The cytokine temporal profile in rat cortex after controlled cortical impact
- 1Department of Anatomy, Physiology and Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 2Department of Neurology, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
Cerebral inflammatory responses may initiate secondary cascades following traumatic brain injury (TBI). Changes in the expression of both cytokines and chemokines may activate, regulate, and recruit innate and adaptive immune cells associated with secondary degeneration, as well as alter a host of other cellular processes. In this study, we quantified the temporal expression of a large set of inflammatory mediators in rat cortical tissue after brain injury. Following a controlled cortical impact (CCI) on young adult male rats, cortical and hippocampal tissue of the injured hemisphere and matching contralateral material was harvested at early (4, 12, and 24 hours) and extended (3 and 7 days) time points post-procedure. Naïve rats that received only anesthesia were used as controls. Processed brain homogenates were assayed for chemokine and cytokine levels utilizing an electrochemiluminescence-based multiplex ELISA platform. The temporal profile of cortical tissue samples revealed a multi-phasic injury response following brain injury. CXCL1, IFN-γ, TNF-α levels significantly peaked at four hours post-injury compared to levels found in naïve or contralateral tissue. CXCL1, IFN-γ, and TNF-α levels were then observed to decrease at least 3-fold by 12 hours post-injury. IL-1β, IL-4, and IL-13 levels were also significantly elevated at four hours post-injury although their expression did not decrease more than 3-fold for up to 24 hours post-injury. Additionally, IL-1β and IL-4 levels displayed a biphasic temporal profile in response to injury, which may suggest their involvement in adaptive immune responses. Interestingly, peak levels of CCL2 and CCL20 were not observed until after four hours post-injury. CCL2 levels in injured cortical tissue were significantly higher than peak levels of any other inflammatory mediator measured, thus suggesting a possible use as a biomarker. Fully elucidating chemokine and cytokine signaling properties after brain injury may provide increased insight into a number of secondary cascade events that are initiated or regulated by inflammatory responses.
Introduction
Traumatic brain injury (TBI) costs the healthcare industry over $6 billion annually in the US, and is a leading cause of death and disability (Finkelstein et al., 2006). Both the primary immediate insult, and the delayed secondary injury cascade of biochemical and metabolic pathological events, cause focal and global neurological dysfunction associated with TBI. The onset of secondary injuries, which include impaired aerobic metabolism, altered calcium homeostasis, disrupted amino acid metabolism, and activated inflammatory responses can be delayed for minutes to hours and persist for weeks to months, resulting in damage far worse than the initial injury (He et al., 2004; Alves et al., 2005; Bartnik et al., 2007; Faria et al., 2007; Deshpande et al., 2008; Lloyd et al., 2008; Sun et al., 2008; Scafidi et al., 2009; Wei et al., 2009; Xing et al., 2009; Cole et al., 2010). Therefore, major research effort has been invested in therapeutic interventions during secondary events (Arcure and Harrison, 2009; Xiong et al., 2009; Adeleye et al., 2010; Ziebell and Morganti-Kossmann, 2010). However, the underlying cause(s) of these complex post-traumatic effects has yet to be completely determined.
Post-traumatic inflammatory responses, including increased cytokine expression and microglial activation, have received increasing attention as both biomarkers and potential drivers of post-TBI. Several investigators described the spatio-temporal accumulation of cytokines, and suggested that the post-traumatic inflammatory response may be a key mediator of the secondary effects. Most recently, using a fluid percussion injury model in rats Mukherjee et al. (2011) reported a comparison of two techniques for quantifying cytokine expression. When quantified 24 hours after injury, seven of the 23 tested cytokines demonstrated significantly elevated concentrations in the ipsilateral cortex (Mukherjee et al., 2011). While their study focused primarily on the comparison of a Bio-Plex assay versus a new MAGPIX assay, these results do support the findings of several other studies. Notably, Harting et al. (2008) described significant increases in the expression of several pro-inflammatory cytokines after a brain injury in rats (Harting et al., 2008). Of the parameters tested, including interleukin-1 alpha (IL-1α), interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-α), all returned to baseline levels within 48 hours of injury. Other researchers have also described a rapid response of cytokines to injury. Notably, interleukin-1 (IL-1) is released within 60 minutes of injury and peaks between three hours and two days later (Fassbender et al., 2000; Kinoshita et al., 2002). Interestingly, the increased expression of IL-1 protein precedes an increase in mRNA expression in injured tissue. This suggests that increases in specific cytokines in the brain may not be exclusively generated locally, but may be part of a systemic response delivering cytokines to the effected regions. Other laboratories have reported a more heterotypic response in brain tissue, with increased IL-1 and IL-6 not being observed until six hours after injury, but with elevations in TNF-α within one hour of injury (Tehranian et al., 2002). In yet another study, interleukin-18 (IL-18) concentrations in brain tissue homogenates reached significantly higher levels seven days post-trauma (Yatsiv et al., 2002). Karaoglan et al., (2011) reported the interesting result that increasing the severity of a controlled cortical impact (CCI) injury resulted in only a minor increase in IL-1β, IL-6, and TNF-α (Karaoglan et al., 2011). However, these data were collected 1, 3, and 5 days after injury. Using human brain microdialysates, Helmy et al. (2009, 2011) provides convincing evidence of the significant contribution of locally generated cytokines, while suggesting that systemic cytokine production plays, at best, a secondary role (Helmy et al., 2009, 2011).
Despite preliminary evidence for potent inflammatory responses following brain injury, it remains unknown whether these responses are protective, neutral, damaging, or a combination over time. Proper characterization of potential mechanisms by which inflammatory responses exacerbate the pathophysiology of TBI requires a thorough understanding of the temporal profile of the expression of key cytokines. While excellent work has been performed using a variety of models, we chose to evaluate the temporal profile of cytokine accumulation in injured rat brain cortex using a technique with greatly improved sensitivity. These results can thus be readily compared to the results of research examining cortical changes in metabolism, calcium homeostasis, protein expression, and a host of other topics. Results of this study, when combined with results of studies examining other post-traumatic changes, may provide insights into underlying causes of secondary injuries.
