Habituation without NMDA receptor-dependent desensitization of Hering–Breuer apnea reflex in a Mecp2+/− mutant mouse model of Rett syndrome
- 1 Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA, USA
- 2 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA
- 3 Whitehead Institute for Biomedical Research, Cambridge, MA, USA
Non-associative learning is a basic neuroadaptive behavior exhibited in almost all animal species and sensory modalities but its functions and mechanisms in the mammalian brain are poorly understood. Previous studies have identified two distinct forms of non-associative learning in the classic Hering–Breuer inflation reflex (HBIR) induced apnea in rats: NMDA receptor (NMDAR)-independent habituation in a primary vagal pathway and NMDAR-dependent desensitization in a secondary pontine pathway. Here, we show that abnormal non-associative learning of the HBIR may underlie the endophenotypic tachypnea in an animal model of Rett syndrome (RTT), an autism-spectrum disorder caused by mutations in the X-linked gene encoding methyl-CpG-binding protein 2 (MECP2). Mecp2+/− symptomatic mice on a mixed-strain background demonstrated significantly increased resting respiratory frequency with shortened expiration and normal inspiratory duration compared with asymptomatic mutants and wild-type controls, a phenotype that is characteristic of girls with RTT. Low-intensity electrical stimulation of the vagus nerve elicited fictive HBIR with time-dependent habituation in both Mecp2+/− and wild-type mice. However, time-dependent desensitization of the HBIR was evidenced only in wild-type controls and asymptomatic mutant mice but was absent or suppressed in Mecp2+/− symptomatic mice or in wild-type mice after blockade of NMDAR with dizocilpine. Remarkably, ∼50% of the Mecp2+/− mice developed these X-linked phenotypes despite somatic mosaicism. Such RTT-like respiratory endophenotypes in mixed-strain Mecp2+/− mice differed from those previously reported in Mecp2-/y mice on pure C57BL/6J background. These findings provide the first evidence indicating that impaired NMDAR-dependent desensitization of the HBIR may contribute to the endophenotypic tachypnea in RTT.
Introduction
Rett syndrome (RTT) is a neurological disorder most frequently caused by sporadic mutations in the X-linked gene encoding methyl-CpG-binding protein 2 (MeCP2; Amir et al., 1999), a transcriptional activator/repressor that regulates the expression of many genes (Chahrour et al., 2008). Homozygous mutation in females is rare (Karall et al., 2007), and hemizygous males (homozygous for a single X-chromosome) usually die shortly after birth except in variant cases (Villard, 2007). Heterozygous females are viable but show rapid developmental regression between ages 1–3 years (Hagberg et al., 1983).
Among the cardinal symptoms of RTT is a highly irregular respiratory rhythm particularly during daytime (Kerr et al., 2001; Hagberg et al., 2002; Julu et al., 2008). Recent studies in these patients reveal a predominantly hyperventilatory pattern with decreased expiratory duration (TE) and increased respiratory frequency; during wakefulness this is also punctuated by frequent episodes of breath-holding/obstructive apnea or Valsalva breathing against closed airways (Julu et al., 2001; Weese-Mayer et al., 2006, 2008). The breath-holding/obstructive apnea phenotype of RTT is often conflated in the clinical literature with central apnea, which has similar physiological effects but fundamentally distinct neural mechanisms (Lugaresi et al., 1985; Cirignotta et al., 1986; Southall et al., 1988; Kerr et al., 1990, 2001; Marcus et al., 1994; Schluter et al., 1995; Rohdin et al., 2007; Stettner et al., 2008b). The irregular breathing pattern in RTT is reproduced in several mutant mouse models to varying degrees but the corresponding respiratory phenotype varies significantly among different mouse strains (Bissonnette and Knopp, 2006; Ogier and Katz, 2008; Katz et al., 2009). In Mecp2tm1.1Jae null (hemizygous) mice on a mixed-strain background (Chen et al., 2001) the principal phenotype is tachypnea along with hyperventilation similar to human RTT patients (Ogier et al., 2007), whereas in Mecp2tm1.1Bird null or heterozygous mice on a pure C57BL/6J background (Guy et al., 2001) the principal phenotype is repetitive spontaneous central apnea (Viemari et al., 2005; Stettner et al., 2007; Abdala et al., 2010).
Remarkably, Mecp2tm1.1Bird null mice are reportedly highly prone to repetitive and prolonged central apneas particularly when vagal and dorsolateral pontine afferent pathways are activated to induce fictive Hering–Breuer inflation reflex (HBIR), a powerful apnea reflex in mammals (Stettner et al., 2007; Abdala et al., 2010). In rats, it has been shown that the HBIR apnea induced by abrupt lung inflation or low-intensity vagal stimulation is typically counteracted centrally by progressive habituation and desensitization, two distinct decrementing forms of non-associative learning (Poon et al., 2000; Siniaia et al., 2000; MacDonald et al., 2007, 2009). Behaviorally, desensitization is distinguished from habituation by the manifestation of a memory trace (engram) of the adaptation effect in a secondary pathway post-stimulation independent from the primary stimulus (Figure 1A; Poon and Young, 2006; Poon and Schmid, 2011). Functionally, desensitization of the HBIR is abolished by lesion of the dorsolateral pontine pneumotaxic center or blockade of NMDA receptor (NMDAR) while the habituation component remains unaffected by these interventions, suggesting that habituation is ascribable to an NMDAR-independent primary afferent pathway that is directly activated by the vagal input, and desensitization to an indirect NMDAR-dependent pontine pathway that is driven by a latent secondary input (Poon et al., 2000; Siniaia et al., 2000; MacDonald et al., 2007). By contrast, the vagal-induced HBIR in the Mecp2tm1.1Bird null mice appeared to exhibit secondary sensitization (rather than desensitization), an incrementing (rather than decrementing) form of non-associative learning characterized by the manifestation of a memory trace of the sensitization effect in a secondary pathway post-stimulation (Figure 1B; Poon and Young, 2006; Poon and Schmid, 2011). Such secondary sensitization effect tended to exacerbate instead of mitigate the HBIR-induced apnea in the Mecp2tm1.1Bird null mice (Poon and Song, 2007).
