Conditional RARα knockout mice reveal acute requirement for retinoic acid and RARα in homeostatic plasticity
- 1Department of Psychiatry and Behavioral Sciences, Stanford Institute of Neuro-Innovation and Translational Neuroscience, Stanford University School of Medicine, Stanford, CA, USA
- 2Department of Molecular and Cell Biology, University of California, Berkeley, CA, USA
All-trans retinoic acid (RA) plays important roles in brain development through regulating gene transcription. Recently, a novel post-developmental role of RA in mature brain was proposed. Specifically, RA rapidly enhanced excitatory synaptic transmission independent of transcriptional regulation. RA synthesis was induced when excitatory synaptic transmission was chronically blocked, and RA then activated dendritic protein synthesis and synaptic insertion of homomeric GluA1 AMPA receptors, thereby compensating for the loss of neuronal activity in a homeostatic fashion. This action of RA was suggested to be mediated by its canonical receptor RARα but no genetic evidence was available. Thus, we here tested the fundamental requirement of RARα in homeostatic plasticity using conditional RARα knockout (KO) mice, and additionally performed a structure-function analysis of RARα. We show that acutely deleting RARα in neurons eliminated RA's effect on excitatory synaptic transmission, and inhibited activity blockade-induced homeostatic synaptic plasticity. By expressing various RARα rescue constructs in RARα KO neurons, we found that the DNA-binding domain of RARα was dispensable for its role in regulating synaptic strength, further supporting the notion that RA and RARα act in a non-transcriptional manner in this context. By contrast, the ligand-binding domain (LBD) and the mRNA-binding domain (F-domain) are both necessary and sufficient for the function of RARα in homeostatic plasticity. Furthermore, we found that homeostatic regulation performed by the LBD/F-domains leads to insertion of calcium-permeable AMPA receptors. Our results confirm with unequivocal genetic approaches that RA and RARα perform essential non-transcriptional functions in regulating synaptic strength, and establish a functional link between the various domains of RARα and their involvement in regulating protein synthesis and excitatory synaptic transmission during homeostatic plasticity.
Introduction
During development, retinoic acid (RA) performs essential functions as a morphogen regulating gene expression. In the nervous system, RA-signaling is involved in neurogenesis and neuronal differentiation. However, several recent lines of evidence support the idea that RA also performs post-developmental functions in the adult brain. First, RA can be rapidly synthesized in various regions of the adult brain (Dev et al., 1993). Second, compromised RA-signaling (through either genetic knockout (KO) of RA receptors or vitamin A deficiency) leads to impaired long-term synaptic plasticity in the hippocampus (Chiang et al., 1998; Misner et al., 2001). The involvement of RA in synaptic plasticity was further supported by deficits in learning and memory tasks observed in mice with a genetic deletion of RA receptors or with vitamin A deficiency (Chiang et al., 1998; Cocco et al., 2002). Third, recent evidence established a role of RA in synaptic signaling in that RA directly potentiates glutamatergic synaptic transmission (Aoto et al., 2008). Blocking synaptic glutamate receptors activates RA synthesis, which in turn promotes local synthesis of various synaptic proteins, including GluA1 subunit of AMPA receptors. Synaptic insertion of homomeric GluA1 AMPA receptors increases synaptic transmission and, therefore, compensates the changes in synaptic activity homeostatically (Aoto et al., 2008). At least the latter action of RA is different from its role in development in that it does not require transcriptional activation (Aoto et al., 2008).
The action of RA is primarily mediated by nuclear retinoid receptor proteins called retinoic acid receptors (RAR-α, -β, -γ) and retinoid “X” receptors (RXR-α, -β, -γ). Like other members of the steroid receptor family, RARs and RXRs are transcription factors. Although structurally similar, the ligand specificity differs between RARs and RXRs in that RARs bind RA with high affinity, whereas RXRs exclusively bind 9-cis-retinoic acid (Soprano et al., 2004). Because 9-cis-RA is undetectable in vivo, the effects of retinoids on gene transcription are presumed to be mediated by RA interactions with RARs. In the mammalian brain, RARα is found in the cortex and hippocampus, while RARβ is highly expressed in the basal ganglia, and RARγ is not detectable (Krezel et al., 1999; Zetterstrom et al., 1999).
The expression and subcellular distribution of RARα in the hippocampus exhibit interesting developmental changes (Huang et al., 2008). Specifically, the expression levels of RARα in hippocampal neurons gradually decrease postnatally, and the subcellular localization of RARα protein shifts from the nucleus to the cytosol. While almost exclusively localized in the nucleus of developing neurons, at least equal amounts of RARα were detected in the cytosol and dendrites in mature hippocampal pyramidal and granule cells (Aoto et al., 2008; Huang et al., 2008; Maghsoodi et al., 2008). Functionally, dendritic RARα acts as an mRNA-binding protein that represses translation of target mRNAs, and thus performs a different role than the transcriptional function of RARα localized in the nucleus (Poon and Chen, 2008). In the dendrites, RA binding to RARα reduces RARα binding to mRNA, and de-represses translation (Poon and Chen, 2008).
The region of RARα that mediates mRNA binding has been determined (Poon and Chen, 2008), but it is unknown whether RA binding by RARα and its mRNA-binding ability are required for its role in regulating synaptic strength. Here, we combine mouse genetics and electrophysiology to rigorously test the role of RA and RARα in homeostatic plasticity, and to systematically examine the contribution of various domains of RARα to synaptic signaling by RA. Consistent with earlier results, we found that RARα is essential for some forms of homeostatic plasticity, and that the carboxyl terminal half of the RARα, which contains the ligand-binding domain (LBD) and the RNA-binding domain (F-domain), are both necessary and sufficient for the function of RARα in the homeostatic regulation of excitatory synaptic strength.
Materials and Methods
Mouse Husbandry and Genotyping
The RARα floxed mouse (C57BL/6 background) is a generous gift from Drs. Pierre Chambon and Norbert Ghyselinck (IGBMC, Strasbourg, France) (Chapellier et al., 2002). Breeding colonies are maintained in the animal facility at Stanford Medical School. Genotyping of the mice was achieved by PCR with the following primers: Primer 1 Fwd 5′-GTGTGTGTGTGTATTCGCGTGC-3′, Primer 2 Rev 5′-ACAAAGCAAGGCTTGTAGATGC-3′ and compared with wild-type (WT) C57BL/6. Following infection with a lentiviral vector expressing Cre recombinase or a truncated and inactive version of Cre, successful lox-P mediated recombination in neuronal cultures was assayed by PCR with Primer 1 and Primer 3: 5′-TACACTAACTACCCTTGACC-3′. Conditions were 30 cycles for 30 s at 92°C, annealing at decreasing temperatures in the range from 62°C to 56°C to increase product specificity and elongation for 30 s at 72°C.
Primary Hippocampal Cultures
Primary hippocampal cultures were prepared from mouse brains at postnatal day 0 and maintained in serum and Neurobasal medium supplemented with Gem21 (Gemini Bioproducts, Sacramento, CA) and Glutamax (GIBCO-Brl, Grand Island, NY) for 2 weeks in vitro (Nam and Chen, 2005). Neurons were infected with a retroviral vector expressing proteins of interest at 4–7 DIV. Manipulations used to induce synaptic scaling in dissociated cultures include: TTX + APV (1 μM TTX + 100 μM APV, 24 h); TTX + APV + DEAB (1 μM TTX + 100 μM APV + DEAB 10 μM, 24 h); RA (1 μM, 30 min followed by 1 h of washout).
