norepinephrine reuptake inhibitor (SNRI) duloxetine 27 (Figure 9) is used in therapeutic off-label treatment of neuropathic pain (Fishbain et al., 2006). The inhibition of NE uptake is essential for the pain efficacy (Leventhal et al., 2007). H3R antagonists Thioperamide 6 and GSK-189254 28 (Figure 9) have been reported to be active in models of pain (Farzin et al., 1994; Medhurst et al., 2008). Using these results Altenbach et al. designed a series of molecules combining pharmacophores of H3R antagonism and NET inhibition in one molecule. An H3R pharmacophore was linked to duloxetine analogs, cf. 28 (Figure 9). Resulting compounds 29–31 (Figure 9) showed low nanomolar affinity at H3R and NET, where 29 additionally had SERT affinity (Ki = 7.6 nM) comparable to that of 28 (Ki = 2.4 nM; Bymaster et al., 2003). This affinity was reduced to Ki > 70 nM in compounds 30, and 31 providing a better selectivity. Compound 29 was also found to be potent in osteoarthritis pain model in rats with efficacies of 70 and 93% at doses of 3 and 10 mg/kg, respectively (Anighoro et al., 2014).
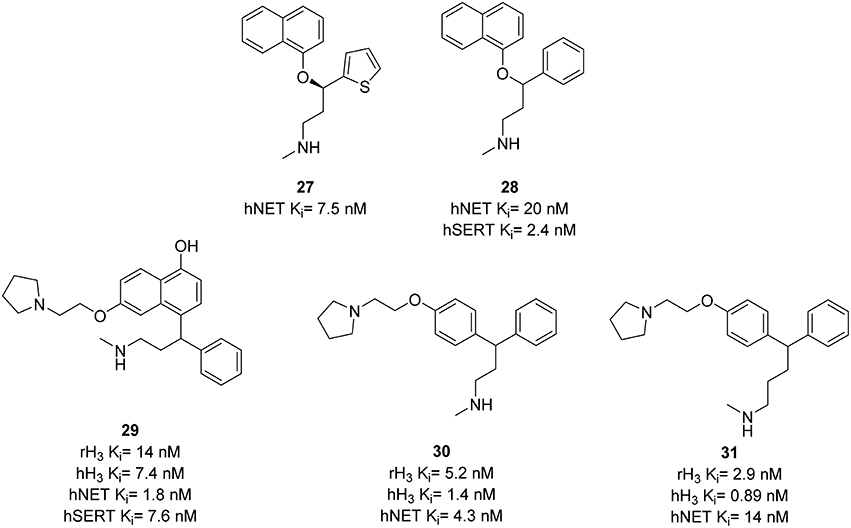
Figure 9. Structures and biological activities of selected H3R/NET ligands.
H3R and Enzymes
Histamine level in the CNS is controlled not only by the receptors but also by the inactivating enzyme histamine N-methyltransferase (HMT; Parsons and Ganellin, 2006). Ligands with dual inhibitory activities on both H3R and HMT could increase intersynaptic histamine levels in the CNS and may lead to beneficial procognitive effects in psychiatric and neurodegenerative diseases (Apelt et al., 2002; Sander et al., 2008). Even if they have low or missing in vivo activity, such ligands could greatly enhance histaminergic neurotransmission via inhibition of histamine H3 auto-receptors and reduce the catabolic rate for histamine degradation via HMT inhibition (Grassmann et al., 2003).
Most of the HMT inhibitors have a 4-aminoquinoline moiety in common (e.g., tacrine, 32, Figure 10). Therefore, the synthetic effort to develop novel and dual H3R\HMT inhibitors started from coupling of different 4-aminoquinolines with different spacers to the piperidine, the basic component that is essential for binding at the H3R. Variation of the spacer structure provides two different series of compounds. The first series have an alkylene spacer separating the basic center from the 4-aminoquinoline. These compounds showed potent HMT inhibitory activities with moderate to high H3R affinity. The second series, which possessed a p-phenoxypropyl spacer, showed a strong inhibitory activity on HMT and the H3R affinity, exceeding that of the first series. One of the compounds, FUB 836 (33, Figure 10), combines a high H3R affinity with a high HMT inhibitory activity and exhibited high H3R selectivity when compared to H1R and H2R (Apelt et al., 2002). Similar approach was applied in designing H3R/HMT dual inhibitors by combining imidazole heterocycle, which is an integral part of potent H3R antagonists, with several aromatic carbo- or heterocyclic structures (e.g., aminoquinoline or tetrahydroacridine moieties) of standard HMT inhibitors by different alkyl and alkenyl spacers. One interesting compound, 34 (Figure 10), showed a high H3R affinity with a high HMT inhibitory activity (Grassmann et al., 2003). Replacing imidazole head with a piperidine ring accompanied by a methylation of the amino functionality improved the inhibitory activity against HMT (e.g., compounds 35 and 36; Figure 10; Grassmann et al., 2004).
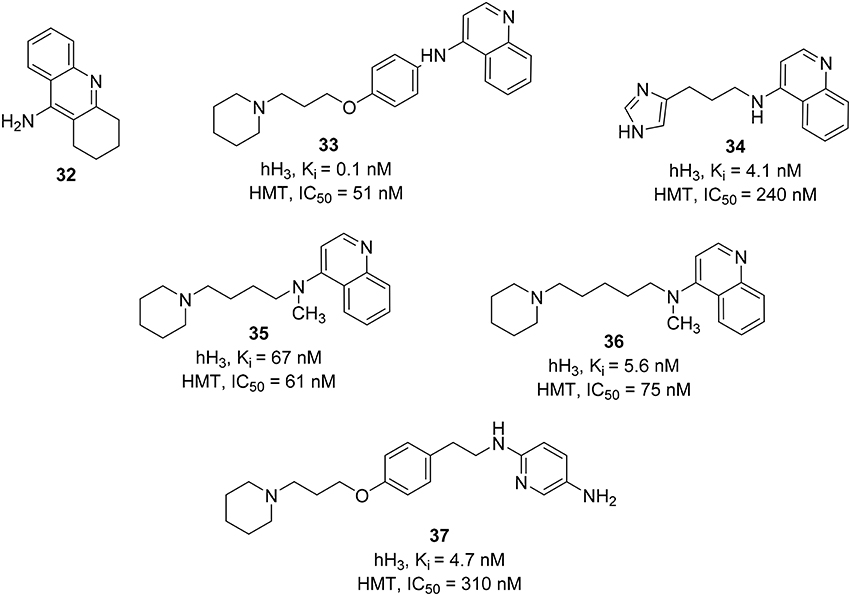
Figure 10. Structures and biological activities of selected H3R/HMT ligands.