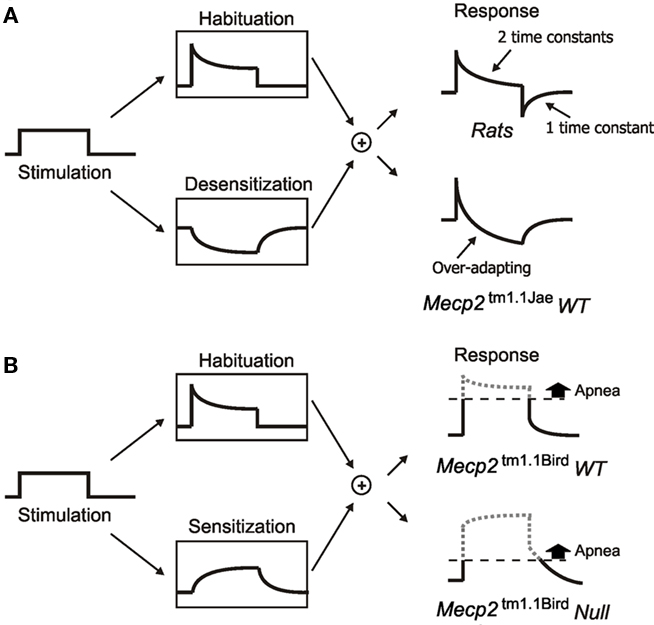
Figure 1. Schematic diagrams illustrating various forms of non-associative learning of HBIR in rats and mice at the behavioral level. (A) Low-intensity vagal stimulation (left panel) in rats elicits fictive HBIR prolongation of expiratory duration (TE), which is continually habituated (upper box, middle) and desensitized (lower box, middle) with different time constants via distinct NMDAR-independent vagal pathway and NMDAR-dependent pontine pathway. Habituation is discernible in the response only during stimulation whereas desensitization is also discernible as a post-stimulation short-term memory resulting in transient shortening of TE (upper box, right). See Siniaia et al. (2000) and MacDonald et al. (2009). In Mecp2tm1.1Jae wild-type mice with mixed-strain background (lower box, right), strong habituation, and/or desensitization result in over-adaptation in the response (see Figure 3A below). (B) In Mecp2tm1.1Bird wild-type mice with pure C57BL/6J background, desensitization is replaced by secondary sensitization with a short-term memory (lower box, middle), resulting in a transient prolongation of TE during the post-stimulation period (upper box, right). The sensitization effect is even stronger in Mecp2tm1.1Bird null mice resulting in prolonged post-stimulation apnea (lower box, right). See Poon and Song (2007). For a historical account of non-associative learning nomenclature in behavioral neuroscience and the contemporary classifications of habituation, desensitization, and primary/secondary sensitization, see Poon and Young (2006) and Poon and Schmid (2011).
Interestingly, wild-type mice with pure C57BL/6J background also demonstrate similar repetitive spontaneous central apneas (Han et al., 2002; Stettner et al., 2007, 2008a,c; Yamauchi et al., 2008) and secondary sensitization of the HBIR albeit to a lesser degree (Poon and Song, 2007; Stettner et al., 2007; Figure 1B). C57BL/6 inbred mouse strains are known to be vulnerable to slight variations in genetic background, such that behavioral phenotypes may vary significantly even among C57BL/6 substrains (Matsuo et al., 2010). A critical question arising is whether the pronounced spontaneous and HBIR-induced central apneas and sensitization of the HBIR in Mecp2tm1.1Bird null mice are intrinsic to the Mecp2 mutation or specific to the mouse strain used. Resolution of this question is crucial in pinpointing the respiratory endophenotypes of Mecp2 mutation in order to elucidate the underlying neural mechanisms or develop proper endophenotype-specific therapeutic strategies for RTT (Katz et al., 2009; Cobb et al., 2010). For example, treatments with certain neurotrophins have been shown to reverse the hyperventilation/tachypnea and other RTT-like symptoms in Mecp2tm1.1Jae null mice (Ogier et al., 2007; Tropea et al., 2009), whereas boosting the levels of certain monoamines and/or GABA have been suggested to effectively suppress spontaneous central apnea in Mecp2tm1.1Bird null mice and prolong their survival (Roux et al., 2007; Zanella et al., 2008; Abdala et al., 2010).
Most previous studies were conducted on Mecp2-/y male mice for their phenotypic homogeneity and early manifestation of respiratory abnormalities (reviewed in Ogier and Katz, 2008; Katz et al., 2009). However, whereas Mecp2-/y mice are viable and may live to adulthood, most human males with MECP2 mutations die perinatally (Hagberg et al., 1983) indicating differential vulnerability of humans and mice to loss of Mecp2. Indeed, mice with a less severe Mecp2 mutation, such as a mutation that results in a truncated protein instead of null mutation, could live even longer (Shahbazian et al., 2002). Recent case studies have shown that male patient survivors with RTT-like phenotypes do not carry pathogenic mutations in the MECP2 gene (Santos et al., 2009). Conversely, there is evidence that MECP2 null mutations in males may be responsible for a wide spectrum of neurological disorders that are distinctly different from RTT (Villard, 2007). In particular, it has been suggested that MeCP2 null males displaying a congenital encephalopathic phenotype, and not females with RTT, represent the human equivalent of the Mecp2tm1.1Bird hemizygous male mouse model (Schule et al., 2008). Here, we show that Mecp2tm1.1Jae mutant mice surprisingly showed Mendelian-like distribution for respiratory symptoms despite reputed somatic mosaicism of the X-linked Mecp2 gene, with heterozygous symptomatic mice demonstrating very different RTT-like breathing patterns at rest and during fictive HBIR than previously reported for hemizygous mice of the Mecp2tm1.1Bird strain. Results reveal distinct abnormalities in the neural pathways mediating NMDAR-dependent desensitization of the HBIR in the Mecp2tm1.1Jae female mouse model of RTT that may be responsible for their tachypneic pattern.
Materials and Methods
Animal Preparation
Heterozygous mice of the Mecp2tm1.1Jae strain (Chen et al., 2001) and their wild-type female littermates were maintained on a mixed background (129Sv, C57BL/6, BALB/c). Genotyping was performed as previously described (Chen et al., 2001). Eight adult heterozygous mice (age = 102 ± 24 days; mean ± SD) and five wild-type littermates (age = 103 ± 31 days) were studied. All of the mutant mice appeared healthy at the time of study and none of them exhibited behavioral and motor abnormalities which typically begin to develop after 5 weeks of age in these animals (Chen et al., 2001). All experimental methods and procedures were as approved by the Animal Care and Use Committee at Massachusetts Institute of Technology and conformed to National Institutes of Health guidelines. Briefly, The mouse was anesthetized with urethane (1.5 g/kg, i.p.), paralyzed with pancuronium bromide (0.1 mg in 0.1 ml, i.p.) and artificial ventilated (Minivent-856, Harvard Apparatus) with humidified and oxygen-enriched medical air (O2 at 40%) through a tracheal cannula. Ventilator tidal volume (80–120 μl) and frequency (100–120 cycles/min) were carefully adjusted to obtain stable phrenic discharge. The animal was kept warm with a heating pad (set at 37.5°C) while lying in supine position. EKG was monitored with subcutaneous needle electrodes. Ringer’s solution (0.5 ml) was subcutaneously injected every hour to keep the animal from dehydration. The depth of anesthetization was regularly checked. Whenever a noxious stimulus (clamping the hind paw) caused changes in respiration and heart rate or elicited a withdrawal reflex, a supplementary dose of urethane (1/10 original dosage) was given (i.p.) to maintain adequate anesthesia.
