Interactions of AChE with Aβ aggregates in Alzheimer’s brain: therapeutic relevance of IDN 5706
- Centro de Envejecimiento y Regeneración (CARE), Departamento de Biología Celular y Molecular, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
Acetylcholinesterase (AChE; EC 3.1.1.7) plays a crucial role in the rapid hydrolysis of the neurotransmitter acetylcholine, in the central and peripheral nervous system and might also participate in non-cholinergic mechanism related to neurodegenerative diseases. Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive deterioration of cognitive abilities, amyloid-β (Aβ) peptide accumulation and synaptic alterations. We have previously shown that AChE is able to accelerate the Aβ peptide assembly into Alzheimer-type aggregates increasing its neurotoxicity. Furthermore, AChE activity is altered in brain and blood of Alzheimer’s patients. The enzyme associated to amyloid plaques changes its enzymatic and pharmacological properties, as well as, increases its resistant to low pH, inhibitors and excess of substrate. Here, we reviewed the effects of IDN 5706, a hyperforin derivative that has potential preventive effects on the development of AD. Our results show that treatment with IDN 5706 for 10 weeks increases brain AChE activity in 7-month-old double transgenic mice (APPSWE–PS1) and decreases the content of AChE associated with different types of amyloid plaques in this Alzheimer’s model. We concluded that early treatment with IDN 5706 decreases AChE–Aβ interaction and this effect might be of therapeutic interest in the treatment of AD.
Introduction
Alzheimer’s disease (AD) is characterized by progressive memory and cognitive impairment and the cerebral accumulation of extracellular amyloid plaques and intra-neuronal neurofibrillary tangles (NFTs) in areas of brain involved in learning and memory (Ballard et al., 2011). Amyloid plaques are extracellular deposits of aggregated amyloid-β (Aβ) peptide, surrounded by dystrophic neurites and reactive glial cells. Aβ peptide is the main constituent of senile plaques and the major neurotoxic agent (Li et al., 2010). Intra-neuronal NFTs consist largely of hyper phosphorylated twisted filaments of the microtubule-associated protein tau (Lee et al., 2001). Synaptic pathology is an early marker of both, AD and aging, with decreased dendritic spine density, degeneration of neurites, neuronal loss, and cortical atrophy (Knobloch and Mansuy, 2008).
Original neurochemical findings in AD brains pointed out to disturbances of acetylcholine metabolism and led to the formulation of the “cholinergic hypothesis” of AD. This hypothesis suggests that there is a loss of cholinergic neurons in the basal forebrain of AD patients (Bartus et al., 1982; Bartus, 2000). The deficiency of cholinergic projections in AD has been linked to the buildup of Aβ and tau. Acetylcholinesterase (AChE; EC 3.1.1.7) and choline acetyltransferase activities decreases, while Na+-dependent high-affinity choline uptake increases, perhaps due to compensatory mechanisms (Slotkin et al., 1994; Bissette et al., 1996; Shinotoh et al., 2000; DeKosky et al., 2002). Presynaptic α7 nicotinic acetylcholine receptors are essential for cognitive processes, and their levels increase in early AD, decreasing later on (Ikonomovic et al., 2009). The levels of muscarinic acetylcholine receptors, or receptor coupling, are reduced in the brains of patients with AD. However, pharmacological stimulation of the postsynaptic muscarinic type 1 acetylcholine receptors activates protein kinase C, favoring the processing of amyloid precursor protein (APP) that does not yield an amyloidogenic fragment (Nitsch, 1996; Farias et al., 2004). According to the cholinergic hypothesis, the impairment of cognitive functions and the behavioral disturbances that affect patients with AD are in part due, to cortical deficiencies in cholinergic neurotransmission (Bartus et al., 1982; Dumas and Newhouse, 2011). AD is associated with an early and severe depletion of cholinergic innervations. AChE activity is lower in most regions of AD brains, but it is increased within and around amyloid plaques (Geula and Mesulam, 1989b; Beach et al., 2000). The different molecular forms of AChE are altered in AD, showing a decrease in the tetrameric AChE G4 isoform localized at central synapses (Xie et al., 2010), while the minor light forms (dimers G2 and monomers G1) increase (Atack et al., 1983; Saez-Valero et al., 1999). Interestingly, the activity of the light forms appears to increase in the most severely affected cases (Arendt et al., 1992). Some studies indicate that the level of an amphiphilic monomeric form of AChE is increased in the brains of transgenic mice which produce the human Aβ protein (Sberna et al., 1998), and in the brain and cerebrospinal fluid (CSF) of rats which received intra-cerebral-ventricular injections of the Aβ peptide (Saez-Valero et al., 2002). So far, the precise nature of this subset of G1 species which increase in AD brains remains unclear, however this minor species can be distinguished from other brain AChE forms (including tetramers but also from other monomeric AChE isoforms), by its unusual lectin-binding pattern and the lack of binding to anti-AChE antibodies (Saez-Valero et al., 2000; Garcia-Ayllon et al., 2007). Some cholinergic deficits have been shown to appear in transgenic mouse model reproducing preclinical and early stages of amyloid pathology. Region-specific modifications in AChE activity were reported in APP696LD (London V642I mutation) transgenic mice with Aβ plaques, being decreased in subiculum but increased in the dentate gyrus, a CA1 sub-region of the hippocampal formation (Bronfman et al., 2000). AChE activity was unchanged in APP751SWE and APP695SWE transgenic mice despite extensive Aβ plaques (Apelt et al., 2002; Boncristiano et al., 2002). However, when specific AChE isoforms were taken into account, the activity of an abnormally glycosylated G1 version increased in cortical extracts of APP695SWE mice, whereas the activity of the tetrameric G4 AChE was unchanged (Fodero et al., 2002).
AChE activity in the blood and plasma has also been measured to assess the pathophysiology of AD. Plasma AChE activity levels are increased in AD patients, which correlates with an increase in the light AChE species (G1 + G2) which are the major species in human plasma, whereas tetramers, that are normally only presents in trace amounts, are slightly decreased in AD plasma (Garcia-Ayllon et al., 2010). Plasma AChE is likely to have multiple cellular origins including cells from the brain. Thus, we can hypothesize that the increase observed in AD plasma may be associated with the particular increase in the light AChE species characterized in AD brain (Arendt et al., 1992; Saez-Valero et al., 1999). Blood AChE and butyrylcholinesterase (BuChE) activities have been studied as markers for Alzheimer’s. AD patients have lower AChE activity in lymphocytes compared to control subjects. In contrast, erythrocyte AChE activity is higher in patients with vascular dementia and is reduced in sporadic AD. Low ChE activity in lymphocytes is the best discriminator for AD. Both globular forms are subnormal. Because it is already low at very early stages of AD, AChE could be helpful as an early biomarker of differential diagnosis for the follow-up of patients during their early stages of cognitive impairment before a clinical dementia is established (Inestrosa et al., 1994; Von Bernhardi et al., 2005). Blood and plasma are easily accessible in comparison to CSF, together with specific and sensitive assays for AChE detection, therefore this enzyme could be used as clinical marker in the development of AD (Garcia-Ayllon et al., 2010).
Is There a Role for AChE in the Pathogenesis of Neurodegenerative Diseases?
