Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation
- Department of Biochemistry, Dartmouth Medical School, Hanover, NH, USA
Ligand-gated ion channels couple the free energy of agonist binding to the gating of selective transmembrane ion pores, permitting cells to regulate ion flux in response to external chemical stimuli. However, the stereochemical mechanisms responsible for this coupling remain obscure. In the case of the ionotropic glutamate receptors (iGluRs), the modular nature of receptor subunits has facilitated structural analysis of the N-terminal domain (NTD), and of multiple conformations of the ligand-binding domain (LBD). Recently, the crystallographic structure of an antagonist-bound form of the receptor was determined. However, disulfide trapping of this conformation blocks channel opening, suggesting that channel activation involves additional quaternary packing arrangements. To explore the conformational space available to iGluR channels, we report here a second, clearly distinct domain architecture of homotetrameric, calcium-permeable AMPA receptors, determined by single-particle electron microscopy of untagged and fluorescently tagged constructs in a ligand-free state. It reveals a novel packing of NTD dimers, and a separation of LBD dimers across a central vestibule. In this arrangement, which reconciles diverse functional observations, agonist-induced cleft closure across LBD dimers can be converted into a twisting motion that provides a basis for receptor activation.
Introduction
Glutamate is the most common excitatory neurotransmitter in the brain, estimated to be responsible for signaling at 80% of cortical synapses (Braitenberg and Schüz, 1998). It activates ionotropic glutamate receptors (iGluRs), a family of ligand-gated ion channels that assemble as functional tetramers. Recently, it has become clear that auxiliary subunits also affect channel properties for some members of this family (Milstein and Nicoll, 2008; Tomita, 2010). However, the core tetrameric complexes provide the central scaffolding that converts the chemical signal of agonist binding into an electrical signal across the membrane. iGluRs have been implicated in synaptic plasticity, a key neurological component of learning and memory (Derkach et al., 2007), and play important roles in pathological processes, particularly those involving the excitotoxicity associated with excess Ca2+ influx (Liu and Zukin, 2007). They also participate in a variety of non-neuronal autocrine and paracrine signaling processes (Hinoi et al., 2004).
Among the iGluRs, the subfamily selective for α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) mediates the fast component of glutamate excitatory signaling. AMPA receptor (AMPAR) subunits are designated GluA1–4 (previously GluRA–D or GluR1–4; Collingridge et al., 2009), and form functional homo- and hetero-tetrameric complexes that conduct Na+, K+, and in some cases Ca2+ ions (Madden, 2002; Cull-Candy et al., 2006). As with all mammalian iGluRs, AMPAR subunits have a modular domain architecture composed of an extracellular N-terminal domain (NTD), a bilobate ligand-binding domain (LBD), a transmembrane domain (TMD), and a cytoplasmic C-terminus (Wo and Oswald, 1995).
Within the TMD, a re-entrant pore loop is found between the first (TM1) and second (TM2) transmembrane helices, with structural homology to the K+ channels (Wo and Oswald, 1995; Panchenko et al., 2001; Kuner et al., 2003). This loop serves as a key ion selectivity determinant in the AMPAR (Verdoorn et al., 1991). The genomic sequences of all four AMPA receptor subunits encode a glutamine (Q) residue located near the tip of this loop, and “Q4” receptors assembled from four Q subunits are Ca2+ permeable. In many neurons, editing of the GluA2 mRNA transcript replaces this Q with an arginine (R) residue, leading to the preferential formation of “Q/R” heteromeric receptors that are selective for monovalent ions (Sommer et al., 1991). Nevertheless, calcium-permeable Q4 receptors are expressed in specific neuronal and glial cell populations and under specific conditions in other neurons, with important physiological and pathological implications (Burnashev et al., 1992; Cull-Candy et al., 2006; Liu and Zukin, 2007; Kelly et al., 2009; Man, 2011).
For more than 10 years, our insights into the molecular mechanisms underlying iGluR function were extrapolated from an extensive series of structural and biochemical studies of the component extracellular domains, along with electrophysiological analysis of intact receptors (Traynelis et al., 2010). The central insight was that agonist binding stabilizes a cleft closure in the LBD (Abele et al., 2000; Armstrong and Gouaux, 2000), but how that cleft closure was coupled to channel opening remained unclear. Recently, the breakthrough crystallization of a tetrameric channel composed of full-length subunits has provided important new insights into the quaternary assembly of this family of receptors (Sobolevsky et al., 2009). The crystal structure of a modified GluA2 Q4 receptor (GluA2cryst) in complex with the competitive antagonist ZK200775 (Turski et al., 1998) confirmed the overall twofold molecular symmetry of the extracellular domains of the four constituent subunits, consistent with previous biochemical and electron microscopic (EM) studies (Ayalon and Stern-Bach, 2001; Mansour et al., 2001; Safferling et al., 2001; Schorge and Colquhoun, 2003; Sobolevsky et al., 2004; Tichelaar et al., 2004; Nakagawa et al., 2006; Midgett and Madden, 2008). It shows that pairs of subunit dimers are held together by cross-over connections between NTD and LBD dimers and illuminated the symmetry mismatch in the coupling between the LBD and the TMD, providing a framework for understanding the process of channel activation (Sobolevsky et al., 2009).
The GluA2cryst structure resembles a “Y” conformation, with the NTD dimers loosely packed across the overall molecular twofold axis, in an arrangement that resembles lattice contacts formed in crystals of the isolated domains (Jin et al., 2009; Sobolevsky et al., 2009). A similar Y-shaped quaternary structure has also been seen in single-particle EM reconstructions of antagonist-stabilized AMPA receptors (Nakagawa et al., 2005; Shanks et al., 2010). In contrast, EM studies of ligand-free homomeric GluA2 receptors reveal a defined O-shaped structure with overall twofold symmetry and a prominent central vestibule for both R4 and Q4 homomers (Tichelaar et al., 2004; Midgett and Madden, 2008). However, the domain architecture of this conformation has never been established, precluding direct comparison with the Y-shaped alternative. As a result, it has been unclear to date how the component domains of the recombinant GluA2 receptors are arranged within the molecular envelope of the O-shaped structure or how they could interact to mediate channel activation.
To address these questions, we performed EM structural analysis on domain-labeled constructs, which revealed a shared core receptor structure in the O-shaped conformation, together with the location of the fusion domains, permitting development of a hybrid model of the GluA2 domain architecture. Analysis of the hybrid model in the presence and absence of agonist provides insights into the coupling of ligand-binding and channel gating in this important family of ligand-gated ion channels.
Materials and Methods
Production of Recombinant Baculoviruses
The baculovirus encoding the Q/R unedited version of the GluA2flop AMPA receptor subunit (GluA2-Q) have been described previously (Midgett and Madden, 2008). Covalently labeled GluA2-Q subunits incorporating a GFP domain at the N-terminus (GFP:GluA2; Braithwaite et al., 2002) or a CFP domain inserted in the NTD (R2Q306c; Sheridan et al., 2006) were cloned into the pFastBac1-derivative pK503-9 baculovirus transfer plasmid (Keinänen et al., 1998). The GluA4 ectodomain constructs were generated by PCR subcloning, also in the pK503-9 backbone. The mature, secreted ectodomain protein contains an N-terminal FLAG tag (DYKDDDDK-RPH), residues 1–507 and 633–792 of the mature R. norvegicus GluA4flip sequence (Uniprot ID P19493-2) joined by a GT linker, and a C-terminal ELSRHHHHHH tag. Expression constructs were verified by DNA sequencing.
Initial virus generation was performed as described (Madden and Safferling, 2007) or using a protocol adopted from Philipps et al. (2005). Sf9 cells (Invitrogen) were grown in a 75-cm2 T-75 flask until they were 80–90% confluent. The cells were suspended and the flask was incubated for 1 h at 27°C in 20 ml of Grace’s insect cell medium (Invitrogen) supplemented with 10% FBS (Invitrogen), 3 μg/ml amphotericin B (Sigma), 10 μg/ml gentamycin (Invitrogen), and 0.1% F-86 (Invitrogen). Thirty microliter of bacmid DNA, and 70 μl of cellfectin (Invitrogen) were incubated in 2 ml of Grace’s medium for 45 min. The cells were washed with 10 ml of Grace’s medium, and 8 ml of Grace’s medium was added to the bacmid/cellfectin mixture, which was then applied to the Sf9 cells. The cells were incubated for 5 h at 27°C. The medium was removed and 20 ml of supplemented Grace’s medium was added to the flask. The cells were then incubated for three additional days at 27°C. On day 3 the cells were resuspended and added to a 225-cm2 T-225 with 50 ml of supplemented medium. Viral supernatants were harvested on day 5 by centrifugation. Large-scale virus generation was performed as described by Madden and Safferling (2007).