Materials and Methods
Animal Subjects
All experiments were performed on adult male (10–12 week old, 250–275 g) Sprague–Dawley rats obtained from Taconic Farms (Germantown, NY). All procedures were approved by the Uniformed Services University of the Health Sciences Institution for Animal Care and Use Committee (IACUC) in accordance with international guidelines on the ethical use of animals. Animals were maintained in a normal 12 hour light:dark cycle, with ad libitum access to both feed and water. The 30 rats were assigned randomly to one of two groups—a control group (n = 5) that was placed in the stereotax and received anesthesia only (control), and a brain injured group (TBI). The TBI group was further distributed randomly to one of five time points with five rats each (4, 12, or 24 hours, and 3 or 7 days) for sacrifice post-surgery and comparison to control animals. Previously, we have shown that a craniotomy itself induces a potent inflammatory response, and so the most appropriate control model, to insure accurate baseline cytokine values is a rat that receives only anesthesia (Cole et al., 2011).
Traumatic Brain Injury Procedure
Rats were induced with 4% isoflurane and maintained with 2% in 98% O2. The vented anesthesia chamber and stereotactic device, which delivered anesthesia via nose cone, were connected to a scrubber. Throughout the following procedure, the depth of sedation was monitored by absent response to toe-pinch. The surgical site was prepared by shaving the top of the animal's head, followed by three successive swab applications of 95% ethanol and betadine. The rat was then placed in the stereotactic device, and head held in a horizontal position by placement of blunt-tipped interaural and incisor bars. The remainder of the procedure used aseptic techniques. A midline scalp incision exposed the skull. After using the stereotactic device to determine the correct location for the craniotomy (+5.2 lambda, +3.5 lateral left), a craniotomy was performed using a 5.2 mm inner diameter trephine bit (Ace Surgical Supply Company, Brockton, MA) attached to a variable speed micrometer drill (Dremel, Racine, WI). After formation of the 6 mm bone flap, the tip of the scalpel was gently placed against the bone flap at an oblique angle to prevent inadvertent damage to the brain, and the flap lifted and removed. A digitally controlled CCI (MyNeuroLab, Richmond, IL) plunger was then moved into position directly above the craniotomy to allow plunger baseline setting and centering, and then impact delivered. The ipsilateral side is defined as the side receiving the impact. The contralateral side is the identical region of cortex in the opposite hemisphere of the brain. The plunger velocity (5 m/s), diameter (5 mm), impact depth (2 mm), and dwell time (100 ms) were previously calibrated to deliver a moderate to severe brain injury. After the bone flap was replaced and secured with bone wax, the incision was sutured using 4-0 nylon monofilament (Ethicon, Somerville, NJ). Alternatively, control (naïve) animals were anesthetized and placed in the stereotax for 22 minutes, which is the same length of time as the average CCI procedure, but did not undergo a scalp incision or receive craniotomy. After the procedure, all animals were returned to a recovery cage and continuously monitored until they were bright, alert and responsive (BAR). Recovery time was typically less than 10 minutes, at which time, the rats were returned to their home cage.
Tissue Collection
At the appropriate time points post-injury, cortical tissues were grossly dissected at the location of cortical impact. Tissue was weighed in the receiving tube and homogenized in 10 volumes per weight of T-Per Extraction Buffer (Pierce Biotechnology, Rockford, IL) with added Halt protease inhibitor (Pierce Biotechnology) utilizing a Biorupter UCD-200 ultrasonic disrupter (Diagenode, Sparta, NH). Homogenates were centrifuged at 10,000 × g for 10 minutes at 4°C. Supernatants were transferred to new tubes and used for analysis. Protein concentration of samples was quantified with a Bradford Protein Assay Kit (Bio-Rad, Hercules, CA).
Electrochemiluminescent Immunoassay Analysis of Chemokines and Cytokines
Analyte levels in cortical homogenates were measured using a combination of singleplex and multiplex electrochemiluminescent immunoassays. Specifically, the Multi-Array Rat Cytokine Ultra-Sensitive 7-Plex Assay, the Rat MCP-1 Ultra-Sensitive Kit, and a Rat MIP-3a Ultra-Sensitive 4-Plex Assay (Meso Scale Discovery, Gaithersburg, MD) were utilized in this study. All assays were performed at the same time with the one aliquot of cortical homogenate sample. Standard curves were present on all plates and performed in triplicate, and blank wells were performed with five technical replicates. Experimental samples were performed in duplicate. During the procedure, plates were washed using the BioTek ELx405 Select automated liquid handling platform. Imaging of the plates was performed using a Sector 6000 Imager (Meso Scale Discovery). A standard curve for each analyte was curve-fitted and allowed determination of the concentration in pg cytokine/mL sample volume in each well, which was normalized to total protein input to calculate analyte amount in pg cytokine/mg total protein. The lower limits of detection for each cytokine were calculated from curve-fitted values in blank wells + 2.5 SDs and are provided in Table 1.

Table 1. Lower limits of detection at 95% confidence.
Statistical Analysis
Collected data were analyzed via ANOVA using SigmaStat (Systat Software, Chicago, IL). When normality tests failed, a Kruskal Wallis ANOVA on Ranks was performed. Significance was based on two-tailed tests, with p < 0.05. For post hoc comparisons, Holm–Sidak tests were used. Concentrations of interferon gamma (IFN-γ) in the control and contralateral tissue were below detectable limits, and so no statistical analyses were performed on this analyte. Data analysis was performed at the conclusion of the project, after all results were collected.
Results
Acute Cytokine Cortical Response to Traumatic Brain Injury
Within four hours of the CCI, eight of the nine cytokines tested for were significantly increased in ipsilateral cortex as compared to levels measured in naïve control cortical tissue. Of these eight, five peaked in expression at four hours, while subsiding to baseline levels within seven days. In TBI ipsilateral cortex, chemokine C-X-C motif ligand 1 (CXCL1) significantly (p < 0.001) increased from 0.74 ± 1.21 pg/mg protein in control cortex to 175.02 ± 20.0 pg/mg protein at four hours after injury, remaining elevated through day three (10.51 ± 3.47 pg/mg protein) post-injury (Figure 1). The contralateral cortex displayed a similar response with a significant increase in expression (p < 0.05) of CXCL1 at four hours (26.60 ± 8.87 pg/mg protein) when compared to control tissue, which returned to normal within 12 hours (4.38 ± 0.48 pg/mg protein post-injury). At 4, 12, and 24 hours, 1, 3, and 7 days after injury, ipsilateral concentrations of CXCL1 were significantly higher (p < 0.05) at all time points compared to the contralateral cortex.