Another approach was attempted on FUB 836 (33) by replacing the aminoquinoline with different heterocycles (e.g., nitro- or amino-substituted pyridines, quinolines, benzothiazole, or pyrroline) in order to improve its dual H3R/HMT affinities. In contrast to the aminoquinoline, the reported compounds showed moderate to good dual affinities. Whereas, some compounds showed potent HMT inhibitors, they only showed a moderate H3R affinity and vice versa (Apelt et al., 2005). The most potent compound in this series was 4-(3-piperidinopropyl)phenylether with substituted alkylaminopyridine (37, Figure 10).
Tacrine (32) mentioned above is an acetylcholinesterase (AChE) inhibitor. Together with the symptomatically acting N-methyl-D-aspartate (NMDA) blocker memantine, tacrine represents the only therapeutic treatment of AD currently available. AD is a complex neurodegenerative disorder and the most common form of dementia. Patients show a degeneration of cholinergic neutrons in the basal forebrain according to cholinergic hypothesis and aggregation of βA through an interaction with the peripheral anionic site (PAS) of the AChE (Davies and Maloney, 1976; Giacobini, 2000). H3R antagonists showed an ability to increase acetylcholine (ACh) but unlike the AChE, H3R antagonist will raise acetylcholine levels mostly in the brain, since H3R is mainly located in the CNS (Clapham and Kilpatrick, 1992; Darras et al., 2014). Therefore, the combination of both activities in a single molecule may offer the desired therapeutic effect with fewer unpleasant side effects considering acetylcholine release in the periphery (Fang et al., 2015; Guzior et al., 2015).
Using available crystal structure information and applying pharmacophore modeling and docking simulations Bembenek et al. proposed compound 38 (Figure 11) and similar structures to have activity on both AChE and H3R. Moreover, the used models suggest a possible interaction for this series of compounds with the PAS of the AChE (Bembenek et al., 2008). Some additional in vitro an in vivo studies with these compounds could be of interest to verify the calculated results. In 2008 Morini et al. introduced a class of symmetric and asymmetric 4,4′−biphenyl H3R antagonists with a moderate ability to inhibit rat brain cholinesterase (Morini et al., 2008). This class is characterized by a rigid biphenyl scaffold and displays nanomolar binding affinities at human and rodent H3R. The compound 39 (Figure 11) showed low nanomolar affinity to the H3R and low micromolar activity to inhibit AChE. Docking the compound 39 into the catalytic cavity of mouse AChE showed similarity to the binding mode, earlier reported for 38, confirming that more rigid and bulky biphenyl scaffolds are tolerated by the AChE active site. Interaction with PAS of the AChE is suggested for 39 as well as for 38. In 2012 Bajda et al. presented a new class of diether derivatives of homo substituted piperidine with 40 (Figure 11) being the most active compound, showing low nanomolar affinity for the hH3R and micromolar inhibitory potency toward both cholinergic receptors (Bajda et al., 2012). In 2014 Darras et al. presented new tetracyclic nitrogen-bridge headed compounds showing balanced affinities as hAChE inhibitor and hH3R antagonist with UW-MD-71 (41, Figure 11). It showed the best activity in two digit nanomolar area for both targets and greater than 200-fold selectivity over the other histamine receptor subtypes. This compound was tested on acquisition, consolidation and retrieval in a model of dizocilpine-induced amnesia. Test results indicated that using multiple targeting ligands lead to pharmacological and behavioral profiles different from interaction with the respective single target ligands. Furthermore, a potential applicability in the modulation of the memory impairment could be shown (Khan et al., 2016).
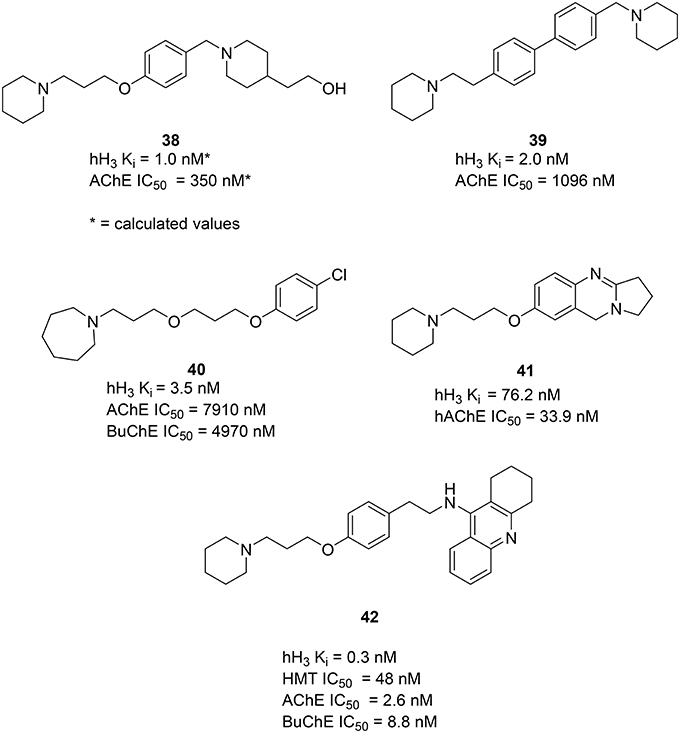
Figure 11. Structures and biological activities of selected H3R/AChE ligands.
In 2006, Petroianu et al. tested several compounds, containing structural features of tacrine (32) for their inhibitory activities on AChE and Butyrylcholinesterase (BuChE; Petroianu et al., 2006). These compounds have previously shown combined H3R antagonist and HMT inhibitory potencies (Apelt et al., 2002; Grassmann et al., 2003). From this series of compounds FUB833 (42, Figure 11) was the most promising four-target compound, showing subnanomolar affinity for hH3R, low nanomolar IC50 values for both cholinesterases and good affinity for HMT. These compounds have shown only moderate effects under in vivo conditions (Apelt et al., 2002). Furthermore, these new compounds might serve as novel important tools for further pharmacological investigations on histaminergic neurotransmission and its regulatory processes.
H3R and No-Releasing Molecules
Nitric oxide (NO) is an endogenous messenger, displaying a variety of actions in our body (Kerwin et al., 1995). NO is a key messenger in cardiovascular, immune, central, and peripheral nervous systems (Szabo, 2010). Released in the CNS after stimulation of excitatory NMDA, it diffuses in the adjacent presynaptic nerve terminal and astrocytes. There it activates the soluble guanylate cyclase (sGC) implying a number of physiological roles like gastro-protective effect, control of food intake and learning and formation of memory. H3R antagonists have also shown positive effects concerning learning and memory (Miyazaki et al., 1997; Komater et al., 2005). Combining H3R antagonists with NO-releasing moiety could synergistically contribute to a curative effect in pathologies like memory and learning disorders. Bertinaria et al. synthesized and tested some H3R antagonists with NO-donor properties by coupling H3R antagonist SKF 91486 (43, Figure 12) with the furoxan system (1,2,5-oxadiazole 2-oxide), which is able to release NO under the action of thiol cofactors like cysteine (Schönafinger, 1999). Resulting compounds had similar or greater potency as SKF 91486 (43). Derivative 44 (Figure 12) showed additional NO-dependent muscle relaxation (Bertinaria, 2003; Bertinaria et al., 2003). Another potent compound 45 is derived from Imoproxifan 46 (Figure 12) by replacing the oxime moiety with a five-membered NO-donor furoxan ring (Tosco et al., 2005). As a further development, a new class of NO-donor H3R antagonists with non-basic (thio)ether linker and furoxan (47) or nitrooxy (48) NO-donor moieties is introduced (Figure 12). These compounds are more appropriate to enter the CNS due to a better lipophilic-hydrophilic balance (Tosco et al., 2004).