Viral Vectors and Virus Production
Lentivirus was produced and purified as described previously (Aoto et al., 2008). Briefly, human embryonic kidney 293T (HEK293T) cells were transfected using calcium phosphate with the transfer vector and three helper plasmids. After 48 h, supernatants were pooled, spun at 25,000 rpm through a sucrose cushion for 1.5 h, and resuspended in PBS. Virus expressing Cre-recombinase or a truncated and inactive version of Cre (ΔCre) [gifts from Dr. Thomas Sudhof's lab, (Kaeser et al., 2011)] was applied overnight to the media of dissociated neuronal cultures generated from RARα floxed mice and washed out the following day. In rescue experiments, Cre-recombinase or ΔCre and RARα were expressed simultaneously from a bicistronic lentiviral transfer vector containing a GFP-Cre 5′ open reading frame immediately followed by an IRES sequence fused to the RARα reading frame RARα or truncated versions of RARα expressing the different domains were inserted in the forward direction into the unique BstXI site located in the MCS region. Neurons were infected at 4–7 DIV and recorded from at 14–16 DIV.
Electrophysiology
Whole-cell patch-clamp recordings were made at room temperature from 14–16 DIV cultured neurons, with 4–6 MΩ patch pipettes filled with an internal solution containing (in mM) 120 CsCl, 2 MgCl2, 5 EGTA, 10 HEPES, 0.3 Na3-GTP, 4 Na2-ATP (pH 7.35). Cultures were continuously superfused with external solution (in mM, 100 NaCl, 26 NaHCO3, 2.5 KCl, 11 glucose, 2.5 CaCl2, 1.3 MgSO4, 1.0 NaH2PO4). For mEPSC recording, tetrodotoxin (TTX) (1 μM) and picrotoxin (100 μM) were included in the external saline. Cells were held at −60 mV. To test for the presence of calcium permeable AMPA receptors on the post-synaptic membrane, 10 μM 1-Naphtylacetylspermine trihydrochloride (NASPM) was bath perfused for 10 min before recording. mEPSCs were collected using Clampex (Axon laboratory) and analyzed with Mini Analysis Program (Synaptosoft).
Statistical Analysis
T-test was used for statistical analysis. Values are presented as mean ± SEM in the figures.
Drugs and Chemicals
RA, Picrotoxin, Naphtyl-acetyl-spermine trihydrochloride, and 4-Diethylaminobenzaldehyde were purchased from Sigma-Aldrich. TTX was purchased from Ascent Scientific (Bristol, UK) and D-(-)-2-Amino-5-phosphonopentanoic acid was purchased from Tocris Biosciences (Ellisville, MO).
Subcellular Localization of RARα Domains
Rat neuronal cultures were co-transfected at 12 DIV using Lipofectamine 2000 with pmCherry-N1, a plasmid that expresses high levels of mCherry to allow neuronal visualization, and pEGFP-N1 expressing various truncated versions of RARα. Cells were then fixed at 14 DIV with 4% para-Formaldehyde, washed with PBS before mounting. Images were acquired using an Olympus FV1000 BX61WI laser-scanning confocal microscope.
Results
Lentivirus-Mediated Conditional Knockout (KO) of RARα in Hippocampal Neurons Cultured from Postnatal RARαfl/fl Mice
We previously investigated the involvement of RARα in activity blockade-induced homeostatic synaptic plasticity (also called synaptic scaling) using shRNA-mediated knockdown (Aoto et al., 2008). However, shRNA-mediated knockdowns suffer from a number of intrinsic limitations, such as potential off-target effects and the fact that no knockdown is ever complete. To test whether the shRNA-dependent results were reliable, we used a previously described conditional floxed RARα mouse (Chapellier et al., 2002). The RARαfl/fl mouse contains loxP sites flanking exon 4 (this exon was called exon 8 at the time of publication and was later corrected; Norbert Ghyselinck, personal communication) (Figure 1A). Cre-mediated recombination leads to excision of exon 4 that encodes a significant part of the DNA-binding domain of RARα (Zelent et al., 1989); after excision of exon 4, the exon 3/5 junction shifts the reading frame, resulting in an RARα null allele (Chapellier et al., 2002) (Figure 1A). Because exon 4 is the first common exon shared by all RARα isoforms (Leroy et al., 1991), its excision leads to a complete deletion of all RARα isoforms.
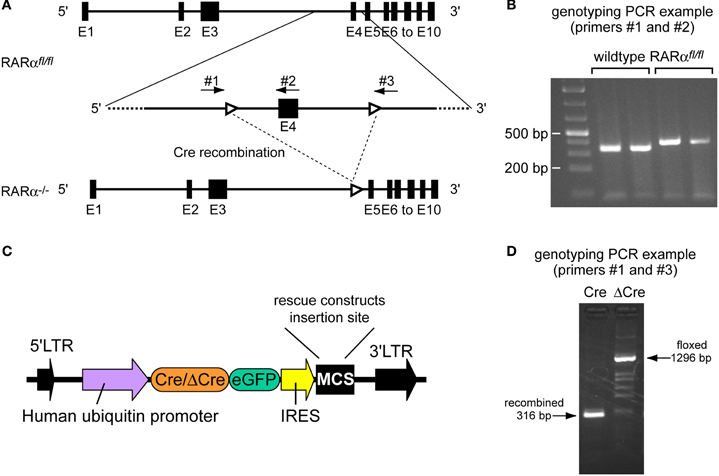
Figure 1. Characterization of the RARα conditional allele. (A) Schematic drawing representing the RARα loxP-flanked allele (RARα fl/fl), as well as the same allele following Cre-mediated excision of exon 4 (RARα−/−). Black boxes represent exons 1–10 (E1–E10). White arrowheads represent loxP sites. The arrows indicate location of primers 1–3 used for characterization of the loxP-flanked allele and the recombined allele. Schematic is based on Gene ID: 19401. (B) Tail DNAs extracted from RARαfl/fl and wild-type C57BL/6 mice were genotyped by PCR using primers number 1 and number 2. Wild-type: 390 bp; floxed allele: 453 bp. (C) Schematic representation of the lentiviral transfer vector used to express Cre-recombinase or ΔCre and RARα rescue constructs. (D) DNA from dissociated hippocampal neuronal cultures infected with either the active recombinase Cre or the inactive form ΔCre was harvested and genotyped by PCR using primers 1 and 3 to test for successful recombination indicated by the presence of a recombined allele (316 bp) or of a floxed allele (1296 bp).
We confirmed the presence of the loxP sites in conditional RARα KO mice by PCR analysis of tail DNA (Figure 1B). To obtain RARα KO neurons, we cultured hippocampal neurons from newborn RARαfl/fl mice and infected the neurons with lentiviruses expressing recently optimized versions of EGFP-tagged active or inactive Cre-recombinase (Cre or DCre) (Kaeser et al., 2011). Rescue experiments were performed by co-expressing various RARα rescue proteins from the same vector as Cre-recombinase via an IRES sequence (Figures 1C,D).