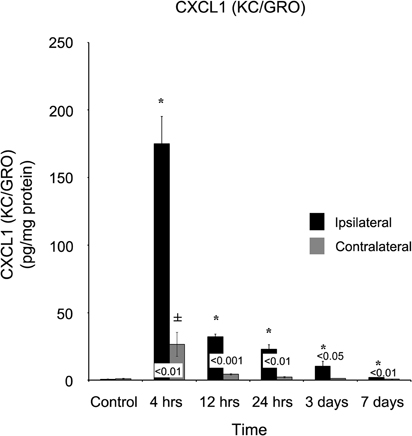
Figure 1. CXCL1 (KC/GRO) concentrations increase in both the ipsilateral and contralateral cortex following a controlled cortical impact. Within four hours of a brain injury, CXCL1 increases (p < 0.05) in both the ipsilateral and contralateral cortex in comparison to control. In comparison to both control and contralateral tissue, the ipsilateral hemisphere continued to express elevated (p < 0.05) concentrations for three days. In contrast, the contralateral cortex only demonstrated an elevated expression (p < 0.05) at four hours, returning to normal levels by 12 hours. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. ±At each time point noted, contralateral differs from control tissue, p < 0.05. Differences between the ipsilateral and contralateral cortices are denoted by placing the appropriate p-value above the two bars.
A dramatic, acute response was observed when IFN-γ was quantified (Figure 2). In CTL, as well as samples 1, 3, and 7 days post-injury, this cytokine was below detectable limits. However, within four hours of injury, IFN-γ levels were approximately (17.59 ± 4.15 pg/mg protein), declining to (3.24 ± 0.87 pg/mg protein) at 12 hours. IFN-γ remained below detectable limits at all time points in the contralateral cortex.
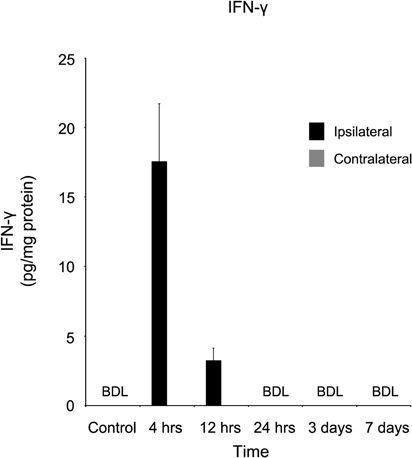
Figure 2. IFN-γ concentration increases in the ipsilateral cortex four and 12 hours following a controlled cortical impact. Within four hours of a brain injury, IFN-γ increases (p < 0.05) in the ipsilateral cortex in comparison to both control and contralateral tissue. At all time points, IFN-γ was below detectable limits (BDL) in both the control and contralateral tissue. Within 24 hours of the injury, IFNγ concentrations returned to below detectable limits. All values are mean ± SEM, n = 5. Since IFNγ in both control and contralateral tissue were below detectable limits, no statistical analyses were performed.
TNF-α also demonstrated a rapid response four hours after injury, increasing to (58.34 ± 7.82 pg/mg protein) in comparison to both the control (4.95 ± 1.34 pg/mg protein) and contralateral (6.66 ± 1.41 pg/mg protein) cortex (Figure 3). At 12 hours after the injury, TNF-α concentration (15.99 ± 1.80 pg/mg protein) remained significantly (p < 0.05) elevated in contrast to control, although due to a non-significant increase in the contralateral cortex (8.00 ± 5.46 pg/mg protein), the difference between the two ipsi- and contralateral cortices became non-significant (p > 0.05).
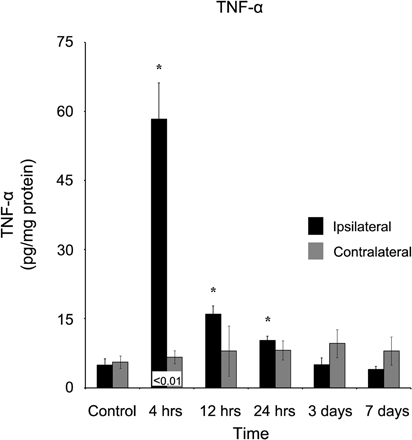
Figure 3. TNF-α concentration increases in the ipsilateral cortex following a controlled cortical impact. Within four hours of a brain injury, TNF-α increases (p < 0.05) in the ipsilateral cortex in comparison to both control and contralateral tissue. TNF-α remained elevated for the first 24 hours following injury, before returning to control levels at three days, post-injury. In contrast, the contralateral cortex did not demonstrate an increase in TNF-α expression at any time point in comparison to control. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. ±At each time point noted, contralateral differ from control tissue, p < 0.05. Differences between the ipsilateral and contralateral cortices are denoted by placing the appropriate p-value above the two bars.
A more spatially generalized response was observed when interleukin-13 (IL-13) was quantified (Figure 4). At four hours, in both ipsilateral (0.68 ± 0.08 pg/mg protein) and contralateral (0.42 ± 0.13 pg/mg protein) cortices, IL-13 was significantly (p < 0.05) elevated in comparison to control tissue, and remained elevated at 12 hours. After one day, IL-13 in the contralateral cortex (0.15 ± 0.07 pg/mg protein) returned to control levels, while slightly elevated IL-13 concentrations (0.22 ± 0.05 pg/mg protein) continued to register in the ipsilateral cortex. At both three and seven days post-injury, neither ipsilateral nor contralateral cortices of the brain contained statistically significantly elevated (p > 0.05) IL-13 levels in comparison to control.
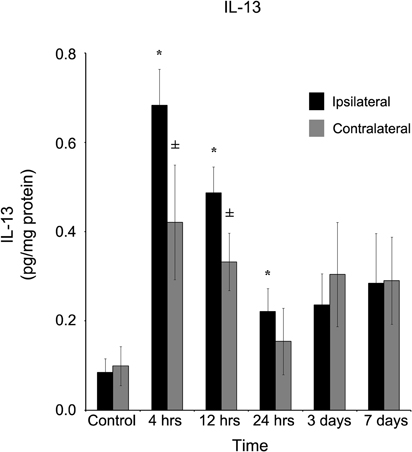
Figure 4. IL-13 concentration increases in both the ipsilateral and contralateral cortex following a controlled cortical impact. Within four hours of a brain injury, TNF-α increases (p < 0.05) in both the ipsilateral and contralateral cortex in comparison to control tissue. There was no difference (p > 0.05) in the ipsilateral and contralateral hemispheres at any time point measured. However, while the contralateral returned to levels no different from control (p > 0.05) within 24 hours, the IL-13 remained elevated (p < 0.05) in comparison to control until three days post-injury. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. ±At each time point noted, contralateral differs from control tissue, p < 0.05.