Electrophysiology
A phrenic nerve and both vagi at the cervical level were isolated from surrounding tissues and severed. The central cut-ends of the phrenic nerve and a vagus nerve were mounted onto bipolar platinum electrodes for recording or electrical stimulation. Phrenic discharges were amplified (CyberAmp 380, Axon Instruments, Union City) and sampled into a Dell PC with LabView (National Instruments, Austin, TX, USA) at a sampling rate of 10 kHz. The phrenic discharges were also integrated with a Paynter filter (time constant 15 ms) and sampled into the computer in a similar fashion. The vagus nerve was stimulated with square-wave pulses (pulse duration 0.1 ms) generated by a voltage pulse generator (A.M.P.I., Master 8) through a stimulus-isolation unit (A.M.P.I., ISO-Flex). The stimulation threshold for each animal was defined as the lowest stimulus current that produced a discernible reflex inhibition of phrenic activity over a 5-s interval. Stimulus pulses (80 Hz) with lowest currents possible (typically between 1.5–2× threshold, or roughly 10–70 μA) were applied repetitively to the vagus nerve for 1 min to evoke fictive HBIR and its resultant habituation and desensitization, as described previously (Siniaia et al., 2000). All exposed nerves were protected from dehydration by immersion in warm paraffin mineral oil pools.
Pharmacology
Dizocilpine (MK-801, Sigma-Aldrich Co., St. Louis, MO, USA), which is a non-competitive NMDAR antagonist that can pass through the blood–brain barrier, was administered i.p. (1.5 mg/kg).
Data Analyses
Inspiratory duration (TI), TE and respiratory frequency were measured for each respiratory cycle from the phrenic discharge. Inspiratory amplitude was measured as the peak of the integrated phrenic signal. Baseline values of these respiratory pattern variables were averaged over 1 min before vagal stimulation. The effects of HBIR were measured by the changes in TE and respiratory frequency in the first respiratory cycle upon abrupt low-intensity vagal stimulation compared with corresponding baseline values. Combined effects of habituation and desensitization of the HBIR were measured by the decreases in TE and respiratory frequency in the last 10 s of the 1-min vagal stimulation compared with those in the first respiratory cycle. The effect of desensitization of the HBIR was measured by the changes in TE and respiratory frequency in the first 5 s upon termination of vagal stimulation compared with corresponding baseline values.
Experiments and data analyses were performed blind to the animal genetic background except during final statistical analysis. Two-tailed Student’s t-test (paired or unpaired where appropriate) was used to determine statistical significance (p < 0.05) of differences in respiratory pattern between animal genetic backgrounds and experimental conditions.
Results
Expiratory-Shortening Phenotype in Mecp2tm1.1Jae Heterozygous Mice
In contrast to the profound spontaneous apnea and periodic breathing in Mecp2tm1.1Bird mutant or wild-type mice (Viemari et al., 2005; Stettner et al., 2007; Abdala et al., 2010), we found no signs of such abnormalities in any of the Mecp2tm1.1Jae wild-type or heterozygous female mice. Half (asymptomatic females, n = 4; age = 102 ± 7 days; mean ± SD) of the Mecp2tm1.1Jae heterozygous mice were found to exhibit breathing patterns similar to the wild-type animals whereas the other half (symptomatic females, n = 4; age = 103 ± 35 days) showed significantly increased respiratory frequency (Figure 2), in agreement with previous results in unanesthetized Mecp2tm1.1Jae null mice (Ogier et al., 2007). Importantly, the increased frequency was attributable mainly to a shortening of TE with little or no change in TI (Figure 2), in exact opposite to the spontaneous apnea or prolongation of TE reported in Mecp2tm1.1Bird heterozygous or null mice (Viemari et al., 2005; Stettner et al., 2007; Abdala et al., 2010).
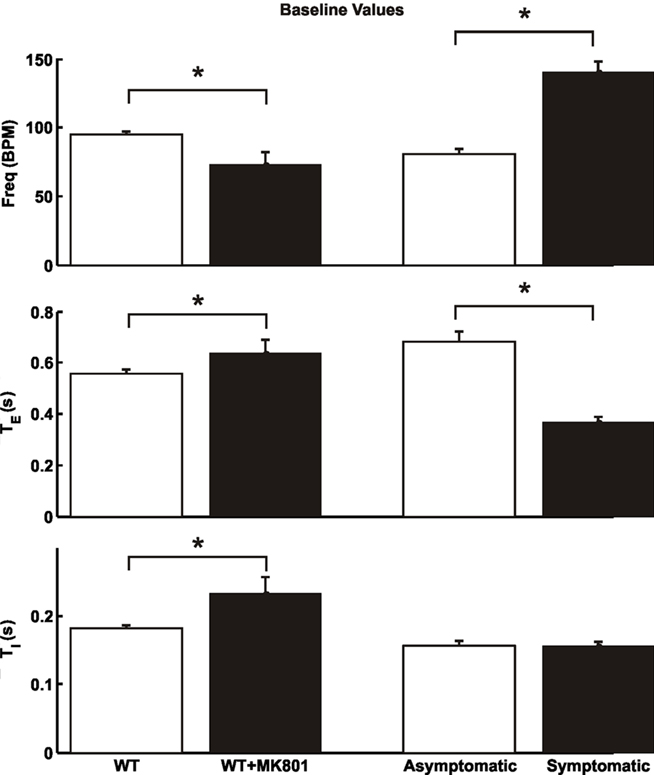
Figure 2. Baseline breathing patterns ofMecp2tm1.1Jae wild-type female mice before and after dizocilpine (MK-801) blockade of NMDARs (*p < 0.05, 2-tailed paired t-test), and of heterozygous asymptomatic and symptomatic mice (*p < 0.05, 2-tailed unpaired t-test). Data are means ± SD.