Synaptic Scaling is Absent in RARα KO Neurons
We first tested whether the genetic KO of RARα impairs homeostatic synaptic plasticity. Under control conditions, Cre-expressing conditional RARα KO neurons did not show changes in miniature excitatory post-synaptic current (mEPSC) amplitude or frequency, suggesting that RARα is not required for the maintenance of basal excitatory synaptic transmission (Figures 2A–D). To induce synaptic scaling, we applied a 24 h treatment of TTX and D-2-amino-5-phosphonovalerate (APV), a well-established activity blockade protocol that activates homeostatic synaptic plasticity (Ju et al., 2004; Sutton et al., 2006; Aoto et al., 2008). TTX + APV treatment induced a robust increase in mEPSC amplitude in uninfected and ΔCre-infected neurons (Figure 2B). Strikingly, deletion of RARα by Cre-recombinase in RARα conditional KO neurons blocked this effect (Figure 2B).
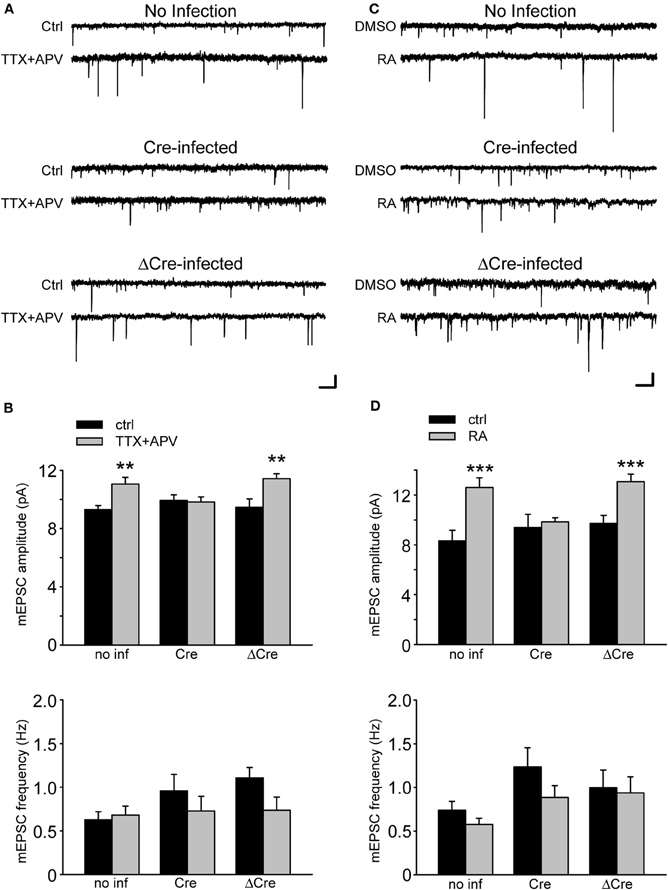
Figure 2. Retinoic Acid Receptor α is required in synaptic scaling induced by activity blockade and by retinoic acid (RA). (A) Representative traces of mEPSCs from RARαfl/fl neurons infected with a lentiviral vector expressing Cre-recombinase or the inactive ΔCre. Neurons have been treated with TTX and APV for 24 h. Scale bars: 10 pA, 0.5 s. (B) Quantification of average mEPSC amplitude (no inf/ctrl: 9.29 ± 0.29 pA; no inf/TTX + APV: 11.05 ± 0.46 pA; Cre/ctrl: 9.94 ± 0.39 pA; Cre/TTX + APV: 9.83 ± 0.34 pA; ΔCre/ctrl: 9.46 ± 0.57 pA; ΔCre/TTX + APV: 11.43 ± 0.33 pA; n = 12–19 from three independent experiments; **p < 0.005) and frequency (no inf/ctrl: 0.63 ± 0.09 Hz; no inf/TTX + APV: 0.68 ± 0.10 Hz; Cre/ctrl: 0.96 ± 0.19 Hz; Cre/TTX + APV: 0.73 ± 0.17 Hz; ΔCre/ctrl: 1.11 ± 0.12 Hz; ΔCre/TTX + APV: 0.74 ± 0.15; n = 12–19 from three independent experiments from experiments; p > 0.5) shown in (A). (C) Representative traces of mEPSCs from RARα KO neurons treated with RA for 30 min followed by 1 h of wash out. Scale bars: 10 pA, 0.5 sec. (D) Quantification of mEPSC amplitude (no inf/ctrl: 8.31 ± 0.86 pA; no inf/RA: 12.60 ± 0.76 pA; Cre/ctrl: 9.37 ± 1.07 pA; Cre/RA: 9.83 ± 0.34 pA; ΔCre/ctrl: 9.72 ± 0.63 pA; ΔCre/RA: 13.06 ± 0.59 pA; n = 12–19, from three independent experiments; ***p < 0.001) and frequency (no inf/ctrl: 0.73 ± 0.10 Hz; no inf/RA: 0.58 ± 0.07 Hz; Cre/ctrl: 1.23 ± 0.22 Hz; Cre/RA: 0.89 ± 0.14 Hz; ΔCre/ctrl: 1.00 ± 0.20 Hz; ΔCre/RA: 0.94 ± 0.18 Hz; n = 12–19, from three independent experiments; p > 0.5) from experiments shown in (C).
Our previous work established a role of RA in mediating activity blockade-induced homeostatic plasticity. Specifically, we found that blocking synaptic activity rapidly induces RA synthesis in neurons, and that the newly synthesized RA increases excitatory synaptic transmission through activation of dendritic protein synthesis (Aoto et al., 2008; Maghsoodi et al., 2008). Consistent with this observation, the conditional deletion of RARα eliminated the ability of RA to increase the mEPSC amplitude (Figures 2C,D), thus confirming the role of RARα in synaptic RA-signaling using a genetic approach.
RARα Mutants Exhibit Distinct Subcellular Localizations
The absence of synaptic scaling and RA-signaling in conditional RARα KO neurons provides a basis for a structure-function analysis of RARα. We, therefore, generated RARα deletion mutants to investigate the contribution of various protein domains to RARα function. Similar to other members of the nuclear receptor superfamily, RARα protein has a modular domain structure, which is comprised of six regions: the N-terminal trans-activation domain (A/B region), the DNA binding domain (DBD or C region), a hinge region (D region), a LBD (or E region) (Evans, 1988; Green and Chambon, 1988; Tora et al., 1988a,b; Tasset et al., 1990), and a C-terminal F-domain whose function is unclear but that we previously implicated in mRNA binding (Poon and Chen, 2008) (Figure 3A).
