GSK3 as a sensor determining cell fate in the brain
- Neurosignalling Group, Garvan Institute of Medical Research, Sydney, NSW, Australia
Glycogen synthase kinase 3 (GSK3) is an unusual serine/threonine kinase that controls many neuronal functions, including neurite outgrowth, synapse formation, neurotransmission, and neurogenesis. It mediates these functions by phosphorylating a wide range of substrates involved in gene transcription, metabolism, apoptosis, cytoskeletal dynamics, signal transduction, lipid membrane dynamics, and trafficking, amongst others. This complicated list of diverse substrates generally follow a more simple pattern: substrates negatively regulated by GSK3-mediated phosphorylation favor a proliferative/survival state, while substrates positively regulated by GSK3 favor a more differentiated/functional state. Accordingly, GSK3 activity is higher in differentiated cells than undifferentiated cells and physiological (Wnt, growth factors) and pharmacological inhibitors of GSK3 promote the proliferative capacity of embryonic stem cells. In the brain, the level of GSK3 activity influences neural progenitor cell proliferation/differentiation in neuroplasticity and repair, as well as efficient neurotransmission in differentiated adult neurons. While defects in GSK3 activity are unlikely to be the primary cause of neurodegenerative diseases, therapeutic regulation of its activity to promote a proliferative/survival versus differentiated/mature functional environment in the brain could be a powerful strategy for treatment of neurodegenerative and other mental disorders.
Glycogen Synthase Kinase 3
Glycogen synthase kinase 3 (GSK3) is a Ser/Thr protein kinase that is expressed in all mammalian tissues and subcellular organelles, but particularly highly in the brain. There are two isoforms encoded by separate genes (GSK3α and GSK3β), although their functions are often indistinguishable. GSK3 is critical for normal function of the central nervous system, where it regulates a variety of neuronal functions, including neurotransmission, neurite outgrowth, growth-cone dynamics, cytoskeletal dynamics, synaptic plasticity, endocytosis, apoptosis, and neurogenesis. Interestingly, it is one of the most unusual kinases in the human genome for three main reasons; (1) Most (if not all) substrates require prior phosphorylation by another kinase before they can be efficiently phosphorylated at Ser/Thr residues by GSK3. This process is known as “priming” and occurs four or five residues C-terminal to the GSK3 target site. (2) GSK3 is highly active in cells under basal conditions. This is partly due to constitutive phosphorylation of a conserved tyrosine residue on the activation loop of the kinase domain (Tyr279 in GSK3α, Tyr216 in GSK3β) that is absolutely required for kinase activity (Cole et al., 2004a). (3) Phosphorylation of GSK3 at an N-terminal serine residue inhibits its kinase activity (Ser21 in GSK3α, Ser9 in GSK3β). This phosphoserine acts as a pseudo-substrate and binds to the phosphate-binding pocket on GSK3, preventing interaction with primed substrates (Frame et al., 2001). Phosphorylation at this site is mediated by members of the AGC family of kinases (e.g., Akt) and commonly occurs downstream of insulin, growth factor, and PI3K signaling. Activation of the canonical Wnt signaling pathway also inhibits GSK3 activity, preventing phosphorylation of β-catenin, although this is not mediated by N-terminal phosphorylation, but by protein–protein interactions (Thomas et al., 1999; Ding et al., 2000).
GSK3 and Alzheimer’s Disease
Glycogen synthase kinase 3 is implicated in the development of Alzheimer’s disease (AD), principally because it phosphorylates Tau and increases its propensity to aggregate into neurofibrillary tangles, which together with β-amyloid plaques are a characteristic lesion of the disease. Transgenic mice displaying increased GSK3 activity develop hyperphosphorylated Tau and other neurological defects (Lucas et al., 2001), while treatment of transgenic mice with a GSK3 inhibitor (lithium) reduces the number of tangles in their brains (Noble et al., 2005). A similar microtubule-binding protein called CRMP2 is also hyperphosphorylated by GSK3 in the brains of AD patients (Cole et al., 2007; Williamson et al., 2011). The amyloid precursor protein has been reported to be a GSK3 substrate (Aplin et al., 1996), while Aβ peptide production is reported to be regulated by GSK3 (Sun et al., 2002; Li et al., 2003; Phiel et al., 2003; Ryder et al., 2003; Su et al., 2004; Sereno et al., 2009), although these observations have been recently disputed (Jaworski et al., 2011). Thus, GSK3 has been implicated in many pathologic processes leading to AD. However, it is unlikely that defects in GSK3 per se are a direct cause of AD, since no mutations, polymorphisms, or dramatic biochemical changes have been consistently detected in AD patients, nor any other types of neurodegenerative, developmental, or psychiatric disorders. Instead, a key function of GSK3 is to act as an “environmental sensor,” by relaying signals from extracellular stimuli (e.g., growth factors, insulin, Wnt) to signaling and transcriptional machinery inside the cell to influence cell fate. This implies that pharmacological manipulation of GSK3 in the brain could be used to selectively promote survival, proliferation, differentiation, neurogenesis, or neuroplasticity in diseased brains. This type of therapy could be used to artificially create an environment in the brain that delays/prevents disease development, or promotes neurogenesis and neuroplasticity to compensate for specific insults. Indeed, encouraging data is now emerging showing chronic lithium treatment improves cognitive function in human patients and mouse models of neurodegeneration and ischemic stroke (for a review, see Chiu and Chuang, 2010). Although GSK3 is not the only in vivo target of lithium (e.g., phosphoinositol phosphatases), these effects are consistent with the known actions of GSK3. It remains to be seen what benefits more selective and potent GSK3 inhibitors might provide.
GSK3 Substrates
In order to fully understand the function of GSK3 in the brain, it is essential to characterize its substrates, since this is the primary function of a kinase and it is the substrates that mediate the functional effects directed by GSK3. Ultimately, all physiological substrates of GSK3 should be cataloged and assigned to particular functions regulated by GSK3 (e.g., neurogenesis, neurite outgrowth, neurotransmission, cytoskeletal regulation). This exercise would delineate the mechanisms by which GSK3 maintains healthy brain function. Importantly, it could identify new therapeutic targets downstream of GSK3 that could be exploited for the treatment of mental and neurodegenerative diseases. Theoretically, these could be more specific with less side effects than targeting GSK3, which is a pleiotropic kinase with many different substrates involved in diverse cellular functions.
So far, over 70 substrates have been identified for GSK3, although caution should be taken since many substrates have been reported with various levels of confidence/evidence (for a full review, see Sutherland, 2011). Reported substrates include a number of cytoskeletal, signaling, and DNA-binding proteins. Interestingly, a pattern emerges whereby many substrates that are negatively regulated by GSK3 are involved in proliferation/survival of cells, whereas substrates that are positively regulated by GSK3 are predominantly expressed and function in mature, differentiated cells. Key substrates that contribute to cellular proliferation, differentiation, and survival are listed in Tables 1 and 2 and discussed below.