Previous studies have demonstrated that AChE and BuChE are present in amyloid plaques in vivo and are associated to a cholinergic deficit (Mesulam and Geula, 1994; Geula and Mesulam, 1995; Figure 1). Moreover, AChE promotes Aβ1–40 fibril formation, in fact, AChE forms macromolecular complexes with the growing amyloid fibrils, and is incorporated into senile-like plaques in vitro (Alvarez et al., 1997, 1998). In this context, studies with synthetic Aβ1–40 in vitro have shown that this peptide aggregates and forms amyloid fibrils similar to the filaments found in the brains of AD patients (Morgan et al., 2004). Different mutations of Aβ were used to see its effect in the formation of aggregates. For example, the single mutation Val18 → Ala induces a significant increase on α-helical content in Aβ, and dramatically diminishes fibrillogenesis (Soto et al., 1995). However, the substitution of Glu22 → Gln found in hereditary cerebral hemorrhage with amyloidosis of the Dutch type, yields a peptide with increased ability to form amyloid fibrils (Soto et al., 1995). In fact, AChE had little effect on the aggregation of the highly amyloidogenic Dutch variant (Inestrosa et al., 1996). However, when the Aβval118 → Ala was incubated with AChE, a significant increase in the amyloid fibrils was observed (Inestrosa et al., 1996; Inestrosa and Alarcon, 1998). Previous investigations have shown that wild-type Aβ1 → 40 is able to bind AChE, while the Dutch variant AβGlu22 → Gln is not (Muñoz and Inestrosa, 1999). These data are correlated with previous observations that indicate that the presence of different types of Aβ peptide differentially affects AChE–Aβ interactions (Inestrosa and Alarcon, 1998). These studies indicated that AChE, but not BuChE increases the final yield of Aβ fibrils. In this context, an in vitro study, demonstrated that BuChE acts as a negative modifier of the Aβ aggregation process, and it is also capable of suppressing the facilitation of amyloid fibril-formation enhanced by AChE. So, BuChE may has acquired an inverse role to that of AChE in the pathogenesis of AD (Diamant et al., 2006).
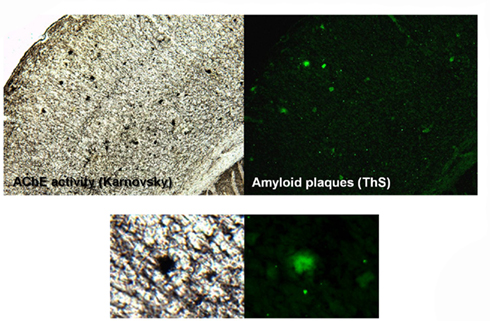
Figure 1. AChE activity in amyloid plaques in the brain of APP–PS1 mice. Studies carried out in the presence of iso-OMPA, an inhibitor of BuChE, in the mouse cerebral cortex. A single amyloid plaque shows its association to AChE activity.
AChE a nucleation factor for Aβ aggregation, and the role of its peripheral anionic site on Aβ aggregation
In 1996, we discovered that AChE was able to accelerate the assembly of Aβ1–40 into Alzheimer’s fibrils by decreasing the lag phase of the peptide aggregation, suggesting a role of AChE as a chaperone for Aβ1–40 assembly into oligomers of a high structural complexity (Inestrosa et al., 1996). These results suggested that the enzyme was acting through two possible mechanisms. First, it might increase the seeds necessary for the nucleation step and second, it may stimulate fibril elongation (Harper et al., 1997; Inestrosa et al., 2005a,b; Figure 2). When the formed amyloid was evaluated with thioflavin-S (ThS) plus AChE activity, it became apparent that the enzyme was strongly associated with amyloid deposits, exactly as described by Mesulam and Geula, for the senile plaques in AD patients (Geula and Mesulam, 1989a). In this case, at least part of the enzyme became tightly associated to the amyloid fibril, as it was shown by electron microscopy, using a monoclonal antibody conjugated with gold particles that nicely decorated growing amyloid fibrils (Reyes et al., 1997). To test this idea in vivo a double transgenic mice which express both human APPSWE and human AChE was generated by Brimijoin, Younkin, and Soreq. In these hybrid transgenic mice, AChE promotes plaque accumulation supporting the notion of its causal involvement with the fibril-formation process (Rees et al., 2003, 2005).
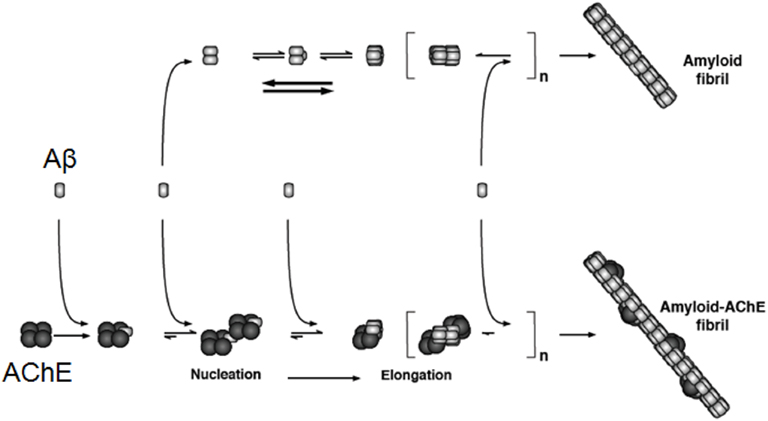
Figure 2. AChE acts on the Aβ aggregation and then in the formation of amyloid fibrils. The incorporation of AChE occurs at early steps of Aβ aggregation, acting as a nucleation factor or seed, then it acts in the elongation of the amyloid fibrils. Evidence for the tight binding of the enzyme with the formed amyloid fibrils was obtained using biochemical as well as immunocytochemical techniques, using an anti-AChE antibody conjugated with gold and examined using a transmission electron microscope. The original data was published by Alvarez et al. (1998).
The kinetic and pharmacological properties of AChE–amyloid complex have been examined, and the data show that the kinetic parameters of the enzyme change (Geula and Mesulam, 1989a). The Km and Vmax values for AChE associated to amyloid were higher than those for the free enzyme. Similarly, for the AChE–AβGlu22 → Gln complex and AChE–AβVal18 → Ala complex, the Km values were elevated compared with the enzyme alone. When kinetic studies were carried out under varying pH conditions, AChE associated to either the wild-type or the mutant Aβ peptides was more resistant to low pH. Similarly, AChE associated in AChE–Aβ complexes was more resistant to the incubation at high substrate concentrations (Alvarez et al., 1998; Inestrosa and Alarcon, 1998; Inestrosa et al., 2005b). Furthermore, biochemical studies have indicated that senile-plaque-associated AChE is only partially extracted using collagenase digestion, heparin, or high-salt buffers plus detergents (Nakamura et al., 1990; Kalaria et al., 1992; Alvarez et al., 1998), indicating that either different molecular forms are involved, or alternative some changes occur in the biochemical properties of the globular subunit.
Pharmacological studies of AChE associated to amyloid showed that AChE in these conditions also appears more resistant to inhibition by anti-AChE agents as observed with both active site inhibitors such as tacrine, edrophonium, and BW284c51, and with peripheral anionic site blockers, such as propidium and gallamine (Inestrosa and Alarcon, 1998). In almost all cases, a higher inhibitor concentration was required to obtain the same level of inhibition observed with the free enzyme. Overall, the AChE–AβVal18 → Ala complex showed the largest differences with respect to the free enzyme, suggesting that it has greater degree of interaction with AChE than the other more amyloidogenic Aβ peptides. Contrastingly the complexes AChE–AβGlu22 → Gln and AChE–Aβ1–42 were the least affected of all complexes studied. These data are consistent with the idea that the association of AChE with Aβ fibrils leads to changes in its enzymatic properties, in the absence of any pathological alteration of the enzyme (Inestrosa and Alarcon, 1998).