Protein Expression and Purification
Tagged and untagged GluA2 receptor proteins were obtained essentially as described (Midgett and Madden, 2008). As previously reported for the untagged receptor, the recombinant fusion proteins were purified to homogeneity by immunoaffinity and size-exclusion chromatography, as assessed by silver stained SDS-PAGE gels. The identity of purified proteins and the presence of the appropriate domain tags were confirmed by Western blotting with antibodies specific for either GluA2/3 (Millipore) or GFP/CFP (Living Colors monoclonal antibody, Clontech). The GFP:GluA2 and R2Q306c receptors reacted to both antibodies while untagged GluA2-Q receptors reacted only to the GluA2/3 antibody. In addition, the tagged receptors showed the expected molecular weight shift due to the tag (Figure A1 in Appendix).
GluR4flip ectodomain expression and purification was performed essentially as reported previously for the S1S2 LBD construct (Madden et al., 2000). Briefly, following concentration and buffer exchange into TBS (140 mM NaCl, 50 mM Tris-base, pH 7.4) by tangential flow filtration, culture medium from baculovirus-infected high five cells (Invitrogen) was supplemented with 1 mM phenylmethylsulfonyl fluoride, 3 mM CaCl2, and 0.02% (w/v) NaN3. The sample was applied to an M1 α-FLAG agarose column (Sigma) for immunoaffinity purification, and eluted with 5 mM EGTA, 1 mM Na-glutamate, 0.02% (w/v) NaN3 in 50 mM Na-acetate buffer, pH 5.0. Fractions containing eluted protein were pooled and dialyzed overnight against 10 mM Tris-base pH 8.8, 40 mM NaCl. After dialysis, the pooled protein was further purified by ion-exchange chromatography using HiTrap Q FF column (GE Healthcare). Eluted protein was pooled, concentrated and dialyzed against TBS containing 1 mM glutamate.
Blue-Native Page
Blue-native PAGE was performed by pre-equilibrating a 4–15% Tris–HCl gel (Biorad) with 50 mM Bis–Tris, 500 mM Tricine as the cathode buffer and 50 mM Bis–Tris as the anode buffer for 3–4 h at 50 V. The cathode buffer was switched to a light-blue buffer 50 mM Bis–Tris, 500 mM Tricine, 0.002% R-250. The samples were loaded in the lanes without any sample buffer and the gel was run at 100 V for 6–8 h at 4°C and then destained.
Electron Microscopy and Image Processing
Receptors were applied to grids as previously described (Midgett and Madden, 2008). When treating the receptors with glutamate, a final concentration of 10 mM glutamate was added to the receptors, and the mixture was incubated for 30 min on ice prior to sample preparation. Micrographs were collected, processed, and particles picked as described (Midgett and Madden, 2008).
Class Averaging
Class averages were created from single-particle images of GFP:GluA2 (2855 particles), R2Q306c (2666 particles), GluA2-Q (5016 particles from a previous data set; Midgett and Madden, 2008), and GluA2-Q treated with glutamate (1170 particles). The particles were masked with a 30 pixel mask, edge-normalized, low-pass filtered with a 30-Å cut-off, and high-pass filtered with a 250-Å cut-off. Class averages were produced by subjecting the filtered images to 10 iterations of K-means classification followed by multi-reference alignment as implemented by refine2d.py from EMAN (Lebart et al., 1984; Ludtke et al., 1999). On average each class contained ~50 particles.
GluA2-Q 3D Reconstruction
The GluA2-Q data set of 5016 particles used previously was reprocessed to correct the contrast transfer function (CTF) without use of the hybrid structure factor curve used in a previous reconstruction (Midgett and Madden, 2008). The CTF was corrected for the particles from each micrograph using CTFFIT from EMAN to fit a theoretical power spectrum to the calculated power spectrum to a resolution of 20 Å. The parameters for the particles were then adjusted until the structure factor curves overlapped to a resolution of 20 Å and the curves continually decreased thereafter. Refinement was carried out as described (Midgett and Madden, 2008). The model was low-pass filtered to a resolution of 28 Å, corresponding to a Fourier-shell correlation of 0.5.
GluA2-Q Domain Architecture
All visualization and distance measurements were performed using Chimera (Pettersen et al., 2004). Images were rendered using POVray 3.6. The NTD dimers were modeled using the GluA2 NTD structure 3H5W (Jin et al., 2009), the LBD dimers using the apo structure 1FTO (Armstrong and Gouaux, 2000), and the pore using the TM regions of the GluA2cryst structure 3KG2 (Sobolevsky et al., 2009). The EM density of the GluA2-Q model was contoured so noise would be minimized. Domain structures were positioned to maximize the overlap between the domain models and EM density while preserving the twofold symmetry of the receptor.
To visualize the channel gating motions of the LBD and TMD, morphing was performed in Chimera. All morphing trajectories were saved as pdb files. For the LBD, a superposition of the open (1FTO) and closed (1FTM; Armstrong and Gouaux, 2000) states was used. For the pore movements of the NaK channel, the closed (2AHY; Shi et al., 2006) and open (3E86; Alam and Jiang, 2009) structures were used. Nineteen N-terminal residues present in the open NaK structure and not present in the closed NaK structure were removed from the open NaK structure before superposition of the open and closed NaK models. The two morphing trajectories of the LBD and one trajectory of the NaK channel were placed in the GluA2-Q EM model based on the hybrid model.
To align the LBD and TMD trajectories, the locations and orientations of the LBD trajectories were held fixed, while rotating the TMD within the hybrid model. To orient the pore properly it was assumed that the distances from the C2 connections to the M2 helices would stay the same throughout the movie, the connections from one LBD dimer would be twofold symmetric in relation to the other LBD dimer, the connections would not cross, finally, the movement of the pore helices would be consistent with the movement of the LBD connections. The C2 to TM2 connections were represented by black lines. The resulting movie was recorded using Chimera. For comparison of different LBD states within the hybrid model, the AMPA-bound (1FTM) and desensitized (2I3W) structures (Armstrong and Gouaux, 2000; Armstrong et al., 2006) were superimposed on the apo structure using the least-squares method as implemented by COOT (Emsley and Cowtan, 2004).
Ectodomain Hydrodynamic Analysis
For analytical ultracentrifugation (AUC) experiments, samples were initially concentrated to an A280 of ~0.25 and then dialyzed into 50 mM Tris, pH 7.4, 140 mM NaCl, 1 mM sodium glutamate, and 0.02% (w/v) NaN3. AUC experiments were performed at 20°C in a Beckman ProteomeLab XL-A centrifuge equipped with an AN-60 rotor and absorbance optics. Sedimentation equilibrium data were recorded for 10 h each at speeds of 7k, 10k, and 14k rpm. Scans were taken at 1 h intervals with a 0.001-cm step size along the radial axis and five replicates/data point. Attainment of sedimentation equilibrium was verified using the program WinMATCH (D. A. Yphantis and J. W. Lary, unpublished). Six-sector cells were loaded with 1×-, 2×-, and 4×-dilutions of ~2.5 μM stock solutions. Curves collected at all three speeds and all three concentrations were globally fit. Protein partial specific volume ( ) and buffer density and viscosity (ρ, η) were calculated using the program SEDNTERP (Laue et al., 1992; J. Philo, D. Hayes, and T. Laue, unpublished). Sedimentation equilibrium data were analyzed using the program SEDANAL (Stafford and Sherwood, 2004), using both single-species and oligomerization models.
) and buffer density and viscosity (ρ, η) were calculated using the program SEDNTERP (Laue et al., 1992; J. Philo, D. Hayes, and T. Laue, unpublished). Sedimentation equilibrium data were analyzed using the program SEDANAL (Stafford and Sherwood, 2004), using both single-species and oligomerization models.
Results
Domain Tagging of Ligand-Free GluA2-Q
Because of the large-scale conformational differences observed, it was not possible to interpret the domain architecture of ligand-free GluA2 receptors (Midgett and Madden, 2008) based directly on the antagonist-bound GluA2cryst structure (Sobolevsky et al., 2009). To obtain experimental constraints on the domain architecture of the ligand-free state, we therefore obtained full-length GluA2-Q receptors with fluorescent protein tags inserted at positions adjacent to the NTD and known to be compatible with the assembly of functional receptors (Braithwaite et al., 2002; Sheridan et al., 2006). The recombinant GFP:GluA2 and R2Q306c fusion proteins were purified to homogeneity (Figures 1A,C).
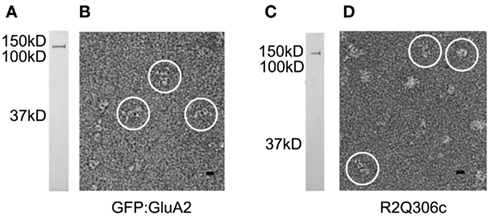
Figure 1. Visualization of GluA2-fusion proteins. Receptors containing GFP:GluA2 (A,B) and R2Q306c (C,D) subunits were visualized by SDS-PAGE and silver stain (A,C), showing a single band of the expected relative molar mass (Mr) for each protein. Following negative staining (B,D), both receptors were visualized as bright, stain-excluding particles (circles) with features characteristic of GluA2 channels. Mr standards are shown to the left of each gel. Scale bar = 10 nm.