Table 1. Substrates involved in proliferation/survival that are negatively regulated by GSK3.
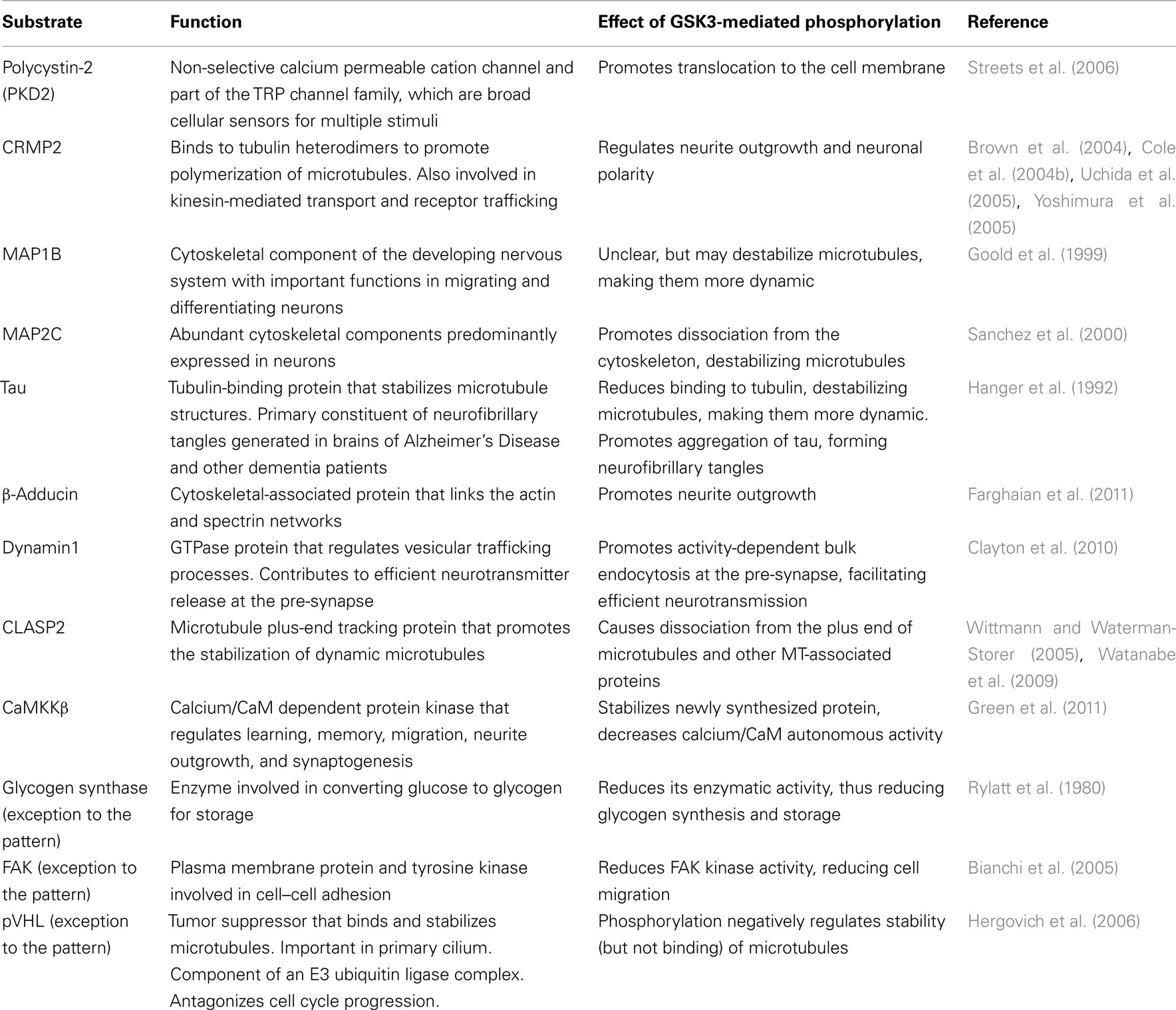
Table 2. Substrates predominantly expressed and functional in mature differentiated cells that are positively regulated by GSK3.
GSK3 and Proliferation
For some time, it has been known that pharmacological inhibition of GSK3 activity maintains the proliferative state of embryonic stem (ES) cells (Sato et al., 2004; Ying et al., 2008). The GSK3 substrates c-myc (Hall et al., 2009) and Klf5 (Jiang et al., 2008) are among several transcription factors that have been used to induce pluripotency (iPS system). GSK3 has also been implicated as a key regulator of adult neurogenesis (generation and incorporation of new neurons into existing circuits of adult brains). Genetic (Eom and Jope, 2009; Kim et al., 2009; Mao et al., 2009) and pharmacological (Sato et al., 2004; Ying et al., 2008; Bone et al., 2009) inhibition of GSK3 activity increases proliferation of neural progenitor cells (NPC’s), but decreases differentiation and incorporation of newborn neurons into the adult brain. Together, these observations demonstrate that low levels of GSK3 activity promote proliferation in ES cells and NPC’s. This correlates with signaling pathways upstream of GSK3 that inhibit GSK3 activity and promote proliferation (e.g., Wnt, growth factors).
Several transcription factors are directly phosphorylated by GSK3 within an [ST]PPx[ST]P or [ST]PxL[ST]P motif. Following priming by another kinase (often a Cdk or MAPK), phosphorylation by GSK3 creates a binding site for E3 ubiquitin ligases that ubiquitinate the protein and target it for proteasome-mediated degradation. Many of these transcription factors have short half-lives, largely due to the actions of GSK3, which is highly active under basal conditions in differentiated cells, including post-mitotic neurons. However, GSK3 activity levels are comparatively lower in ES cells and NPC’s, induced by persistent growth factor and Wnt signaling to maintain the proliferative capacity of these cells (Cartwright et al., 2005). Here, phosphorylation and ubiquitination of transcription factors by GSK3 is reduced, thus stabilizing the proteins (prolonging their half-lives) and contributing to stem/precursor cell proliferation. Such GSK3 targets include well-known proliferative factors, such as c-myc, c-jun, β-catenin, cyclin E1, and Klf5 (Tables 1 and 2; Figure 1). Recent studies suggest that attenuating GSK3-mediated degradation of β-catenin, a key effector of the Wnt signaling pathway, is vital for maintaining ES proliferation and pluripotency (Mao et al., 2009; Kelly et al., 2011; Wray et al., 2011). Interestingly, a viral oncogenic form of c-jun (v-jun) is mutated at the GSK3 target site (Ser239). This prevents phosphorylation by GSK3 and subsequent ubiquitination, thus stabilizing the protein and driving uncontrolled proliferation in tumourigenesis (Wei et al., 2005). Similarly, the GSK3 phosphosite (Thr58) is mutated in the viral oncogenic form of c-myc (v-myc; Pulverer et al., 1994). While it is established that low GSK3 activity levels are required for maintaining the proliferative capacity of ES cells and NPC’s, there are many DNA-binding substrates of GSK3 implicated in this process and their precise roles and relative importance are only beginning to be clarified.