Pronounced Habituation and Desensitization of Fictive HBIR in Wild-Type Mice
To test whether the reported sensitization of HBIR prolongation of TE in the Mecp2tm1.1Bird wild-type mice (Poon and Song, 2007; Stettner et al., 2007) was specific to the C57BL/6J strain, we examined the use-dependent learning and memory of the HBIR in the mixed-strain Mecp2tm1.1Jae wild-type mice using an established protocol that has been shown to reproduce the habituation and desensitization of the HBIR induced by sustained lung inflation in rats (MacDonald et al., 2009). Low-intensity vagal stimulation (10–70 μA) in Mecp2tm1.1Jae wild-type mice elicited the classic HBIR response characterized by immediate cessation of phrenic discharge and prolongation of TE (Figure 3A). Thereafter, rhythmic phrenic discharge gradually reappeared and TE gradually shortened in adaptation to the initial reflex apnea response. As a result, respiratory frequency exhibited an initial abrupt decrease followed by gradual adaptive increase. As the stimulation continued, the shortening of TE and increase in respiratory frequency eventually over-adapted and went beyond their baseline levels. By the end of the 1-min vagal stimulation, TE was 13–39% below (−17.0 ± 3.5%, mean ± SD) its pre-stimulation value and respiratory frequency was 18–57% above (25.3 ± 4.2%). Upon cessation of vagal stimulation, the over-adaptations in TE and respiratory frequency persisted with a short-term memory indicating desensitization, and gradually returned to baselines in ∼20 s. The time-dependent adaptations of TE and respiratory frequency during and after low-intensity vagal stimulation (Figure 3A) are characteristic of, and even stronger than, the habituation and desensitization of the HBIR seen in rats (Figure 1A).
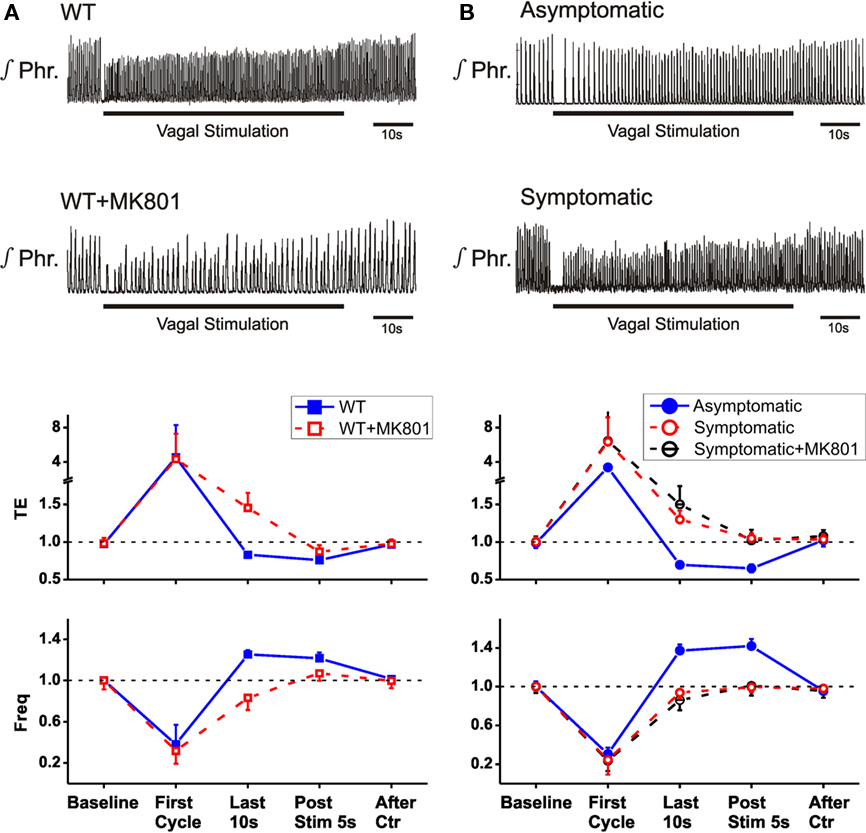
Figure 3. Non-associative learning of HBIR inMecp2tm1.1Jae mice. Top two panels are integrated phrenic recordings in a representative animal. Bottom panels are group averaged data (means ± SD) for TE and respiratory frequency normalized with respect to corresponding baseline values, plotted at selected time points before, during, and after vagal stimulation. (A) Wild-type female mice before and after dizocilpine treatment. (B) Heterozygous asymptomatic and symptomatic female mice. Dizocilpine had no effect in symptomatic mice (p > 0.1, 2-tailed paired t-test).
Dizocilpine Suppresses Desensitization of Fictive HBIR in Wild-Type Mice
Next, we tested the effects of blockade of NMDARs by systemic administration of dizocilpine (MK-801). Fifteen minutes after systemic administration (1.5 mg/kg, i.p.) of this chemical in wild-type mice of the Mecp2tm1.1Jae strain, respiratory frequency was significantly decreased and TI was significantly increased (Figure 2), as previously reported in vagotomized rats and mice (Cassus-Soulanis et al., 1995). Under dizocilpine, low-intensity vagal stimulation still elicited fictive HBIR response with immediate cessation of phrenic discharge and prolongation of TE (Figure 3A). However, the ensuing time-dependent adaptations of TE and respiratory frequency responses during vagal stimulation were much weaker than before dizocilpine treatment as measured in the last 10 s of vagal stimulation (p < 0.05, 2-tailed paired t-test). Importantly, the responses in TE and respiratory frequency within the first 5-s post-stimulation were not significantly different from the corresponding baseline values (p > 0.1) indicating suppressed desensitization of the HBIR, although possible attenuation of the habituation component cannot be ruled out (Figure 3A).
Absence of Sensitization/Desensitization of Fictive HBIR in Mecp2tm1.1Jae Heterozygous Mice
To investigate whether the Mecp2 mutation demonstrated similar effects as dizocilpine, we applied the above low-intensity vagal stimulation protocol to Mecp2tm1.1Jae heterozygous mice. In heterozygous asymptomatic mutants the HBIR responses in TE and respiratory frequency (Figure 3B) were similar to those of their wild-type littermates (Figure 3A). In heterozygous symptomatic mutants characterized by decreased TE and increased respiratory frequency (Figure 2), low-intensity vagal stimulation also elicited immediate HBIR prolongation of TE and decrease of respiratory frequency. However, the ensuing time-dependent adaptations of TE and respiratory frequency were much weaker (p < 0.01, 2-tailed unpaired t-test) and never exceeded the corresponding baselines. Neither of these variables exhibited post-stimulation short-term memory such that responses within the first 5-s post-stimulation were indistinguishable from corresponding baseline values (p > 0.1), indicating that the adaptations are now ascribable solely to habituation without desensitization or sensitization (Figure 3B). These effects were similar before or after dizocilpine application (p > 0.1, Figure 3B) and resembled those resulting from dizocilpine in wild-type animals (Figure 3A).