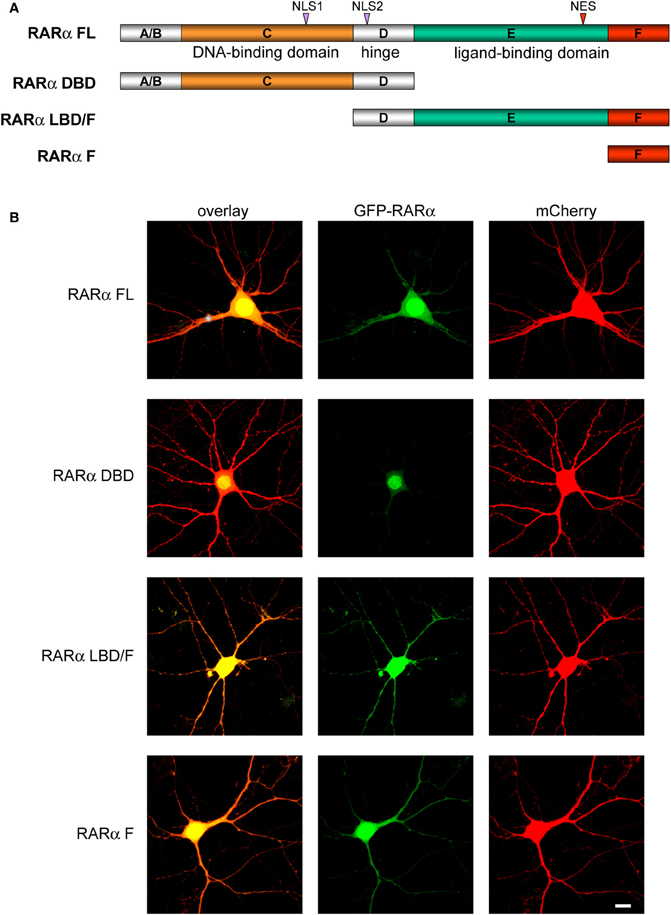
Figure 3. Subcellular localization of RARα and its deletion mutants. (A) Schematic of RARα deletion constructs. Recombinant full-length and truncated RARα domains were encoded as N-Terminal eGFP fusion proteins (FL, Full-length; DBD, N-terminal DNA Binding Domain; LBD; F, C-Terminal F-domain). NLS, Nuclear Localization Signal; NES, Nuclear Export Signal. (B) Subcellular distribution of full-length or truncated RARα proteins (green). mCherry (red) was co-expressed to visualize neuronal morphology. Scale bar: 10 μm.
We generated three deletion mutants of RARα: RARα DBD, RARα LBD/F (LBD and F-domain), and RARα F (F-domain containing the terminal α-helix of LBD-H12) (Figure 3A). When expressed in cultured neurons as GFP-tagged proteins, the different RARα proteins exhibited distinct subcellular distributions. Compared to full-length RARα which was present in both the nucleus and the cytosol, RARα DBD was confined to the nucleus, presumably because of its two nuclear localization signals (NLS) (Hamy et al., 1991) (Figure 3B). By contrast, significant amounts of both the RARα LBD/F and the F-domain protein were present outside of the nucleus and present at high levels in dendrites (Figure 3B).
Rescue of Synaptic Scaling in RARα KO Neurons
Individual domains of RARα are associated with different functions. The two most important domains for mediating RA's effect on protein translation are the LBD and the F-domain—the F-domain is required for RARα's ability to bind to mRNA and to repress translation, while the LBD binds to RA and acts as a switch to allow translational activation (or de-repression) by RA (Poon and Chen, 2008). To directly examine the role of various RARα domains in synaptic scaling, we expressed RARα mutant proteins in neurons with RARα deletion. To achieve this, the various RARα mutants were expressed by inserting their coding sequences after the IRES sequence into the same lentiviral vector that we used for generating RARα KO neurons (Figure 1C). Co-expression of Cre and RARα in the same neurons allows us to examine the effect of rescue by the various RARα deletion mutants in the RARα KO background.
We examined the ability of various RARα mutants to restore RA-induced increase in synaptic strength and to rescue TTX + APV-induced synaptic scaling in RARα KO neurons by co-expressing RARα with Cre in RARαfl/fl neurons. As expected, expression of RARα FL completely rescued synaptic scaling, induced by RA or TTX + APV, in RARα KO neurons (Figures 4B,C). Expression of RARα DBD failed to rescue synaptic scaling by either RA or TTX + APV, which is consistent with its role in DNA- but not RNA-binding (Figures 4B,C). Importantly, RARα LBD/F fully restored synaptic scaling (Figures 4A,B,C). This indicates that the LBD and F-domain, which convey the ligand-binding and RNA-binding ability, respectively, works independently from the DBD in mediating RA's effect at the synapses and is sufficient to restore synaptic scaling when expressed at appropriate levels. This result also confirms the notion that RA and RARα function in a transcription-independent manner in synaptic scaling as the RARα LBD/F lacks the ability to bind DNA. Not surprisingly, when only the F-domain is expressed in RARα KO neurons, RA failed to increase synaptic strength (Figures 4A,B). Likewise, synaptic scaling induced by TTX + APV was absent when only F-domain is expressed (Figure 4C), demonstrating that activation of RARα by RA is a required step in synaptic scaling. Moreover, when RA synthesis is blocked by 4-diethylamino-benzaldehyde (DEAB), an inhibitor for RALDH (Russo et al., 1988), TTX + APV-induced synaptic scaling was completely blocked in RARα LBD/F-expressing neurons (Figures 5A,B).
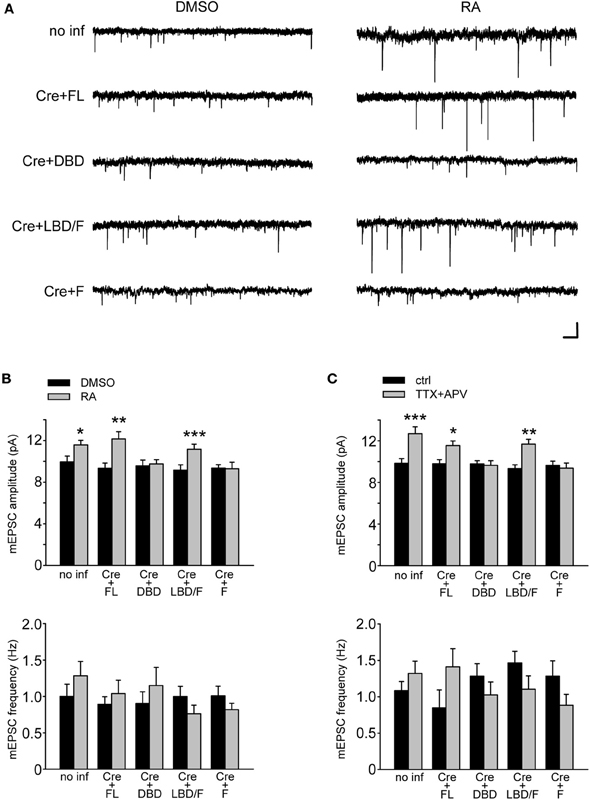
Figure 4. Functional rescue of synaptic scaling in RARα KO neurons RARα expression. (A) Representative traces of mEPSCs from RARαfl/fl hippocampal neurons infected with a bicistronic lentiviral vector coding for Cre-recombinase and either FL RARα or truncated RARα proteins. Neurons were treated with RA or with DMSO as control. Scale bars: 10 pA, 0.5 s. (B) mEPSCs amplitude and frequency were quantified showing rescue of RA-induced synaptic scaling in neurons expressing FL or LBD/F RARα. Amplitude: no inf/DMSO: 9.94 ± 0.56 pA; no inf/RA: 11.58 ± 0.45 pA; Cre + FL/DMSO: 9.34 ± 0.50 pA; Cre + FL/RA: 12.15 ± 0.70 pA; Cre + DBD/DMSO: 9.55 ± 0.57 pA; Cre + DBD/RA: 9.75 ± 0.4 pA; Cre + LBD-F/DMSO: 9.15 ± 0.52 pA; Cre + LBD-F/RA: 11.15 ± 0.50 pA; Cre + F/DMSO: 9.34 ± 0.34 pA; Cre + F/RA: 9.30 ± 0.62 pA; n= 12–19, from three independent experiments; *p < 0.05; **p < 0.005; ***p < 0.001. Frequency: no inf/DMSO: 1.00 ± 0.17 Hz; no inf/RA: 1.28 ± 0.20 Hz; Cre + FL/DMSO: 