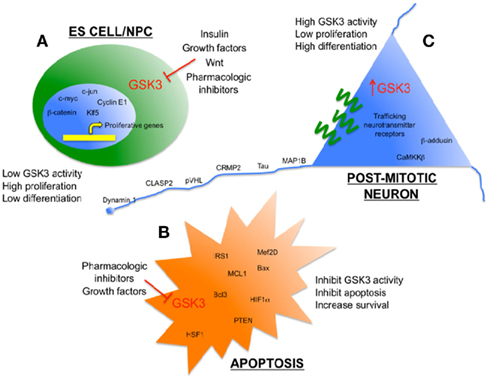
Figure 1. Glycogen synthase kinase 3 as an enzymatic sensor for determining cell fate in the brain. (A) Low levels of GSK3 activity in ES cells/NPC’s are maintained by persistent growth factor/Wnt signaling to promote proliferation. Some pro-proliferation transcription factors that are direct targets of GSK3 are shown. (B) Low levels of GSK3 activity inhibit apoptosis and promote survival when cells are exposed to toxic stimuli, such as hypoxia and amyloid peptides. Some pro-survival substrates of GSK3 are shown. (C) Relatively high levels of GSK3 activity help to promote differentiation and efficient function of mature, post-mitotic neurons, including several cytoskeleton-associated proteins that maintain neuronal morphology and neurotransmission.
GSK3 and Differentiation
Not only does low GSK3 activity promote proliferation, it also prevents differentiation. GSK3α/β double knockout ES cells are severely compromised in their ability to differentiate, largely due to hyperactivation of the Wnt signaling pathway (Doble et al., 2007), while conditional deletion of both isoforms in NPC’s in mice suppressed the generation of post-mitotic neurons (Kim et al., 2009). Also, expression of mutant GSK3 and RNAi-mediated knockdown impairs neuronal polarization in cultured primary neurons (Jiang et al., 2005; Yoshimura et al., 2005; Kim et al., 2009). GSK3 knockin mice expressing GSK3α/β (Ser21/9Ala) that are insensitive to growth factor-induced inhibition exhibited reduced neurogenesis and behavioral defects, despite normal NPC proliferation (Eom and Jope, 2009; Ackermann et al., 2010), suggesting defective differentiation/maturation of NPC’s. In contrast, mice expressing mutant DISC1 (mutated in schizophrenia patients) caused increased GSK3 activity, inhibition of the Wnt signaling pathway, and decreased NPC proliferation (Mao et al., 2009). This suggests that inhibition of GSK3 by the Wnt signaling pathway promotes NPC proliferation, while inhibition of GSK3 by growth factor signaling promotes differentiation of NPC’s into post-mitotic neurons.
Candidate substrates for promoting differentiation include the zinc-finger transcriptional regulator Gli3 (mammalian homolog of Ci155 in the fly), an effector of the hedgehog pathway that is critical for efficient embryo patterning and neural tube formation. In the absence of hedgehog, Gli3 is phosphorylated by GSK3 and CK1 (following priming by PKA), which targets it for ubiquitination and proteolysis, generating a truncated repressor form lacking the C-terminal activation domains (Jia et al., 2002; Price and Kalderon, 2002; Pan et al., 2006; Tempe et al., 2006; Wang and Li, 2006). But in the presence of hedgehog, phosphorylation and processing of Gli3 is inhibited, leading to transactivation by the full-length protein. It has recently been shown that the truncated repressor form of Gli3 is important for differentiation of cortical neurons from neural progenitors, while the full-length active form of Gli3 helps to maintain progenitors in the cell cycle (Wang et al., 2011). This correlates with GSK3 activity, with low GSK3 activity in undifferentiated ES cells increasing the amount of active, full-length Gli3 for maintaining proliferation in ES cells, and high GSK3 activity promoting proteolysis of Gli3 and formation of the truncated repressor form, facilitating differentiation into mature neurons.
Another zinc-finger transcriptional repressor called snail regulates transition of epithelial cells into fibroblast-like mesenchymal cells during development and tumor metastasis, essentially a form of “de-differentiation.” Snail suppresses the expression of E-cadherin, a cell–cell adhesion molecule that is critical for maintaining epithelial cell identity. Relatively high activity of GSK3 in epithelial cells promotes phosphorylation and ubiquitin/proteasome-mediated degradation of snail (Zhou et al., 2004). However, in fibroblast/mesenchymal-like cells of human breast tumors where GSK3 activity is lower, snail is stabilized and suppresses E-cadherin expression (Zhou et al., 2004; Yook et al., 2006). Pharmacological inhibition of GSK3 activity in epithelial cells reduces E-cadherin expression and induces a more-mesenchymal-like morphology via increased snail activity (Bachelder et al., 2005). These observations demonstrate that Snail is an example of a GSK3 substrate controlling cellular differentiation. It does not appear to regulate neuronal differentiation (Murray and Gridley, 2006), although it has been shown to regulate cell fate in glioblastoma cells (Han et al., 2011).
Myocardin is a transcription factor that is required for cardiac and skeletal muscle cell differentiation during development. Again, it is not expressed in neurons or glial cells, but interestingly, it is upregulated in vascular smooth muscle cells in the brains of AD patients, where it enhances accumulation of Aβ in blood vessel walls (Bell et al., 2009). Myocardin is phosphorylated by GSK3, targeting it for ubiquitin, and proteasome-mediated degradation (Badorff et al., 2005; Xie et al., 2009; Hu et al., 2010), however it is not yet clear if upregulation of myocardin levels is due to reduced GSK3-mediated phosphorylation and degradation. In summary, several substrates of GSK3 regulating cell differentiation have been identified, although mostly in non-neural cell types and neuron-specific differentiation factors await identification.
GSK3 and Survival
Glycogen synthase kinase 3 promotes intrinsic apoptotic signaling in neurons, and overexpression of GSK3 is sufficient to induce apoptosis in cultured cells (Pap and Cooper, 1998; Bijur et al., 2000) and in mouse brain (Lucas et al., 2001). Deletion of the GSK3β isoform in mice causes severe liver degeneration during mid-gestation due to excessive tumor necrosis factor-induced apoptosis (Hoeflich et al., 2000). In contrast, numerous studies have demonstrated that genetic or pharmacologic inhibition of GSK3 protects neurons from a wide range of environmental stresses, including hypoxia and amyloid toxicity, which may be relevant for treatment of stroke and AD patients, respectively (for a review, see Mines et al., 2011).