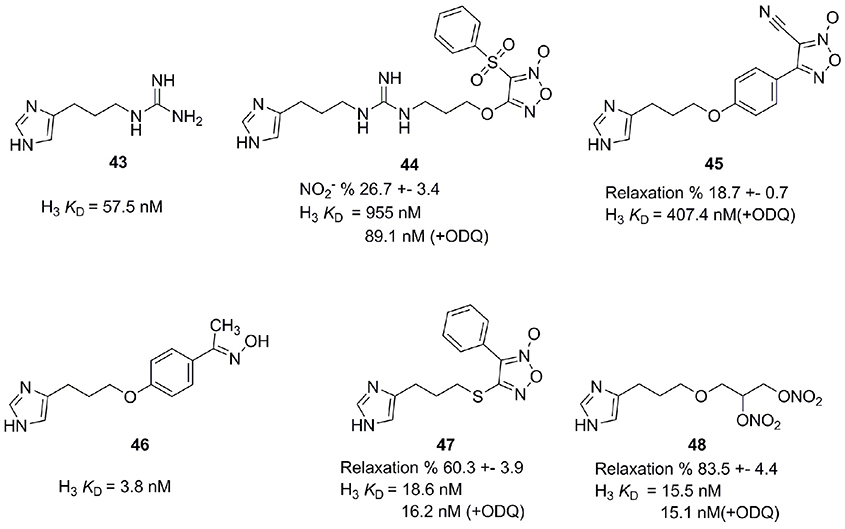
Figure 12. Structures and biological activities of selected H3R/NO-donor ligands.
H3R and Different Antiseizure Pharmacophores
Epilepsy is a common human brain disorder, affecting more than 60 million people worldwide. There is a need to discover an effective and safer antiepileptic drugs (AED) since Phenytoin (49) and recent AEDs like Loreclezole (50), Remacemide (51), and Safinamide (52) (Figure 13) only show efficacy within a maximum of 60–80% of patients and are responsible for many unwanted side-effects, such as headache, nausea, anorexia, ataxia, hepatotoxicity, drowsiness, gastrointestinal disturbance, gingival hyperplasia, attention deficit, und cognitive problems leading to additional discomfort (Sadek et al., 2014). There are indices for histamine receptors to improve the development of convulsions (Kasteleijn-Nolst Trenité et al., 2013). Seizure threshold can be increased and seizure susceptibility to electrically and chemically induced seizures can be decreased via activation of the central histaminergic system (Zhu et al., 2007; Bhowmik et al., 2012). Pitolisant has been tested in clinical trial phase II for patients suffering from photosensitive epilepsy. Supported by these results Sadek et al. designed some multiple-target ligands by combining the known 3-piperidinopropoxy or (3-piperidinopropoxy)aryl H3R pharmacophore with different AEDs on the market (49–52) leading to a small series of compounds (53–56, Figure 13; Sadek et al., 2014). These compounds showed moderate to good affinity to H3R with Ki values in the range of 562–0.24 nM and were tested in vivo for their anticonvulsive effect against maximum electroshock (MES)-induced and pentylenetetrazole (PTZ)-kindled convulsions in rats having phenytoin (55) as the reference AED. Surprisingly the compound with the lowest in vitro potency (55) was the only one to show the ability to reduce convulsions in both in vivo models being administered at 10 mg/kg intraperitoneally. Still the results are controversial and need new epilepsy models to elucidate the pharmacological profile of the current multiple targeting class in order to develop suitable and clinically useful AEDs (Bertinaria, 2003).
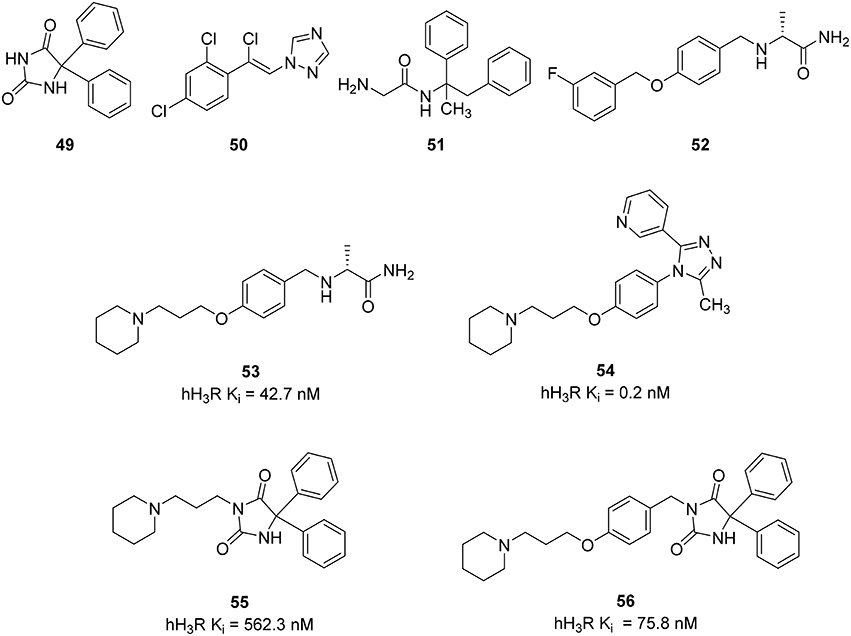
Figure 13. Structures and biological activities of selected H3R/antiseizure ligands.
Conclusion
Several combinations of different H3R pharmacophores with pharmacophoric elements of other histamine subtypes, other aminergic GPCRs, other transporters, other enzymes, and other disease-modifying elements have been described. The increasing knowledge on the complex interaction of the different signaling pathways as well as on the complex mechanism of central disorders, give promises for the development of optimized drugs with synergistic pharmacological properties at multiple targets and also reduced side effects. The different leads for MTDLs described here, are very early or at best preclinical candidates. Therefore, a lot of work on improvements has to be performed before these designed multiple targeting approaches will get into clinical trials.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support was kindly provided by the EU COST Actions CM1103, CM1207, and CA15135 as well by DFG INST 208/664-1 FUGG and Ol16112039.