Discussion
The foregoing results demonstrate that respiratory abnormalities are different in adult Mecp2tm1.1Jae symptomatic female mice (∼P100 of age) than previously reported in hemizygous male or heterozygous female mice of the Mecp2tm1.1Bird strain (P40 to 14 months of age). A salient respiratory symptom of the Mecp2tm1.1Jae heterozygous mutant mice was a shortening of TE and resultant increase in respiratory frequency, which is diametrically opposite to the repetitive spontaneous central apnea or prolongation of TE in the Mecp2tm1.1Bird null mice (Viemari et al., 2005; Stettner et al., 2007; Abdala et al., 2010). The tachypnea phenotype in the Mecp2tm1.1Jae heterozygous mutants under anesthesia and bivagotomy is consistent with a previously reported increase in mean respiratory frequency in Mecp2tm1.1Jae null mice (P35 of age) during wakefulness (Ogier et al., 2007) and more importantly, it is in agreement with the documented tachypneic breathing pattern with a shortened TE in girls with RTT during sleep (Weese-Mayer et al., 2008) as well as during wakefulness amidst intermittent breath-holding/obstructive apnea episodes (Weese-Mayer et al., 2006). As with RTT patients during sleep, breath-holding/obstructive apnea and Valsalva maneuvers were not observed in these anesthetized animals.
Another important, albeit more subtle, respiratory endophenotype of the Mecp2tm1.1Jae heterozygous symptomatic mice was a degenerate non-associative learning characterized by a significant habituation of the HBIR but absent the NMDAR-dependent desensitization that is found in wild-type or asymptomatic female mice. None of these wild-type and heterozygous mutant mice evidenced the sensitization of HBIR seen in the Mecp2tm1.1Bird null and wild-type mice (Figure 1B). This suggests that secondary sensitization of the HBIR is specific to the Mecp2tm1.1Bird strain.
To put these findings in perspective, a current working model (Siniaia et al., 2000; Poon, 2004; Song and Poon, 2004; MacDonald et al., 2007, 2009) postulates that the HBIR is mediated by two parallel vagally modulated afferent pathways acting in concert to modulate TE: a primary (NMDAR-independent) habituation-prone direct pathway via the nucleus tractus solitarius (NTS), and a secondary (NMDAR-dependent) desensitization-prone indirect pathway via both the NTS and the Kölliker-Fuse/parabrachial complex (“pneumotaxic center;” Lumsden, 1923; Song et al., 2006) in dorsolateral pons (Figure 4A). In the absence of an extrinsic vagal input (such as post-bivagotomy), the secondary pathway provides an intrinsic (pontine) expiratory-promoting signal that facilitates the inspiratory off-switch, perhaps via pontine post-inspiratory activity (Dutschmann and Herbert, 2006) which has been postulated to suppress inspiratory rhythm generation in the preBötzinger complex (Wittmeier et al., 2008) and has been shown to correlate with prolonged apneas in the Mecp2tm1.1Bird null mice (Stettner et al., 2007). A strong lung volume-related vagal input elicits the HBIR by providing an extrinsic (vagal) expiratory-promoting signal and simultaneously triggering the intrinsic expiratory-promoting signal. The ensuing HBIR is continuously attenuated in a time-dependent manner by use-dependent habituation and desensitization; the latter is manifested as a post-stimulation short-term memory (Figure 1A). Under this scheme, inactivation of the pneumotaxic center by pontine lesion or NMDAR blockade disrupts the intrinsic expiratory-promoting signal and its desensitization but not the extrinsic expiratory-promoting signal and its habituation, as observed experimentally in rats (Poon et al., 2000; Siniaia et al., 2000; MacDonald et al., 2007).
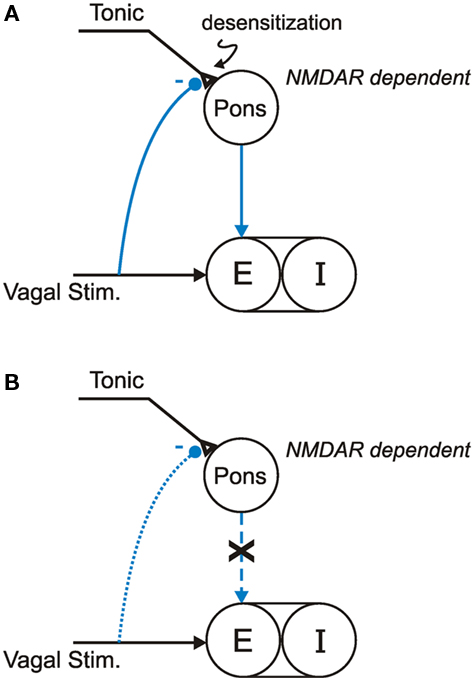
Figure 4. Working models of non-associative learning of HBIR in: (A) In Mecp2tm1.1Jaewild-type mice, HBIR is moderated by NMDAR-dependent desensitization in the pontine pathway; (B) In Mecp2tm1.1Jae heterozygous symptomatic mice, NMDAR-dependent pontine desensitization is impaired.
In the present study, use-dependent non-associative learning modulation of the HBIR was seen to be similar, and even stronger, in the wild-type mice compared to rats. Further, as has been observed in rats, administration of dizocilpine diminished the desensitization without affecting the habituation of HBIR in wild-type mice, indicating similar vagal-pontine neural organization of the HBIR and its non-associative learning in these species (Figure 4A). Accordingly, the absence of desensitization in the HBIR response presently found in the Mecp2tm1.1Jae heterozygous symptomatic mice suggests that the intrinsic NMDAR-dependent expiratory-promoting mechanism in the pneumotaxic center was impaired in these animals (Figure 4B). This is further supported by the lack of effect of dizocilpine in the symptomatic mice (Figure 3B). Such a correlation between Mecp2 mutation and disruption of NMDAR-dependent pontine desensitization provides a mechanistic explanation of the significant decrease in TE with no change in TI presently observed in the symptomatic mutant mice, as with RTT patients.
Given the general variability of phenotypes in X-linked genetic mutations such as in RTT due to somatic mosaicism, it is remarkable that Mecp2+/− mutants expressed the respiratory phenotypes with ∼50% probability. To understand this, it should be noted that X-chromosome inactivation takes place early in embryonic development before gastrulation, not in individual somatic cells. Type-specific neurons with lineage to the same post-gastrulation progenitor cell inherit the X-inactivation state of the progenitor cell that does not change throughout life (Dvash and Fan, 2009). Therefore, although the X-inactivation state may vary between different cell types giving rise to somatic mosaicism, the X-inactivation state within the same cell type could remain uniform. An endophenotype has a 50% chance to express if it is dependent on the genotype of a single cell type. This is in contrast to phenotypes (non-endophenotypes) that are dependent on multiple cell types or tissues (or even multiple organs), where the phenotype-genotype relationship is less distinct and more susceptible to somatic mosaicism. The distinct 50–50 expression ratio of NMDAR-dependent desensitization of the HBIR in asymptomatic-symptomatic mutants therefore implies that disruption of NMDAR signaling in the pons is the single respiratory-related endophenotype in Mecp2+/− symptomatic mutants. This also explains the consistent expression of the hyperventilation and shortened TE phenotypes in RTT girls as opposed to the variability of other phenotypes in these patients (Julu et al., 2001; Weese-Mayer et al., 2006, 2008). The distinct respiratory phenotype-genotype relationship in Mecp2+/− symptomatic mutants provides a unique opportunity for studying the effect of Mecp2 mutation in the mammalian brain.