Several GSK3 substrates have been implicated in regulation of apoptosis. Bax is a pro-apoptotic member of the Bcl2 family that oligomerizes at the mitochondrial outer membrane, forming a pore to release cytochrome c, inducing cell death. Phosphorylation of Bax at Ser163 by GSK3 promotes translocation to the mitochondria, whereas inhibition of phosphorylation using lithium reduced Bax translocation and cytochrome c release, thus antagonizing apoptosis (Somervaille et al., 2001; Linseman et al., 2004). VDAC1 is a voltage-dependent anion channel in the mitochondrial outer membrane that also mediates cytochrome c release during apoptosis and is a direct substrate of GSK3 (Pastorino et al., 2005), although the effect of phosphorylation on cytochrome c release from the mitochondria and apoptosis is not yet clear. In contrast, MCL1 is an anti-apoptotic, pro-survival member of the Bcl2 family, and phosphorylation by GSK3 targets it for degradation by the ubiquitin–proteasome-mediated pathway (Maurer et al., 2006). Thus, low GSK3 activity would reduce phosphorylation and degradation of MCL1, favoring cell survival. Several transcription factor substrates of GSK3 have also been implicated in the balance between apoptosis and cell survival by regulating transcription of pro-apoptotic or pro-survival genes, including the pro-survival factors HIF1α, HSF1, Mef2D, and BCL3. GSK3 phosphorylation of each of these substrates targets them for ubiquitin and proteasome-mediated degradation. In summary, many apoptosis-related GSK3 substrates identified so far are pro-survival, and when GSK3 activity is low (e.g., undifferentiated or pharmacologically treated cells), reduced phosphorylation of substrates protects them against ubiquitin and proteasome-mediated degradation, promoting survival of the cell.
GSK3 and Neuronal Morphology
GSK3 is an important regulator of neuronal morphology and synapse formation in mature, post-mitotic neurons. Pharmacologic inhibition of GSK3 activity reduces the rate of axon elongation in hippocampal neurons, increases the size of growth cones (Owen and Gordon-Weeks, 2003), and disturbs polarity, leading to the formation of multiple axon-like processes (Gartner et al., 2006; Garrido et al., 2007). Treatment of cerebellar granule cells with a GSK3 inhibitor increased the number of synapses on mossy fibers (Hall et al., 2000), whereas inactivation of the Drosophila homolog of GSK3, shaggy, promoted synapse formation at neuromuscular junctions by increasing the number of synaptic boutons (Franco et al., 2004). Assuming these interventions were selective, then taken together they demonstrate that GSK3 regulates synapse formation. Accordingly, neurotrophin and growth factor stimuli (e.g., BDNF, NGF, IGF-1) that inhibit GSK3 activity, promote neurite outgrowth, and synapse formation.
Several groups recently demonstrated that the actin-capping protein β-adducin is critical for synapse stability and turnover, underlying learning and memory in flies and mammals (Bednarek and Caroni, 2011; Pielage et al., 2011; Ruediger et al., 2011). Moreover, dynamic disassembly and reassembly of synapses by β-adducin is regulated by phosphorylation at its C-terminal region by PKC. This domain is also targeted by GSK3 following priming by Cdk5 and phosphorylation by these kinases is necessary for β-adducin’s ability to promote neurite outgrowth in cultured primary neurons (Farghaian et al., 2011). Therefore, it will be interesting to see if GSK3-mediated phosphorylation of β-adducin also regulates synapse formation and stability. Several other cytoskeleton-associated proteins are phosphorylated by GSK3 in mature, post-mitotic neurons, in particular the tubulin-binding proteins Tau, MAP1B, MAP2, CRMP2, CLASP2, and pVHL. Phosphorylation of these substrates causes their dissociation from tubulin, destabilizing the microtubule structure. In post-mitotic neurons where GSK3 activity levels are relatively high, this would reduce interactions between GSK3 substrates and tubulin, resulting in destabilization of microtubules. Accordingly, non-phospho-mutant forms of substrates (e.g., CRMP2, MAP1B) increase the stability of microtubules, causing impaired neurite outgrowth, and polarity in cultured primary neurons (Cole et al., 2004b; Trivedi et al., 2005; Yoshimura et al., 2005). In theory, high levels of phosphorylation of these substrates would promote their dissociation from microtubules, favoring dynamic remodeling of the cytoskeleton, and enhancing neuroplasticity, although this is yet to be proven in vivo.
GSK3 and Neurotransmission
A systematic screen of Ser/Thr kinases using a panel of pharmacological inhibitors revealed that GSK3 was the only kinase among 58 Ser/Thr kinases that was required for induction of NMDA-induced long-term depression (LTD) in hippocampal CA1 pyramidal neurons (Peineau et al., 2009). LTD increases GSK3 activity via decreased phosphorylation of Ser21/9 at its N-terminus, while NMDA-induced long-term potentiation (LTP) reduces GSK3 activity by increasing Ser21/9 phosphorylation (Hooper et al., 2007; Peineau et al., 2007). Meanwhile, GSK3 inhibitors do not affect baseline synaptic transmission (Peineau et al., 2007; Zhu et al., 2007; Li et al., 2009). GSK3 regulates transmission at both the pre- and post-synapse. For example, high GSK3 activity reduces glutamate release from the pre-synapse, inhibiting LTP (Hooper et al., 2007; Zhu et al., 2007, 2010), while retrieval of synaptic vesicles at the pre-synapse by endocytosis requires GSK3 (Clayton et al., 2010). Dynamin 1 is a large GTPase that regulates vesicle endocytosis at the pre-synapse. Phosphorylation by GSK3 at Ser774 is required for re-uptake of neurotransmitters during times of elevated neuronal activity (Clayton et al., 2010). Thus, relatively high GSK3 activity in differentiated neurons would be expected to activate Dynamin 1 and facilitate efficient recycling of neurotransmitters at the synapse. At the post-synapse, pharmacological inhibition of GSK3 decreases surface expression of NMDA and AMPA receptors (Chen et al., 2007; Wei et al., 2010). CRMP2 is a GSK3 substrate that has been implicated in trafficking of transmembrane proteins to the cell surface (Nishimura et al., 2003; Brittain et al., 2011), although the effect that phosphorylation has on this process has not yet been determined. Together, these observations demonstrate a clear requirement for GSK3 at the synapse, although the synaptic substrates that mediate these effects remain to be fully uncovered.