It is well know that AChE possesses two binding sites for the neurotransmitter acetylcholine, the active center site that is located at the bottom of a 20-Å gorge and the peripheral anionic binding site (PAS) that is rich in hydrophobic residues and is located at the rim of the gorge on the surface of the enzyme (Sussman et al., 1991). When aggregation experiments were carried and repeated in the presence of AChE inhibitors (AChEIs) directed against the two different sites, it turned out that only the PAS inhibitors were able to block the effect of AChE on amyloid formation (Alvarez et al., 1998). The PAS inhibitors, propidium and fasciculin, were able to prevent the effect of AChE on Aβ aggregation process (Bartolini et al., 2003; Inestrosa et al., 2008). On the other hand, the amyloid aggregation in the presence of edrophonium, an active site inhibitor of AChE, showed no effect on the role of AChE in this capacity to accelerate Aβ assembly into Alzheimer’s fibrils (Inestrosa et al., 2008). Further studies, we were able to identify a 3.5-kDa peptide located close the PAS region which was able to mimic effect of the whole AChE enzyme in its capacity to stimulate the Aβ aggregation (De Ferrari et al., 2001). Moreover, structural studies of AChE showed how the regulation of catalysis by PAS ligands (propidium, decidium, and gallamine) offers information on the residues that interact with other molecules and which could participate in the nucleation process of amyloid fibrils (Bartolini et al., 2003; Inestrosa et al., 2008). To understand the mechanism of the AChE–Aβ interaction Vaux and co-workers have studied a 14 residue peptide named AChE586–599, which corresponds to a region within the C-terminal oligomerization domain of human AChE (Jean et al., 2008). The region encompassing AChE586–599 shares homology with Aβ and possesses high propensity for conversion to non-native β-strand, a property associated to amyloidogenicity (Cottingham et al., 2002; Greenfield et al., 2008; Belli et al., 2011). Analysis of stabilizing or destabilizing effects of residue substitutions on the amyloid assembly of AChE586–599 has provided evidence for the critical role of specific side-chain interactions in the stabilization of nascent aggregates and for the position dependence of these side-chains upon polymerization and fibril formation. Consistently with the experimental observations and assembly models for other amyloid systems, they have proposed a model for AChE586–599 assembly in which a steric-zipper formed through specific interactions (hydrophobic, electrostatic, cation-π, SH-aromatic, metal chelation, and polar–polar) would maintain the β-sheets together. The dissection of the specific molecular recognition driving AChE586–599 amyloid assembly has provided further knowledge on such poorly understood and complicated process, which could be applied to protein folding and the targeting of amyloid diseases (Belli et al., 2011).
Dimeric type of AChE inhibitors directed against the active and the Aβ site of the enzyme
The current standard of care for mild to moderate AD includes treatment with AChEIs to improve cognitive function (Hardy and Selkoe, 2002; Terry and Buccafusco, 2003; Ballard et al., 2011). Several classes of AChEIs such as donepezil, rivastigmine, and galantamine were developed to treat AD (Colombres et al., 2004), and currently constitute the only FDA approved therapeutic approach. The NMDA antagonist memantine, has also been shown to improve cognitive function and reached the market in 2004 (Cummings et al., 2006). Nevertheless AChEIs, even valuable in improving the patient’s quality of life, represent only symptomatic and palliative tools that slow down the progression of the disease. Blockade of PAS by specific inhibitors has emerged as promising disease-modifying therapeutic strategies for AD. Based on these assumptions, the dual binding AChEIs, that are molecules able to interact simultaneously with both, the catalytic and the peripheral binding sites of the enzyme, emerged as valuable tools to pursue a disease-modifying approach (Colombres et al., 2004; Muñoz-Torrero, 2008). In this regard, several classes of dual binding site AChEIs have been developed and proved to be endowed with a strong inhibitory activity due to the increased capability to interact with both bindings sites of the target (Muñoz-Torrero and Camps, 2006). Some recent examples include benzophenone-based derivatives bearing a [benzyl(methyl)amino]methyl moiety (Belluti et al., 2009), Xanthostigmine derivatives (Belluti et al., 2005) and novel huprine derivatives with inhibitory activity toward Aβ aggregation and formation (Viayna et al., 2010). Owing to the simultaneous activity against AChE and amyloid formation and aggregation, dimeric type of AChEIs might attack AD on multiple fronts, with a better therapeutic outcome. Together with coumarin derivatives (Piazzi et al., 2008), and tacrine based heterobivalent ligands (Camps et al., 2009) they are able to act both at the acetylcholine site and at the amyloid formation triggering site. To further support this strategy, Shen and co-workers reported the discovery of novel dual inhibitors of AChE and BACE-1, which demonstrated not only in vitro enzyme inhibitory potency and cellular activity, but, more importantly, in vivo functional efficacy (Zhu et al., 2009). This strategy, embodied by single chemical entities able to simultaneously modulate multiple targets involved in the neurodegenerative cascade, has proven particularly fruitful in recent years and has led to the discovery of several promising anti-AD drug candidates.
AChE Induces the Aggregation of the Cellular Prion Protein
Prion disease, such as the Creutzfeldt–Jakob disease (CJD) in human and bovine spongiform encephalopathy (BSE), can be transmitted by an infectious process which involves the prion protein (PrP). The most remarkable feature of PrP is its ability to be folded into two isoforms, PrPC (C, cellular form) being the native protein and PrPSc (Sc, scrapie form) being the pathological conformation (Prusiner, 1998; Varela-Nallar et al., 2006). During the pathogenesis of prion disease there is a conformational conversion from PrPC to PrPSc consisting of a drastic alteration of the structure, as well as of the biochemical properties of the protein. Aβ-positive senile plaques in AD brains commonly contain PrP deposits; while sporadically Aβ-positive senile plaques have also been identified in prion diseases such as CJD and Gerstmann–Sträussler–Scheinker (GSS) disease (Miyazono et al., 1992; Hainfellner et al., 1998). On the other hand, a decrease in the in CFS levels of AChE from patients with CJD has been demonstrated, suggesting that an alteration in the cholinergic system also occurs in some prion diseases (Silveyra et al., 2006). Based on the common features between PrP and Aβ, it has been shown that AChE is able to induce the aggregation of the peptide deduced from PrP sequence spanning residues 106–126 (PrP106–126), the hydrophobic segment involved in PrP protein aggregation as has been previously described in a similar way for Aβ protein (Pera et al., 2006), through the PAS region of AChE (Inestrosa et al., 2008). The role of the peripheral site of AChE accelerating the assembly of PrP82–146 was demonstrated using propidium iodide (Pera et al., 2009), a specific inhibitor of the PAS region of AChE (Inestrosa et al., 1996; Bartolini et al., 2003). It has been extensively demonstrated that propidium iodide can also inhibits the AChE-induced Aβ aggregation. This study showed that AChE acts as a nucleating factor increasing not only the formation of new oligomers, but also fibril formation. A similar effect has been observed with huprine derivatives X, Y, and Z (Clos et al., 2006; Pera et al., 2006), which is spite of being active site AChEIs have been shown to interfere with the binding of ligands to the peripheral site of the enzyme (Camps et al., 2000). Therefore, inhibitors of the PAS region of AChE could be relevant as potential anti-Aβ and PrP aggregation drugs.