Following preparation in negative stain, the domain-labeled receptors were visualized by EM as well-dispersed, stain-excluding particles (Figures 1B,D). Single-particle data sets were collected for each of the tagged receptor constructs. As observed previously for the untagged receptors, the fusion proteins adopt a variety of orientations on the EM carbon support film. In general, compared to the untagged GluA2 receptors (Midgett and Madden, 2008), the tagged proteins appear more elongated (Figures 1B,D), consistent with the location of the fusion domains at the membrane-distal end of the receptor.
Single-particle images of the tagged receptors were aligned, classified, and averaged to obtain characteristic projection views of the corresponding receptor constructs. To facilitate direct comparison with the untagged receptor, images of the GluA2-Q particles used in a previous reconstruction (Midgett and Madden, 2008) were reprocessed using the same strategy. Consistent with the appearance of the individual single-particle images, several class averages of the tagged receptors showed clear features adjacent to the donut-shaped structure at the top of Figures 2A,B. Neither of these features is observed in the GluA2-Q class averages (Figure 2C), suggesting that they represent the fusion domains and confirming the identification of the shared GluA2 receptor core.
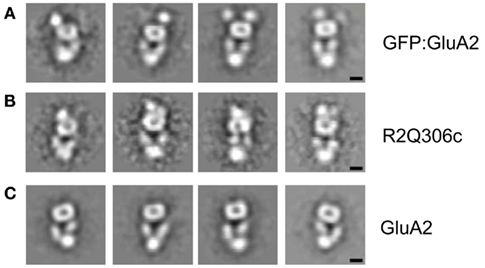
Figure 2. GluA2-Q domain tagging. Four representative class averages are shown for GFP:GluA2 (A), R2Q306c (B), and GluA2-Q (C) receptors. Additional stain-excluding regions are visualized at the top of the projection views of both the GFP:GluA2 (A) and R2Q306c (B) receptors, adjacent to the donut-shaped feature also seen in the untagged GluA2-Q receptors (C). Scale bar = 5 nm.
The position of the additional features at one end of the class averages is consistent with this assignment. The GFP tag is attached to the N-terminus of each GFP:GluA2 subunit. On the basis of NTD homology models, the R2Q306c CFP tag was originally thought to lie at the junction between the NTD and the LBD, and thus on the opposite side of the NTD from the N-terminus (Sheridan et al., 2006). However, when the sequence of the fusion construct is mapped to the experimental GluA2 NTD structure (Jin et al., 2009), it is actually found to be on the same surface as the N-terminus itself (Figure A2 in Appendix). Thus, both domain labels are predicted to be attached to the N-terminal face of the NTD, extending the long axis of the receptor, as observed (Figures 2A,B).
The differential spacing and flexibility of the domains relative to the receptor core also supports this interpretation. The fusion domain in the GFP:GluA2 construct is attached by a single, nine amino-acid linker to the GluA2 N-terminus. In the EM images, it appears somewhat distinct from the rest of the structure (Figure 2A). Analysis of component images also shows some variability in the position of the domain relative to the receptor. In comparison, the fusion domain of the R2Q306c construct, which is covalently attached to the receptor by short linkers at both ends, is more tightly and uniformly connected to the GluA2 framework (Figure 2B). Together, the positions of the fusion domains in our two domain-labeled constructs unambiguously permit assignment of the NTD to the donut-shaped feature at the top of the projection images as presented in Figure 2C.
Oligomeric Status of Imaged GluA2-Q Receptors
Parallel three-dimensional reconstructions of the domain-labeled and the unlabeled GluA2-Q receptors confirmed the presence of a shared structure corresponding to the core receptor domain (Figure 3A), similar to that previously reported for homomeric channels composed of either edited (R4) or unedited (Q4) GluA2 subunits (Midgett and Madden, 2008). However, only two additional molecular features are observed attached to receptor core in images of the domain-labeled GluA2 receptors (Figures 2A,B). We therefore wished to assess the oligomeric state of the GluA2 receptors by analyzing both their hydrodynamic and structural characteristics. The untagged GluA2-Q receptors eluted from the size-exclusion column between the apoferritin (440 kDa) and thyroglobulin (669 kDa) standard proteins, corresponding to the size expected for a tetrameric assembly (420 kDa + micelle). As an additional control for the oligomeric state of the GluA2-Q receptors, blue-native PAGE was performed (Schägger et al., 1994). Under native conditions, purified GluA2-Q receptors migrated at a molecular weight consistent with that of a tetramer (Figure 4A). Following treatment with SDS, dissociated GluA2-Q proteins migrated at distinct positions, primarily corresponding to monomers, as has been seen in other reports (Greger et al., 2003). Finally, purified GluA2-Q receptors remain functionally competent: when reconstituted into lipid bilayers, they yield glutamate-activated and NBQX-sensitive currents (J. Baranovic and J. Ryan, unpublished).
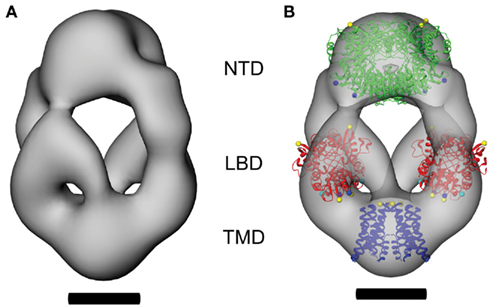
Figure 3. The domain architecture of ligand-free GluA2-Q receptors. The CTF-corrected molecular envelope determined from class averages of untagged GluA2-Q receptors is shown (A), together with a representation in which crystal structures of two NTD dimers (green, PDB entry 3H5W; Jin et al., 2009), two LBD dimers (red, PDB entry 1FTO; Armstrong and Gouaux, 2000), and a TMD tetramer (blue, PDB entry 3KG2; Sobolevsky et al., 2009) have been fitted to determine the domain architecture of the receptor (B). Scale bars = 5 nm.
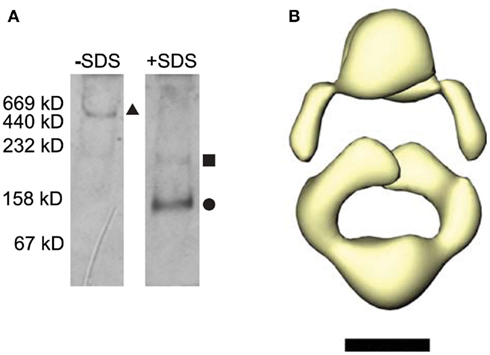
Figure 4. Tetrameric assembly of receptors visualized by EM. (A) Purified GluA2-Q receptors were concentrated and run on a blue-native PAGE gel with thyroglobulin, apo-ferretin, catalase, aldolase, and albumin as molecular weight standards. The relative molar masses are shown left of the gel. Native receptors were run in the first lane, and receptors treated with 1% SDS were run in the second lane. The size corresponding to GluA2-Q tetramers, dimers, and monomers are marked by a triangle, square, and circle, respectively. (B) The 3D molecular of the receptor was contoured to yield a mass equivalent to a subunit dimer, yielding a hollow, attenuated structure with dimensions similar to the molecular envelope shown in Figure 3B. Scale bar = 5 nm.
To evaluate the formal possibility that the GluA2-Q receptors imaged in negative stain could represent dimeric subassemblies captured during the staining process, we raised the contouring threshold of the three-dimensional reconstruction such that the enclosed molecular volume corresponded to that of a receptor dimer (250 kDa). If the particles imaged were indeed subunit dimers, the resulting molecular envelope should contract to a smaller, continuous core, reflecting the underlying dimeric structure. If the particles are tetramers, the high-contour map should instead show an attenuated or fragmented structure distributed over a total volume corresponding to the four component subunits. As predicted for intact receptor tetramers, the reduced-volume molecular envelope is hollowed out, but preserves the outline and dimensions of the tetrameric structure (Figure 4B). This result is clearly incompatible with a dimeric state of the imaged protein. Instead, it appears that the four GFP domains in the tetrameric fusion constructs have associated pairwise to form two dimers, each of which is visualized as a single feature at the resolution of the reconstructions. This result is consistent with the inherent dimerization affinity of the GFP domain (Tsien, 1998) and the high local concentration imposed by tethering them to adjacent subunits in the receptor.
The Ectodomain Architecture of a Ligand-Free Conformation
Having confirmed the oligomeric integrity of the molecular envelope and having located the fusion domains at one end of the molecular envelope, we can confidently locate the NTD at the adjacent end of the receptor core. The threshold for the 3D reconstruction was set to enclose a volume with minimal noise (Verschoor et al., 1984). In the front view shown in Figure 3A, the volume at the top of the three-dimensional reconstruction of GluA2-Q corresponds to the donut-shaped feature seen next to the fluorescent protein domain tags in the class averages (Figures 2A,B).