Conclusion
When analyzing the substrates of GSK3, a pattern emerges whereby those that are negatively regulated by GSK3 are commonly involved in promoting proliferation and/or survival, while substrates that are positively regulated by phosphorylation are predominantly expressed in differentiated post-mitotic neurons and are required for efficient function of mature neurons. The former substrates include pro-proliferation transcription factors or pro-survival proteins targeted for ubiquitin-mediated degradation by GSK3, while the latter are often cytoskeleton-associated proteins. Thus, low GSK3 activity levels are conducive to proliferative ES cells and NPC’s, while higher GSK3 activity is required for efficient function of differentiated neurons. This pattern implies that pharmacologic manipulation of GSK3 activity can be used to influence cell fate between proliferative/undifferentiated and mature/differentiated states, as has already been successfully demonstrated for ES cells. In the brain, inhibition of GSK3 would promote proliferation of NPC’s, while high levels of GSK3 would promote neuronal differentiation and efficient function of post-mitotic neurons. Also, it is possible that high GSK3 activity in post-mitotic neurons could promote neuroplasticity, learning, and memory via increased dynamics of the cytoskeleton. Manipulation of GSK3 activity may be of great therapeutic benefit for neurodegenerative and other mental disorders. In AD, the use of pharmacologic inhibitors of GSK3 has been proposed to decrease phosphorylation of Tau, reducing its aggregation and formation of neurofibrillary tangles. This strategy has shown some success in mouse models of AD (Perez et al., 2003; Nakashima et al., 2005; Noble et al., 2005; Leroy et al., 2010). In elderly humans and AD patients, chronic (but not acute) treatment with GSK3 inhibitors reduced decline in cognitive and memory function (Nunes et al., 2007; Chiu and Chuang, 2010; Kessing et al., 2010; Forlenza et al., 2011). These studies have been performed using lithium, a relatively weak and non-specific inhibitor of GSK3, so it is necessary to advance these studies using more potent and specific inhibitors of GSK3.
Another exciting potential therapeutic use of GSK3 inhibitors in the clinic is to maintain neuron survival under stressful conditions, including neurodegenerative diseases and acute injuries, such as stroke. Since GSK3 inhibitors are such effective inhibitors of neuronal apoptosis (at least in vitro), rapid administration of these drugs could help to prevent neuronal loss during the immediate period following injury. By keeping these neurons alive, one might expect an improved prognosis for functional recovery. It might also promote proliferation of NPC’s that could later be induced to differentiate into functional post-mitotic neurons to compensate for damages incurred at the site of injury. So far, several groups have elegantly demonstrated that lithium treatment effectively protects neurons and even promotes migration of stem cells to affected regions (Chiu and Chuang, 2010; Tsai et al., 2011).
Of course, there is the danger that inhibition of GSK3 activity could impede the basic function of post-mitotic neurons. However, it should be remembered that very few drugs inhibit kinases 100%, therefore any treatments are likely to reduce GSK3 activity, not completely inhibit it. Also, GSK3 substrates that are relatively resistant to phosphatases are beginning to be discovered [e.g., β-adducin (Farghaian et al., 2011), CRMP2 (Cole et al., 2008)] and moderate reduction of GSK3 activity is unlikely to affect the stoichiometry of phosphorylation of these substrates. This provides another good reason for identifying and characterizing each individual substrate of GSK3 in the brain. Importantly, downstream targets of GSK3 that are specifically involved in a particular neuronal process (e.g., neurogenesis, neurotransmission) may prove to be better therapeutic targets than GSK3, being more potent and selective with fewer side effects. Therefore, the full catalog of GSK3 substrates and their physiological functions needs to be completed.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thank you to Dr. Calum Sutherland and Dr. Ritchie Williamson, University of Dundee, Scotland for advice, and comments on this review. Adam R. Cole is supported by an Australian NHMRC Peter Doherty Fellowship (#454886).
Abbreviations
AD, Alzheimer’s disease; ES, embryonic stem (cell); GSK3, glycogen synthase kinase 3; NPC, neural progenitor cell.
References
Ackermann, T. F., Kempe, D. S., Lang, F., and Lang, U. E. (2010). Hyperactivity and enhanced curiosity of mice expressing PKB/SGK-resistant glycogen synthase kinase-3 (GSK-3). Cell. Physiol. Biochem. 25, 775–786.
Al-Khouri, A. M., Ma, Y., Togo, S. H., Williams, S., and Mustelin, T. (2005). Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J. Biol. Chem. 280, 35195–35202.
Aplin, A. E., Gibb, G. M., Jacobsen, J. S., Gallo, J. M., and Anderton, B. H. (1996). In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J. Neurochem. 67, 699–707.
Bachelder, R. E., Yoon, S. O., Franci, C., De Herreros, A. G., and Mercurio, A. M. (2005). Glycogen synthase kinase-3 is an endogenous inhibitor of snail transcription: implications for the epithelial-mesenchymal transition. J. Cell Biol. 168, 29–33.
Badorff, C., Seeger, F. H., Zeiher, A. M., and Dimmeler, S. (2005). Glycogen synthase kinase 3beta inhibits myocardin-dependent transcription and hypertrophy induction through site-specific phosphorylation. Circ. Res. 97, 645–654.
Bednarek, E., and Caroni, P. (2011). beta-Adducin is required for stable assembly of new synapses and improved memory upon environmental enrichment. Neuron 69, 1132–1146.
Bell, R. D., Deane, R., Chow, N., Long, X., Sagare, A., Singh, I., Streb, J. W., Guo, H., Rubio, A., Van Nostrand, W., Miano, J. M., and Zlokovic, B. V. (2009). SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 11, 143–153.
Bianchi, M., De Lucchini, S., Marin, O., Turner, D. L., Hanks, S. K., and Villa-Moruzzi, E. (2005). Regulation of FAK Ser-722 phosphorylation and kinase activity by GSK3 and PP1 during cell spreading and migration. Biochem. J. 391, 359–370.
Bijur, G. N., De Sarno, P., and Jope, R. S. (2000). Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J. Biol. Chem. 275, 7583–7590.
Bone, H. K., Damiano, T., Bartlett, S., Perry, A., Letchford, J., Ripoll, Y. S., Nelson, A. S., and Welham, M. J. (2009). Involvement of GSK-3 in regulation of murine embryonic stem cell self-renewal revealed by a series of bisindolylmaleimides. Chem. Biol. 16, 15–27.