AChE increases the neurotoxicity of Aβ aggregates
Considering that the presence of senile plaques in the brain of aging individuals does not necessarily lead to symptoms of AD (Katzman et al., 1988), the presence of AChE in some critically located amyloid plaques could play a key role in triggering the cytotoxic events that occur around mature plaques in AD (Mesulam, 2004). In vitro assays on PC12 cells showed that aggregates of AChE–Aβ1–40 complexes were more toxic than those of Aβ1–40 and that neurotoxicity depends on the amount of AChE bound to the complexes, suggesting that AChE may play a key role in the neurodegenerative changes observed in Alzheimer brain (Muñoz and Inestrosa, 1999). In this context, previous results showed that Aβ–AChE complexes are more toxic than the Aβ fibrils alone on rat hippocampal neurons. In fact, neurons treated with Aβ–AChE complexes showed a much disrupted neurite network compared to neurons treated with Aβ (Alvarez et al., 1998). Other in vivo study showed that the hippocampal injection of AChE–Aβ complexes results in the appearance of some features reminiscent of Alzheimer-like lesions in rat brain (Reyes et al., 2004). The early events triggered in neurons in response to Aβ peptide have been largely studied. Aβ oligomers/fibrils induce intracellular calcium deregulation that leads to apoptosis through mitochondrial dysfunction, by direct interaction with isolated mitochondria or by indirect association with the neuronal membrane (Kim et al., 2002; Abramov et al., 2004). One of the earliest effect of Aβ–AChE complexes was the increase in intracellular calcium, which leads to the loss of the mitochondrial membrane potential (Dinamarca et al., 2010). Disruption of intracellular homeostasis of Ca2+ by channels opening has been extensively proposed as a mechanism of Aβ neurotoxicity (Mattson et al., 1992; Laferla, 2002). Aβ–AChE complexes and Aβ treatment have different effects over the mitochondrial membrane potential. Our studies indicated that Aβ–AChE complexes affected ΔΨmit more than Aβ alone; also, we observed that the mitochondrial membrane potential was compromised in a non-reversible manner even when the calcium increase was reversed after wash out. On the other hand, previous studies from our laboratory indicated that lithium (a pharmacological activator of Wnt signaling) protects hippocampal neurons against Aβ peptide and Aβ–AChE complex neurotoxicity (Dinamarca et al., 2010). Additionally, we found that pre-incubation with the Wnt-7a ligand prevents the increase in cytosolic calcium induced by Aβ (Quintanilla et al., 2005). These studies suggest that the activation of Wnt signaling prevent the toxic effects of Aβ–AChE complexes (Inestrosa et al., 2008), consistent with this possibility a “synaptic form” of AChE induces tau phosphorylation and activation of glycogen synthase kinase-3β (GSK-3β, a component of the Wnt/β-catenin signaling pathway). These effects were prevented by GSK-3β and AChE inhibition (Toiber et al., 2008). In this context it is interesting to mention that Huperzine A, a lycopodium alkaloid extracted from the chinese folk medicine, Huperzia serrata, a reversible and selective inhibitor of AChE, activates Wnt/β-catenin signaling and enhances the non-amyloidogenic pathway in a transgenic mouse model of AD (Wang et al., 2011).
IDN 5706 a Potential Drug Against AD
Previously, we have shown that hyperforin, the active molecule for the anti-depressant activity of St. John’s Wort (Hypericum perforatum; Griffith et al., 2010), reduces the behavioral alteration induced by intra-hippocampal injection of Aβ aggregates, an acute rat model of AD (Dinamarca et al., 2006). Tetrahydrohyperforin (IDN 5706), a semi synthetic derivative of hyperforin with higher stability and increased oral bioavailability (Cerpa et al., 2010) also shown some neuroprotective properties. Previous studies in our laboratory indicated that IDN 5706 was able to reduce memory impairments, as well as neuropathological markers in 12-month-old APP–PS1 mice treated with 2 mg/kg IDN 5706 for 1 month (Cerpa et al., 2010). Even more, a reduction in the size of ThS positive plaques was observed by this treatment. Interestingly, we have previously demonstrated that IDN 5706: (a) releases AChE from the Aβ aggregates, and (b) inhibits AChE–Aβ interaction in vitro and in vivo (12-month-old APP–PS1 mice treated with 2 mg/kg IDN 5706; Cerpa et al., 2010). In young APP–PS1 mice, IDN 5706 improves memory and prevents the impairment of synaptic plasticity, inducing a recovery of long-term potentiation, prevented the decrease in synaptic proteins in hippocampus and cortex, decreased levels of tau hyperphosphorylation, astrogliosis and total forms of Aβ (Inestrosa et al., 2011). Moreover, we have shown that in vitro, hyperforin is able to disaggregate pre-formed fibrils into protofibrils and amorphous material (Dinamarca et al., 2006). Taking in consideration our previous study in AD aged and young mice (Cerpa et al., 2010; Inestrosa et al., 2011), it is apparent that IDN 5706 has anti-amyloidogenic actions in vitro and in vivo.
Effect of IDN 5706 on the esterase activity present in Aβ plaques in a mouse model of AD
A major issue in AD research is to find some new therapeutic drugs which decrease Aβ aggregation and inhibit AChE with dual specificity, being directed to both the active and “peripheral” sites (De Ferrari et al., 2001; Inestrosa et al., 2008). For these reasons, we investigate the effect of IDN 5706, in those activities. IDN 5706 also inhibits the aggregation of Aβ1–40, delaying the nucleation phase. When we checked the amount of soluble Aβ peptide after the aggregation assay, IDN 5706 decreased the amount of Aβ peptide in the sediment fraction increasing the amount of soluble Aβ. Furthermore, we evaluated whether the anti-aggregation property of IDN 5706 was stronger or weaker than the pro-aggregating effect of the AChE. To explore this point we evaluated the stability of the Aβ fibrils formed in the absence or the presence of the AChE incubated with IDN 5706. The hyperforin derivative was able to disassemble a 50% of the Aβ fibrils, but only a 15% of the AChE–Aβ fibrils after 5 h incubation, suggesting that the fibrils formed in the presence of AChE have a more stable arrangement (Dinamarca et al., 2008). Then, the effect of this compound in the AChE activity was evaluated. Toward this aim, aliquots were taken at different time points and the pellet (fibrils) was separated from the soluble fraction by centrifugation. As expected, at initial time of incubation with IDN 5706, most of the enzyme activity is found in the pellet fraction, but after 2 h of incubation of the AChE–Aβ aggregates with IDN 5706, the enzyme decreases in the pellet fraction and started to increase in the soluble fraction. After 4 h incubation the enzyme is found in both pellet and soluble fraction in similar amount (Dinamarca et al., 2008). This data showed that IDN 5706 is able to disaggregate the AChE from the AChE–amyloid complexes in vitro.
Presence of AChE in amyloid plaques of a double APPSWE + PS1 transgenic mice
A decrease of AChE in the brain appears to be a consistent finding in AD brain (DeKosky and Scheff, 1990). We observed similar finding in APP–PS1 mice brains compared with wild-type mice. Moreover, IDN 5706 treatment increases AChE activity in the brains of APP–PS1 mice injected with 4 and 6 mg/kg (Figure 3A) and also increases the BuChE activity of APP–PS1 brain in mice injected with 6 mg/kg of IDN 5706 (Table 1). The specific neuroprotective effect of IDN 5706 might be related to the increases in the AChE activity in total brain protein extracts, suggesting a neuroprotective effect on cholinergic and cholinoceptive neurons. Moreover, we observed AChE activity associated to amyloid plaques (Figure 3B, left panel) visualized by the Karnovsky reaction for AChE (Figure 3B, right panel) in APP–PS1 mice treated with iso-OMPA an inhibitor for BuChE activity. Such amyloid plaques from mice brain are similar to those observed in patients with AD (Tago et al., 1986; Geula et al., 1994).
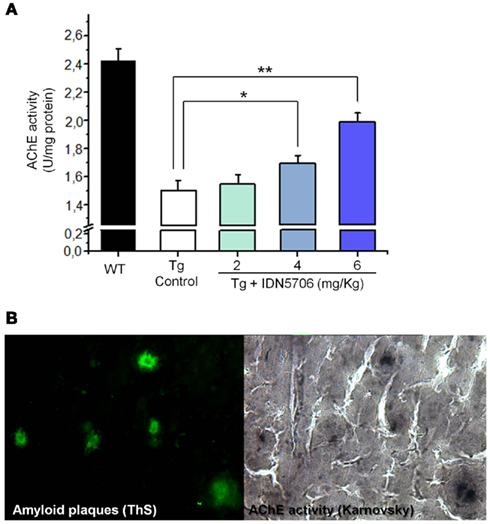
Figure 3. IDN 5706 increases AChE activity in whole brain extract of APP–PS1 transgenic mice. Effects of IDN 5706 on AChE activity from brains of transgenic mice injected i.p. with vehicle solution or 2, 4, and 6 mg/kg IDN 5706 three times a week per 10 weeks (A). AChE activity in amyloid plaques from brain of APP–PS1 mice in the presence of iso-OMPA (B). *p < 0.05; **p < 0.01.