At 4 (1.01 ± 0.14 pg/mg protein), 12 (0.53 ± 0.06 pg/mg protein), and 24 (0.63 ± 0.17 pg/mg protein) hours after injury, interleukin-4 (IL-4) was elevated (Figure 5, p < 0.05) in the ipsilateral, but not (p > 0.05) contralateral cortex at those same time points (0.64 ± 0.18, 0.31 ± 0.03, and 0.51 ± 0.08 pg/mg protein, respectively). Within three days of injury, IL-4 concentrations in the ipsilateral cortex were not statistically significantly different from concentrations in control cortex.
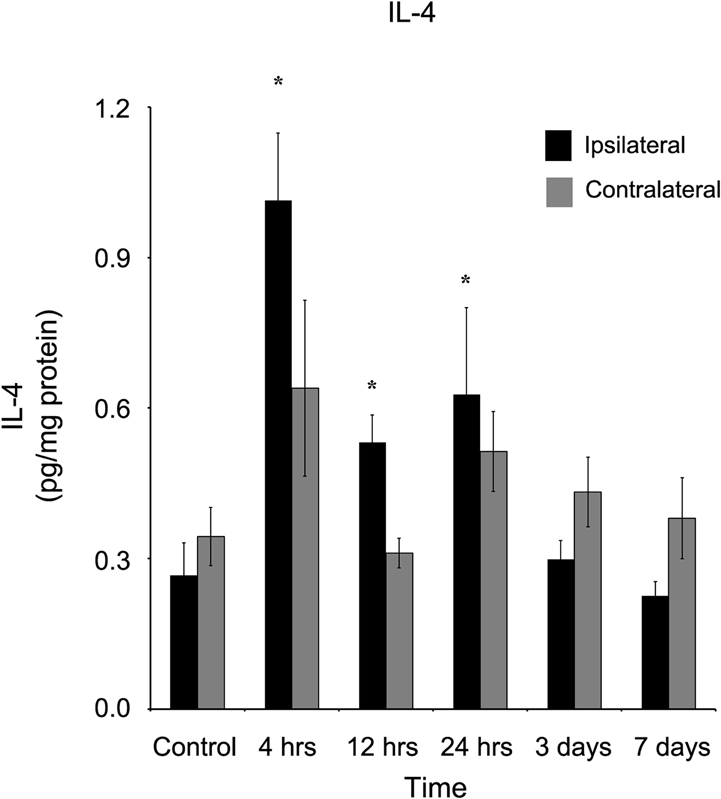
Figure 5. IL-4 concentration increases in the ipsilateral cortex following a controlled cortical impact. Within four hours of a brain injury, and persisting for 24 hours, IL-4 increases (p < 0.05) in the ipsilateral cortex in comparison to control tissue. There was no difference (p > 0.05) between the ipsilateral and contralateral hemispheres at any time point measured. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05.
Delayed Cytokine Cortical Response to Traumatic Brain Injury
In contrast to the acute response observed (within four hours of brain injury), the appearance of several cytokines took considerably longer to develop. Within four hours of injury, chemokine C-C motif ligand 2 (CCL2) was elevated (p < 0.05; 913.91 ± 91.49 pg/mg protein) in comparison to both control (14.68 ± 0.96 pg/mg protein) and the contralateral (188.21 ± 77.56 pg/mg protein) cortex (Figure 6). However, the peak concentration of CCL2 did not occur until 12 hours post-injury (1333.39 ± 154.48 pg/mg protein) and remained elevated throughout the remainder of the study. At every time point measured, the ipsilateral cortex contained a greater concentration of CCL2 than the contralateral cortex. The contralateral cortex was elevated four hours after injury, displayed no distinct peak, and returned to levels not statistically significantly different from control levels at three days after injury.
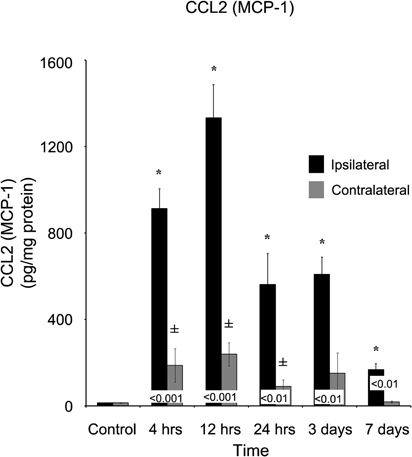
Figure 6. CCL2 concentrations increase in both the ipsilateral and contralateral cortex following a controlled cortical impact. Within four hours of a brain injury, CCL2 increases (p < 0.05) in both the ipsilateral and contralateral cortex in comparison to control. In comparison to both control and contralateral tissue, the ipsilateral hemisphere continued to express elevated (p < 0.05) concentrations for seven days. In contrast, the contralateral cortex only demonstrated an elevated expression in comparison to control (p < 0.05) at 4, 12, and 24 hours, returning to normal levels by three days after injury. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. ±At each time point noted, contralateral differs from control tissue, p < 0.05. Differences between the ipsilateral and contralateral cortices are denoted by placing the appropriate p-value above the two bars.
Chemokine C-C motif ligand 20 (CCL20) also demonstrated a delayed onset of maximum concentration (Figure 7). Within four hours of injury, the ipsilateral cortex (13.62 ± 2.04 pg/mg protein) was significantly elevated (p < 0.05) in comparison to both the control (1.14 ± 0.16 pg/mg protein) and contralateral (3.99 ± 1.91 pg/mg protein) cortex. However, peak concentration was not observed in the ipsilateral cortex until one day post-injury (55.82 ± 14.22 pg/mg protein). At no time point did the contralateral cortex express elevated CCL20 (p > 0.05).
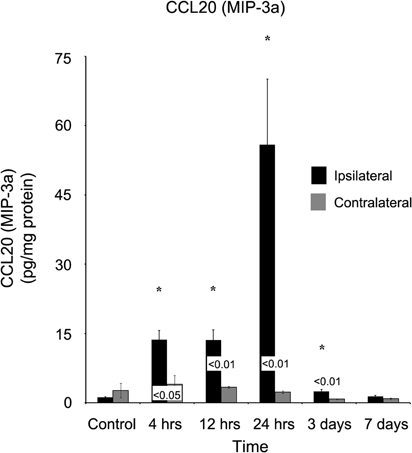
Figure 7. CCL20 concentrations increase in the ipsilateral cortex following a controlled cortical impact. Within four hours of a brain injury, CCL20 increases (p < 0.05) in the ipsilateral cortex in comparison to both control and contralateral tissue. The expression of CCL20 peaked 24 hours after injury, and did not return to levels observed in control or contralateral tissue until seven days post-injury. At no time, was the expression of CCL20 observed to be elevated (p > 0.05) in the contralateral cortex in comparison to control. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. Differences between the ipsilateral and contralateral cortices are denoted by placing the appropriate p-value above the two bars.