0.89 ± 0.11 Hz; Cre + FL/RA: 1.04 ± 0.18 Hz; Cre + DBD/DMSO: 0.90 ± 0.16 Hz; Cre + DBD/RA: 1.15 ± 0.25 Hz; Cre + LBD-F/DMSO: 1.00 ± 0.14 Hz; Cre + LBD-F/RA: 0.76 ± 0.12 Hz; Cre + F/DMSO: 1.01 ± 0.13 Hz; Cre + F/RA: 0.82 ± 0.09 Hz; n= 12–19, from three independent experiments; p > 0.5. (C) Synaptic scaling induced by activity blockade is rescued by LBD/F-domain expression in neurons treated with TTX and APV for 24 h. Amplitude: no inf/ctrl: 9.83 ± 0.45 pA; no inf/TTX + APV: 12.69 ± 0.65 pA; Cre + FL/ctrl: 9.79 ± 0.39 pA; Cre + FL/TTX + APV: 11.55 ± 0.43 pA; Cre + DBD/ctrl: 9.78 ± 0.30 pA; Cre + DBD/TTX + APV: 9.63 ± 0.44 pA; Cre + LBD-F/ctrl: 9.33 ± 0.35 pA; Cre + LBD-F/TTX + APV: 11.67 ± 0.48 pA; Cre + F/ctrl: 9.64 ± 0.40 pA; Cre + F/TTX + APV: 9.36 ± 0.50 pA; n= 12–19, from three independent experiments; *p < 0.05; **p < 0.005; ***p < 0.001. Frequency: no inf/ctrl: 1.09 ± 0.12 Hz; no inf/TTX + APV: 1.32 ± 0.17 Hz; Cre + FL/ctrl: 0.85 ± 0.25 Hz; Cre + FL/TTX + APV: 1.41 ± 0.25 Hz; Cre + DBD/ctrl: 1.28 ± 0.17 Hz; Cre + DBD/TTX + APV: 1.03 ± 0.18 Hz; Cre + LBD-F/ctrl: 1.46 ± 0.16 Hz; Cre + LBD-F/TTX + APV: 1.1 ± 0.18 Hz; Cre + F/ctrl: 1.28 ± 0.21 Hz; Cre + F/TTX + APV: 0.88 ± 0.15 Hz; n = 12–19, from three independent experiments; p > 0.5.
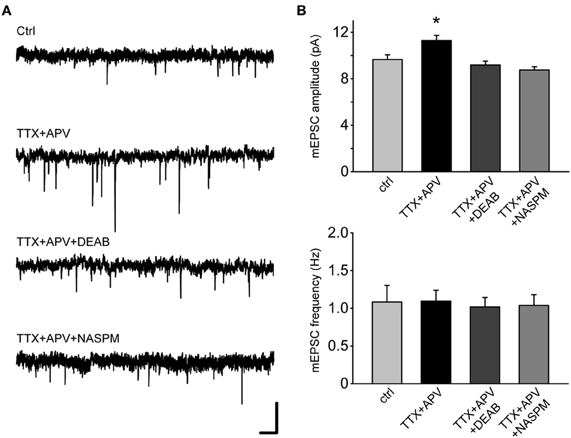
Figure 5. Synaptic scaling mediated by LBD/F-domain depends on RA synthesis and leads to synaptic insertion of calcium permeable AMPA receptors.(A) Representative traces from RARα KO neurons expressing the LBD/F-domain. To verify the dependence of synaptic scaling mediated by the LBD/F-domain on RA synthesis, neurons were co-treated for 24 h with TTX, APV and DEAB. In addition, to test whether synaptic scaling mediated by the LBD/F-domain manifests as the insertion of calcium permeable AMPA receptors, neurons were pretreated with TTX and APV for 24 h and NASPM was bath applied during mEPSCs recording. Scale bars: 10 pA, 0.5 s. (B) mEPSCs amplitude and frequency analysis of (A). Amplitude: ctrl: 9.65 ± 0.40 pA; TTX + APV: 11.29 ± 0.45 pA; TTX + APV + DEAB: 9.18 ± 0.32 pA; TTX + APV + NASPM: 8.74 ± 0.27 pA (n = 16–18, from three independent experiments; *p < 0.05). Frequency: ctrl: 1.08 ± 0.22 Hz; TTX + APV: 1.09 ± 0.14 Hz; TTX + APV + DEAB: 1.02 ± 0.13 Hz; TTX + APV + NASPM: 1.04 ± 0.14 Hz (n = 16–18, from three independent experiments, p > 0.5).
Results from above establish that two RARα activities primarily mediate synaptic scaling: the RNA binding by the F-domain, and RA binding by the LBD. While the former interaction silences translation of target mRNAs, the latter interaction acts as a switch to turn on translation of substrate mRNAs in neuronal dendrites (Poon and Chen, 2008). Our previous work demonstrated that one of the essential targets of RA/RARα regulation of protein translation in the context of synaptic scaling is the AMPA receptor subunit GluA1 (Poon and Chen, 2008). As a result, the newly inserted synaptic AMPA receptors after TTX + APV treatment are calcium-permeable GluA1 homomeric receptors that are sensitive to polyamine blockers (Aoto et al., 2008). We, therefore, asked whether synaptic scaling rescued in RARα LBD/F-expressing neurons is also mediated by synaptic insertion of GluA1 homomeric receptors. 1-naphthyl acetyl spermine (NASPM), a synthetic analog of Joro Spider toxin in the polyamine toxin family, completely reversed the increase in mEPSC amplitude in TTX + APV-treated neurons (Figures 5A,B), indicating that RARα LBD/F likely activates the signaling pathway identical to that of full-length RARα upon activity blockade and is fully capable of mediating RA-signaling in the context of synaptic scaling.
Validation of shRNA-Based Knockdown and Rescue Approach to Study RARα Function
We previously suggested a critical involvement of RARα in homeostatic synaptic plasticity by using a shRNA-based knockdown approach (Aoto et al., 2008). Although accepted as a useful and less time-consuming method in understanding the function(s) of a particular protein, the RNAi method also faced skepticism given the potential off-target effects inherent to the approach. We, therefore, decided to go back to this system and compare the results obtained with the knockdown-rescue approach with that of the genetic approach. We performed standard knockdown-rescue experiments by transfecting cultured hippocampal neurons with plasmids expressing both a shRNA against RARα and a truncated version of the RARα that is also resistant to the shRNA (rescue), and subject these neurons to 24 h TTX + APV treatment. The results thus obtained completely corroborate those obtained with the genetic KO-rescue experiments (Figures 6A–C), demonstrating that the shRNA approach is a valid approach for studying RARα functions in mature neurons.