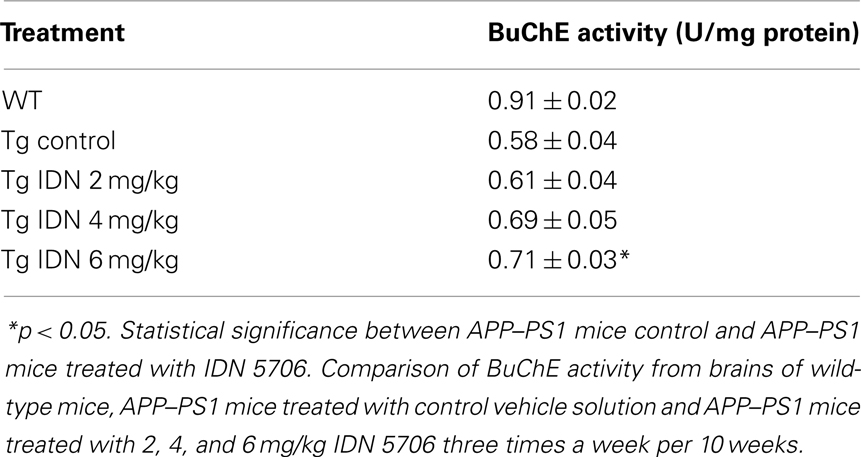
Table 1. IDN 506 increases BuChE activity in brains of double transgenic APP–PS1 mice.
Effect of IDN 5706 on AChE activity associated to amyloid plaques
Since AChE associated with amyloid and its activity correlates with amyloid plaque toxicity (Alvarez et al., 1998; Reyes et al., 2004; Dinamarca et al., 2010), we carried out an analysis of amyloid plaques positive for AChE activity. Brains from control and treated APP–PS1 animals were stained for AChE with the method of Karnovsky (Tago et al., 1986) and amyloid plaques were revealed by ThS staining. In APP–PS1 mice most of the amyloid plaques were positive for AChE in cortex (Figure 4A), however, the percentage of AChE-positive plaques in relation to the total amount of ThS positive plaques in the cortex were decreased in IDN 5706 treated mice (Figure 4B), suggesting that in addition to the decreased number of amyloid plaques, there is a decrease in the association of AChE with the amyloid plaques present in IDN 5706 treated APP–PS1 mice.
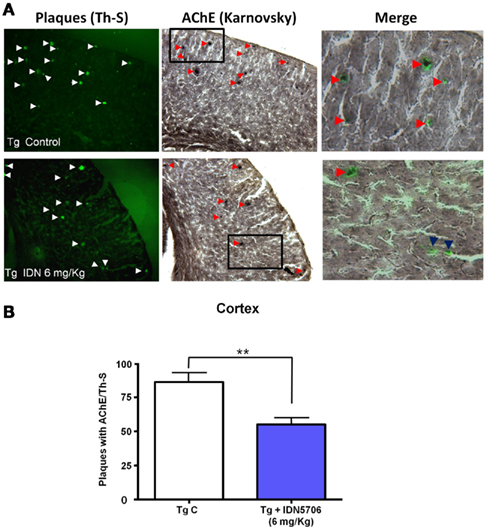
Figure 4. IDN 5706 decreases amyloid plaques positive for AChE activity in APP–PS1 mice. (A) Cortical slices obtained from APP–PS1 mice stained for ThS for amyloid plaques and AChE activity with the Karnovsky method. (B) Quantification of the percentage of amyloid plaques with AChE activity in the cortex of APP–PS1 brain of mice injected i.p. with vehicle solution or 6 mg/kg IDN 5706 three times a week per 10 weeks. *p < 0.05.
We have also determined AChE activity in two types of amyloid plaques, the diffuse (Type I) and compact (Type II) plaques present in control transgenic mice (Figure 5A, left panels), as well as in mice treated with IDN 5706. Under this condition AChE activity was reduced (Figure 5A, right panels). Quantification of the number of different type of amyloid plaques positive for AChE with the Karnovsky reaction revealed that treatment with IDN 5706 decreases the amount of AChE activity in type II plaques however, no effect was observed in type I plaques (Figure 5B). These preliminary results suggest a rather specific effect of IDN 5706 on the association of AChE with Aβ aggregates. Previous studies from our laboratory indicate a key role for AChE in the neurotoxicity of amyloid plaques (Alvarez et al., 1998; Chacon et al., 2003; Reyes et al. 2004; Dinamarca et al. 2010). Taken together, our data indicates that IDN 5706 might be considered as a possible therapeutic agent for AD treatment.
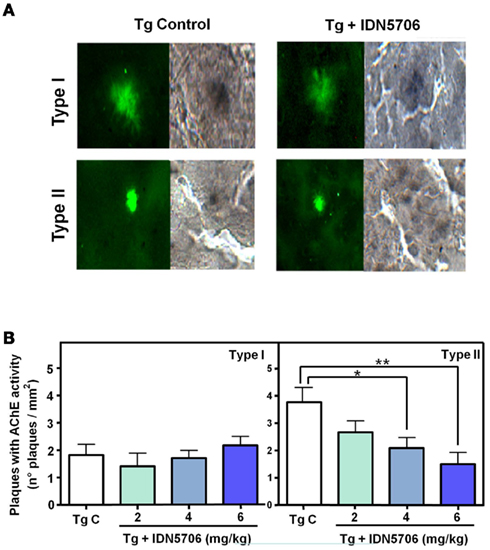
Figure 5. IDN 5706 reduces the amount of different types of amyloid plaques containing AChE activity. (A) Detection of amyloid plaques (ThS) positives for AChE activity in vehicle and IDN 5706 (6 mg/kg) treated APP–PS1 mice brain. Plaques were morphologically classified as Type I (diffuse plaques) and Type II (compact plaques). (B) Quantification of the total number of the different types of plaques positive for AChE activity in brain from APP–PS1 mice injected i.p. with vehicle solution or 2, 4, and 6 mg/kg IDN 5706 three times a week per 10 weeks. *p < 0.05; **p < 0.01.
Conclusion
As discussed, AChE is able to accelerate amyloid formation of at least two different macromolecules: the Aβ peptide and the PrP. In addition pro-aggregating effect of the enzyme depends on the intrinsic amyloidogenic properties of the peptide used. In AD patients, AChE activity is altered in the brain and in the blood, and co-localized with senile plaques. AChE associated to amyloid plaques showed changes in biochemical and pharmacological properties, as well as an increase in the neurotoxicity of the AChE–Aβ complexes. The AChE effect on amyloid aggregation is sensitive to drugs that block the PAS of the enzyme, suggesting that new and specific AChEIs might well provide an attractive therapeutic possibility for AD treatment. IDN 5706, an hyperforin derivative, prevents the development of the disease in a transgenic mice model of AD (Inestrosa et al. 2011). Interestingly, we have previously demonstrated that IDN 5706 releases AChE from the Aβ aggregates and inhibit AChE–Aβ interactions in vitro and in vivo. In summary, our findings indicate that IDN 5706 decreases AChE–Aβ interaction and this effect might be of therapeutic interest for the treatment of AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the Inestrosa laboratory for discussion and scientific support during the course of this project. This work was supported by grants from FONDEF (N^ D07I1052) and the Basal Center of Excellence in Aging and Regeneration (CONICYT – PFB 12/2007) to Nibaldo C. Inestrosa. Part of this work will be presented by Francisco Javier Carvajal as a partial requirement for the Biochemist degree at the Catholic University of Valparaiso.