References
Aluisio, L., Lord, B., Barbier, A. J., Fraser, I. C., Wilson, S. J., Boggs, J., et al. (2008). In-vitro and in-vivo characterization of JNJ-7925476, a novel triple monoamine uptake inhibitor. Eur. J. Pharmacol. 587, 141–146. doi: 10.1016/j.ejphar.2008.04.008
Anighoro, A., Bajorath, J., and Rastelli, G. (2014). Polypharmacology:challenges and opportunities in epigenetic drug discovery. J. Med. Chem. 57, 7874–7887. doi: 10.1021/jm5006463
Apelt, J., Grassmann, S., Ligneau, X., Pertz, H. H., Ganellin, C. R., Arrang, J. M., et al. (2005). Search for histamine H3 receptor antagonists with combined inhibitory potency at N-tau-methyltransferase: Ether derivatives. Pharmazie 60, 97–106.
Apelt, J., Ligneau, X., Pertz, H. H., Arrang, J. M., Ganellin, C. R., Schwartz, J. C., et al. (2002). Development of a new class of nonimidazole histamine H(3) receptor ligands with combined inhibitory histamine N-methyltransferase activity. J. Med. Chem. 45, 1128–1141. doi: 10.1021/jm0110845
Arrang, J. M., Garbarg, M., and Schwartz, J. C. (1983). Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302, 832–837. doi: 10.1038/302832a0
Bajda, M., Kuder, K. J., Lażewska, D., Kieć-Kononowicz, K., Więckowska, A., Ignasik, M., et al. (2012). Dual-acting diether derivatives of piperidine and homopiperidine with histamine H3 receptor antagonistic and anticholinesterase activity. Arch. Pharm. (Weinheim) 345, 591–597. doi: 10.1002/ardp.201200018
Bakker, R. A., Timmerman, H., and Leurs, R. (2002). Histamine receptors: Specific ligands, receptor biochemistry, and signal transdution. Clin. Allergy Immunol. 17, 27–64.
Barbier, A. J., Aluisio, L., Lord, B., Qu, Y., Wilson, S. J., Boggs, J. D., et al. (2007). Pharmacological characterization of JNJ-28583867, a histamine H(3) receptor antagonist and serotonin reuptake inhibitor. Eur. J. Pharmacol. 576, 43–54. doi: 10.1016/j.ejphar.2007.08.009
Bembenek, S. D., Keith, J. M., Letavic, M. A., Apodaca, R., Barbier, A. J., Dvorak, L., et al. (2008). Lead identification of acetylcholinesterase inhibitors-histamine H3 receptor antagonists from molecular modeling. Bioorg. Med. Chem. 16, 2968–2973. doi: 10.1016/j.bmc.2007.12.048
Berlin, M., Boyce, C. W., and de Lera Ruiz, M. (2011). Histamine H3 receptor as a drug discovery target. J. Med. Chem. 54, 26–53. doi: 10.1021/jm100064d
Bertinaria, M. (2003). H3 receptor ligands: new imidazole H3-antagonists endowed with NO-donor properties. Farmaco 58, 279–283. doi: 10.1016/S0014-827X(03)00023-5
Bertinaria, M., Stilo, A. D., Tosco, P., Sorba, G., Poli, E., Pozzoli, C., et al. (2003). [3-(1H-Imidazol-4-yl)propyl]guanidines containing furoxan moieties. Bioorg. Med. Chem. 11, 1197–1205. doi: 10.1016/S0968-0896(02)00651-X
Bhowmik, M., Khanam, R., and Vohora, D. (2012). Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: a systemic consideration of recent progress and perspectives. Br. J. Pharmacol. 167, 1398–1414. doi: 10.1111/j.1476-5381.2012.02093.x
Bongers, G., Bakker, R. A., and Leurs, R. (2007). Molecular aspects of the histamine H3 receptor. Biochem. Pharmacol. 73, 1195–1204. doi: 10.1016/j.bcp.2007.01.008
Brioni, J. D., Esbenshade, T. A., Garrison, T. R., Bitner, S. R., and Cowart, M. D. (2011). Discovery of histamine H3 antagonists for the treatment of cognitive disorders and Alzheimer's disease. J. Pharmacol. Exp. Ther. 336, 38–46. doi: 10.1124/jpet.110.166876
Bymaster, F. P., Beedle, E. E., Findlay, J., Gallagher, P. T., Krushinski, J. H., Mitchell, S., et al. (2003). Duloxetine (Cymbalta), a dual inhibitor of serotonin and norepinephrine reuptake. Bioorg. Med. Chem. Lett. 13, 4477–4480. doi: 10.1016/j.bmcl.2003.08.079
Ceras, J., Cirauqui, N., Pérez-Silanes, S., Aldana, I., Monge, A., and Galiano, S. (2012). Novel sulfonylurea derivatives as H3 receptor antagonists. Preliminary SAR studies. Eur. J. Med. Chem. 52, 1–13. doi: 10.1016/j.ejmech.2012.02.049
Chazot, P. L. (2010). Therapeutic potential of histamine H3 receptor antagonists in dementias. Drug News Perspect. 2, 99–103. doi: 10.1358/dnp.2010.23.2.1475899
Chojnacki, J. E., Liu, K., Yan, X., Toldo, S., Selden, T., Estrada, M., et al. (2014). Discovery of 5-(4-Hydroxyphenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide as a neuroprotectant for Alzheimer's disease by hybridizayion of curcumin and melatonin. Chem. Neurosci. 5, 690–699. doi: 10.1021/cn500081s
Clapham, J., and Kilpatrick, G. J. (1992). Histamine H3 receptors modulate the release of [3H]-acetylcholine from slices of rat entorhinal cortex: evidence for the possible existence of H3 receptor subtypes. Br. J. Pharmacol. 107, 919–923. doi: 10.1111/j.1476-5381.1992.tb13386.x
Corrêa, M. F., and Fernandes, J. P. D. S. (2015). Histamine H4 receptor ligands: future applications and state of art. Chem. Biol. Drug Des. 85, 461–480. doi: 10.1111/cbdd.12431
Cowart, M., Altenbach, R., Black, L., Faghih, R., Zhao, C., and Hancock, A. A. (2004). Medicinal chemistry and biological properties of non-imidazole histamine H3 antagonists. Mini Rev. Med. Chem. 4, 979–992. doi: 10.2174/1389557043403215
Dagli, M., Goksu, N., Eryilmaz, A., Mocan Kuzey, G., Bayazit, Y., Gun, B. D., et al. (2008). Expression of histamine receptors (H1, H2, and H3) in the rabbit endolymphatic sac: an immunohistochemical study. Am. J. Otolaryngol. 29, 20–23. doi: 10.1016/j.amjoto.2006.12.003
Daley-Yates, P., Ambery, C., Sweeney, L., Watson, J., Oliver, A., and McQuade, B. (2012). The efficacy and tolerability of two novel H(1)/H(3) receptor antagonists in seasonal allergic rhinitis. Int. Arch. Allergy Immunol. 158, 84–98. doi: 10.1159/000329738
Darras, F. H., Pockes, S., Huang, G., Wehle, S., Strasser, A., Wittmann, H. J., et al. (2014). Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem. Neurosci. 5, 225–242. doi: 10.1021/cn4002126
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 308, 1403.