Since the NTD forms robust homodimers (Kuusinen et al., 1999; Jin et al., 2009), modeling the tetrameric assembly requires placing two copies of the dimer within the molecular envelope. The domain structures of the GluA2 NTD dimer (PDB entry 3H5W; Jin et al., 2009) were positioned to maximize the overlap of the domains with the volume of the EM reconstruction and to maintain the overall twofold symmetry of the receptor. The envelope in this region is continuous across the molecular symmetry axis, and together with the steric outline, drives the formation of a compact dimer-of-dimers assembly (Figure 3B). A few modest excursions of the model outside the molecular envelope are observed in the region of a dimple associated with the prominent stain feature that forms the “donut hole” in projection images, and may reflect staining artifacts in this region. Otherwise, the two dimers fit well within the constraints of the envelope.
The positioning of the NTDs largely fills the volume at the top of the model. As a result, based on domain connectivity, the adjacent LBD dimers must be located below the NTDs in the two arms that are located on either side of the central vestibule, connecting the upper and lower densities (Figure 3B). Small-angle X-ray scattering data (Madden et al., 2005) have shown that the full-length glycosylated LBD (Dmax ≥ 105 Å) is substantially more elongated than the GluA2 LBD core that has been crystallized (Dmax ≤ 65 Å; Armstrong and Gouaux, 2000). This suggests that the full-length linker peptides and carbohydrate moieties missing from the crystallized LBD core construct are likely positioned at one or both ends of the domain. These moieties are all present in the GluA2-Q tetramer, so the LBD dimers (PDB entry 1FTO; Armstrong and Gouaux, 2000) were placed within a ~100 Å column located between the NTDs and the TMD (Figure 3B). Independent of the exact vertical placement of the LBD dimers within this column, they are separated from each other by the central vestibule, such that LBD dimer–dimer contacts would be restricted to the vicinity of the membrane.
Complementary LBD and NTD Dimerizations Stabilize a Tetrameric Assembly
The placement of the LBDs fixes the positions of the dimers on either side of the central vestibule. However, at the resolution of our EM reconstruction, the NTD dimers could be placed in two different orientations with similar qualities of fit (Figure A3 in Appendix). The first orientation has the dimer interfaces of the NTDs and LBDs parallel to each other in a “cooperative” arrangement; the second has the NTD and LBD dimer interfaces orthogonal to each other in a “complementary” configuration (Gill and Madden, 2006). The structure of the antagonist-bound GluA2cryst tetramer reveals cross-over NTD–LBD connections between pairs of subunit dimers, consistent with the complementary orientation (Sobolevsky et al., 2009). However, the NTD–LBD linker was truncated in the crystallized construct to reduce glycosylation. Also, based on the EM reconstruction, there are substantial conformational differences between the NTD tetramerizations in the antagonist-bound and ligand-free conformations. As a result, we designed an experiment to test the NTD–LBD connectivity in our expression system.
For these studies, we monitored the oligomerization of a soluble protein construct that consists of the NTD and LBD of the homologous GluA4flip subunit in a tandem array, corresponding to the entire extracellular moiety of the subunit. A similar construct was previously shown to dimerize strongly (Kuusinen et al., 1999). Since the isolated NTD also dimerizes strongly, but the wild-type LBD does not, it appears that the WT ectodomain dimers are stabilized primarily by NTD:NTD interactions. To test the relative orientation of the LBD dimerization, we introduced the L484Y mutation into the GluA4 ectodomain constructs. This mutation is located at the LBD dimer interface and blocks desensitization by enhancing the affinity of LBD dimerization (Stern-Bach et al., 1998; Sun et al., 2002; Gill et al., 2008). As a result, if the LBD dimer interface were formed within the NTD-stabilized dimer, the L484Y mutation should enhance dimer stability. If the LBD dimer interface is instead formed between NTD-stabilized dimers, it should favor a secondary dimerization, leading to the generation of ectodomain tetramers.
To evaluate these alternatives experimentally, we monitored the hydrodynamic behavior of the WT and mutant ectodomain constructs by analytical ultracentrifugation. As shown in Figure 5A, the sedimentation equilibrium data for the WT ectodomain can be well modeled by a monomer:dimer equilibrium with a dimerization Kd of 0.3 μM (95% confidence interval: 0.06–0.53 μM). Fitting with a monomer:dimer:tetramer equilibrium did not improve the WT fit, and could not be distinguished from a pure monomer:dimer equilibrium at the 95% confidence level. In contrast, the equilibrium for the L484Y ectodomain cannot be fit by a monomer:dimer equilibrium without systematic residuals (Figure 5B, dashed line). Instead, it is well fit by a monomer:dimer:tetramer equilibrium (Figure 5B, solid line) with a dimerization Kd of 1.5 μM (95% CI: 0.1–2.9 μM) and a tetramerization Kd of 1.1 μM (95% CI: 0.2–4.0 μM). These data thus favor a complementary orientation of the NTD and LBD dimerization interfaces, such that LBD dimerization can stabilize a pairwise interaction between subunit dimers that are held together at the NTD. This is the orientation of the domains shown in Figure 3B.
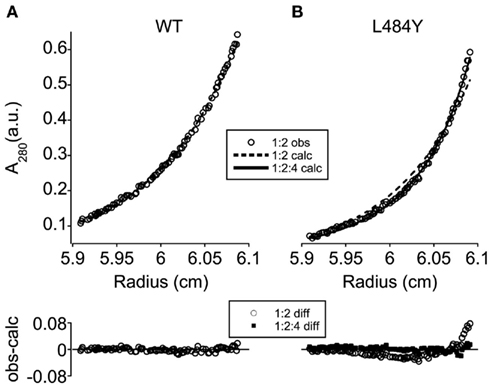
Figure 5. Ligand-binding domain–N-terminal domain cross-over drives ectodomain tetramerization. Representative experimental sedimentation equilibrium absorbance profiles (A280, open circles) are shown for WT (A) and L484Y (B) ectodomain constructs, together with curves predicted by a three-speed, three-channel global fit using monomer:dimer (dashed line) and monomer:dimer:tetramer [solid line (B) only] equilibrium models. The deviation between observed and calculated A280 values is shown below each profile for the monomer:dimer (open circles) and monomer:dimer:tetramer (filled squares) models. While the WT data were well fit by a monomer:dimer equilibrium (A), systematic deviations between the monomer:dimer predictions and the experimental data were observed for the L484Y mutant in the absence of a secondary dimerization reaction (B).
A Model of AMPAR Activation Constrains TMD Orientation
Based on the positioning of the NTD and LBD dimers, the TMD can be assigned to the cone-shaped density at the bottom of the receptor (Figure 3B). The transmembrane helices of all four GluA2cryst subunits are resolved in the crystal structure of the antagonist-bound conformation of intact GluA2 (Sobolevsky et al., 2009), permitting us to model this region of the molecular envelope (Figure 3B). Alignment of the GluA2 and TMD symmetry axes, together with the steric constraints of the molecular envelope and the position of the LBD, provide strong constraints on the position of the center of the TMD. However, the rotational orientation of the TMD around the axis was not clearly defined by these steric and symmetry considerations. To resolve this issue and to clarify the structural basis of AMPAR activation, we compared conformational changes in the LBD and the TMD that are coupled within the intact receptor at the S1-TM1 and TM2-S2 junctions.
The LBD conformational changes associated with gating were assessed by comparing the apo (open cleft) and AMPA-bound (closed-cleft) states (PDB entries 1FTO and 1FTM, respectively; Armstrong and Gouaux, 2000). In the LBD dimers, the residues that connect to the TM linkers (C1–C3, representing the core residues closest in sequence to TM1–TM3, respectively) are arranged roughly in lines running parallel to the LBD dimer interface (Figure 6A). The transition from the apo to the AMPA-bound state increases the C1 and C2 inter-protomer distances, while the C3 inter-protomer distance remains unchanged. The C2 spacing increases the most (from 29 to 39 Å), followed by that of C1 (32–36 Å; Table A1 in Appendix). The result is a pseudo-rotation around C3, displacing C2 more than C1 (Figure A4 in Appendix). Within the hybrid model, this motion results in a scissoring motion away from the C3–C3 axis in each dimer. Based on the placement of the dimers on either side of the central vestibule, the resulting motion involves both radial and tangential components around the symmetry axis of the channel (Figure 6B).
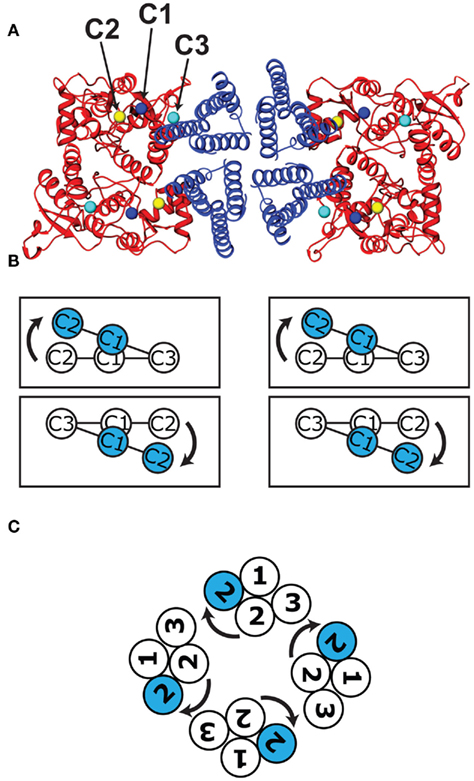
Figure 6. Movements of LBD–TMD attachment points during channel activation. (A) A view of the hybrid model from the cytoplasmic face, showing the central TMD (blue), and behind it to the left and right, the LBD dimers (red) as positioned within the arms of the molecular envelope (Figure 3B). The LBD connection points (C1–C3) that attach to the three corresponding transmembrane helices (TM1–TM3) are marked with blue, yellow, and cyan spheres, respectively. (B) The attachment points for each of the three monomers are shown schematically in the apo (white) and AMPA-bound (blue) configurations, showing the scissoring motion described in Figure A4 in Appendix. (C) Following rotations of the TMD about the channel symmetry axis to align the LBD and TMD activation trajectories, the four sets of three helices are oriented as shown schematically, as viewed from the outside of the cell. The prominent motion of the TMD helix associated with channel opening (blue) is also shown.