IL-1b similar to IL-4, was elevated (p < 0.05) at four hours (17.17 ± 2.37 pg/mg protein) post-injury and remained elevated (p < 0.05) at both 12 hours (12.09 ± 1.64 pg/mg protein) and one day (17.44 ± 4.49 pg/mg protein) in comparison to control (Figure 8). At each of these time points, the ipsilateral cortex was significantly elevated (p < 0.05) compared to the contralateral cortex. IL-1b was elevated at four hours in the contralateral cortex (3.41 ± 0.58 pg/mg protein), but by 12 hours, the level (0.97 ± 0.26 pg/mg protein) was not significantly different from observed levels in naïve controls.
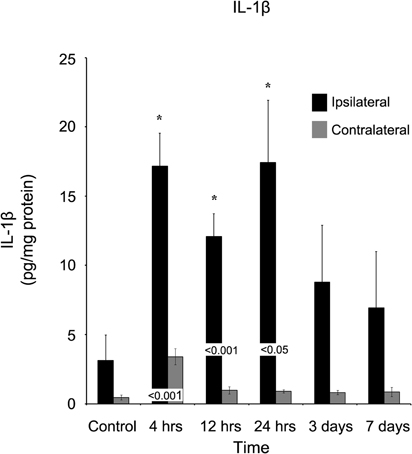
Figure 8. IL-1β concentrations increase in the ipsilateral cortex following a controlled cortical impact. Within four hours of a brain injury, IL-1β increases (p < 0.05) in the ipsilateral cortex in comparison to both control and contralateral tissue. The expression of IL-1β remained elevated for the first 24 hours after injury, and did not return to levels observed in control or contralateral tissue (p > 0.05) until three days post-injury. At no time, was the expression of IL-1β observed to be elevated (p > 0.05) in the contralateral cortex in comparison to control. All values are mean ± SEM, n = 5. *At each time point noted, ipsilateral differs from control tissue, p < 0.05. Differences between the ipsilateral and contralateral cortices are denoted by placing the appropriate p-value above the two bars.
Interleukin-5 (IL-5) concentration levels, in contrast to all other tested cytokines, was not statistically significantly different in either ipsilateral cortical tissue as compared to contralateral tissue, or in either side as compared to naïve cortical tissue at all time points measured during this study (Figure 9).
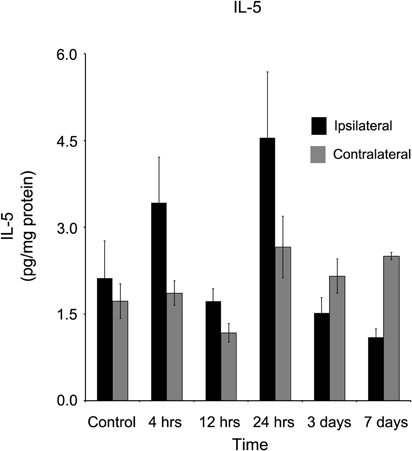
Figure 9. IL-5 concentrations in the ipsilateral and contralateral cortex were not affected by a controlled cortical impact. At no time point after the brain injury were elevated concentrations of IL-5 observed in either the ipsilateral or contralateral cortex in comparison to control tissue (p > 0.05). All values are mean ± SEM, n = 5.
Discussion
We report here that TBI via CCI on one brain hemisphere results in early and later responses in specific chemokines and cytokines in cortical tissue. Furthermore, we find that there is a differential discrimination with respect to the responses in the damaged ipsilateral versus the undamaged contralateral cortex. This is consistent with our hypothesis that combinations of beneficial, neutral, and deleterious inflammatory responses occur in distinct temporal and anatomical-dependent manners after TBI. In general, we observed significant increases in eight of the nine analytes studied, compared to levels measured in the uninjured naïve cortical tissue. More specifically, TBI caused early activation of TNF-α, CXCL1, and IFN-γ, with a later activation of CCL2 and CCL-20. Significantly, specific inflammatory chemokines and cytokines peaked at time points consistent with their known roles in a pathogen-mediated inflammatory cascade. For example, CXCL1 (KC/GRO) and TNF-α are early stimuli responsive pro-inflammatory mediators, and their peak levels (increasing ∼236-fold and ∼18-fold compared to control concentrations, respectively) occur at the earliest time point in this study (four hours post-injury).
TBI Causes Early Activation of CXCL1, IFN-γ, TNF-α, and IL-13
Functionally, both CXCL1 and TNF-α have biological relevance to outcomes in TBI. CXCL1, CXCL2, and CXCL8 are related ligands of the CXCR2 receptor, which are known to be significantly expressed by neutrophils. Not surprisingly, rapid increase in CXCR2 ligands after TBI in rodent studies is associated with early robust infiltration of neutrophils into the area of brain tissue injury (Bye et al., 2007). Consistently, recent work in CXCR2 knockout animals have demonstrated a massive reduction in neutrophil influx into injured brain tissue, with a correlated decrease in lesion volume, as compared to wild-type controls (Semple et al., 2010b). The prompt elevation of CXCL1, the rat equivalent of CXCL8 in humans, is reminiscent of the report that CXCL8 levels in the CSF of patients correlates with mortality in patients with severe TBI (Whalen et al., 2000). Clearly, it would be of translational importance to understand CXCR ligand upregulation in brain tissue after injury.
IFN-γ is produced by microglia and brain macrophages upon pathogen stimulation or cellular activation (Suzuki et al., 2005; Kawanokuchi et al., 2006). Therefore, the brief increase in IFN-γ observed only at the earliest time point in this study suggests that primary TBI effects may be the cause for IFN-γ production by brain innate immune cells. Interestingly, IFN-γ plays a functional role in early innate immunity by activation of leukocyte adhesion and antigen presentation by microglia and macrophages, as well as delayed adaptive immunity by promoting a Th1 cellular response. Interestingly, blockade of IFN-γ signaling did not alter outcome in a stab injury model study (Rostworowski et al., 1997). Therefore, analysis of delayed outcome measures in a TBI model with IFN-γ blockade or knockout is warranted.