References
Abramov, A. Y., Canevari, L., and Duchen, M. R. (2004). β-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 24, 565–575.
Alvarez, A., Alarcon, R., Opazo, C., Campos, E. O., Muñoz, F. J., Calderon, F. H., Dajas, F., Gentry, M. K., Doctor, B. P., De Mello, F. G., and Inestrosa, N. C. (1998). Stable complexes involving acetylcholinesterase and amyloid-β peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J. Neurosci. 18, 3213–3223.
Alvarez, A., Opazo, C., Alarcon, R., Garrido, J., and Inestrosa, N. C. (1997). Acetylcholinesterase promotes the aggregation of amyloid-β-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 272, 348–361.
Apelt, J., Kumar, A., and Schliebs, R. (2002). Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human β-amyloid precursor protein. Brain Res. 953, 17–30.
Arendt, T., Bruckner, M. K., Lange, M., and Bigl, V. (1992). Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development – a study of molecular forms. Neurochem. Int. 21, 381–396.
Atack, J. R., Perry, E. K., Bonham, J. R., Perry, R. H., Tomlinson, B. E., Blessed, G., and Fairbairn, A. (1983). Molecular forms of acetylcholinesterase in senile dementia of Alzheimer type: selective loss of the intermediate (10S) form. Neurosci. Lett. 40, 199–204.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Bartolini, M., Bertucci, C., Cavrini, V., and Andrisano, V. (2003). β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 65, 407–416.
Bartus, R. T. (2000). On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 163, 495–529.
Bartus, R. T., Dean, R. L. III, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414.
Beach, T. G., Kuo, Y. M., Spiegel, K., Emmerling, M. R., Sue, L. I., Kokjohn, K., and Roher, A. E. (2000). The cholinergic deficit coincides with Aβ deposition at the earliest histopathologic stages of Alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 308–313.
Belli, M., Ramazzotti, M., and Chiti, F. (2011). Prediction of amyloid aggregation in vivo. EMBO Rep. 12, 657–663.
Belluti, F., Piazzi, L., Bisi, A., Gobbi, S., Bartolini, M., Cavalli, A., Valenti, P., and Rampa, A. (2009). Design, synthesis, and evaluation of benzophenone derivatives as novel acetylcholinesterase inhibitors. Eur. J. Med. Chem. 44, 1341–1348.
Belluti, F., Rampa, A., Piazzi, L., Bisi, A., Gobbi, S., Bartolini, M., Andrisano, V., Cavalli, A., Recanatini, M., and Valenti, P. (2005). Cholinesterase inhibitors: xanthostigmine derivatives blocking the acetylcholinesterase-induced β-amyloid aggregation. J. Med. Chem. 48, 4444–4456.
Bissette, G., Seidler, F. J., Nemeroff, C. B., and Slotkin, T. A. (1996). High affinity choline transporter status in Alzheimer’s disease tissue from rapid autopsy. Ann. N. Y. Acad. Sci. 777, 197–204.
Boncristiano, S., Calhoun, M. E., Kelly, P. H., Pfeifer, M., Bondolfi, L., Stalder, M., Phinney, A. L., Abramowski, D., Sturchler-Pierrat, C., Enz, A., Sommer, B., Staufenbiel, M., and Jucker, M. (2002). Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 22, 3234–3243.
Bronfman, F. C., Moechars, D., and Van Leuven, F. (2000). Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein London mutant transgenic mice. Neurobiol. Dis. 7, 152–168.
Camps, P., Cusack, B., Mallender, W. D., El Achab, R. E., Morral, J., Muñoz-Torrero, D., and Rosenberry, T. L. (2000). Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharmacol. 57, 409–417.
Camps, P., Formosa, X., Galdeano, C., Munoz-Torrero, D., Ramirez, L., Gomez, E., Isambert, N., Lavilla, R., Badia, A., Clos, M. V., Bartolini, M., Mancini, F., Andrisano, V., Arce, M. P., Rodriguez-Franco, M. I., Huertas, O., Dafni, T., and Luque, F. J. (2009). Pyrano[3,2-c]quinoline-6-chlorotacrine hybrids as a novel family of acetylcholinesterase- and β-amyloid-directed anti-Alzheimer compounds. J. Med. Chem. 52, 5365–5379.
Cerpa, W., Hancke, J. L., Morazzoni, P., Bombardelli, E., Riva, A., Marin, P. P., and Inestrosa, N. C. (2010). The hyperforin derivative IDN5706 occludes spatial memory impairments and neuropathological changes in a double transgenic Alzheimer’s mouse model. Curr. Alzheimer Res. 7, 126–133.
Chacon, M. A., Reyes, A. E., and Inestrosa, N. C. (2003). Acetylcholinesterase induces neuronal cell loss, astrocyte hypertrophy and behavioral deficits in mammalian hippocampus. J. Neurochem. 87, 195–204.
Clos, M. V., Pera, M., Ratia, M., Roman, S., Camps, P., Muñoz-Torrero, D., Colombo, L., Salmona, M., and Badia, A. (2006). Effect of acetylcholinesterase inhibitors on AChE-induced PrP106-126 aggregation. J. Mol. Neurosci. 30, 89–90.
Colombres, M., Sagal, J. P., and Inestrosa, N. C. (2004). An overview of the current and novel drugs for Alzheimer’s disease with particular reference to anti-cholinesterase compounds. Curr. Pharm. Des. 10, 3121–3130.
Cottingham, M. G., Hollinshead, M. S., and Vaux, D. J. (2002). Amyloid fibril formation by a synthetic peptide from a region of human acetylcholinesterase that is homologous to the Alzheimer’s amyloid-β peptide. Biochemistry 41, 13539–13547.
Cummings, J. L., Schneider, E., Tariot, P. N., and Graham, S. M. (2006). Behavioral effects of memantine in Alzheimer disease patients receiving donepezil treatment. Neurology 67, 57–63.
De Ferrari, G. V., Canales, M. A., Shin, I., Weiner, L. M., Silman, I., and Inestrosa, N. C. (2001). A structural motif of acetylcholinesterase that promotes amyloid β-peptide fibril formation. Biochemistry 40, 10447–10457.
DeKosky, S. T., Ikonomovic, M. D., Styren, S. D., Beckett, L., Wisniewski, S., Bennett, D. A., Cochran, E. J., Kordower, J. H., and Mufson, E. J. (2002). Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 51, 145–155.
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464.
Diamant, S., Podoly, E., Friedler, A., Ligumsky, H., Livnah, O., and Soreq, H. (2006). Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. U.S.A. 103, 8628–8633.
Dinamarca, M. C., Arrazola, M., Toledo, E., Cerpa, W. F., Hancke, J., and Inestrosa, N. C. (2008). Release of acetylcholinesterase (AChE) from β-amyloid plaques assemblies improves the spatial memory impairments in APP-transgenic mice. Chem. Biol. Interact. 175, 142–149.
Dinamarca, M. C., Cerpa, W., Garrido, J., Hancke, J. L., and Inestrosa, N. C. (2006). Hyperforin prevents β-amyloid neurotoxicity and spatial memory impairments by disaggregation of Alzheimer’s amyloid- β-deposits. Mol. Psychiatry 11, 1032–1048.
Dinamarca, M. C., Sagal, J. P., Quintanilla, R. A., Godoy, J. A., Arrazola, M. S., and Inestrosa, N. C. (2010). Amyloid-β-Acetylcholinesterase complexes potentiate neurodegenerative changes induced by the Aβ peptide. Implications for the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 5, 4.
Dumas, J. A., and Newhouse, P. A. (2011). The cholinergic hypothesis of cognitive aging revisited again: cholinergic functional compensation. Pharmacol. Biochem. Behav. 99, 254, 261.