To visualize the associated conformational change in the TMD, we generated additional models of the TMD using crystal structures of the NaK sodium/potassium channel pore. The NaK channel is a member of a family that shares sequence similarity with TM1, TM2, and the pore loop of the AMPAR (Wo and Oswald, 1995; Panchenko et al., 2001; Kuner et al., 2003). It been crystallized in both the closed and open channel conformations (PDB entries 2AHY and 3E86, respectively; Shi et al., 2006; Alam and Jiang, 2009). As a result, we can compare the channel opening trajectories of the TM1 and TM2 attachment points to the corresponding trajectories of the LBD connection points C1 and C2.
The azimuthal orientation of the NaK TMD around the central axis was optimized to align the trajectories of the corresponding LBD and TMD helix attachment points and to avoid linker conformations that would cross during channel opening. This led to a pore orientation in which the tops of TM2 form a diamond shape (Figure 6C) relative to the LBDs (Figure 6B). As for the LBDs, the motions of the TM helix attachment points involve a combination of rotational and translational movements, with the TM2 helix undergoing the larger displacement. When agonist binds to the receptor, C1 and C2 pivot around C3 pulling on the linkers, causing TM1 and TM2 to rotate relative to TM3, and opening the pore (Movie in Supplementary Material). The resulting model of AMPAR gating suggests that the agonist-induced motions of the separated LBD dimers are fundamentally compatible with those of the potassium and sodium channels.
The model also shows that for both the LBDs and the TMD, the conformational changes associated with agonist activation are readily accommodated within the GluA2-Q molecular envelope (Movie in Supplementary Material). To characterize the magnitude of the conformational changes associated with channel activation directly, we performed two additional comparisons. First, we incubated GluA2-Q receptors with saturating concentrations of glutamate before imaging them in negative stain. The resulting projection images were indistinguishable from those of the untreated receptors (Figures 2C and 7A). Second, LBD dimers were modeled into the single-particle molecular envelope in the apo, AMPA-bound, and desensitized states (PDB entries 1FTO, 1FTM, and 2I3W, respectively; Armstrong and Gouaux, 2000; Armstrong et al., 2006). All three states were accommodated in the arms surrounding the central vestibule (Figures 7B–D).
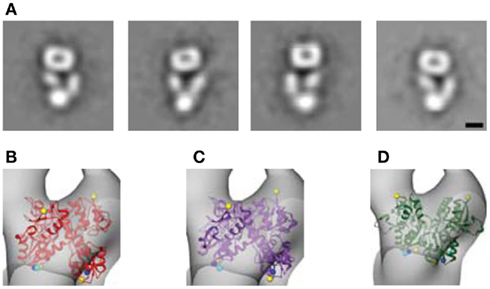
Figure 7. Magnitude of conformational changes associated with channel activation. (A) To identify conformational changes associated with channel activation or desensitization, GluA2-Q receptors were treated with 10 mM glutamate prior to EM sample preparation. Following negative staining and EM imaging, class averages were computed, which closely resemble those seen for the ligand-free receptor (Figure 2C). (B–D) To assess the ability of the ligand-free molecular envelope to accommodate the extent of LBD conformational changes associated with agonist binding, we modeled the apo (B), AMPA-bound (C), and desensitized (D) structures of the LBD dimer (PDB entries 1FTO, 1FTM, and 2I3W, respectively; Armstrong and Gouaux, 2000; Armstrong et al., 2006) into the molecular envelope. All three structures are accommodated. Scale bar = 5 nm.
Discussion
Despite previous reports of AMPAR structural heterogeneity in the absence of antagonist (Nakagawa et al., 2005; Shanks et al., 2010), in this and in earlier studies (Tichelaar et al., 2004; Midgett and Madden, 2008) we have identified a specific, well-defined conformation for both GluA2 Q4 and R4 homomeric receptors purified and imaged without antagonist. Filter binding studies have shown that these receptors are essentially free of any tightly bound ligands in the agonist binding site (Madden et al., 2000; Safferling et al., 2001). Furthermore, the diversity of single-particle orientations contributing to the 3D reconstruction (Tichelaar et al., 2004) means that the receptor contacts the EM carbon support film through a variety of interfaces, in each case yielding projection images consistent with a single 3D structure. As a result, it is also unlikely that this structure represents a non-native conformation that happens to be selectively stabilized by each of these highly variable substrate interactions (Mayer, 2011).
Based on these considerations, we propose that the resulting structure reflects a predominant conformation of the receptor in its ligand-free state. In the present study, we have determined the molecular architecture of this conformation for the calcium-permeable AMPA receptor formed by GluA2 Q4 homotetramers, building on crystal structures of the component domains. Our model reveals that the two NTD dimers are packed together at the membrane-distal end of the receptor, with a large central vestibule separating two arms that each contain an LBD dimer. At the membrane-proximal end of the receptor, the LBD arms reconnect to a single compact volume that contains the four TMDs, arranged around the central symmetry axis of the channel (Figure 3B). The resulting O-shaped arrangement stands in contrast to the Y-shaped structure of antagonist-bound channels, in which the two NTD dimers are splayed apart, whereas the LBDs are more closely packed (Nakagawa et al., 2005; Sobolevsky et al., 2009; Shanks et al., 2010). Nevertheless, as seen in a side-by-side comparison (Figure 8), both conformations result in a layered arrangement of domains with respect to the membrane, with similar spacings and overall vertical dimensions. Furthermore, as for the Y-shaped GluA2cryst structure (Sobolevsky et al., 2009), the symmetry of the O-shaped particle is twofold, reflecting the dimer-of-dimers assembly that has been proposed for the iGluR family (Ayalon and Stern-Bach, 2001; Schorge and Colquhoun, 2003).
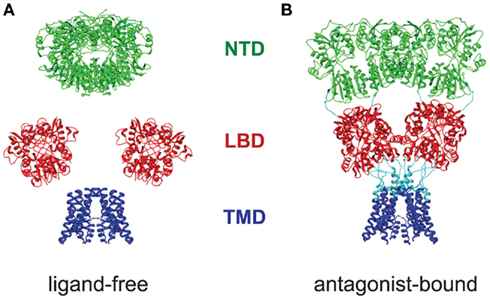
Figure 8. Comparison of the domain architectures of ligand-free and antagonist-bound GluA2. The NTD (green), LBD (red), and TMD (blue) domains of ligand-free (A) and antagonist-bound (B) (PDB entry 3KG2; Sobolevsky et al., 2009) GluA2 receptors are viewed parallel to the plane of a hypothetical cell membrane. Although the horizontal dimer–dimer packing of the NTD and LBD differs noticeably, the overall vertical dimensions and domain layering of two models is conserved.
The domain architecture presented here for the O-shaped conformation is consistent with data on multiple aspects of AMPA receptor function. At the level of the LBD, although dimer-of-dimers packing models were originally proposed (Sun et al., 2002; Jin and Gouaux, 2003), extensive mutagenesis failed to identify modulatory residues in the candidate LBD dimer–dimer interfaces (Horning and Mayer, 2004; see also discussion of disulfide trapping experiments below). A defining feature of the O-shaped conformation is the vestibule located between the LBD dimers (Figure 3A), which would account for the lack of evidence for substantial functional cross-talk between LBD dimers. The presence of the vestibule suggests that the two LBD dimers in a receptor will make largely independent contributions to ion channel desensitization (Mansour et al., 2001). It also provides a firm structural basis for observations that the dimer forms a fundamental functional unit within the receptor (Sun et al., 2002; Schorge and Colquhoun, 2003; Horning and Mayer, 2004; Furukawa et al., 2005).
The domain packing differences between the O- and Y-shaped conformations are also consistent with the ability of NTD ligands to modulate iGluR electrophysiological characteristics allosterically (Hansen et al., 2010). The Y-shaped conformation appears to be strongly stabilized by competitive antagonists (Nakagawa et al., 2005; Sobolevsky et al., 2009; Shanks et al., 2010). Thus, the O–Y transition associated with antagonist binding simultaneously brings together the LBD dimers and separates the NTD dimers (Figure 8). This suggests an allosteric coupling between the ligand/antagonist binding cleft and the packing of both pairs of dimers, consistent with substantial data showing a reciprocal influence of NTD ligands on NMDA receptor characteristics (Hansen et al., 2010). Indeed, recent structural data suggest that AMPAR NTD may also exhibit conformational flexibility that could support similar allosteric effects (Sukumaran et al., 2011). This NTD–LBD coupling presumably reflects the extensive interactions between the domains, which cross-over to drive secondary dimerization independent of the TMD (Figure 4).