Deng, C., Weston-Green, K., and Huang, X. F. (2010). The role of histaminergic H1 and H3 receptors in food intake: a mechanism for atypical antipsychotic-induced weight gain? Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1–4. doi: 10.1016/j.pnpbp.2009.11.009
Esbenshade, T. A., Browman, K. E., Bitner, R. S., Strakhova, M., Cowart, M. D., and Brioni, J. D. (2008). The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol. 154, 1166–1181. doi: 10.1038/bjp.2008.147
Fang, J., Li, Y., Liu, R., Pang, X., Li, C., Yang, R., et al. (2015). Discovery of multitarget-directed ligands against Alzheimer' s disease through systematic prediction of chemical -protein interactions. J. Chem. Inf. Model. 55, 149–164. doi: 10.1021/ci500574n
Farzin, D., Asghari, L., and Nowrouzi, M. (1994). Rodent antinociception following acute treatment with different histamine receptor agonists and antagonists. Pharmacol. Biochem. Behav. 111, 751–760.
Ferrada, C., Ferré, S., Casadó, V., Cortés, A., Justinova, Z., Barnes, C., et al. (2008). Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55, 190–197. doi: 10.1016/j.neuropharm.2008.05.008
Fishbain, D., Berman, K., and Kajdasz, D. K. (2006). Duloxetine for neuropathic pain based on recent clinical trials. Curr. Pain Headache Rep. 10, 199–204. doi: 10.1007/s11916-006-0046-7
Fredrickson, A., Snyder, P. J., Cromer, J., Thomas, E., Lewis, M., and Maruff, P. (2008). The use of effect sizes to characterize the nature of cognitive change in psychopharmacological studies: an example with scopolamine. Hum. Psychopharmacol. 23, 425–436. doi: 10.1002/hup.942
Gemkow, M. J., Davenport, A. J., Harich, S., Ellenbroek, B. A., Cesura, A., and Hallett, D. (2009). The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov. Today 14, 509–515. doi: 10.1016/j.drudis.2009.02.011
Ghasemi, J. B., and Tavakoli, H. (2012). Improvement of the prediction power of the CoMFA and CoMSIA models on histamine H3 antagonists by different variable selection methods. Sci. Pharm. 80, 547–566. doi: 10.3797/scipharm.1204-19
Giacobini, E. (2000). Cholinesterase inhibitors stabilize Alzheimer's disease. Ann. N.Y. Acad. Sci. 920, 321–327. doi: 10.1111/j.1749-6632.2000.tb06942.x
Grassmann, S., Apelt, J., Ligneau, X., Pertz, H. H., Arrang, J. M., Ganellin, C. R., et al. (2004). Search for histamine H(3) receptor ligands with combined inhibitory potency at histamine N-methyltransferase: omega-piperidinoalkanamine derivatives. Arch. Pharm. 337, 533–545. doi: 10.1002/ardp.200400897
Grassmann, S., Apelt, J., Sippl, W., Ligneau, X., Pertz, H. H., Zhao, Y. H., et al. (2003). Imidazole derivatives as a novel class of hybrid compounds with inhibitory histamine N-methyltransferase potencies and histamine hH3 receptor affinities. Bioorg. Med. Chem. 11, 2163–2174. doi: 10.1016/S0968-0896(03)00120-2
Guzior, N., Wieckowska, A., Panek, D., and Malawska, B. (2015). Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer's disease. Curr. Med. Chem. 22, 373–404. doi: 10.2174/0929867321666141106122628
Héron, A., Rouleau, A., Cochois, V., Pillot, C., Schwartz, J. C., and Arrang, J. M. (2001). Expression analysis of the histamine H3 receptor in developing rat tissues. Mech. Dev. 105, 167–173. doi: 10.1016/S0925-4773(01)00389-6
Herrmann, N., Chau, S. A., and Kircanski, L. K. (2011). Current and emerging drug treatment options for Alzheimer's disease: a systematic review. Drugs 71, 2031–2065. doi: 10.2165/11595870-000000000-00000
Hey, J. A., Del Prado, M., Egan, R. W., Kreutner, W., and Chapman, R. W. (1992). Inhibition of sympathetic hypertensive responses in the guinea-pig by prejunctional histamine H3-receptors. Br. J. Pharmacol. 107, 347–351. doi: 10.1111/j.1476-5381.1992.tb12749.x
Hopkins, A. L. (2008). Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4, 682–690. doi: 10.1038/nchembio.118
Hotte, M., Dauphin, F., Freret, T., Boulouard, M., and Levellat, G. (2012). A biphasic and brain-region selective down-regulation of cyclic adenosine monophosphate concentrations supports object recognition in the rat. PLoS ONE 7:e32244. doi: 10.1371/journal.pone.0032244
Huang, G., Nimczick, M., and Decker, M. (2015). Rational modification of the biological profile of GPCR ligands through combination with other biologically active moieties: GPCR ligand combinations. Arch. Pharm. (Weinheim) 348, 531–540. doi: 10.1002/ardp.201500079
Humbert-Claude, M., Morisset, S., Gbahou, F., and Arrang, J. M. (2007). Histamine H3 and dopamine D2 receptor-mediated [35S]GTPγ[S] binding in rat striatum: evidence for additive effects but lack of interactions. Biochem. Pharmacol. 73, 1172–1181. doi: 10.1016/j.bcp.2007.01.006
Jadhav, H. R., and Singh, M. (2013). Histamine H3 receptor function and ligands: recent developments. Mini Rev. Med. Chem. 13, 47–57. doi: 10.2174/138955713804484695
Jin, C. Y., and Panula, P. (2005). The laminar histamine receptor system in human prefrontal cortex suggests multiple levels of histaminergic regulation. Neuroscience 132, 137–149. doi: 10.1016/j.neuroscience.2004.12.017
Kagermeier, N., Werner, K., Keller, M., Baumeister, P., Seifert, R., Buschauer, A., et al. (2015). Dimeric carbamoylguanidine-type histamine H2 receptor ligands: a new class of potent and selective agonists. Bioorg. Med. Chem. 23, 3957–3969. doi: 10.1016/j.bmc.2015.01.012
Kasteleijn-Nolst Trenité, D., Parain, D., Genton, P., Masnou, P., Schwartz, J.-C., and Hirsch, E. (2013). Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 28, 66–70. doi: 10.1016/j.yebeh.2013.03.018
Keith, J. M., Gomez, L. A., Barbier, A. J., Wilson, S. J., Boggs, J. D., Lord, B., et al. (2007a). Pyrrolidino-tetrahydroisoquinolines bearing pendant heterocycles as potent dual H3 antagonist and serotonin transporter inhibitors. Bioorg. Med. Chem. Lett. 17, 4374–4377. doi: 10.1016/j.bmcl.2007.03.043