The present results corroborate the notion that the spontaneous apnea and prolonged HBIR-induced apnea previously reported in the Mecp2tm1.1Bird null mice are mediated by an over-expression of the intrinsic expiratory-promoting signal, as indicated by the pronounced pontine post-inspiratory activity in those animals (Stettner et al., 2007). Rather than desensitize, the intrinsic post-inspiratory activity in the secondary pathway appears to sensitize upon vagal stimulation instead – thereby sustaining and prolonging the induced apnea, overshadowing any habituation of the HBIR in the primary pathway (Poon and Song, 2007). The cause of such divergent mal-adaptations of the HBIR in the Mecp2tm1.1Bird null mice and the Mecp2tm1.1Jae heterozygous mice is not clear. However, the fact that Mecp2tm1.1Bird null and wild-type mice displayed similar propensity for spontaneous apnea and secondary sensitization of the HBIR albeit to varying degrees suggests that such abnormalities may be intrinsic to the C57BL/6J strain and are exacerbated in the Mecp2tm1.1Bird null mice. Indeed, the C57BL/6J mouse strain is known to be predisposed to spontaneous deletion mutation in the gene encoding nicotinamide nucleotide transhydrogenase, an inner mitochondrial membrane transmembrane proton-translocating protein involved in regenerating intramitochondrial NADPH (Freeman et al., 2006; Huang et al., 2006; Rydstrom, 2006), which plays an important role in mitochondrial metabolism of reactive oxygen species (ROS; Andreyev et al., 2005; Kowaltowski et al., 2009). Recent evidence reveals that Mecp2tm1.1Bird null mice are susceptible to many other mitochondrial abnormalities that may further promote mitochondrial production of ROS (Kriaucionis et al., 2006). The latter has been shown to contribute to the expression of long-term facilitation of carotid body chemosensory activity and phrenic nerve respiratory motor activity in normal animals after exposure to intermittent hypoxia (Peng et al., 2003; MacFarlane and Mitchell, 2009). We speculate that such ROS-induced long-term facilitation of chemoreflex afferent and efferent signaling may be intrinsic to the C57BL/6J mice and Mecp2tm1.1Bird null mice, effectively increasing the respiratory controller gain in these animals. The resultant amplifications in respiratory system loop gain could potentially leave these animals with ROS excess at high risk of periodic breathing and spontaneous central apnea (Khoo, 2000). Alternatively, there is evidence that increased ROS production may directly modulate the respiratory rhythm in preBötzinger complex neurons (Garcia et al., 2011). Further studies are needed to investigate whether the intermittent apnea in the Mecp2tm1.1Bird null mice and the C57BL/6J wild-type strain represents ROS-induced chemoreflex instability in the respiratory system or abnormal rhythmogenesis in pacemaker neurons.
To our knowledge, this is the first experimental demonstration of abnormal non-associative learning caused by a specific genetic mutation that is linked to a well-defined clinical phenotype of a congenital neurological disease. Although the cellular bases of non-associative learning paradigms such as habituation and sensitization have been extensively studied in invertebrate sensorimotor systems (Kandel, 1978; Glanzman, 2009; Ardiel and Rankin, 2010), details of their counterparts in mammalian brain systems are only beginning to emerge recently (Siniaia et al., 2000; MacDonald et al., 2009; Wilson, 2009; Schmid et al., 2010). The present results provide a novel mammalian model of studying the structure-function correlations of two contrasting forms of non-associative learning (sensitization and desensitization) from genetic to behavioral levels.
In conclusion, we have shown that mutation in the Mecp2 gene may lead to disparate respiratory endophenotypes in the Mecp2tm1.1Jae and Mecp2tm1.1Bird mouse strains, indicating possible interaction of the Mecp2 gene with animal genetic background. Whereas the Mecp2tm1.1Bird male mice provide an excellent animal model of spontaneous central apnea and possibly obstructive apnea (Voituron et al., 2010), the present study confirmed that a clinically relevant RTT endophenotype – tachypnea with shortened TE – is more faithfully reproduced in Mecp2tm1.1Jae female mice. Importantly, the shortening of TE was found to correlate with the lack of NMDAR-dependent desensitization of the HBIR in these mixed-strain heterozygous mutants compared to wild-type mice, in sharp contrast to the abnormal prolongation of TE and secondary sensitization of the HBIR reported in the Mecp2tm1.1Bird null mice. These findings shed new light on the mechanisms of disordered breathing in RTT and corroborate a working model of non-associative learning in the mammalian brain. This non-associative learning perspective provides a new dimension for further investigation of the pathogenesis of breathing abnormalities in these mutant animals with impaired methylated DNA binding or those with DNA hypomethylation (Fan et al., 2001), and in patients with RTT caused by mutations of the MECP2 gene.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. C. Beard, G. Fan and R. Jaenisch for useful discussions on the genetic aspect of the manuscript, Drs. C. Marcus and D. Weese-Mayer on the clinical aspect, and Dr. K. Strohl on the physiological aspect. We also thank Dr. R. Jaenisch for providing the mutant and wild-type animals used in this study. This work was supported by National Institutes of Health grants HL093225 (GS), HL067966, HL072849, HL079503, and RR028241 (CSP).
References
Abdala, A. P., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. (2010). Correction of respiratory disorders in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 18208–18213.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry Mosc. 70, 200–214.
Ardiel, E. L., and Rankin, C. H. (2010). An elegant mind: learning and memory in Caenorhabditis elegans. Learn. Mem. 17, 191–201.
Bissonnette, J. M., and Knopp, S. J. (2006). Separate respiratory phenotypes in methyl-CpG-binding protein 2 (Mecp2) deficient mice. Pediatr. Res. 59, 513–518.
Cassus-Soulanis, S., Foutz, A. S., and Denavit-Saubie, M. (1995). Involvement of NMDA receptors in inspiratory termination in rodents: effects of wakefulness. Brain Res. 679, 25–33.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., and Zoghbi, H. Y. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Cirignotta, F., Lugaresi, E., and Montagna, P. (1986). Breathing impairment in Rett syndrome. Am. J. Med. Genet. Suppl. 1, 167–173.