Brittain, J. M., Duarte, D. B., Wilson, S. M., Zhu, W., Ballard, C., Johnson, P. L., Liu, N., Xiong, W., Ripsch, M. S., Wang, Y., Fehrenbacher, J. C., Fitz, S. D., Khanna, M., Park, C. K., Schmutzler, B. S., Cheon, B. M., Due, M. R., Brustovetsky, T., Ashpole, N. M., Hudmon, A., Meroueh, S. O., Hingtgen, C. M., Brustovetsky, N., Ji, R. R., Hurley, J. H., Jin, X., Shekhar, A., Xu, X. M., Oxford, G. S., Vasko, M. R., White, F. A., and Khanna, R. (2011). Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2) channel complex. Nat. Med. 17, 822–829.
Brown, M., Jacobs, T., Eickholt, B., Ferrari, G., Teo, M., Monfries, C., Qi, R. Z., Leung, T., Lim, L., and Hall, C. (2004). Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. 24, 8994–9004.
Cartwright, P., Mclean, C., Sheppard, A., Rivett, D., Jones, K., and Dalton, S. (2005). LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885–896.
Chen, P., Gu, Z., Liu, W., and Yan, Z. (2007). Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51.
Chen, Y., Yue, S., Xie, L., Pu, X. H., Jin, T., and Cheng, S. Y. (2011). Dual phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. J. Biol. Chem. 286, 13502–13511.
Chiu, C. T., and Chuang, D. M. (2010). Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol. Ther. 128, 281–304.
Chu, B., Zhong, R., Soncin, F., Stevenson, M. A., and Calderwood, S. K. (1998). Transcriptional activity of heat shock factor 1 at 37 degrees C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Calpha and Czeta. J. Biol. Chem. 273, 18640–18646.
Clayton, E. L., Sue, N., Smillie, K. J., O’Leary, T., Bache, N., Cheung, G., Cole, A. R., Wyllie, D. J., Sutherland, C., Robinson, P. J., and Cousin, M. A. (2010). Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat. Neurosci. 13, 845–851.
Cole, A., Frame, S., and Cohen, P. (2004a). Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377, 249–255.
Cole, A. R., Knebel, A., Morrice, N. A., Robertson, L. A., Irving, A. J., Connolly, C. N., and Sutherland, C. (2004b). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180.
Cole, A. R., Noble, W., Van Aalten, L., Plattner, F., Meimaridou, R., Hogan, D., Taylor, M., Lafrancois, J., Gunn-Moore, F., Verkhratsky, A., Oddo, S., Laferla, F., Giese, K. P., Dineley, K. T., Duff, K., Richardson, J. C., Yan, S. D., Hanger, D. P., Allan, S. M., and Sutherland, C. (2007). Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 103, 1132–1144.
Cole, A. R., Soutar, M. P., Rembutsu, M., Van Aalten, L., Hastie, C. J., Mclauchlan, H., Peggie, M., Balastik, M., Lu, K. P., and Sutherland, C. (2008). Relative resistance of Cdk5-phosphorylated CRMP2 to dephosphorylation. J. Biol. Chem. 283, 18227–18237.
Ding, V. W., Chen, R. H., and Mccormick, F. (2000). Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J. Biol. Chem. 275, 32475–32481.
Doble, B. W., Patel, S., Wood, G. A., Kockeritz, L. K., and Woodgett, J. R. (2007). Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 12, 957–971.
Eldar-Finkelman, H., and Krebs, E. G. (1997). Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. U.S.A. 94, 9660–9664.
Eom, T. Y., and Jope, R. S. (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 66, 494–502.
Farghaian, H., Turnley, A. M., Sutherland, C., and Cole, A. R. (2011). Bioinformatic prediction and confirmation of beta-adducin as a novel substrate of glycogen synthase kinase 3. J. Biol. Chem. 286, 25274–25283.
Forlenza, O. V., Diniz, B. S., Radanovic, M., Santos, F. S., Talib, L. L., and Gattaz, W. F. (2011). Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br. J. Psychiatry 198, 351–356.
Frame, S., Cohen, P., and Biondi, R. M. (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7, 1321–1327.
Franco, B., Bogdanik, L., Bobinnec, Y., Debec, A., Bockaert, J., Parmentier, M. L., and Grau, Y. (2004). Shaggy, the homolog of glycogen synthase kinase 3, controls neuromuscular junction growth in Drosophila. J. Neurosci. 24, 6573–6577.
Fuentealba, L. C., Eivers, E., Ikeda, A., Hurtado, C., Kuroda, H., Pera, E. M., and De Robertis, E. M. (2007). Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993.
Garrido, J. J., Simon, D., Varea, O., and Wandosell, F. (2007). GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 581, 1579–1586.
Gartner, A., Huang, X., and Hall, A. (2006). Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3beta) independently of Akt/PKB serine phosphorylation. J. Cell. Sci. 119, 3927–3934.
Goold, R. G., Owen, R., and Gordon-Weeks, P. R. (1999). Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell. Sci. 112(Pt 19), 3373–3384.
Green, M. F., Scott, J. W., Steel, R., Oakhill, J. S., Kemp, B. E., and Means, A. R. (2011). Ca2+/calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J. Biol. Chem. 286, 28066–28079.
Hall, A. C., Lucas, F. R., and Salinas, P. C. (2000). Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 100, 525–535.
Hall, J., Guo, G., Wray, J., Eyres, I., Nichols, J., Grotewold, L., Morfopoulou, S., Humphreys, P., Mansfield, W., Walker, R., Tomlinson, S., and Smith, A. (2009). Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 5, 597–609.
Han, S. P., Kim, J. H., Han, M. E., Sim, H. E., Kim, K. S., Yoon, S., Baek, S. Y., Kim, B. S., and Oh, S. O. (2011). SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell. Mol. Neurobiol. 31, 489–496.
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62.
Hergovich, A., Lisztwan, J., Thoma, C. R., Wirbelauer, C., Barry, R. E., and Krek, W. (2006). Priming-dependent phosphorylation and regulation of the tumor suppressor pVHL by glycogen synthase kinase 3. Mol. Cell. Biol. 26, 5784–5796.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Hu, G., Wang, X., Saunders, D. N., Henderson, M., Russell, A. J., Herring, B. P., and Zhou, J. (2010). Modulation of myocardin function by the ubiquitin E3 ligase UBR5. J. Biol. Chem. 285, 11800–11809.
Ikeda, S., Kishida, S., Yamamoto, H., Murai, H., Koyama, S., and Kikuchi, A. (1998). Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384.
Jaworski, T., Dewachter, I., Lechat, B., Gees, M., Kremer, A., Demedts, D., Borghgraef, P., Devijver, H., Kugler, S., Patel, S., Woodgett, J. R., and Van Leuven, F. (2011). GSK-3α/β kinases and amyloid production in vivo. Nature 480, E4–E5.