TNF-α also plays a role in the initial response to tissue damage, by rapidly increasing after TBI (Shohami et al., 1997). In the present study, TNF-α peaked at four hours post-injury, but was markedly decreased by 12 hours. From studies on direct administration of recombinant TNF-α protein into the brain, the functional consequence is compromise of blood-brain barrier integrity, induction of inflammatory cascades, and promotion of immune cell infiltration into brain parenchyma (Ramilo et al., 1990; Kim et al., 1992). Presumably, all three consequences are visited on the damaged cortex by the rapid increase in endogenous TNF-α. Consistently, agents that act to inhibit TNF-α activity, all result in improved neurological outcome after TBI (Shohami et al., 1997).
The anti-inflammatory cytokine IL-13, which also peaks within four hours of injury, is reported to reduce the production of pro-inflammatory mediators from activated microglia and macrophages. Macrophages reduce the expression of iNOS protein and mRNA following pre-treatment of IL-13, by a mechanism that is antagonized by IFN-γ (Bogdan et al., 1997). Paludan et al., (1997) also confirmed these results (Paludan et al., 1997). This is surprising that two cytokines, IL-13 and IFN-γ, with opposing effects would be so potently increased following a brain injury. To understand this process, however, will require additional investigation.
TBI Causes Temporary Increases in anti-Inflammatory Cytokine IL-4
Of the inflammatory mediators analyzed in this study, IL-4 is the primary regulatory cytokine that possess anti-inflammatory functions. We observed modest but significant increases in injured tissue for an early time window (4–12 hours post-injury). This finding was unexpected as previous studies of brain injury have shown IL-4 to be below detectable levels or unchanged (Takamiya et al., 2007). However, it is known that both astrocytes and microglia have the capacity to produce IL-4, and this production is increased upon activation (Hulshof et al., 2002; Ponomarev et al., 2007). Since IL-4 has been recently appreciated to signal alternative neuroprotective activation of microglia (Zhao et al., 2006), the short duration increase of IL-4 after rat TBI may play an autoregulatory role to reduce inflammatory-related damage. Other anti-inflammatory mediators should be analyzed and evaluated in future rodent TBI studies.
TBI Causes a Delayed Activation of CCL2 and CCL20
We also observed robust but delayed changes in the levels of CCL2 (MCP-1) and CCL20 chemokines following early CXCL1 and TNF-α responses after CCI injury. CCL2 (MCP-1) peak levels were ∼90-fold higher than uninjured healthy tissue at 12 hours post-injury and absolute CCL2 levels of approximately 1300 pg/mg protein were significantly higher than all other analytes measured. This observation supports previous information that CCL2 is routinely measurable in brain tissue (Glabinski et al., 1996) and cerebrospinal fluid (Semple et al., 2010a) due to both high basal concentrations and to sensitivity to injury (Muessel et al., 2000; Rancan et al., 2001). Our results also found that CCL2 levels remain significantly greater in injured cortex, than in uninjured control and contralateral tissue throughout all time points measured (seven days). These data agree also with repeated measurements for CCL2 in CSF of patients with TBI, where levels are significantly greater than in control patients at each day post-injury up to day 9 (Semple et al., 2010a). Furthermore, (Semple et al., 2010b) investigated the potential pathological role of CCL2 following a TBI in CCL2 knockout and wild-type mice (Semple et al., 2010a). In support of a positive functional role for CCL2 during the secondary phase of TBI processes, the study did not observe differences in lesion size or cell death within seven days after brain injury, but found significant decreases in lesion volume, reactive gliosis, macrophage infiltration, and neurological outcome in CCL2−/− mice as compared to wild-type controls in subjects at second and fourth weeks. Despite these data demonstrating that lost CCL2 signaling is beneficial, it is also confirming that CCL2 acts in chemotaxis of peripheral macrophages and the activation of both microglia and macrophages. Therefore, the role of these two cell populations in the removal of cellular debris may also serve to protect the brain from injury at specific temporal windows. The combined impact of these reports on the response of CCL2 to TBI suggests that CCL2 merits further testing of potential therapeutics in preclinical rodent models of TBI.
A delayed peak level after CCI injury was also observed in analysis of CCL20 (MIP-3a). There is less known about the role of this chemokine in the context of TBI. Of translational relevance is the fact that CCL2 levels have been observed to be sustained in CSF of TBI patients (Semple et al., 2010a). However, in studies of experimental autoimmune encephalomyelitis, a model for demyelinating multiple sclerosis, astrocytes have been shown to be the primary source of CCL20. Furthermore, the increase in CCL20 is associated with recruitment of highly inflammatory autoimmune-related Th17 cells (Ambrosini et al., 2003). Additionally, CCL20 is responsible for B cell lymphocyte maturation and antigen-mediated differentiation (Krzysiek et al., 2000). Thus, the delayed expression of CCL20 may suggest a recruitment of the adaptive immune response and potential autoimmunity at this time point after CCI injury. Interestingly, CCL20 is the sole known ligand for CCR6 and thus may provide a therapeutic target for preclinical studies in TBI where minimal off-target effects exist.
Selection of ECL-Based Immunoassay Technology for Brain Homogenate Analysis
For this study, we selected and utilized commercially available electrochemiluminescent-based singleplex and multiplex immunoassays for profiling inflammatory chemokines and cytokines in rat brain homogenates. The selection of this type of assay was due to advantages in linear dynamic range, precision, lower limits of detection, and low sample volume requirements as compared to the other surveyed assays (conventional ELISA, reverse phase capture array, bead-based multiplex assay). Comparative analysis of this assay technology with others surveyed has been described in previous studies by other laboratories with similar conclusions (Chowdhury et al., 2009; Fu et al., 2010).
In summary, this study provides a framework for determining the levels of inflammatory mediators in rat brain tissue homogenates, provides key information to aid in the selection of analytes as an early and delayed phase outcome measure in preclinical studies of TBI, and supports the hypothesis that rapid and potent inflammatory responses in injured brain tissue are also associated with delayed and robust responses that could mediate recruitment of the adaptive immune response. Whether activation of the adaptive immune response aids in brain tissue repair or further damage is not clear. There is potential for the use of select chemokines and cytokines for biomarkers in clinical TBI and, therefore, further study of the modulation and functional role of these inflammatory mediators in rodent models (including genetic knockout subjects) in TBI is warranted. Finally, a thorough review of the literature reveals a number of excellent studies examining changes in cytokine expression at 24 hours or more after injury, including a recent study confirming observed increases in TNF-α and CCL-2 levels using two independent techniques. These authors suggest that by neglecting earlier time points, within just hours of injury, potentially invaluable data is not being captured given the transient response in cytokine expression observed here. Our analysis of cytokine expression using an independent high sensitivity two antibody electrochemiluminescence-based ELISA methodology provides further validation that these changes are occurring in the brain parenchyma after injury.