Farias, G. G., Godoy, J. A., Hernandez, F., Avila, J., Fisher, A., and Inestrosa, N. C. (2004). M1 muscarinic receptor activation protects neurons from β-amyloid toxicity. A role for Wnt signaling pathway. Neurobiol. Dis. 17, 337–348.
Fodero, L. R., Saez-Valero, J., Mclean, C. A., Martins, R. N., Beyreuther, K., Masters, C. L., Robertson, T. A., and Small, D. H. (2002). Altered glycosylation of acetylcholinesterase in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J. Neurochem. 81, 441–448.
Garcia-Ayllon, M. S., Riba-Llena, I., Serra-Basante, C., Alom, J., Boopathy, R., and Saez-Valero, J. (2010). Altered levels of acetylcholinesterase in Alzheimer plasma. PLoS ONE 5, e8701.
Garcia-Ayllon, M. S., Silveyra, M. X., Andreasen, N., Brimijoin, S., Blennow, K., and Saez-Valero, J. (2007). Cerebrospinal fluid acetylcholinesterase changes after treatment with donepezil in patients with Alzheimer’s disease. J. Neurochem. 101, 1701–1711.
Geula, C., Greenberg, B. D., and Mesulam, M. M. (1994). Cholinesterase activity in the plaques, tangles and angiopathy of Alzheimer’s disease does not emanate from amyloid. Brain Res. 644, 327–330.
Geula, C., and Mesulam, M. (1989a). Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 498, 185–189.
Geula, C., and Mesulam, M. M. (1989b). Cortical cholinergic fibers in aging and Alzheimer’s disease: a morphometric study. Neuroscience 33, 469–481.
Geula, C., and Mesulam, M. M. (1995). Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 9(Suppl. 2), 23–28.
Greenfield, S. A., Zimmermann, M., and Bond, C. E. (2008). Non-hydrolytic functions of acetylcholinesterase. The significance of C-terminal peptides. FEBS J. 275, 604–611.
Griffith, T. N., Varela-Nallar, L., Dinamarca, M. C., and Inestrosa, N. C. (2010). Neurobiological effects of Hyperforin and its potential in Alzheimer’s disease therapy. Curr. Med. Chem. 17, 391–406.
Hainfellner, J. A., Wanschitz, J., Jellinger, K., Liberski, P. P., Gullotta, F., and Budka, H. (1998). Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 96, 116–122.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Harper, J. D., Lieber, C. M., and Lansbury, P. T. Jr. (1997). Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem. Biol. 4, 951–959.
Ikonomovic, M. D., Wecker, L., Abrahamson, E. E., Wuu, J., Counts, S. E., Ginsberg, S. D., Mufson, E. J., and DeKosky, S. T. (2009). Cortical α7 nicotinic acetylcholine receptor and β-amyloid levels in early Alzheimer disease. Arch. Neurol. 66, 646–651.
Inestrosa, N. C., and Alarcon, R. (1998). Molecular interactions of acetylcholinesterase with senile plaques. J. Physiol. Paris 92, 341–344.
Inestrosa, N. C., Alarcon, R., Arriagada, J., Donoso, A., Alvarez, J., and Campos, E. O. (1994). Blood markers in Alzheimer disease: subnormal acetylcholinesterase and butyrylcholinesterase in lymphocytes and erythrocytes. J. Neurol. Sci. 122, 1–5.
Inestrosa, N. C., Alvarez, A., Dinamarca, M. C., Perez-Acle, T., and Colombres, M. (2005a). Acetylcholinesterase-amyloid-β-peptide interaction: effect of Congo Red and the role of the Wnt pathway. Curr. Alzheimer Res. 2, 301–306.
Inestrosa, N. C., Sagal, J. P., and Colombres, M. (2005b). Acetylcholinesterase interaction with Alzheimer amyloid β. Subcell. Biochem. 38, 299–317.
Inestrosa, N. C., Alvarez, A., Perez, C. A., Moreno, R. D., Vicente, M., Linker, C., Casanueva, O. I., Soto, C., and Garrido, J. (1996). Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891.
Inestrosa, N. C., Dinamarca, M. C., and Alvarez, A. (2008). Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 275, 625–632.
Inestrosa, N. C., Tapia-Rojas, C., Griffith, T. N., Carvajal, F. J., Benito, M. J., Rivera-Dictter, A., Alvarez, A. R., Serrano, F. G., Hancke, J. L., Burgos, P. V., Parodi, J., and Varela-Nallar, L. (2011). Tetrahydrohyperforin prevents cognitive deficit, Aβ deposition, tau phosphorylation and synaptotoxicity in the APPswe/PSEN1Δ9 model of Alzheimer’s disease: a possible effect on APP procesing. Transl. Psychiatr. 1, e20.
Jean, L., Lee, C. F., Shaw, M., and Vaux, D. J. (2008). Structural elements regulating amyloidogenesis: a cholinesterase model system. PLoS ONE 3, e1834.
Kalaria, R. N., Kroon, S. N., Grahovac, I., and Perry, G. (1992). Acetylcholinesterase and its association with heparan sulphate proteoglycans in cortical amyloid deposits of Alzheimer’s disease. Neuroscience 51, 177–184.
Katzman, R., Terry, R., Deteresa, R., Brown, T., Davies, P., Fuld, P., Renbing, X., and Peck, A. (1988). Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 23, 138–144.
Kim, H. S., Lee, J. H., Lee, J. P., Kim, E. M., Chang, K. A., Park, C. H., Jeong, S. J., Wittendorp, M. C., Seo, J. H., Choi, S. H., and Suh, Y. H. (2002). Amyloid β peptide induces cytochrome C release from isolated mitochondria. Neuroreport 13, 1989–1993.
Knobloch, M., and Mansuy, I. M. (2008). Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol. Neurobiol. 37, 73–82.
Laferla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 3, 862–872.
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159.
Li, S., Shankar, G. M., and Selkoe, D. J. (2010). How do soluble oligomers of amyloid β-protein impair hippocampal synaptic plasticity? Front. Cell. Neurosci. 4:5.
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389.
Mesulam, M. (2004). The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn. Mem. 11, 43–49.
Mesulam, M. M., and Geula, C. (1994). Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann. Neurol. 36, 722–727.
Miyazono, M., Kitamoto, T., Iwaki, T., and Tateishi, J. (1992). Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Straussler syndrome. Acta Neuropathol. 83, 333–339.
Morgan, C., Colombres, M., Nunez, M. T., and Inestrosa, N. C. (2004). Structure and function of amyloid in Alzheimer’s disease. Prog. Neurobiol. 74, 323–349.
Muñoz, F. J., and Inestrosa, N. C. (1999). Neurotoxicity of acetylcholinesterase amyloid β-peptide aggregates is dependent on the type of Aβ peptide and the AChE concentration present in the complexes. FEBS Lett. 450, 205–209.
Muñoz-Torrero, D. (2008). Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr. Med. Chem. 15, 2433–2455.
Muñoz-Torrero, D., and Camps, P. (2006). Dimeric and hybrid anti-Alzheimer drug candidates. Curr. Med. Chem. 13, 399–422.
Nakamura, S., Kawashima, S., Nakano, S., Tsuji, T., and Araki, W. (1990). Subcellular distribution of acetylcholinesterase in Alzheimer’s disease: abnormal localization and solubilization. J. Neural Transm. Suppl. 30, 13–23.
Nitsch, R. M. (1996). From acetylcholine to amyloid: neurotransmitters and the pathology of Alzheimer’s disease. Neurodegeneration 5, 477–482.
Pera, M., Martinez-Otero, A., Colombo, L., Salmona, M., Ruiz-Molina, D., Badia, A., and Clos, M. V. (2009). Acetylcholinesterase as an amyloid enhancing factor in PrP82-146 aggregation process. Mol. Cell. Neurosci. 40, 217–224.