At the level of the LBD–TMD interconnection, the dual scissoring model of receptor activation that provides orientational constraints on the TMD (Figure 6 and Movie in Supplementary Material) is also consistent with functional data on channel opening. Based on the relative shifts of the TM attachment points in the two LBD dimers, in this model TM1 and TM2 rotate around TM3 to open the pore, maintaining a relatively fixed separation between the C3 attachment points within and between dimers. Consistent with this model, scanning cysteine accessibility studies of NMDA receptors showed that similar sets of residues were accessible on TM3 whether the channel was open or closed (Sobolevsky et al., 2002), suggesting that it is relatively static during gating. The model also indicates that the largest motions are associated with TM2, again mirroring scanning cysteine mutagenesis data for NMDA receptors and broadly consistent with the model proposed on the basis of the antagonist-bound GluA2cryst structure (Sobolevsky et al., 2002, 2009).
While our model is thus consistent with a number of functional observations, there are also a few aspects that must be reconciled with biochemical data. At the level of the TMD, the motion of the NaK TM2 helix is larger in magnitude than the corresponding motion of the C2 attachment point on the LBD (Movie in Supplementary Material). This most likely reflects the limitations of using the NaK channel as a surrogate for GluA2 TMD gating motions, which have not yet been experimentally resolved but which must accommodate a two- to four-fold symmetry transition not present in NaK. As seen in the GluA2cryst structure, the TM2-S2 linker peptides adopt two distinct conformations, accommodating the strongly twofold symmetry of the LBD attachment points and the opposing pivot motions of the C1 and C2 linkers around C3 (Sobolevsky et al., 2009). Similarly, some twofold character has also been proposed for the TMD of both NMDA (Sobolevsky et al., 2004, 2007) and kainate receptors (Kaczor et al., 2008). It is therefore likely that the motions observed in the fourfold symmetrical NaK channel only approximate those occurring in the iGluR family. In addition, it is possible that the linker peptides are not themselves static, but contribute actively or passively to the activation mechanism, consistent with mutations in the linker regions that have striking effects on channel activation (Klein and Howe, 2004).
The compact NTD dimer-of-dimers architecture defined by our model has not been observed in lattice-packing contacts of published GluA2 NTD structures (Clayton et al., 2009; Jin et al., 2009; Rossmann et al., 2011) nor in the GluA2cryst:ZK200775 complex (Sobolevsky et al., 2009). Indeed, fitting of the extended NTD dimer-of-dimers within our EM reconstruction leaves a large void within the molecular envelope and positions two of the NTD monomers substantially outside of it. As a result, it is clear that repacking of the NTD dimers must occur in the transition between the Y- and O-shaped conformations. It is possible that shifts in the relative disposition of the LBD dimers drives offsetting changes in the packing of the NTD dimers, reflecting distinct functional states of the receptor. Unfortunately, our negatively stained single-particle reconstruction does not provide sufficient resolution to engineer cysteine probes of the closely packed NTD interface, but higher-resolution studies should facilitate the investigation of alternative NTD packing models.
Perhaps the most important consideration is the previous demonstration that cysteine mutants engineered at the LBD dimer–dimer interface observed in the Y-shaped conformation can form inter-subunit dimers, indicating that this conformation is accessible to GluA2cryst homomers in the cellular context. However, the I664C mutation almost completely blocks channel activation unless disulfide bonds are released by treatment with reducing agents (Sobolevsky et al., 2009). Thus, if the LBD tetramer is locked in the Y-shaped conformation, the channels appear unable to open. To what extent the dimer–dimer interface must separate or rearrange to permit activation is unclear, but the earlier inability to identify mutations that modulate, rather than block, activation (Horning and Mayer, 2004) suggests that gating is driven from relatively independent LBD dimers. Thus, it is possible that the LBD hyperextension observed upon ZK200775 binding (Sobolevsky et al., 2009) permits disulfide stabilization of a conformation in which the LBD are more closely packed than in the resting or activated states.
The differences in the NTD and LBD dimer–dimer interfaces observed in the Y- and O-shaped conformations (Figure 8) indicate that the receptor is able to undergo large-scale conformational rearrangements in response to antagonist binding. Higher-resolution characterization of these differences may facilitate the development of non-competitive pharmacological reagents that exploit the substantially different domain and subunit interfaces associated with distinct functional states. However, a major question concerns the extent to which such macroscopic motions are required for iGluR activation and desensitization. In conformationally unstable AMPAR, application of agonist was observed to drive large-scale rearrangements (Nakagawa et al., 2005), suggesting that negative staining was compatible with agonist binding. In contrast, in our samples pretreatment of the O-shaped conformation with agonist did not cause substantial conformational changes. Although we cannot experimentally confirm the presence of agonist in the stain-embedded receptors shown in Figure 7A, the spectrum of LBD conformations that have been seen crystallographically are also accommodated similarly within the O-shaped molecular envelope (Figures 7B–D). Modest excursions are seen for all the fitted LBD, but these may reflect technical limitations of the molecular envelope and/or conformational distortions. Ultimately, detergent-solubilized GluA2 receptors or recombinant domain structures cannot be strictly correlated with specific functional states or electrophysiological intermediates (Zhang et al., 2008; Poon et al., 2010; Prieto and Wollmuth, 2010; Traynelis et al., 2010), and may therefore reflect incommensurate functional states. However, the comparison highlights the limited scale of LBD cleft closures relative to the quaternary structure of the fully assembled channel. Similarly, only small displacements are required to open the channel gate. The open AMPAR pore is estimated to be roughly 7 Å in diameter (Burnashev et al., 1996; Kuner et al., 2001). The closed pore itself may be as large as 4–5 Å in diameter, which would still prevent passage of hydrated K+ or Na+ ions (Mullins, 1960). Each subunit therefore only has to contribute a change of at most 3.5 Å to the radius of the pore, and potentially as little as 1 Å. Thus, particularly at the resolution of single-particle EM structures, large-scale quaternary rearrangements may not be required by any of the component motions associated with channel activation.
Overall, the domain architecture model proposed here reconciles a wide variety of structural, functional, and biochemical observations. The strongly twofold nature of the molecular envelope and the separation of LBD dimers across a prominent central vestibule confirm the idea that subunit dimers are the fundamental modules of glutamate receptor activation. The trajectories of channel opening at the level of the LBD and the TMD attachment points also provide a conformationally parsimonious model for agonist-mediated iGluR gating, potentially requiring only modest rearrangements at the level of quaternary structure. And while the roles of the distinct conformations visualized in the ligand-free and antagonist-bound states remain to be clarified, both models provide insights into channel gating that are broadly compatible with biochemical data sampled from the three major iGluR subfamilies. Thus, it is likely that fundamental features of the gating process are conserved among iGluRs, and will ultimately be revealed by structural and functional studies that provide additional snapshots of the associated conformational transitions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/molecular_neuroscience/10.3389/fnmol.2011.00056/abstract
Acknowledgments
We thank Dr. Charles Daghlian, and Lousia Howard of the Rippel EM facility for help in using the JEOL 1010 microscope and Dr. Amanda Birdsey-Benson for helpful discussions. In addition, we would like to thank Dr. Thom Hughes (University of Montana) for the AMPAR fusion construct R2Q306c, Dr. Kari Keinänen (University of Helsinki) for the GFP:GluA2 construct, and Dr. Teresa Ruiz and Dr. Michael Radermacher (University of Vermont) and Dr. Steven Ludtke (Baylor College of Medicine) for assistance with electron microscopy. Financial support was provided in part by the Hitchcock Foundation and the Libby Cass Library Memorial Fund.
References
Abele, R., Keinänen, K., and Madden, D. R. (2000). Agonist-induced isomerization in a glutamate receptor ligand-binding domain: a kinetic and mutagenetic analysis. J. Biol. Chem. 275, 21355–21363.
Alam, A., and Jiang, Y. (2009). High-resolution structure of the open NaK channel. Nat. Struct. Mol. Biol. 16, 30–34.
Armstrong, N., Jasti, J., Beich-Frandsen, M., and Gouaux, E. (2006). Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127, 85–97.
Armstrong, N. A., and Gouaux, E. (2000). Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181.
Ayalon, G., and Stern-Bach, Y. (2001). Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron 13, 103–113.
Braitenberg, V., and Schüz, A. (1998). Cortex: Statistics and Geometry of Neuronal Connectivity. Berlin: Springer Verlag.
Braithwaite, S. P., Xia, H., and Malenka, R. C. (2002). Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc. Natl. Acad. Sci. U.S.A. 99, 7096–7101.