Disclaimer
The opinions expressed herein belong solely to the authors. They do not nor should they be interpreted as representative of or endorsed by the Uniformed Services University of the Health Sciences, US Army, US Navy, Department of Defense or any other agency of the federal government. Products mentioned by name within this study do not imply endorsement of these products nor condemnation of similar products not used.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the Comprehensive National Neuroscience Program (CNNP, award number W81XWH-07-0679).
References
Adeleye, A., Shohami, E., Nachman, D., Alexandrovich, A., Trembovler, V., Yaka, R., Shoshan, Y., Dhawan, J., and Biegon, A. (2010). D-cycloserine improves functional outcome after traumatic brain injury with wide therapeutic window. Eur. J. Pharmacol. 629, 25–30.
Alves, O. L., Bullock, R., Clausen, T., Reinert, M., and Reeves, T. M. (2005). Concurrent monitoring of cerebral electrophysiology and metabolism after traumatic brain injury: an experimental and clinical study. J. Neurotrauma 22, 733–749.
Ambrosini, E., Columba-Cabezas, S., Serafini, B., Muscella, A., and Aloisi, F. (2003). Astrocytes are the major intracerebral source of macrophage inflammatory protein-3alpha/CCL20 in relapsing experimental autoimmune encephalomyelitis and in vitro. Glia 41, 290–300.
Arcure, J., and Harrison, E. E. (2009). A review of the use of early hypothermia in the treatment of traumatic brain injuries. J. Spec. Oper. Med. 9, 22–25.
Bartnik, B. L., Lee, S. M., Hovda, D. A., and Sutton, R. L. (2007). The fate of glucose during the period of decreased metabolism after fluid percussion injury: a 13C NMR study. J. Neurotrauma 24, 1079–1092.
Bogdan, C., Thuring, H., Dlaska, M., Rollinghoff, M., and Weiss, G. (1997). Mechanism of suppression of macrophage nitric oxide release by IL-13: influence of the macrophage population. J. Immunol. 159, 4506–4513.
Bye, N., Habgood, M. D., Callaway, J. K., Malakooti, N., Potter, A., Kossmann, T., and Morganti-Kossmann, M. C. (2007). Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 204, 220–233.
Chowdhury, F., Williams, A., and Johnson, P. (2009). Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J. Immunol. Methods 340, 55–64.
Cole, J. T., Mitala, C. M., Kundu, S., Verma, A., Elkind, J. A., Nissim, I., and Cohen, A. S. (2010). Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc. Natl. Acad. Sci. U.S.A. 107, 366–371.
Cole, J. T., Yarnell, A., Kean, W. S., Gold, E., Lewis, B., Ren, M., McMullen, D. C., Jacobowitz, D. M., Pollard, H. B., O'Neill, J. T., Grunberg, N. E., Dalgard, C. L., Frank, J. A., and Watson, W. D. (2011). Craniotomy: true sham for traumatic brain injury, or a sham of a sham? J. Neurotrauma 28, 359–369.
Deshpande, L. S., Sun, D. A., Sombati, S., Baranova, A., Wilson, M. S., Attkisson, E., Hamm, R. J., and DeLorenzo, R. J. (2008). Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 441, 115–119.
Faria, M. H. G., Muniz, L. R. F., and De Vasconcelos, P. R. L. (2007). Ketone bodies metabolism during ischemic and reperfusion brain injuries following bilateral occlusion of common carotid arteries in rats. Acta Cir. Bras. 22, 125–129.
Fassbender, K., Schneider, S., Bertsch, T., Schlueter, D., Fatar, M., Ragoschke, A., Kuhl, S., Kischka, U., and Hennerici, M. (2000). Temporal profile of release of interleukin-1beta in neurotrauma. Neurosci. Lett. 284, 135–138.
Finkelstein, E., Corso, P., Miller, T., and Associates. (2006). The Incidence and Economic Burden of Injuries in the United States. New York, NY: Oxford University Press.
Fu, Q., Zhu, J., and Van Eyk, J. E. (2010). Comparison of multiplex immunoassay platforms. Clin. Chem. 56, 314–318.
Glabinski, A. R., Balasingam, V., Tani, M., Kunkel, S. L., Strieter, R. M., Yong, V. W., and Ransohoff, R. M. (1996). Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 156, 4363–4368.
Harting, M. T., Jimenez, F., Adams, S. D., Mercer, D. W., and Cox, C. S. Jr. (2008). Acute, regional inflammatory response after traumatic brain injury: implications for cellular therapy. Surgery 144, 803–813.
He, X. S., Xiang, Z., Zhou, F., Fu, L. A., and Shuang, W. (2004). Calcium overloading in traumatic axonal injury by lateral head rotation: a morphological evidence in rat model. J. Clin. Neurosci. 11, 402–407.
Helmy, A., Carpenter, K. L., Menon, D. K., Pickard, J. D., and Hutchinson, P. J. (2011). The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. 31, 658–670.
Helmy, A., Carpenter, K. L., Skepper, J. N., Kirkpatrick, P. J., Pickard, J. D., and Hutchinson, P. J. (2009). Microdialysis of cytokines: methodological considerations, scanning electron microscopy, and determination of relative recovery. J. Neurotrauma 26, 549–561.
Hulshof, S., Montagne, L., De Groot, C. J., and Van Der Valk, P. (2002). Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia 38, 24–35.
Karaoglan, A., Akdemir, O., Cinar, N., Cal, M. A., Kelten, B., Uzun, H., and Colak, A. (2011). Correlation between leptin and pro-inflammatory cytokines in cortical contusion injury model. Ulus. Travma Acil Cerrahi Derg. 17, 298–302.
Kawanokuchi, J., Mizuno, T., Takeuchi, H., Kato, H., Wang, J., Mitsuma, N., and Suzumura, A. (2006). Production of interferon-gamma by microglia. Mult. Scler. 12, 558–564.
Kim, K. S., Wass, C. A., Cross, A. S., and Opal, S. M. (1992). Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 11, 293–298.