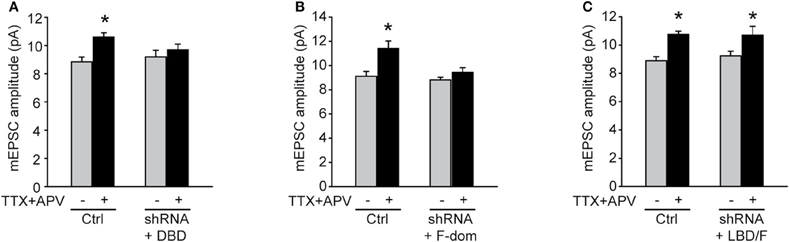
Figure 6. LDB/F-domain is required for activity blockade induced synaptic scaling. Rat primary hippocampal cultures were co-transfected with shRNA against RARα and three different truncated forms of RARα spanning (A) the DBD, (B) the F-domain, and (C) the LBD/F-domains. Synaptic scaling induced by activity blockade with TTX and APV for 24 h was rescued by co-expression of an RNAi resistant form of the LBD/F-domain as shown by quantitative analysis of mEPSCs amplitude (A: ctrl/no treatment: 8.85 ± 0.32 pA; ctrl/TTX + APV: 10.60 ± 0.29 pA; shRNA + DBD/no treatment: 9.20 ± 0.47 pA; shRNA + DBD/TTX + APV: 9.70 ± 0.40 pA. B: ctrl/no treatment: 9.11 ± 0.40 pA; ctrl/TTX + APV: 11.40 ± 0.61 pA; shRNA + F/no treatment: 8.80 ± 0.22 pA; shRNA + F/TTX + APV: 9.43 ± 0.40 pA. C: ctrl/no treatment: 8.90 ± 0.28 pA; ctrl/TTX + APV: 10.75 ± 0.25 pA; shRNA + LBD-F/no treatment: 9.24 ± 0.32 pA; shRNA + LBD-F/TTX + APV: 10.70 ± 0.62 pA. n= 9–12, from two independent experiments; *p < 0.05).
Synaptic Scaling in Neurons Overexpressing Various RARα Mutants
Results from the rescue experiments done in RARα KO or KD neurons provided important functional information regarding the contribution of different RARα domains to synaptic RA-signaling. We were curious whether any of the RARα deletion mutants could act dominant negatively in neurons expressing normal levels of endogenous RARα. To achieve this, we simply co-expressed ΔCre with various RARα fragments, using the same lentiviral vector that delivered Cre and RARα in the rescue experiments.
Interestingly, when challenged with RA treatment, neurons overexpressing RARα full-length protein failed to up-regulate mEPSC amplitude to a similar level as the uninfected neurons (Figures 7A,B), suggesting that increasing expression levels of RARα compromises a neuron's ability to undergo up-regulation of synaptic strength by RA. This result is somewhat unexpected as it cannot be explained by a simple dominant negative scenario, and is not likely due to general side effects associated with viral infection because RARα FL rescued synaptic scaling when expressed in RARα KO neurons.
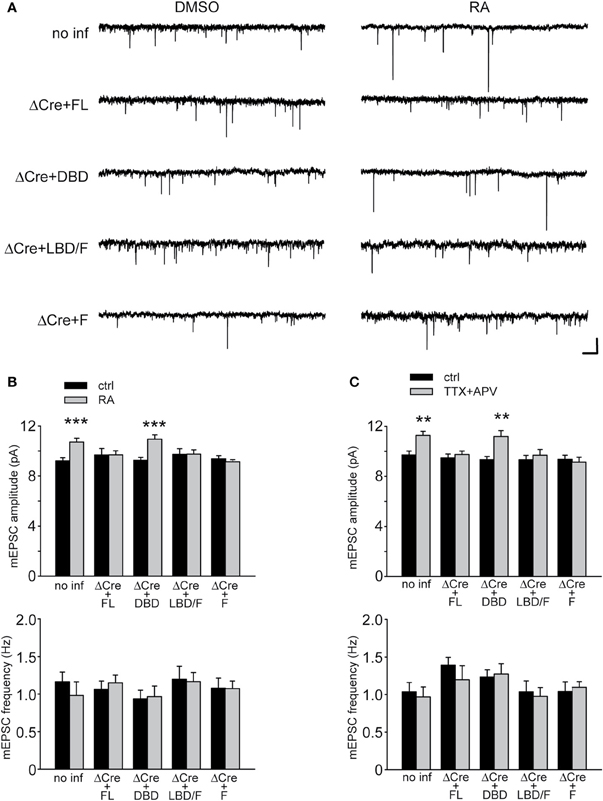
Figure 7. Synaptic scaling in neurons overexpressing RARα domains. (A) Representative traces from RARαfl/fl hippocampal neurons infected with lentiviral vectors co-expressing ΔCre and either full-length or deletion mutants of RARα. Neurons were treated with RA or with DMSO as control and mEPSCs recorded at −60 mV. Scale bars: 10 pA, 0.5 s. (B) Quantitative analysis of mEPSCs amplitude and frequency in neurons treated with RA. Amplitude: no inf/DMSO: 9.20 ± 0.25 pA; no inf/RA: 10.70 ± 0.31 pA; ΔCre + FL/DMSO: 9.67 ± 0.51 pA; ΔCre + FL/RA: 9.67 ± 0.33 pA; ΔCre + DBD/DMSO: 9.25 ± 0.23 pA; ΔCre + DBD/RA: 10.92 ± 0.34 pA; ΔCre + LBD-F/DMSO: 9.71 ± 0.46 pA; ΔCre + LBD-F/RA: 9.74 ± 0.34 pA; ΔCre + F/DMSO: 9.36 ± 0.25 pA; ΔCre + F/RA: 9.13 ± 0.16 pA; n= 16–18, from three independent experiments; ***p < 0.001. Frequency: no inf/DMSO: 1.17 ± 0.13 Hz; no inf/RA: 0.98 ± 0.18 Hz; ΔCre + FL/DMSO: 1.06 ± 0.11 Hz; ΔCre + FL/RA: 1.15 ± 0.11 Hz; ΔCre + DBD/DMSO: 0.94 ± 0.11 Hz; ΔCre + DBD/RA: 0.97 ± 0.14 Hz; ΔCre + LBD-F/DMSO: 1.20 ± 0.18 Hz; ΔCre + LBD-F/RA: 1.17 ± 0.12 Hz; ΔCre + F/DMSO: 1.08 ± 0.13 Hz; ΔCre + F/RA: 1.07 ± 0.10 Hz; n= 16–18, from three independent experiments; p > 0.5. (C) Quantitative analysis of mEPSCs amplitude and frequency recorded in RARα KO hippocampal neurons upon treatment with TTX and APV for 24 h. Amplitude: no inf/ctrl: 9.71 ± 0.31 pA; no inf/TTX + APV: 11.27 ± 0.33 pA; ΔCre + FL/ctrl: 9.48 ± 0.31 pA; ΔCre + FL/TTX + APV: 9.74 ± 0.28 pA; ΔCre + DBD/ctrl: 9.34 ± 0.25 pA; ΔCre + DBD/TTX + APV: 11.19 ± 0.48 pA; ΔCre + LBD-F/ctrl: 9.33 ± 0.35 pA; ΔCre + LBD-F/TTX + APV: 9.69 ± 0.46 pA; ΔCre + F/ctrl: 9.36 ± 0.33 pA; ΔCre + F/TTX + APV: 9.14 ± 0.39 pA; n= 16–18, from three independent experiments; **p < 0.005. Frequency: no inf/ctrl: 1.04 ± 0.12 Hz; no inf/TTX + APV: 0.97 ± 0.13 Hz; ΔCre + FL/ctrl: 1.39 ± 0.11 Hz; ΔCre + FL/TTX + APV: 1.20 ± 0.19 Hz; ΔCre + DBD/ctrl: 1.23 ± 0.10 Hz; ΔCre + DBD/TTX + APV: 1.27 ± 0.14 Hz; ΔCre + LBD-F/ctrl: 1.04 ± 0.14 Hz; ΔCre + LBD-F/TTX + APV: 0.98 ± 0.12 Hz; ΔCre + F/ctrl: 1.04 ± 0.13 Hz; ΔCre + F/TTX + APV: 1.10 ± 0.07 Hz; n= 16–18, from three independent experiments; p > 0.5.