Keith, J. M., Gomez, L. A., Letavic, M. A., Ly, K. S., Jablonowski, J. A., Seierstad, M., et al. (2007b). Dual serotonin transporter/histamine H3 ligands: optimization of the H3 pharmacophore. Bioorg. Med. Chem. Lett. 17, 702–706. doi: 10.1016/j.bmcl.2006.10.089
Keith, J. M., Gomez, L. A., Wolin, R. L., Barbier, A. J., Wilson, S. J., Boggs, J. D., et al. (2007c). Pyrrolidino-tetrahydroisoquinolines as potent dual H3 antagonist and serotonin transporter inhibitors. Bioorg. Med. Chem. Lett. 17, 2603–2607. doi: 10.1016/j.bmcl.2007.01.106
Kerwin, J. F., Lancaster, J. R., and Feldman, P. L. (1995). Nitric oxide: a new paradigm for second messengers. J. Med. Chem. 38, 4343–4362. doi: 10.1021/jm00022a001
Khan, N., Saad, A., Nurulain, S. M., Darras, F. H., Decker, M., and Sadek, B. (2016). The dual-acting H3 receptor antagonist and AChE inhibitor UW-MD-71 dose-dependently enhances memory retrieval and reverses dizocilpine-induced memory impairment in rats. Behav. Brain. Res. 297, 155–164. doi: 10.1016/j.bbr.2015.10.022
Klinkenberg, I., and Blokland, A. (2011). The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci. Biobehav. Rev. 34, 1307–1350. doi: 10.1016/j.neubiorev.2010.04.001
Komater, V. A., Buckley, M. J., Browman, K. E., Pan, J. B., Hancock, A. A., Decker, M. W., et al. (2005). Effects of histamine H3 receptor antagonists in two models of spatial learning. Behav. Brain Res. 159, 295–300. doi: 10.1016/j.bbr.2004.11.008
Kottke, T., Sander, K., Weizel, L., Schneider, E. H., Seifert, R., and Stark, H. (2011). Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur. J. Pharmacol. 654, 200–208. doi: 10.1016/j.ejphar.2010.12.033
Kroeze, W. K., Hufeisen, S. J., Popadak, B. A., Renock, S. M., Steinberg, S., Ernsberger, P., et al. (2003). H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28, 519–526. doi: 10.1038/sj.npp.1300027
Lelong, V., Dauphin, F., and Boulouard, M. (2001). RS 67333 and D-cycloserine accelerate learning acquisition in the rat. Neuropharmacology 41, 517–522. doi: 10.1016/S0028-3908(01)00085-5
Lelong, V., Lhonneur, L., Dauphin, F., and Boulouard, M. (2003). BIMU 1 and RS 67333, two 5-HT4 receptor agonists, modulate spontaneous alternation deficits induced by scopolamine in the mouse. Naunyn Schmiedebergs Arch. Pharmacol. 367, 621–628. doi: 10.1007/s00210-003-0743-2
Leon, R., Garcia, A. G., and Marco-Contelles, J. (2013). Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer's disease. Med. Res. Rev. 33, 139–189. doi: 10.1002/med.20248
Lepailleur, A., Freret, T., Lemaître, S., Boulouard, M., Dauphin, F., Hinschberger, A., et al. (2014). Dual histamine H3R/serotonin 5-HT4R ligands with antiamnesic properties: pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 54, 1773–1784. doi: 10.1021/ci500157n
Letavic, M. A., Barbier, A. J., Dvorak, C. A., and Carruthers, N. I. (2006). Recent medicinal chemistry of the histamine H3 receptor. Prog. Med. Chem. 44, 181–206. doi: 10.1016/S0079-6468(05)44405-7
Letavic, M. A., Keith, J. M., Jablonowski, J. A., Stocking, E. M., Gomez, L. A., Ly, K. S., et al. (2007a). Novel tetrahydroisoquinolines are histamine H3 antagonists and serotonin reuptake inhibitors. Bioorg. Med. Chem. Lett. 17, 1047–1051. doi: 10.1016/j.bmcl.2006.11.036
Letavic, M. A., Stocking, E. M., Barbier, A. J., Bonaventure, P., Boggs, J. D., Lord, B., et al. (2007b). Benzylamine histamine H(3) antagonists and serotonin reuptake inhibitors. Bioorg. Med. Chem. Lett. 17, 4799–4803. doi: 10.1016/j.bmcl.2007.06.061
Levallet, G., Hotte, M., Boulouard, M., and Dauphin, F. (2009). Increased particulate phosphodiesterase 4 in the prefrontal cortex supports 5-HT4 receptor-induced improvement of object recognition memory in the rat. Psychopharmacology 202, 125–139. doi: 10.1007/s00213-008-1283-8
Leventhal, L., Smith, V., Hornby, G., Andree, T. H., Brandt, M. R., and Rogers, K. E. (2007). Differential and synergistic effects of selective norepinephrine and serotonin reuptake inhibitors in rodent models of pain. J. Pharmacol. Exp. Ther. 320, 1178–1185. doi: 10.1124/jpet.106.109728
Lezoualc'h, F. (2007). 5-HT4 receptor and Alzheimer's disease: the amyloid connection. Exp. Neurol. 205, 325–329. doi: 10.1016/j.expneurol.2007.02.001
Lian, J., Huang, X. F., Pai, N., and Deng, C. (2014). Betahistine ameliorates olanzapine-induced weight gain through modulation of histaminergic, NPY and AMPK pathways. Psychoneuroendocrinology 48, 77–86. doi: 10.1016/j.psyneuen.2014.06.010
Lian, J., Huang, X. F., Pai, N., and Deng, C. (2016). Ameliorating antipsychotic-induced weight gain by betahistine: mechanisms and clinical implications. Pharmacol. Res. 106, 51–63. doi: 10.1016/j.phrs.2016.02.011
Ligneau, X., Landais, L., Perrin, D., Piriou, J., Uguen, M., Denis, E., et al. (2007). Brain histamine and schizophrenia: potential therapeutic applications of H3-receptor inverse agonists studied with BF2.649. Biochem. Pharmacol. 73, 1215–1224. doi: 10.1016/j.bcp.2007.01.023
Lim, H. D., Istyastono, E. P., van de Stolpe, A., Romeo, G., Gobbi, S., Schepers, M., et al. (2009). Clobenpropit analogs as dual activity ligands for the histamine H3 and H4 receptors: synthesis, pharmacological evaluation, and cross-target QSAR studies. Bioorg. Med. Chem. 17, 3987–3994. doi: 10.1016/j.bmc.2009.04.007
Lin, J., Sergeeva, O. A., and Haas, H. L. (2011). Histamine H3 receptors and sleep-wake regulation. J. Pharmacol. Exp. Ther. 336, 17–23. doi: 10.1124/jpet.110.170134
Ly, K. S., Letavic, M. A., Keith, J. M., Miller, J. M., Stocking, E. M., Barbier, A. J., et al. (2008). Synthesis and biological activity of piperazine and diazepane amides that are histamine H3 antagonists and serotonin reuptake inhibitors. Bioorg. Med. Chem. Lett. 18, 39–43. doi: 10.1016/j.bmcl.2007.11.016