Kinoshita, K., Chatzipanteli, K., Vitarbo, E., Truettner, J. S., Alonso, O. F., and Dietrich, W. D. (2002). Interleukin-1beta messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: importance of injury severity and brain temperature. Neurosurgery 51, 195–203; discussion 203.
Krzysiek, R., Lefevre, E. A., Bernard, J., Foussat, A., Galanaud, P., Louache, F., and Richard, Y. (2000). Regulation of CCR6 chemokine receptor expression and responsiveness to macrophage inflammatory protein-3alpha/CCL20 in human B cells. Blood 96, 2338–2345.
Lloyd, E., Somera-Molina, K., Van Eldik, L. J., Watterson, D. M., and Wainwright, M. S. (2008). Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J. Neuroinflammation 5, 28.
Muessel, M. J., Berman, N. E., and Klein, R. M. (2000). Early and specific expression of monocyte chemoattractant protein-1 in the thalamus induced by cortical injury. Brain Res. 870, 211–221.
Mukherjee, S., Katki, K., Arisi, G. M., Foresti, M. L., and Shapiro, L. A. (2011). Early TBI-induced cytokine alterations are similarly detected by two distinct methods of multiplex assay. Front. Mol. Neurosci. 4, 21. doi: 10.3389/fnmol.2011.00021
Paludan, S. R., Lovmand, J., Ellermann-Eriksen, S., and Mogensen, S. C. (1997). Effect of IL-4 and IL-13 on IFN-gamma-induced production of nitric oxide in mouse macrophages infected with herpes simplex virus type 2. FEBS Lett. 414, 61–64.
Ponomarev, E. D., Maresz, K., Tan, Y., and Dittel, B. N. (2007). CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714–10721.
Ramilo, O., Saez-Llorens, X., Mertsola, J., Jafari, H., Olsen, K. D., Hansen, E. J., Yoshinaga, M., Ohkawara, S., Nariuchi, H., and McCracken, G. H. Jr. (1990). Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J. Exp. Med. 172, 497–507.
Rancan, M., Otto, V. I., Hans, V. H., Gerlach, I., Jork, R., Trentz, O., Kossmann, T., and Morganti-Kossmann, M. C. (2001). Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J. Neurosci. Res. 63, 438–446.
Rostworowski, M., Balasingam, V., Chabot, S., Owens, T., and Yong, V. W. (1997). Astrogliosis in the neonatal and adult murine brain post-trauma: elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-gamma. J. Neurosci. 17, 3664–3674.
Scafidi, S., O'Brien, J., Hopkins, I., Robertson, C., Fiskum, G., and McKenna, M. (2009). Delayed cerebral oxidative glucose metabolism after traumatic brain injury in young rats. J. Neurochem. 109(Suppl. 1), 189–197.
Semple, B. D., Bye, N., Rancan, M., Ziebell, J. M., and Morganti-Kossmann, M. C. (2010a). Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J. Cereb. Blood Flow Metab. 30, 769–782.
Semple, B. D., Bye, N., Ziebell, J. M., and Morganti-Kossmann, M. C. (2010b). Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol. Dis. 40, 394–403.
Shohami, E., Gallily, R., Mechoulam, R., Bass, R., and Ben-Hur, T. (1997). Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J. Neuroimmunol. 72, 169–177.
Sun, D. A., Deshpande, L. S., Sombati, S., Baranova, A., Wilson, M. S., Hamm, R. J., and DeLorenzo, R. J. (2008). Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur. J. Neurosci. 27, 1659–1672.
Suzuki, Y., Claflin, J., Wang, X., Lengi, A., and Kikuchi, T. (2005). Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int. J. Parasitol. 35, 83–90.
Takamiya, M., Fujita, S., Saigusa, K., and Aoki, Y. (2007). Simultaneous detections of 27 cytokines during cerebral wound healing by multiplexed bead-based immunoassay for wound age estimation. J. Neurotrauma 24, 1833–1844.
Tehranian, R., Andell-Jonsson, S., Beni, S. M., Yatsiv, I., Shohami, E., Bartfai, T., Lundkvist, J., and Iverfeldt, K. (2002). Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. Neurotrauma 19, 939–951.
Wei, H. H., Lu, X. C., Shear, D. A., Waghray, A., Yao, C., Tortella, F. C., and Dave, J. R. (2009). NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. J. Neuroinflammation 6, 19.
Whalen, M. J., Carlos, T. M., Kochanek, P. M., Wisniewski, S. R., Bell, M. J., Clark, R. S., DeKosky, S. T., Marion, D. W., and Adelson, P. D. (2000). Interleukin-8 is increased in cerebrospinal fluid of children with severe head injury. Crit. Care Med. 28, 929–934.
Xing, G., Ren, M., Watson, W. A., O'Neil, J. T., and Verma, A. (2009). Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: a mechanism of dysregulated glucose metabolism. Neurosci. Lett. 454, 38–42.
Xiong, Y., Chopp, M., and Lee, C. P. (2009). Erythropoietin improves brain mitochondrial function in rats after traumatic brain injury. Neurol. Res. 31, 496–502.
Yatsiv, I., Morganti-Kossmann, M. C., Perez, D., Dinarello, C. A., Novick, D., Rubinstein, M., Otto, V. I., Rancan, M., Kossmann, T., Redaelli, C. A., Trentz, O., Shohami, E., and Stahel, P. F. (2002). Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J. Cereb. Blood Flow Metab. 22, 971–978.
Zhao, W., Xie, W., Xiao, Q., Beers, D. R., and Appel, S. H. (2006). Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J. Neurochem. 99, 1176–1187.
Keywords: traumatic brain injury, cytokines, inflammation, IL-8, inflammatory response
Citation: Dalgard CL, Cole JT, Kean WS, Lucky JJ, Sukumar G, McMullen DC, Pollard HB and Watson WD (2012) The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci. 5:6. doi: 10.3389/fnmol.2012.00006
Received: 09 November 2011; Accepted: 12 January 2012;
Published online: 25 January 2012.
Edited by:
Lashuel Hilal, Ecole Polytechnique Federale De Lausanne, SwitzerlandCopyright: © 2012 Dalgard, Cole, Kean, Lucky, Sukumar, McMullen, Pollard and Watson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jeffrey T. Cole, Department of Neurology, Uniformed Services University of the Health Sciences, B-3012, 4301 Jones Bridge Road, Bethesda, MD 20814, USA. e-mail: jeffrey.cole@usuhs.mil
† These authors contributed equally to this manuscript.