Jia, J., Amanai, K., Wang, G., Tang, J., Wang, B., and Jiang, J. (2002). Shaggy/GSK3 antagonizes Hedgehog signalling by regulating cubitus interruptus. Nature 416, 548–552.
Jiang, H., Guo, W., Liang, X., and Rao, Y. (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135.
Jiang, J., Chan, Y. S., Loh, Y. H., Cai, J., Tong, G. Q., Lim, C. A., Robson, P., Zhong, S., and Ng, H. H. (2008). A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 10, 353–360.
Kelly, K. F., Ng, D. Y., Jayakumaran, G., Wood, G. A., Koide, H., and Doble, B. W. (2011). beta-Catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 8, 214–227.
Kessing, L. V., Forman, J. L., and Andersen, P. K. (2010). Does lithium protect against dementia? Bipolar Disord. 12, 87–94.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Leroy, K., Ando, K., Heraud, C., Yilmaz, Z., Authelet, M., Boeynaems, J. M., Buee, L., De Decker, R., and Brion, J. P. (2010). Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J. Alzheimers Dis. 19, 705–719.
Li, B., Ryder, J., Su, Y., Zhou, Y., Liu, F., and Ni, B. (2003). FRAT1 peptide decreases Abeta production in swAPP(751) cells. FEBS Lett. 553, 347–350.
Li, Y. C., Xi, D., Roman, J., Huang, Y. Q., and Gao, W. J. (2009). Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563.
Liberman, Z., and Eldar-Finkelman, H. (2005). Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 280, 4422–4428.
Linseman, D. A., Butts, B. D., Precht, T. A., Phelps, R. A., Le, S. S., Laessig, T. A., Bouchard, R. J., Florez-Mcclure, M. L., and Heidenreich, K. A. (2004). Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 24, 9993–10002.
Litovchick, L., Chestukhin, A., and Decaprio, J. A. (2004). Glycogen synthase kinase 3 phosphorylates RBL2/p130 during quiescence. Mol. Cell. Biol. 24, 8970–8980.
Liu, N., Li, H., Li, S., Shen, M., Xiao, N., Chen, Y., Wang, Y., Wang, W., Wang, R., Wang, Q., Sun, J., and Wang, P. (2010). The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J. Biol. Chem. 285, 18858–18867.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
Maccario, H., Perera, N. M., Davidson, L., Downes, C. P., and Leslie, N. R. (2007). PTEN is destabilized by phosphorylation on Thr366. Biochem. J. 405, 439–444.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Maurer, U., Charvet, C., Wagman, A. S., Dejardin, E., and Green, D. R. (2006). Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760.
Mines, M. A., Beurel, E., and Jope, R. S. (2011). Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int. J. Alzheimers Dis. 2011, 861072.
Moberg, K. H., Mukherjee, A., Veraksa, A., Artavanis-Tsakonas, S., and Hariharan, I. K. (2004). The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr. Biol. 14, 965–974.
Mottet, D., Dumont, V., Deccache, Y., Demazy, C., Ninane, N., Raes, M., and Michiels, C. (2003). Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 278, 31277–31285.
Murray, J. T., Campbell, D. G., Morrice, N., Auld, G. C., Shpiro, N., Marquez, R., Peggie, M., Bain, J., Bloomberg, G. B., Grahammer, F., Lang, F., Wulff, P., Kuhl, D., and Cohen, P. (2004). Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488.
Murray, S. A., and Gridley, T. (2006). Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 10300–10304.
Nakashima, H., Ishihara, T., Suguimoto, P., Yokota, O., Oshima, E., Kugo, A., Terada, S., Hamamura, T., Trojanowski, J. Q., Lee, V. M., and Kuroda, S. (2005). Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556.
Nishimura, T., Fukata, Y., Kato, K., Yamaguchi, T., Matsuura, Y., Kamiguchi, H., and Kaibuchi, K. (2003). CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat. Cell Biol. 5, 819–826.
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., Lafrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., and Duff, K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995.
Nunes, P. V., Forlenza, O. V., and Gattaz, W. F. (2007). Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br. J. Psychiatry 190, 359–360.
Owen, R., and Gordon-Weeks, P. R. (2003). Inhibition of glycogen synthase kinase 3beta in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol. Cell. Neurosci. 23, 626–637.
Pan, Y., Bai, C. B., Joyner, A. L., and Wang, B. (2006). Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377.
Pap, M., and Cooper, G. M. (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932.
Pastorino, J. G., Hoek, J. B., and Shulga, N. (2005). Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554.
Peineau, S., Nicolas, C. S., Bortolotto, Z. A., Bhat, R. V., Ryves, W. J., Harwood, A. J., Dournaud, P., Fitzjohn, S. M., and Collingridge, G. L. (2009). A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol Brain 2, 22.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Perez, M., Hernandez, F., Lim, F., Diaz-Nido, J., and Avila, J. (2003). Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimers Dis. 5, 301–308.
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439.
Pielage, J., Bulat, V., Zuchero, J. B., Fetter, R. D., and Davis, G. W. (2011). Hts/Adducin controls synaptic elaboration and elimination. Neuron 69, 1114–1131.
Price, M. A., and Kalderon, D. (2002). Proteolysis of the Hedgehog signaling effector cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108, 823–835.
Pulverer, B. J., Fisher, C., Vousden, K., Littlewood, T., Evan, G., and Woodgett, J. R. (1994). Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 9, 59–70.
Ruediger, S., Vittori, C., Bednarek, E., Genoud, C., Strata, P., Sacchetti, B., and Caroni, P. (2011). Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature 473, 514–518.
Ryder, J., Su, Y., Liu, F., Li, B., Zhou, Y., and Ni, B. (2003). Divergent roles of GSK3 and CDK5 in APP processing. Biochem. Biophys. Res. Commun. 312, 922–929.
Rylatt, D. B., Aitken, A., Bilham, T., Condon, G. D., Embi, N., and Cohen, P. (1980). Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur. J. Biochem. 107, 529–537.
Sanchez, C., Perez, M., and Avila, J. (2000). GSK3beta-mediated phosphorylation of the microtubule-associated protein 2C (MAP2C) prevents microtubule bundling. Eur. J. Cell Biol. 79, 252–260.
Sapkota, G., Alarcon, C., Spagnoli, F. M., Brivanlou, A. H., and Massague, J. (2007). Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441–454.
Sato, N., Meijer, L., Skaltsounis, L., Greengard, P., and Brivanlou, A. H. (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10, 55–63.
Sereno, L., Coma, M., Rodriguez, M., Sanchez-Ferrer, P., Sanchez, M. B., Gich, I., Agullo, J. M., Perez, M., Avila, J., Guardia-Laguarta, C., Clarimon, J., Lleo, A., and Gomez-Isla, T. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367.
Somervaille, T. C., Linch, D. C., and Khwaja, A. (2001). Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood 98, 1374–1381.