Cobb, S., Guy, J., and Bird, A. (2010). Reversibility of functional deficits in experimental models of Rett syndrome. Biochem. Soc. Trans. 38, 498–506.
Dutschmann, M., and Herbert, H. (2006). The Kolliker-Fuse nucleus gates the postinspiratory phase of the respiratory cycle to control inspiratory off-switch and upper airway resistance in rat. Eur. J. Neurosci. 24, 1071–1084.
Dvash, T., and Fan, G. (2009). Epigenetic regulation of X-inactivation in human embryonic stem cells. Epigenetics 4, 19–22.
Fan, G., Beard, C., Chen, R. Z., Csankovszki, G., Sun, Y., Siniaia, M., Biniszkiewicz, D., Bates, B., Lee, P. P., Kuhn, R., Trumpp, A., Poon, C., Wilson, C. B., and Jaenisch, R. (2001). DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 21, 788–797.
Freeman, H. C., Hugill, A., Dear, N. T., Ashcroft, F. M., and Cox, R. D. (2006). Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 55, 2153–2156.
Garcia, A. J., Dean, J. B., and Ramirez, J. M. (2011). Reactive oxygen species production and modulation of rhythmogenesis from VRG neurons. FASEB J. 25,1074.3.
Glanzman, D. L. (2009). Habituation in Aplysia: the Cheshire cat of neurobiology. Neurobiol. Learn. Mem. 92, 147–154.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479.
Hagberg, B., Hanefeld, F., Percy, A., and Skjeldal, O. (2002). An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 6, 293–297.
Han, F., Subramanian, S., Price, E. R., Nadeau, J., and Strohl, K. P. (2002). Periodic breathing in the mouse. J. Appl. Physiol. 92, 1133–1140.
Huang, T. T., Naeemuddin, M., Elchuri, S., Yamaguchi, M., Kozy, H. M., Carlson, E. J., and Epstein, C. J. (2006). Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum. Mol. Genet. 15, 1187–1194.
Julu, P. O., Engerstrom, I. W., Hansen, S., Apartopoulos, F., Engerstrom, B., Pini, G., Delamont, R. S., and Smeets, E. E. (2008). Cardiorespiratory challenges in Rett’s syndrome. Lancet 371, 1981–1983.
Julu, P. O., Kerr, A. M., Apartopoulos, F., Al-Rawas, S., Engerstrom, I. W., Engerstrom, L., Jamal, G. A., and Hansen, S. (2001). Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch. Dis. Child. 85, 29–37.
Kandel, E. R. (1978). A Cell-Biological Approach to Learning. Bethesda, MD: Society for Neuroscience.
Karall, D., Haberlandt, E., Scholl-Bürgi, S., Baumgartner, S., Naudó, M., and Martorell, L. (2007). Homozygosity for MECP2 gene in a girl with classical Rett syndrome. Eur. J. Med. Genet. 50, 465–468.
Katz, D. M., Dutschmann, M., Ramirez, J. M., and Hilaire, G. (2009). Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir. Physiol. Neurobiol. 168, 101–108.
Kerr, A., Southall, D., Amos, P., Cooper, R., Samuels, M., Mitchell, J., and Stephenson, J. (1990). Correlation of electroencephalogram, respiration and movement in the Rett syndrome. Brain Dev. 12, 61–68.
Kerr, A. M., Nomura, Y., Armstrong, D., Anvret, M., Belichenko, PV., Budden, S., Cass, H., Christodoulou, J., Clarke, A., Ellaway, C., d’Esposito, M., Francke, U., Hulten, M., Julu, P., Leonard, H., Naidu, S., Schanen, C., Webb, T., Engerstrom, IW., Yamashita, Y., and Segawa, M. (2001). Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev. 23, 208–211.
Khoo, M. C. (2000). Determinants of ventilatory instability and variability. Respir. Physiol. 122, 167–182.
Kowaltowski, A. J., de Souza-Pinto, N. C., Castilho, R. F., and Vercesi, A. E. (2009). Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 47, 333–343.
Kriaucionis, S., Paterson, A., Curtis, J., Guy, J., Macleod, N., and Bird, A. (2006). Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell. Biol. 26, 5033–5042.
Lugaresi, E., Cirignotta, F., and Montagna, P. (1985). Abnormal breathing in the Rett syndrome. Brain Dev. 7, 329–333.
Lumsden, T. (1923). Observations on the respiratory centres in the cat. J. Physiol. (Lond.) 57, 153–160.
MacDonald, S. M., Song, G., and Poon, C. S. (2007). Nonassociative learning promotes respiratory entrainment to mechanical ventilation. PLoS ONE 2, e865. doi: 10.1371/journal.pone.0000865
MacDonald, S. M., Tin, C., Song, G., and Poon, C. S. (2009). Use-dependent learning and memory of the Hering–Breuer inflation reflex in rats. Exp. Physiol. 94, 269–278.
MacFarlane, P. M., and Mitchell, G. S. (2009). Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J. Physiol. 587, 5469–5481.
Marcus, C. L., Carroll, J. L., McColley, S. A., Loughlin, G. M., Curtis, S., Pyzik, P., and Naidu, S. (1994). Polysomnographic characteristics of patients with Rett syndrome. J. Pediatr. 125, 218–224.
Matsuo, N., Takao, K., Nakanishi, K., Yamasaki, N., Tanda, K., and Miyakawa, T. (2010). Behavioral profiles of three C57BL/6 substrains. Front. Behav. Neurosci. 4:29. doi: 10.3389/fnbeh.2010.00029
Ogier, M., and Katz, D. M. (2008). Breathing dysfunction in Rett syndrome: understanding epigenetic regulation of the respiratory network. Respir. Physiol. Neurobiol. 164, 55–63.
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., and Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci. 27, 10912–10917.
Peng, Y. J., Overholt, J. L., Kline, D., Kumar, G. K., and Prabhakar, N. R. (2003). Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc. Natl. Acad. Sci. U.S.A. 100, 10073–10078.
Poon, C. S. (2004). Organization of central pathways mediating the Hering–Breuer reflex and carotid chemoreflex. Adv. Exp. Med. Biol. 551, 95–100.
Poon, C. S., and Song, G. (2007). Habituation, desensitization and sensitization of the Hering Breuer reflex in normal and Mecp2 /y knockout mice. J. Physiol. 584, 359–360; author reply 361.
Poon, C. S., and Young, D. L. (2006). Nonassociative learning as gated neural integrator and differentiator in stimulus-response pathways. Behav. Brain Funct. 2, 29.