Burnashev, N., Monyer, H., Seeburg, P. H., and Sakmann, B. (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8, 189–198.
Burnashev, N., Villarroel, A., and Sakmann, B. (1996). Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J. Physiol. 496, 165–173.
Clayton, A., Siebold, C., Gilbert, R. J., Sutton, G. C., Harlos, K., Mcilhinney, R. A., Jones, E. Y., and Aricescu, A. R. (2009). Crystal structure of the GluR2 amino-terminal domain provides insights into the architecture and assembly of ionotropic glutamate receptors. J. Mol. Biol. 392, 1125–1132.
Collingridge, G. L., Olsen, R. W., Peters, J., and Spedding, M. (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology. 56, 2–5.
Cull-Candy, S., Kelly, L., and Farrant, M. (2006). Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr. Opin. Neurobiol. 16, 288–297.
Derkach, V. A., Oh, M. C., Guire, E. S., and Soderling, T. R. (2007). Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113.
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132.
Furukawa, H., Singh, S. K., Mancusso, R., and Gouaux, E. (2005). Subunit arrangement and function in NMDA receptors. Nature 438, 185–192.
Gill, A., Birdsey-Benson, A., Jones, B. L., Henderson, L. P., and Madden, D. R. (2008). Correlating AMPA receptor activation and cleft closure across subunits: crystal structures of the GluR4 ligand-binding domain in complex with full and partial agonists. Biochemistry 47, 13831–13841.
Gill, A., and Madden, D. R. (2006). “Glutamate receptor ion channels: structural insights into molecular mechanisms,” in Structural Biology of Membrane Proteins, eds R. Grisshammer, and S. Buchanan (Cambridge, UK: Royal Society of Chemistry), 349–372.
Greger, I. H., Khatri, L., Kong, X., and Ziff, E. B. (2003). AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774.
Hansen, K. B., Furukawa, H., and Traynelis, S. F. (2010). Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol. Pharmacol. 78, 535–549.
Hinoi, E., Takarada, T., Ueshima, T., Tsuchihashi, Y., and Yoneda, Y. (2004). Glutamate signaling in peripheral tissues. Eur. J. Biochem. 271, 1–13.
Horning, M. S., and Mayer, M. L. (2004). Regulation of AMPA receptor gating by ligand binding core dimers. Neuron 41, 379–388.
Jin, R., and Gouaux, E. (2003). Probing the function, confomational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry 42, 5201–5213.
Jin, R., Singh, S. K., Gu, S., Furukawa, H., Sobolevsky, A. I., Zhou, J., Jin, Y., and Gouaux, E. (2009). Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J. 28, 1812–1823.
Kaczor, A. A., Kijkowska-Murak, U. A., and Matosiuk, D. (2008). Theoretical studies on the structure and symmetry of the transmembrane region of glutamatergic GluR5 receptor. J. Med. Chem. 51, 3765–3776.
Keinänen, K., Jouppila, A., and Kuusinen, A. (1998). Characterization of the kainate-binding domain of the glutamate receptor GluR-6 subunit. Biochem. J. 330, 1461–1467.
Kelly, L., Farrant, M., and Cull-Candy, S. G. (2009). Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat. Neurosci. 12, 593–601.
Klein, R. M., and Howe, J. R. (2004). Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J. Neurosci. 24, 4941–4951.
Kuner, T., Beck, C., Sakmann, B., and Seeburg, P. H. (2001). Channel-lining residues of the AMPA receptor M2 segment: structural environment of the Q/R site and identification of the selectivity filter. J. Neurosci. 21, 4162–4172.
Kuner, T., Seeburg, P. H., and Guy, H. R. (2003). A common architecture for K+ channels and ionotropic glutamate receptors? Trends Neurosci. 26, 27–32.
Kuusinen, A., Abele, R., Madden, D. R., and Keinänen, K. (1999). Oligomerization and ligand-binding properties of the ectodomain of the AMPA receptor subunit GluRD. J. Biol. Chem. 274, 28937–28943.
Laue, T. M., Shah, B. D., Ridgeway, T. M., and Pelletier, S. L. (1992). “Computer-aided interpretation of analytical sedimentation data for proteins,” in Analytical Ultracentrifugation in Biochemistry and Polymer Sciences, eds S. E. Harding, A. J. Rowe, and J. C. Horton (Cambridge, UK: Royal Society for Chemistry), 90–125.
Lebart, L., Morineau, A., and Warwick, K. M. (1984). “Automatic classification-clustering techniques used with principal axes methods,” in Multivariate Descriptive Statistical Analysis, ed. E. M. Berry (New York: John Wiley and Sons), 109–145.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Ludtke, S. J., Baldwin, P. R., and Chiu, W. (1999). EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97.
Madden, D. R. (2002). The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci. 3, 91–101.
Madden, D. R., Abele, R., Andersson, A., and Keinänen, K. (2000). Large-scale expression and thermodynamic characterization of a glutamate receptor agonist-binding domain. Eur. J. Biochem. 267, 4281–4289.
Madden, D. R., Armstrong, N., Svergun, D., Pérez, J., and Vachette, P. (2005). Solution-scattering evidence for agonist- and antagonist-induced modulation of cleft closure in a glutamate receptor ligand-binding domain. J. Biol. Chem. 280, 23637–23642.
Madden, D. R., and Safferling, M. (2007). Baculoviral expression of an integral membrane protein for structural studies. Methods Mol. Biol. 363, 39–57.
Man, H. Y. (2011). GluA2-lacking, calcium-permeable AMPA receptors – inducers of plasticity? Curr. Opin. Neurobiol. 21, 291–298.
Mansour, M., Nagarajan, N., Nehring, R., Clements, J., and Rosenmund, C. (2001). Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron 32, 841–853.
Mayer, M. L. (2011). Structure and mechanism of glutamate receptor ion channel assembly, activation and modulation. Curr. Opin. Neurobiol. 21, 283–290.
Midgett, C. R., and Madden, D. R. (2008). The quaternary structure of a calcium-permeable AMPA receptor: conservation of shape and symmetry across functionally distinct subunit assemblies. J. Mol. Biol. 382, 578–584.
Milstein, A. D., and Nicoll, R. A. (2008). Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol. Sci. 29, 333–339.
Mullins, L. J. (1960). An analysis of pore size in excitable membranes. J. Gen. Physiol. 43, 105–117.
Nakagawa, T., Cheng, Y., Ramm, E., Sheng, M., and Walz, T. (2005). Structure and different conformational states of native AMPA receptor complexes. Nature 433, 545–549.
Nakagawa, T., Cheng, Y., Sheng, M., and Walz, T. (2006). Three-dimensional structure of an AMPA receptor without associated stargazin/TARP proteins. Biol. Chem. 387, 179–187.
Panchenko, V. A., Glasser, C. R., and Mayer, M. L. (2001). Structural similarities between glutamate receptor channels and K+ channels examined by scanning mutagenesis. J. Gen. Physiol. 117, 345–359.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., and Ferrin, T. E. (2004). UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612.
Philipps, B., Rotmann, D., Wicki, M., Mayr, L. M., and Forstner, M. (2005). Time reduction and process optimization of the baculovirus expression system for more efficient recombinant protein production in insect cells. Protein Expr. Purif. 42, 211–218.
Poon, K., Nowak, L. M., and Oswald, R. E. (2010). Characterizing single-channel behavior of GluA3 receptors. Biophys. J. 99, 1437–1446.
Prieto, M. L., and Wollmuth, L. P. (2010). Gating modes in AMPA receptors. J. Neurosci. 30, 4449–4459.
Rossmann, M., Sukumaran, M., Penn, A. C., Veprintsev, D. B., Babu, M. M., and Greger, I. H. (2011). Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J. 30, 959–971.
Safferling, M., Tichelaar, W., Kümmerle, G., Jouppila, A., Kuusinen, A., Keinänen, K., and Madden, D. R. (2001). First images of a glutamate receptor ion channel: oligomeric state and molecular dimensions of GluRB homomers. Biochemistry 40, 13948–13953.
Schägger, H., Cramer, W. A., and Von Jagow, G. (1994). Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217, 220–230.
Schorge, S., and Colquhoun, D. (2003). Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J. Neurosci. 23, 1151–1158.
Shanks, N. F., Maruo, T., Farina, A. N., Ellisman, M. H., and Nakagawa, T. (2010). Contribution of the global subunit structure and stargazin on the maturation of AMPA receptors. J. Neurosci. 30, 2728–2740.
Sheridan, D. L., Robert, A., Cho, C. H., Howe, J. R., and Hughes, T. E. (2006). Regions of α-amino-5-methyl-3-hydroxy-4-isoxazole propionic acid receptor subunits that are permissive for the insertion of green fluorescent protein. Neuroscience 141, 837–849.
Shi, N., Ye, S., Alam, A., Chen, L., and Jiang, Y. (2006). Atomic structure of a Na+- and K+-conducting channel. Nature 440, 570–574.
Sobolevsky, A. I., Beck, C., and Wollmuth, L. P. (2002). Molecular Rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron 33, 75–85.