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P., and Kivipelto, M. (2010). Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716. doi: 10.1016/S1474-4422(10)70119-8
McLeod, R. L., Mingo, G. G., Herczku, C., DeGennaro-Culver, F., Kreutner, W., Egan, R. W., et al. (1999). Combined Histamine H1 and H3 receptor blockade produces nasal degongestion in an experimental model of nasal congestion. Am. J. Rhinol. 13, 391–399. doi: 10.2500/105065899781367483
Medhurst, S. J., Collins, S. D., Billinton, A., Bingham, S., Dalziel, R. G., Brass, A., et al. (2008). Novel histamine H3 receptor antagonists GSK189254 and GSK334429 are efficacious in surgically-induced and virally-induced rat models of neuropathic pain. Pain 138, 61–69. doi: 10.1016/j.pain.2007.11.006
Medina, V. A., and Rivera, E. S. (2010). Histamine receptors and cancer pharmacology. Br. J. Pharmacol. 161, 755–767. doi: 10.1111/j.1476-5381.2010.00961.x
Menza, M. A., Kaufman, K. R., and Castellanos, A. (2000). Modafinil augmentation of antidepressant treatment in depression. J. Clin. Psychiatry 61, 378–381. doi: 10.4088/JCP.v61n0510
Millan, M. J. (2014). On “polypharmacy” and multi-target agents, complementary strategies for improving the treatment of depression: a comparative appraisal. Int. J. Neuropsychopharmacol. 17, 1009–1037. doi: 10.1017/S1461145712001496
Miyazaki, S., Onodera, K., Imaizumi, M., and Timmerman, H. (1997). Effects of clobenpropit (VUF-9153), a histamine H3-receptor antagonist, on learning and memory, and on cholinergic and monoaminergic systems in mice. Life Sci. 61, 355–361. doi: 10.1016/S0024-3205(97)00406-2
Møller, M. N., Kirkeby, S., Vikeså, J., Nielsen, F. C., and Caye-Thomasen, P. (2015). Expression of histamine receptors in the human endolymphatic sac: the molecular rationale for betahistine use in Menieres disease. Eur. Arch. Otorhinolaryngol. doi: 10.1007/s00405-015-3731-5. [Epub ahead of print].
Morini, G., Comini, M., Rivara, M., Rivara, S., Bordi, F., Plazzi, P. V., et al. (2008). Synthesis and structure-activity relationships for biphenyl H3 receptor antagonists with moderate anti-cholinesterase activity. Bioorg. Med. Chem. 16, 9911–9924. doi: 10.1016/j.bmc.2008.10.029
Neumann, D., Beermann, S., Burhenne, H., Glage, S., Hartwig, C., and Seifert, R. (2013). The dual H3/4R antagonist thioperamide does not fully mimic the effects of the “standard” H4R antagonist JNJ 7777120 in experimental murine asthma. Naunyn Schmiedebergs Arch. Pharmacol. 386, 983–990. doi: 10.1007/s00210-013-0898-4
Nikolic, K., Filipic, S., Agbaba, D., and Stark, H. (2014). Procognitive properties of drugs with single and multitargeting H 3 receptor antagonist activities. CNS Neurosci. Ther. 20, 613–623. doi: 10.1111/cns.12279
Norman, P. (2011). New H1/H3 antagonists for treating allergic rhinitis: WO2010094643. Expert. Opin. Ther. Pat. 21, 425–429. doi: 10.1517/13543776.2011.536533
Pala, D., Scalvini, L., Lodola, A., Mor, M., Flammini, L., Barocelli, E., et al. (2014). Synthesis and characterization of new bivalent agents as melatonin- and histamine H3-ligands. Int. J. Mol. Sci. 15, 16114–16133. doi: 10.3390/ijms150916114
Panula, P., Chazot, P. L., Cowart, M., Gutzmer, R., Leurs, R., Liu, W. L., et al. (2015). International union of basic and clinical pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 67, 601–655. doi: 10.1124/pr.114.010249
Parsons, M. E., and Ganellin, C. R. (2006). Histamine and its receptors. Br. J. Pharmacol. 147, S127–S135. doi: 10.1038/sj.bjp.0706440
Peters, J. U. (2013). Polypharmacology - Foe or friend? J. Med. Chem. 56, 8955–8971. doi: 10.1021/jm400856t
Petroianu, G., Arafat, K., Sasse, B. C., and Stark, H. (2006). Multiple enzyme inhibitions by histamine H3 receptor antagonists as potential procognitive agents. Pharmazie 61, 179–182.
Raddatz, R., Tao, M., and Hudkins, R. L. (2010). Histamine H3 antagonists for treatment of cognitive deficits in CNS diseases. Curr. Top. Med. Chem. 10, 153–169. doi: 10.2174/156802610790411027
Remington, G. (2003). Understanding antipsychotic “atypicality”: a clinical and pharmacological moving target. J. Psychiatry Neurosci. 28, 275–284.
Sabbagh, M. N. (2009). Drug development for Alzheimer's disease: where are we now and where are we headed? Am. J. Geriatr. Pharmacother. 7, 167–185. doi: 10.1016/j.amjopharm.2009.06.003
Sadek, B., Schwed, J. S., Subramanian, D., Weizel, L., Walter, M., Adem, A., et al. (2014). Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur. J. Med. Chem. 77, 269–279. doi: 10.1016/j.ejmech.2014.03.014
Sander, K., Kottke, T., and Stark, H. (2008). Histamine H3 receptor antagonists go to clinics. Biol. Pharm. Bull. 31, 2163–2181. doi: 10.1248/bpb.31.2163
Schlicker, E., Betz, R., and Göthert, M. (1998). Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedebergs Arch. Pharmacol. 337, 588–590.
Schlicker, E., Fink, K., Detzner, M., and Göthert, M. (1993). Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J. Neural Transm. Gen. Sect. 93, 1–10. doi: 10.1007/BF01244933
Schlicker, E., Fink, K., Göthert, M., Hoyer, D., Molderings, G., Roschke, I., et al. (1989). The pharmacological properties of the presynaptic serotonin autoreceptor in the pig brain cortex conform to the 5-HT1D receptor subtype. Naunyn Schmiedebergs Arch. Pharmacol. 340, 45–51. doi: 10.1007/BF00169206
Schlicker, E., Schunack, W., and Göthert, M. (1990). Histamine H3 receptor-mediated inhibition of noradrenaline release in pig retina discs. Naunyn Schmiedeberg Arch. Pharmacol. 342, 497–501. doi: 10.1007/BF00169035
Schönafinger, K. (1999). Heterocyclic NO prodrugs. Farmaco 54, 316–320. doi: 10.1016/S0014-827X(99)00031-2
Schwartz, J. C., Arrang, J. M., Garbarg, M., Pollard, H., and Ruat, M. (1991). Histaminergic transmission in the mammalian brain. Physiol. Rev. 71, 1–21.