We then examined RA-induced scaling in neurons overexpressing the RARα DBD. The RARα DBD localizes primarily to the nucleus and does not participate in RNA binding or RA binding. Overexpression of RARα DBD did not affect RA-induced synaptic scaling because endogenous RARα is sufficient to mediate RA's action (Figures 7A,B). This again rules out the possibility of viral infection-induced side effects in general, and suggests instead that the expression levels of RARα affect its function in synaptic signaling. Indeed, similar to RARα full-length overexpression result, overexpression of RARα LBD/F also impaired synaptic scaling by RA (Figures 7A,B), although this mutant RARα is able to bind to mRNA and respond to RA (Poon and Chen, 2008). Interestingly, overexpression of F-domain in neurons also prevented the increase in mEPSC amplitude after RA treatment (Figures 7A,B). Because F-domain mediates RNA-binding, but does not mediate RA-induced translational switch, it can act dominant negatively when expressed alone to compete with endogenous RARα in RNA-binding and sequester RNA substrates.
Given the critical role of RA in activity blockade-induced synaptic scaling, we next examined the effect of overexpression of various RARα mutants in TTX + APV-induced synaptic scaling with the prediction that those mediate by normal RA-induced synaptic scaling should also support TTX + APV-induced synaptic scaling. Indeed, neurons overexpressing RARα DBD did not interfere with the function of endogenous RARα in TTX + APV-induced synaptic scaling, but overexpression of RARα FL, LBD/F, and F-domain blocked synaptic scaling (Figure 7C). These results suggest that RARα expression levels in neurons are tightly coupled to its function, and may be strictly regulated in both developing and mature neurons.
Discussion
The involvement of RARα in RA-mediated homeostatic synaptic plasticity was demonstrated previously using a shRNA-based knockdown method (Aoto et al., 2008). Although rescue experiments with a shRNA-resistant version of full-length RARα was done in that study, the complexity of RNAi experiments does not allow us to completely exclude the possibility of an off-target effect. We intended to achieve two goals in the current study: to validate the shRNA results using a genetic approach, and to perform structure-function analysis of RARα. Indeed, results from the RARα KO neurons confirmed our previous findings, validating the conclusion that RARα is required for homeostatic up-regulation of synaptic strength. Importantly, co-expression of full-length RARα together with the Cre-recombinase successfully rescued synaptic scaling, making this an ideal system for subsequent structure-function analyses.
Our results provide strong evidence for a non-genomic role of RARα in regulating excitatory synaptic strength. Specifically, we show that knocking out RARα in mature neurons acutely blocks homeostatic up-regulation of synaptic strength, a process mediated by RA. The rescue experiments with various forms of mutant RARα revealed that individual RARα domains perform differential functions in RA-mediated homeostatic synaptic plasticity—the DNA-binding activity of RARα was dispensable, while the RNA-binding activity of the F-domain and the RA-binding activity of the LBD were both required. The LBD and F-domains of the receptor do not participate in DNA-binding, and, therefore, are not known to be directly mediating the transcriptional regulation by RARα. Instead, our previous work demonstrated that the F-domain has mRNA binding abilities and that binding occurs in a sequence specific manner. The consensus sequences for binding are potentially present in many dendritically localized mRNAs, in particular the ones encoding proteins known to be involved in synaptic scaling, such as the mRNA coding for the GluA1 subunit of AMPA receptors (Poon and Chen, 2008). In vitro studies suggested that binding of the LBD/F-domain to mRNA regulates translation in an RA-dependent manner so that addition of RA leads to translational de-repression and increased GluA1 protein levels (Poon and Chen, 2008). Recent evidence from our lab and others indicate that RARα protein is not restricted in the nucleus in mature neurons, but can be found also in neuronal dendrites (Huang et al., 2008; Maghsoodi et al., 2008), further supporting a possible non-genomic function of RARα in mature brains. Taken together, we hypothesize that direct binding of RARα to GluA1 mRNA through the F-domain and the ability of RARα to localize to dendrites provides a repertoire of dendritic GluA1 mRNA that is translationally dormant under basal synaptic activity. Another important piece of the puzzle is the regulation of RA synthesis by changes in synaptic activity. We have shown that blocking excitatory synaptic transmission leads to rapid up-regulation of RA synthesis (Aoto et al., 2008), a process that is tightly controlled by dendritic calcium levels (Wang et al., 2011). The RA thus made binds to the LBD domain of RARα and reduces its mRNA-binding affinity, allowing rapid increase of dendritic GluA1 synthesis through translational de-repression (Poon and Chen, 2008). Synaptic insertion of newly synthesized GluA1 homomeric AMPA receptors compensates the decrease in excitatory synaptic transmission. Calcium-permeable AMPA receptors have been implicated in many forms of synaptic plasticity and diseases (Isaac et al., 2007; Liu and Zukin, 2007). In this case, the calcium-permeable nature of these receptors sends a negative feedback signal to reduce and eventually halt RA synthesis, thus stabilizing synaptic strength. In this context, findings from this study on the critical role of the RARα LBD/F-domains in synaptic scaling provide an important functional correlate to the observations previously made in vitro, and also suggest a basic mechanism for translational regulation that allows neurons to quickly respond to changes in activity with increased protein levels.
An unexpected finding of our study is the impact of RARα expression levels on synaptic scaling—synaptic scaling is fully rescued when full-length RARα or the LBD/F-domain of the RARα is expressed in RARα KO neurons, but is impaired by expression of full-length RARα or the LBD/F-domains in WT neurons containing endogenous RARα (Figures 4 and 5). A possible explanation for this observation is that there may be two limiting factors in dendrites for synaptic RA-signaling: the amount of RA produced during activity blockade-induced synthesis, and the amount of mRNAs in dendrites that serve as RARα substrates. When RARα is expressed at higher levels, these two factors can limit synaptic scaling through two non-mutually exclusive mechanisms. If the amount of GluA1 mRNA is limiting, there will be a fraction of RARα that is not mRNA-bound. Additionally, a large part of GluA1 mRNA may fail to localize to dendrites because of their association with somatically localized RARα due to high RARα expression levels. If the amount of RA is limiting, on the other hand, part of dendritic RARα that is GluA1 mRNA-bound may not be activated by RA produced during activity blockade. Additionally, the mRNA-free RARα will further exacerbate the situation by competing with GluA1-bound RARα for RA binding. One or both limiting factors could explain the lack of synaptic scaling upon TTX + APV treatment in the presence of excess RARα. The fact that synaptic scaling induced by direct application of exogenous RA is impaired in RARα full-length and LBD/F-domain expressing WT neurons argues that simply supplying more RA is not enough, and that dendritically localized GluA1 mRNA may be the other limiting factor. The developmentally regulated reduction of RARα expression as neurons become mature (Huang et al., 2008) supports this notion, and suggests that protein expression levels can be tightly coupled with switches of function during development.