Pera, M., Roman, S., Ratia, M., Camps, P., Muñoz-Torrero, D., Colombo, L., Manzoni, C., Salmona, M., Badia, A., and Clos, M. V. (2006). Acetylcholinesterase triggers the aggregation of PrP 106-126. Biochem. Biophys. Res. Commun. 346, 89–94.
Piazzi, L., Cavalli, A., Colizzi, F., Belluti, F., Bartolini, M., Mancini, F., Recanatini, M., Andrisano, V., and Rampa, A. (2008). Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorg. Med. Chem. Lett. 18, 423–426.
Quintanilla, R. A., Munoz, F. J., Metcalfe, M. J., Hitschfeld, M., Olivares, G., Godoy, J. A., and Inestrosa, N. C. (2005). Trolox and 17 β-estradiol protect against amyloid β-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J. Biol. Chem. 280, 11615–11625.
Rees, T., Hammond, P. I., Soreq, H., Younkin, S., and Brimijoin, S. (2003). Acetylcholinesterase promotes β-amyloid plaques in cerebral cortex. Neurobiol. Aging 24, 777–787.
Rees, T. M., Berson, A., Sklan, E. H., Younkin, L., Younkin, S., Brimijoin, S., and Soreq, H. (2005). Memory deficits correlating with acetylcholinesterase splice shift and amyloid burden in doubly transgenic mice. Curr. Alzheimer Res. 2, 291–300.
Reyes, A. E., Chacon, M. A., Dinamarca, M. C., Cerpa, W., Morgan, C., and Inestrosa, N. C. (2004). Acetylcholinesterase-β complexes are more toxic than Aβ fibrils in rat hippocampus: effect on rat β-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am. J. Pathol. 164, 2163–2174.
Reyes, A. E., Perez, D. R., Alvarez, A., Garrido, J., Gentry, M. K., Doctor, B. P., and Inestrosa, N. C. (1997). A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem. Biophys. Res. Commun. 232, 652–655.
Saez-Valero, J., De Ceballos, M. L., Small, D. H., and De Felipe, C. (2002). Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid β-peptide. Neurosci. Lett. 325, 199–202.
Saez-Valero, J., Mok, S. S., and Small, D. H. (2000). An unusually glycosylated form of acetylcholinesterase is a CSF biomarker for Alzheimer’s disease. Acta Neurol. Scand. Suppl. 176, 49–52.
Saez-Valero, J., Sberna, G., Mclean, C. A., and Small, D. H. (1999). Molecular isoform distribution and glycosylation of acetylcholinesterase are altered in brain and cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurochem. 72, 1600–1608.
Sberna, G., Saez-Valero, J., Li, Q. X., Czech, C., Beyreuther, K., Masters, C. L., Mclean, C. A., and Small, D. H. (1998). Acetylcholinesterase is increased in the brains of transgenic mice expressing the C-terminal fragment (CT100) of the β-amyloid protein precursor of Alzheimer’s disease. J. Neurochem. 71, 723–731.
Shinotoh, H., Namba, H., Fukushi, K., Nagatsuka, S., Tanaka, N., Aotsuka, A., Tanada, S., and Irie, T. (2000). Brain acetylcholinesterase activity in Alzheimer disease measured by positron emission tomography. Alzheimer Dis. Assoc. Disord. 14(Suppl. 1), S114–S118.
Silveyra, M. X., Garcia-Ayllon, M. S., Calero, M., and Saez-Valero, J. (2006). Altered glycosylation of acetylcholinesterase in the Creutzfeldt-Jakob cerebrospinal fluid. J. Mol. Neurosci. 30, 65–66.
Slotkin, T. A., Nemeroff, C. B., Bissette, G., and Seidler, F. J. (1994). Overexpression of the high affinity choline transporter in cortical regions affected by Alzheimer’s disease. Evidence from rapid autopsy studies. J. Clin. Invest. 94, 696–702.
Soto, C., Castaño, E. M., Frangione, B., and Inestrosa, N. C. (1995). The α-helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amyloid formation. J. Biol. Chem. 270, 3063–3067.
Sussman, J. L., Harel, M., Frolow, F., Oefner, C., Goldman, A., Toker, L., and Silman, I. (1991). Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 253, 872–879.
Tago, H., Kimura, H., and Maeda, T. (1986). Visualization of detailed acetylcholinesterase fiber and neuron staining in rat brain by a sensitive histochemical procedure. J. Histochem. Cytochem. 34, 1431–1438.
Terry, A. V. Jr., and Buccafusco, J. J. (2003). The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 306, 821–827.
Toiber, D., Berson, A., Greenberg, D., Melamed-Book, N., Diamant, S., and Soreq, H. (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS ONE 3, e3108.
Varela-Nallar, L., Gonzalez, A., and Inestrosa, N. C. (2006). Role of copper in prion diseases: deleterious or beneficial? Curr. Pharm. Des. 12, 2587–2595.
Viayna, E., Gomez, T., Galdeano, C., Ramirez, L., Ratia, M., Badia, A., Clos, M. V., Verdaguer, E., Junyent, F., Camins, A., Pallas, M., Bartolini, M., Mancini, F., Andrisano, V., Arce, M. P., Rodriguez-Franco, M. I., Bidon-Chanal, A., Luque, F. J., Camps, P., and Muñoz-Torrero, D. (2010). Novel huprine derivatives with inhibitory activity toward β-amyloid aggregation and formation as disease-modifying anti-Alzheimer drug candidates. ChemMedChem 5, 1855–1870.
Von Bernhardi, R., Alarcon, R., Mezzano, D., Fuentes, P., and Inestrosa, N. C. (2005). Blood cells cholinesterase activity in early stage Alzheimer’s disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 19, 204–212.
Wang, C. Y., Zheng, W., Wang, T., Xie, J. W., Wang, S. L., Zhao, B. L., Teng, W. P., and Wang, Z. Y. (2011). Huperzine A activates Wnt/β-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 36, 1073–1089.
Xie, H. Q., Leung, K. W., Chen, V. P., Chan, G. K., Xu, S. L., Guo, A. J., Zhu, K. Y., Zheng, K. Y., Bi, C. W., Zhan, J. Y., Chan, W. K., Choi, R. C., and Tsim, K. W. (2010). PRiMA directs a restricted localization of tetrameric AChE at synapses. Chem. Biol. Interact. 187, 78–83.
Zhu, Y., Xiao, K., Ma, L., Xiong, B., Fu, Y., Yu, H., Wang, W., Wang, X., Hu, D., Peng, H., Li, J., Gong, Q., Chai, Q., Tang, X., Zhang, H., and Shen, J. (2009). Design, synthesis and biological evaluation of novel dual inhibitors of acetylcholinesterase and β-secretase. Bioorg. Med. Chem. 17, 1600–1613.
Keywords: Alzheimer’s disease, amyloid plaques, acetylcholinesterase, AChE–Aβ interactions, Aβ neurotoxicity, AChE activity, APP–PS1 transgenic mice, IDN 5706
Citation: Carvajal FJ and Inestrosa NC (2011) Interactions of AChE with Aβ aggregates in Alzheimer’s brain: therapeutic relevance of IDN 5706. Front. Mol. Neurosci. 4:19. doi: 10.3389/fnmol.2011.00019
Received: 28 June 2011; Accepted: 21 August 2011;
Published online: 14 September 2011.
Edited by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelReviewed by:
Javier Saez-Valero, Universidad Miguel Hernandez, SpainDiego Muñoz-Torrero, Universitat de Barcelona, Spain
Stephen Brimijoin, Mayo Clinic, USA
Copyright: © 2011 Carvajal and Inestrosa. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Nibaldo C. Inestrosa, Basal CARE Center, P. Catholic University of Chile, 8331150 Santiago, Chile. e-mail: ninestrosa@bio.puc.cl