Slack, R. J., Russell, L. J., Hall, D. A., Luttmann, M. A., Ford, A. J., Saunders, K. A., et al. (2011). Pharmacological characterization of GSK1004723, a novel, long-acting antagonist at histamine H1 and H3 receptors. Br. J. Pharmacol. 164, 1627–1641. doi: 10.1111/j.1476-5381.2011.01285.x
Snyder, P. J., Bednar, M. M., Cromer, J. R., and Maruff, P. (2005). Reversal of scopolamine-induced deficits with a single dose of donepezil, an acetylcholinesterase inhibitor. Alzheimers Dement. 1, 126–135. doi: 10.1016/j.jalz.2005.09.004
Srinivasan, V., Pandi-Perumal, S. R., Cardinali, D. P., Poeggeler, B., and Hardeland, R. (2006). Melatonin in Alzheimer's disease and other neurodegenerative disorders. Behav. Brain. Funct. 2, 1–23. doi: 10.1186/1744-9081-2-15
Stahl, M. (2001). “Commentary on the limitation of antidepressants in current use,” in Antidepressants: Milestones in Drug Therapy MDT, ed B. E. Leonard (Basel; Boston, MA; Berlin: Birkhäuser Basel), 31–43.
Stark, H., Kathmann, M., Schlicker, E., Schunack, W., Schlegel, B., and Sippl, W. (2004). Medicinal chemical and pharmacological aspects of imidazole-containing histamine H3 receptor antagonists. Mini Rev. Med. Chem. 4, 965–977. doi: 10.2174/1389557043403107
Stocking, E. M., Miller, J. M., Barbier, A. J., Wilson, S. J., Boggs, J. D., McAllister, H. M., et al. (2007). Synthesis and biological evaluation of diamine-based histamine H3 antagonists with serotonin reuptake inhibitor activity. Bioorg. Med. Chem. Lett. 17, 3130–3135. doi: 10.1016/j.bmcl.2007.03.034
Szabo, C. (2010). Gaseotransmitters: new frontiers for translational science. Sci. Transl. Med. 2, 1–7. doi: 10.1126/scitranslmed.3000721
Thurmond, R. L., Gelfand, E. W., and Dunford, P. J. (2008). The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat. Rev. Drug. Discov. 7, 41–53. doi: 10.1038/nrd2465
Tosco, P., Bertinaria, M., Di Stilo, A., Cena, C., Sorba, G., Fruttero, R., et al. (2005). Furoxan analogues of the histamine H3-receptor antagonist imoproxifan and related furazan derivatives. Bioorg. Med. Chem. 13, 4750–4759. doi: 10.1016/j.bmc.2005.05.004
Tosco, P., Bertinaria, M., Di Stilo, A., Marini, E., Rolando, B., Sorba, G., et al. (2004). A new class of NO-donor H3-antagonists. Farmaco 59, 359–371. doi: 10.1016/j.farmac.2003.12.008
Von Coburg, Y., Kottke, T., Weizel, L., Ligneau, X., and Stark, H. (2009). Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett. 19, 538–542. doi: 10.1016/j.bmcl.2008.09.012
Vuyyuru, L., Schubert, M. L., Harrington, L., Arimura, A., and Makhlouf, G. M. (1995). Dual inhibitory pathways link antral somatostatin and histamine secretion in human, dog, and rat stomach. Gastroenterology 109, 1566–1574. doi: 10.1016/0016-5085(95)90645-2
Walczyński, K., Guryn, R., Zuiderveld, O. P., and Timmerman, H. (1999). Non-imidazole histamine H3 ligands. Part, I. Synthesis of 2-(1-piperazinyl)- and 2-(hexahydro-1H-1,4-diazepin-1-yl)benzothiazole derivatives as H3-antagonists with H1 blocking activities. Farmaco 54, 684–694. doi: 10.1016/S0014-827X(99)00081-6
Wicek, M., Kottke, T., Ligneau, X., Schunack, W., Seifert, R., Stark, H., et al. (2011). N-Alkenyl and cycloalkyl carbamates as dual acting histamine H3 and H4 receptor ligands. Bioorganic. Med. Chem. 19, 2850–2858. doi: 10.1016/j.bmc.2011.03.046
Wieland, K., Laak, A. M., Smit, M. J., Kuhne, R., Timmerman, H., and Leurs, R. (1999). Mutational analysis of the antagonist-binding site of the histamine H1 receptor. J. Biol. Chem. 274, 29994–30000. doi: 10.1074/jbc.274.42.29994
Witkin, J. M., and Nelson, D. L. (2004). Selective histamine H3 receptor antagonists for treatment of cognitive deficiencies and other disorders of the central nervous system. Pharmacol. Ther. 103, 1–20. doi: 10.1016/j.pharmthera.2004.05.001
Wong, D. T., Bymaster, F. P., and Engleman, E. A. (1995). Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci. 57, 411–441. doi: 10.1016/0024-3205(95)00209-O
Ye, N., Song, Z., and Zhang, A. (2014). Dual ligands targeting dopamine D2 and serotonin 5-HT1A receptors as new antipsychotical or anti-Parkinsonian agents. Curr. Med. Chem. 21, 437–457. doi: 10.2174/09298673113206660300
Youdim, M. B., and Buccafusco, J. J. (2005). Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 26, 27–35. doi: 10.1016/j.tips.2004.11.007
Zhou, L., Zhou, W., Zhang, S., Liu, B., Leng, Y., Zhou, R., et al. (2013). Changes in histamine receptors (H1, H2, and H3) expression in rat medial vestibular nucleus and flocculus after unilateral labyrinthectomy: histamine receptors in vestibular compensation. PLoS ONE 8:e66684. doi: 10.1371/journal.pone.0066684
Keywords: multiple targeting, GPCR, enzymes, NO, histamine, transporter
Citation: Khanfar MA, Affini A, Lutsenko K, Nikolic K, Butini S and Stark H (2016) Multiple Targeting Approaches on Histamine H3 Receptor Antagonists. Front. Neurosci. 10:201. doi: 10.3389/fnins.2016.00201
Received: 05 February 2016; Accepted: 25 April 2016;
Published: 30 May 2016.
Edited by:
Giuseppe Di Giovanni, University of Malta, MaltaReviewed by:
Peter McCormick, University of Barcelona, SpainWladyslaw Lason, Polish Academy of Sciences, Poland
Copyright © 2016 Khanfar, Affini, Lutsenko, Nikolic, Butini and Stark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger Stark, stark@hhu.de
†These authors have contributed equally to this work.