Sobolevsky, A. I., Prodromou, M. L., Yelshansky, M. V., and Wollmuth, L. P. (2007). Subunit-specific contribution of pore-forming domains to NMDA receptor channel structure and gating. J. Gen. Physiol. 129, 509–525.
Sobolevsky, A. I., Rosconi, M. P., and Gouaux, E. (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756.
Sobolevsky, A. I., Yelshansky, M. V., and Wollmuth, L. P. (2004). The outer pore of the glutamate receptor channel has 2-fold rotational symmetry. Neuron 41, 367–378.
Sommer, B., Köhler, M., Sprengel, R., and Seeburg, P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19.
Stafford, W. F., and Sherwood, P. J. (2004). Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys. Chem. 108, 231–243.
Stern-Bach, Y., Russo, S., Neuman, M., and Rosenmund, C. (1998). A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron 21, 907–918.
Sukumaran, M., Rossmann, M., Shrivastava, I., Dutta, A., Bahar, I., and Greger, I. H. (2011). Dynamics and allosteric potential of the AMPA receptor N-terminal domain. EMBO J. 30, 972–982.
Sun, Y., Olson, R., Horning, M., Armstrong, N., Mayer, M., and Gouaux, E. (2002). Mechanism of glutamate receptor desensitization. Nature 417, 245–253.
Tichelaar, W., Safferling, M., Keinänen, K., Stark, H., and Madden, D. R. (2004). The three-dimensional structure of an ionotropic glutamate receptor reveals a dimer-of-dimers assembly. J. Mol. Biol. 344, 435–442.
Tomita, S. (2010). Regulation of ionotropic glutamate receptors by their auxiliary subunits. Physiology (Bethesda) 25, 41–49.
Traynelis, S. F., Wollmuth, L. P., Mcbain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., and Dingledine, R. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Turski, L., Huth, A., Sheardown, M., Mcdonald, F., Neuhaus, R., Schneider, H. H., Dirnagl, U., Wiegand, F., Jacobsen, P., and Ottow, E. (1998). ZK200775: a phosphonate quinoxalinedione AMPA antagonist for neuroprotection in stroke and trauma. Proc. Natl. Acad. Sci. U.S.A. 95, 10960–10965.
Verdoorn, T. A., Burnashev, N., Monyer, H., Seeburg, P. H., and Sakmann, B. (1991). Structural determinants of ion flow through recombinant glutamate receptor channels. Science 252, 1715–1718.
Verschoor, A., Frank, J., Radermacher, M., Wagenknecht, T., and Boublik, M. (1984). Three-dimensional reconstruction of the 30 S ribosomal subunit from randomly oriented particles. J. Mol. Biol. 178, 677–698.
Wo, Z. G., and Oswald, R. E. (1995). Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 18, 161–168.
Zhang, W., Howe, J. R., and Popescu, G. K. (2008). Distinct gating modes determine the biphasic relaxation of NMDA receptor currents. Nat. Neurosci. 11, 1373–1375.
Appendix

Table A1. Intradimer distances and displacements of TM connection points in the open- and closed-cleft states of the LBD dimer.
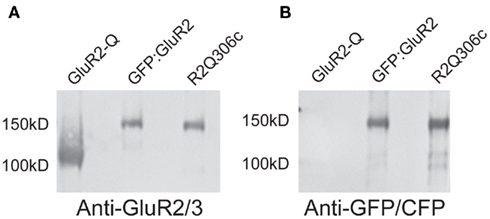
Figure A1. Western blots confirm the identity of GFP:GluA2, R2Q306c, and GluA2-Q receptors. To ensure that the GFP and CFP tags were incorporated purified receptors, GluA2-Q, GFP:GluR, and R2Q306c, were run on gel and blotted with either α-GluA2/3 (A) or α-GFP/CFP (B) antibodies. All receptor constructs interacted with the α-GluA2/3 antibody. Only the tagged receptors reacted with the anti-GFP/CFP antibody, as well as having the expected molecular weight shift demonstrating incorporation of the tags.
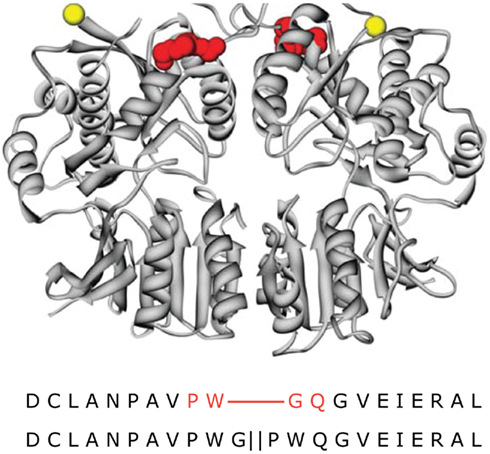
Figure A2. Mapping the CFP domain in the R2Q306c receptor. The site of the insertion of the CFP into the R2Q306c receptor was mapped using the GluA2 NTD structure (PDB entry 3H5W; Jin et al., 2009). The location of the N-terminus of the structure is shown in yellow and the residues surrounding the CFP insertion site are in red, top. Below the NTD structure is the primary sequence of the R2Q306c receptor (bottom) aligned with the GluA2 NTD primary sequence (top). The GluA2 NTD residues flanking the insertion site are shown in red.
Figure A3. Alternative orientations of the NTD dimers within the steric constraints of the ligand-free GluA2-Q molecular envelope. The crystal structure of the GluA2 NTD dimer (PDB entry 3H5W; Jin et al., 2009) is shown in two orientations. The “cooperative” orientation is shown in (A), in which an NTD dimer is oriented parallel to one of the LBD dimers. The “complementary” orientation is shown in (B), in which an NTD dimer connects to both LBD dimers by spanning the central vestibule. Scale bar = 5 nm.
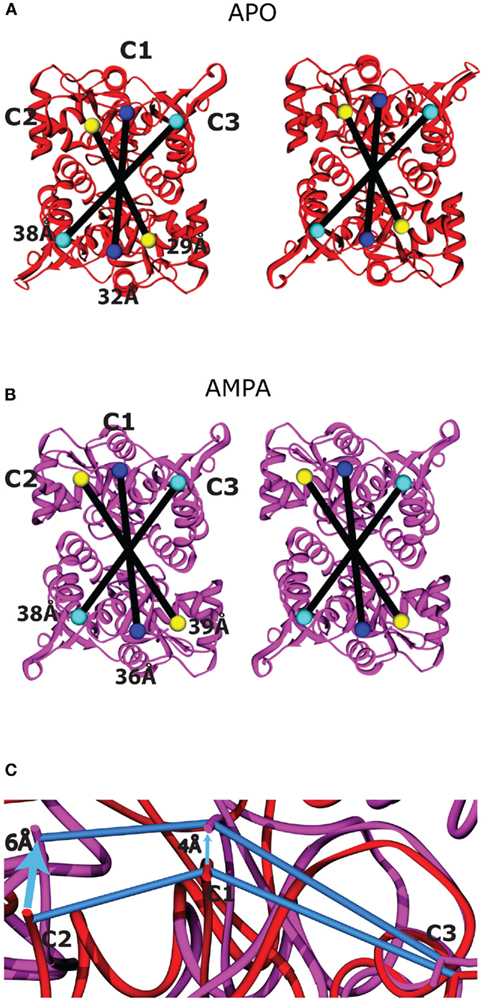
Figure A4. Ligand-binding domain movements between the apo and AMPA-bound structures. The membrane-proximal face of the two LBD dimers is shown as viewed from the cytoplasm following superposition of the apo [(A), red] and AMPA-bound [(B), purple] states, modeled using PDB entries 1FTO and 1FTM, respectively (Armstrong and Gouaux, 2000). The residues on the LBD that connect to the TMD are colored according to the TM helix (TM1–TM3) to which they connect: C1 = Lys 506, blue; C2 = Pro 632, yellow; and C3 = Cys 773, cyan. The connecting residues are twofold symmetrical across the dimer interface and are linearly arranged. The intermonomer distances as measured from the Cα positions of the connecting residues are shown as black lines within each dimer. The C3 intermonomer distance similar in the apo and AMPA-bound structures. (C) To highlight the scissoring motion within each monomer, Lobe I of the apo (red) and AMPA-bound (purple) domains was superimposed, and the displacement of the three attachment points (connected by blue lines in each structure) are highlighted by arrows. The displacement is greatest at C2, intermediate at C1, and minimal at C3, which is closest to the hinge axis.
Keywords: ligand-gated ion channels, receptor activation, ionotropic glutamate receptors, protein structure, electron microscopy, single-particle reconstruction
Citation: Midgett CR, Gill A and Madden DR (2012) Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation. Front. Mol. Neurosci. 4:56. doi: 10.3389/fnmol.2011.00056
Received: 30 August 2011;
Accepted: 10 December 2011;
Published online: 02 January 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USACopyright: © 2012 Midgett, Gill and Madden. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Dean R. Madden, Department of Biochemistry, Dartmouth Medical School, 7200 Vail Building, Hanover, NH 03755, USA. e-mail: drm0001@dartmouth.edu