In summary, we have in the present study pursued two goals: first, to use rigorous genetic tools to test our hypothesis that RARα, previously only known to function as a nuclear RA-activated transcription factor, leads a double life as a dendritic repressor of protein translation whose repressive activity is reversed by RA (Aoto et al., 2008); and second, to examine whether the same protein domains of RARα are required for this double life, or whether there is a dissociation in domain requirements for its two functions. Our data confirm RARα's double life, validating that this RA-regulated protein exhibits an amazing and unexpected versatility in which RA activates transcription of genes but inactivates repression of protein translation. Moreover, together with previous studies on the structure/function relationship of RARα as a transcription factor (Evans, 1988; Green and Chambon, 1988; Tora et al., 1988a,b; Tasset et al., 1990), our data reveal that the two functional lives of RARα depend on distinct protein domains, whereby RA-binding to the LBD serves as the central event that regulates either an N-terminal transcription activity (via the DBD that is not needed for the translational function of RARα ) or a C-terminal translation repression activity (via the F-domain). Thus, the architecture of RARα exhibits an exquisite symmetry in which the same central regulatory domain (the LBD) acts either on a transcriptional or translational control element, both of which affect gene expression as the final readout but operate by completely different mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320.
Chapellier, B., Mark, M., Garnier, J. M., LeMeur, M., Chambon, P., and Ghyselinck, N. B. (2002). A conditional floxed (loxP-flanked) allele for the retinoic acid receptor alpha (RARalpha) gene. Genesis 32, 87–90.
Chiang, M. Y., Misner, D., Kempermann, G., Schikorski, T., Giguere, V., Sucov, H. M., Gage, F. H., Stevens, C. F., and Evans, R. M. (1998). An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron 21, 1353–1361.
Cocco, S., Diaz, G., Stancampiano, R., Diana, A., Carta, M., Curreli, R., Sarais, L., and Fadda, F. (2002). Vitamin A deficiency produces spatial learning and memory impairment in rats. Neuroscience 115, 475–482.
Dev, S., Adler, A. J., and Edwards, R. B. (1993). Adult rabbit brain synthesizes retinoic acid. Brain Res. 632, 325–328.
Green, S., and Chambon, P. (1988). Nuclear receptors enhance our understanding of transcription regulation. Trends Genet. 4, 309–314.
Hamy, F., Verwaerde, P., Helbecque, N., Formstecher, P., and Henichart, J. P. (1991). Nuclear targeting of a viral-cointernalized protein by a short signal sequence from human retinoic acid receptors. Bioconjug. Chem. 2, 375–378.
Huang, H., Wei, H., Zhang, X., Chen, K., Li, Y., Qu, P., Chen, J., Liu, Y., Yang, L., and Li, T. (2008). Changes in the expression and subcellular localization of RARalpha in the rat hippocampus during postnatal development. Brain Res. 1227, 26–33.
Isaac, J. T., Ashby, M. C., and McBain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C. C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253.
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., and Sudhof, T. C. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295.
Krezel, W., Kastner, P., and Chambon, P. (1999). Differential expression of retinoid receptors in the adult mouse central nervous system. Neuroscience 89, 1291–1300.
Leroy, P., Krust, A., Zelent, A., Mendelsohn, C., Garnier, J. M., Kastner, P., Dierich, A., and Chambon, P. (1991). Multiple isoforms of the mouse retinoic acid receptor alpha are generated by alternative splicing and differential induction by retinoic acid. EMBO J. 10, 59–69.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Maghsoodi, B., Poon, M. M., Nam, C. I., Aoto, J., Ting, P., and Chen, L. (2008). Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 105, 16015–16020.
Misner, D. L., Jacobs, S., Shimizu, Y., de Urquiza, A. M., Solomin, L., Perlmann, T., De Luca, L. M., Stevens, C. F., and Evans, R. M. (2001). Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 98, 11714–11719.
Nam, C. I., and Chen, L. (2005). Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl. Acad. Sci. U.S.A. 102, 6137–6142.
Poon, M. M., and Chen, L. (2008). Retinoic acid-gated sequence-specific translational control by RARalpha. Proc. Natl. Acad. Sci. U.S.A. 105, 20303–20308.
Russo, J. E., Hauguitz, D., and Hilton, J. (1988). Inhibition of mouse cytosolic aldehyde dehydrogenase by 4-(diethylamino)benzaldehyde. Biochem. Pharmacol. 37, 1639–1642.
Soprano, D. R., Qin, P., and Soprano, K. J. (2004). Retinoic acid receptors and cancers. Annu. Rev. Nutr. 24, 201–221.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Tasset, D., Tora, L., Fromental, C., Scheer, E., and Chambon, P. (1990). Distinct classes of transcriptional activating domains function by different mechanisms. Cell 62, 1177–1187.
Tora, L., Gaub, M. P., Mader, S., Dierich, A., Bellard, M., and Chambon, P. (1988a). Cell-specific activity of a GGTCA half-palindromic oestrogen-responsive element in the chicken ovalbumin gene promoter. EMBO J. 7, 3771–3778.
Tora, L., Gronemeyer, H., Turcotte, B., Gaub, M. P., and Chambon, P. (1988b). The N-terminal region of the chicken progesterone receptor specifies target gene activation. Nature 333, 185–188.
Wang, H. L., Zhang, Z., Hintze, M., and Chen, L. (2011). Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. J. Neurosci. 31, 17764–17771.
Zelent, A., Krust, A., Petkovich, M., Kastner, P., and Chambon, P. (1989). Cloning of murine alpha and beta retinoic acid receptors and a novel receptor gamma predominantly expressed in skin. Nature 339, 714–717.
Keywords: retinoic acid, retinoic acid receptor alpha, homeostatic synaptic plasticity, synaptic scaling, mRNA binding, calcium-permeable AMPA receptor, dendritic protein translation, conditional RARalpha knockout
Citation: Sarti F, Schroeder J, Aoto J and Chen L (2012) Conditional RARα knockout mice reveal acute requirement for retinoic acid and RARα in homeostatic plasticity. Front. Mol. Neurosci. 5:16. doi: 10.3389/fnmol.2012.00016
Received: 12 October 2011; Paper pending published: 24 November 2011;
Accepted: 02 February 2012; Published online: 15 February 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Roger L. Papke, University of Florida School of Medicine, USAGong Chen, The Pennsylvania State University, USA
Copyright: © 2012 Sarti, Schroeder, Aoto and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lu Chen, Department of Psychiatry and Behavioral Sciences, Stanford University, Lorry Lokey Stem Cell Research Building, 265 Campus Drive, Room G1034B, Stanford, CA 94305-5453, USA. e-mail: luchen1@stanford.edu