Poon, C. S., Young, D. L., and Siniaia, M. S. (2000). High-pass filtering of carotid-vagal influences on expiration in rat: role of N-methyl-D-aspartate receptors. Neurosci. Lett. 284, 5–8.
Poon, C.-S., and Schmid, S. (2011). “Nonassociative learning,” in Encyclopedia of the Sciences of Learning, ed. N. M. Seel (New York: Springer), in press.
Rohdin, M., Fernell, E., Eriksson, M., Albage, M., Lagercrantz, H., and Katz-Salamon, M. (2007). Disturbances in cardiorespiratory function during day and night in Rett syndrome. Pediatr. Neurol. 37, 338–344.
Roux, J. C., Dura, E., Moncla, A., Mancini, J., and Villard, L. (2007). Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur. J. Neurosci. 25, 1915–1922.
Rydstrom, J. (2006). Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726.
Santos, M., Temudo, T., Kay, T., Carrilho, I., Medeira, A., Cabral, H., Gomes, R., Lourenco, M. T., Venancio, M., Calado, E., Moreira, A., Oliveira, G., and Maciel, P. (2009). Mutations in the MECP2 gene are not a major cause of Rett syndrome-like or related neurodevelopmental phenotype in male patients. J. Child Neurol. 24, 49–55.
Schluter, B., Aguigah, G., Buschatz, D., Trowitzsch, E., and Aksu, F. (1995). Polysomnographic recordings of respiratory disturbances in Rett syndrome. J. Sleep Res. 4, 203–207.
Schmid, S., Brown, T., Simons-Weidenmaier, N., Weber, M., and Fendt, M. (2010). Group III metabotropic glutamate receptors inhibit startle-mediating giant neurons in the caudal pontine reticular nucleus but do not mediate synaptic depression/short-term habituation of startle. J. Neurosci. 30, 10422–10430.
Schule, B., Armstrong, D. D., Vogel, H., Oviedo, A., and Francke, U. (2008). Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin. Genet. 74, 116–126.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R., and Zoghbi, H. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254.
Siniaia, M. S., Young, D. L., and Poon, C. S. (2000). Habituation and desensitization of the Hering–Breuer reflex in rat. J. Physiol. 523 (Pt 2), 479–491.
Song, G., and Poon, C. S. (2004). Functional and structural models of pontine modulation of mechanoreceptor and chemoreceptor reflexes. Respir. Physiol. Neurobiol. 143, 281–292.
Song, G., Yu, Y., and Poon, C. S. (2006). Cytoarchitecture of pneumotaxic integration of respiratory and nonrespiratory information in the rat. J. Neurosci. 26, 300–310.
Southall, D. P., Kerr, A. M., Tirosh, E., Amos, P., Lang, M. H., and Stephenson, J. B. (1988). Hyperventilation in the awake state: potentially treatable component of Rett syndrome. Arch. Dis. Child. 63, 1039–1048.
Stettner, G. M., Huppke, P., Brendel, C., Richter, D. W., Gartner, J., and Dutschmann, M. (2007). Breathing dysfunctions associated with impaired control of postinspiratory activity in Mecp2-/y knockout mice. J. Physiol. 579, 863–876.
Stettner, G. M., Zanella, S., Hilaire, G., and Dutschmann, M. (2008a). 8-OH-DPAT suppresses spontaneous central apneas in the C57BL/6J mouse strain. Respir. Physiol. Neurobiol. 161, 10–15.
Stettner, G. M., Huppke, P., Gartner, J., Richter, D. W., and Dutschmann, M. (2008b). Disturbances of breathing in Rett syndrome: results from patients and animal models. Adv. Exp. Med. Biol. 605, 503–507.
Stettner, G. M., Zanella, S., Huppke, P., Gartner, J., Hilaire, G., and Dutschmann, M. (2008c). Spontaneous central apneas occur in the C57BL/6J mouse strain. Respir. Physiol. Neurobiol. 160, 21–27.
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., Flannery, R., Jaenisch, R., and Sur, M. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034.
Viemari, J. C., Roux, J. C., Tryba, A. K., Saywell, V., Burnet, H., Pena, F., Zanella, S., Bevengut, M., Barthelemy-Requin, M., Herzing, L. B., Moncla, A., Mancini, J., Ramirez, J. M., Villard, L., and Hilaire, G. (2005). Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J. Neurosci. 25, 11521–11530.
Voituron, N., Menuet, C., Dutschmann, M., and Hilaire, G. (2010). Physiological definition of upper airway obstructions in mouse model for Rett syndrome. Respir. Physiol. Neurobiol. 173, 146–156.
Weese-Mayer, D. E., Lieske, S. P., Boothby, C. M., Kenny, A. S., Bennett, H. L., and Ramirez, J. M. (2008). Autonomic dysregulation in young girls with Rett Syndrome during nighttime in-home recordings. Pediatr. Pulmonol. 43, 1045–1060.
Weese-Mayer, D. E., Lieske, S. P., Boothby, C. M., Kenny, A. S., Bennett, H. L., Silvestri, J. M., and Ramirez, J. M. (2006). Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr. Res. 60, 443–449.
Wilson, D. A. (2009). Olfaction as a model system for the neurobiology of mammalian short-term habituation. Neurobiol. Learn. Mem. 92, 199–205.
Wittmeier, S., Song, G., Duffin, J., and Poon, C. S. (2008). Pacemakers handshake synchronization mechanism of mammalian respiratory rhythmogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 18000–18005.
Yamauchi, M., Ocak, H., Dostal, J., Jacono, F. J., Loparo, K. A., and Strohl, K. P. (2008). Post-sigh breathing behavior and spontaneous pauses in the C57BL/6J (B6) mouse. Respir. Physiol. Neurobiol. 162, 117–125.
Keywords: Rett syndrome, Mecp2, NMDA receptor, non-associative learning, habituation, sensitization, desensitization, X-chromosome inactivation
Citation: Song G, Tin C, Giacometti E and Poon C-S (2011) Habituation without NMDA receptor-dependent desensitization of Hering–Breuer apnea reflex in a Mecp2+/− mutant mouse model of Rett syndrome. Front. Integr. Neurosci. 5:6. doi: 10.3389/fnint.2011.00006
Received: 14 January 2011;
Accepted: 12 April 2011;
Published online: 02 May 2011.
Edited by:
John J. Foxe, Albert Einstein College of Medicine, USAReviewed by:
Mathias Dutschmann, Universtiy of Leeds, UKLisa M. Monteggia, UT Southwestern Medical Center, USA
Copyright: © 2011 Song, Tin, Giacometti and Poon. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Chi-Sang Poon, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Bldg. E25-250, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. e-mail: cpoon@mit.edu