Streets, A. J., Moon, D. J., Kane, M. E., Obara, T., and Ong, A. C. (2006). Identification of an N-terminal glycogen synthase kinase 3 phosphorylation site which regulates the functional localization of polycystin-2 in vivo and in vitro. Hum. Mol. Genet. 15, 1465–1473.
Su, Y., Ryder, J., Li, B., Wu, X., Fox, N., Solenberg, P., Brune, K., Paul, S., Zhou, Y., Liu, F., and Ni, B. (2004). Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 43, 6899–6908.
Sun, X., Sato, S., Murayama, O., Murayama, M., Park, J. M., Yamaguchi, H., and Takashima, A. (2002). Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 321, 61–64.
Sutherland, C. (2011). What are the bona fide GSK3 substrates? Int. J. Alzheimers Dis. 2011, 505607.
Tempe, D., Casas, M., Karaz, S., Blanchet-Tournier, M. F., and Concordet, J. P. (2006). Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol. Cell. Biol. 26, 4316–4326.
Thomas, G. M., Frame, S., Goedert, M., Nathke, I., Polakis, P., and Cohen, P. (1999). A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS Lett. 458, 247–251.
Trivedi, N., Marsh, P., Goold, R. G., Wood-Kaczmar, A., and Gordon-Weeks, P. R. (2005). Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell. Sci. 118, 993–1005.
Tsai, L. K., Wang, Z., Munasinghe, J., Leng, Y., Leeds, P., and Chuang, D. M. (2011). Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 42, 2932–2939.
Uchida, Y., Ohshima, T., Sasaki, Y., Suzuki, H., Yanai, S., Yamashita, N., Nakamura, F., Takei, K., Ihara, Y., Mikoshiba, K., Kolattukudy, P., Honnorat, J., and Goshima, Y. (2005). Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 10, 165–179.
Viatour, P., Dejardin, E., Warnier, M., Lair, F., Claudio, E., Bureau, F., Marine, J. C., Merville, M. P., Maurer, U., Green, D., Piette, J., Siebenlist, U., Bours, V., and Chariot, A. (2004). GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol. Cell 16, 35–45.
Wang, B., and Li, Y. (2006). Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. U.S.A. 103, 33–38.
Wang, H., Ge, G., Uchida, Y., Luu, B., and Ahn, S. (2011). Gli3 is required for maintenance and fate specification of cortical progenitors. J. Neurosci. 31, 6440–6448.
Wang, X., She, H., and Mao, Z. (2009). Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3beta in neuronal apoptosis. J. Biol. Chem. 284, 32619–32626.
Watanabe, T., Noritake, J., Kakeno, M., Matsui, T., Harada, T., Wang, S., Itoh, N., Sato, K., Matsuzawa, K., Iwamatsu, A., Galjart, N., and Kaibuchi, K. (2009). Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J. Cell. Sci. 122, 2969–2979.
Wei, J., Liu, W., and Yan, Z. (2010). Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 285, 26369–26376.
Wei, W., Jin, J., Schlisio, S., Harper, J. W., and Kaelin, W. G. Jr. (2005). The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8, 25–33.
Welcker, M., Orian, A., Jin, J., Grim, J. E., Harper, J. W., Eisenman, R. N., and Clurman, B. E. (2004). The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9085–9090.
Welcker, M., Singer, J., Loeb, K. R., Grim, J., Bloecher, A., Gurien-West, M., Clurman, B. E., and Roberts, J. M. (2003). Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 12, 381–392.
Welsh, G. I., and Proud, C. G. (1993). Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J. 294 (Pt 3), 625–629.
Williamson, R., Van Aalten, L., Mann, D. M., Platt, B., Plattner, F., Bedford, L., Mayer, J., Howlett, D., Usardi, A., Sutherland, C., and Cole, A. R. (2011). CRMP2 hyperphosphorylation is characteristic of Alzheimer’s disease and not a feature common to other neurodegenerative diseases. J. Alzheimers Dis. 27, 615–625.
Wittmann, T., and Waterman-Storer, C. M. (2005). Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J. Cell Biol. 169, 929–939.
Wray, J., Kalkan, T., Gomez-Lopez, S., Eckardt, D., Cook, A., Kemler, R., and Smith, A. (2011). Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat. Cell Biol. 13, 838–845.
Xie, P., Fan, Y., Zhang, H., Zhang, Y., She, M., Gu, D., Patterson, C., and Li, H. (2009). CHIP represses myocardin-induced smooth muscle cell differentiation via ubiquitin-mediated proteasomal degradation. Mol. Cell. Biol. 29, 2398–2408.
Yada, M., Hatakeyama, S., Kamura, T., Nishiyama, M., Tsunematsu, R., Imaki, H., Ishida, N., Okumura, F., Nakayama, K., and Nakayama, K. I. (2004). Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125.
Ying, Q. L., Wray, J., Nichols, J., Batlle-Morera, L., Doble, B., Woodgett, J., Cohen, P., and Smith, A. (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523.
Yook, J. I., Li, X. Y., Ota, I., Hu, C., Kim, H. S., Kim, N. H., Cha, S. Y., Ryu, J. K., Choi, Y. J., Kim, J., Fearon, E. R., and Weiss, S. J. (2006). A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 8, 1398–1406.
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A., and Kaibuchi, K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149.
Zhao, D., Zheng, H. Q., Zhou, Z., and Chen, C. (2010). The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer Res. 70, 4728–4738.
Zhou, B. P., Deng, J., Xia, W., Xu, J., Li, Y. M., Gunduz, M., and Hung, M. C. (2004). Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940.
Zhu, L. Q., Liu, D., Hu, J., Cheng, J., Wang, S. H., Wang, Q., Wang, F., Chen, J. G., and Wang, J. Z. (2010). GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J. Neurosci. 30, 3624–3633.
Keywords: GSK3, phosphorylation, kinase, substrate, proliferation, differentiation, neural progenitor, Alzheimer’s disease
Citation: Cole AR (2012) GSK3 as a sensor determining cell fate in the brain. Front. Mol. Neurosci. 5:4. doi: 10.3389/fnmol.2012.00004
Received: 03 November 2011;
Accepted: 10 January 2012;
Published online: 09 February 2012.
Edited by:
Jean-Martin Beaulieu, Université Laval, CanadaReviewed by:
Jesus Avila, Centro de Biología Molecular Severo Ochoa CSIC–UAM, SpainPeter S. Klein, University of Pennsylvania, USA
Copyright: © 2012 Cole. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Adam R. Cole, Neurosignalling Group, Garvan Institute of Medical Research, 384 Victoria Street, Darlinghurst, Sydney, NSW 2010, Australia. e-mail: a.cole@garvan.org.au