The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer's disease—old wine in new bottles
- 1Molecular and Cellular Neuroscience Laboratory, Department Biochemistry and Molecular Biology, Southern Research Institute, Birmingham, AL, USA
- 2Guest group In vitro-Electrophysiology, Department of Neurophysiology, Medical Faculty, Ruhr-University Bochum, Bochum, Germany
The neuronal Ca2+-sensor (NCS) proteins VILIP-1 and VILIP-3 have been implicated in the etiology of Alzheimer's disease (AD). Genome-wide association studies (GWAS) show association of genetic variants of VILIP-1 (VSNL1) and VILIP-3 (HPCAL1) with AD+P (+psychosis) and late onset AD (LOAD), respectively. In AD brains the expression of VILIP-1 and VILIP-3 protein and mRNA is down-regulated in cortical and limbic areas. In the hippocampus, for instance, reduced VILIP-1 mRNA levels correlate with the content of neurofibrillary tangles (NFT) and amyloid plaques, the pathological characteristics of AD, and with the mini mental state exam (MMSE), a test for cognitive impairment. More recently, VILIP-1 was evaluated as a cerebrospinal fluid (CSF) biomarker and a prognostic marker for cognitive decline in AD. In CSF increased VILIP-1 levels correlate with levels of Aβ, tau, ApoE4, and reduced MMSE scores. These findings tie in with previous results showing that VILIP-1 is involved in pathological mechanisms of altered Ca2+-homeostasis leading to neuronal loss. In PC12 cells, depending on co-expression with the neuroprotective Ca2+-buffer calbindin D28K, VILIP-1 enhanced tau phosphorylation and cell death. On the other hand, VILIP-1 affects processes, such as cyclic nucleotide signaling and dendritic growth, as well as nicotinergic modulation of neuronal network activity, both of which regulate synaptic plasticity and cognition. Similar to VILIP-1, its interaction partner α4β2 nicotinic acetylcholine receptor (nAChR) is severely reduced in AD, causing severe cognitive deficits. Comparatively little is known about VILIP-3, but its interaction with cytochrome b5, which is part of an antioxidative system impaired in AD, hint toward a role in neuroprotection. A current hypothesis is that the reduced expression of visinin-like protein (VSNLs) in AD is caused by selective vulnerability of subpopulations of neurons, leading to the death of these VILIP-1-expressing neurons, explaining its increased CSF levels. While the Ca2+-sensor appears to be a good biomarker for the detrimental effects of Aβ in AD, its early, possibly Aβ-induced, down-regulation of expression may additionally attenuate neuronal signal pathways regulating the functions of dendrites and neuroplasticity, and as a consequence, this may contribute to cognitive decline in early AD.
Introduction
In the old scriptures it is said: neither do men put new wine into old bottles, else the bottles break, and the wine runneth out, and the bottles perish, but they put new wine into new bottles, and both are preserved (Matthew 9:17). In science the phrase “To put old wine in new bottles” is often used when we aim to put old knowledge in the context of new findings, in the hope to create a good tasting wine. Recent findings on the implication of neuronal Ca2+-sensor (NCS) proteins in the etiology of Alzheimer's disease (AD), particularly the role of VILIP-1 as cerebrospinal fluid (CSF) biomarker for AD, its correlation with MMSE scores and predictive value for cognitive decline in healthy individuals (Lee et al., 2008; Craig-Schapiro et al., 2009; Tarawneh et al., 2011), provoke new questions about what role these Ca2+-sensors play in early cognitive impairment in AD. Some of our previous knowledge about these proteins may help to find answers for these questions. Thus, in this review I will focus on two members of the visinin-like protein (VSNL)-subfamily of NCS proteins, VILIP-1 and VILIP-3, reiterate some of the background information about these Ca2+-signaling proteins, and summarize the current knowledge about their effects on neuronal signaling, which may be of potential relevance for the understanding of their link to disease severity and early cognitive decline in AD.
The Visinin-Like Proteins
Multiple Ca2+-sensing proteins have been identified in the central nervous system (CNS) over the last decades, reflecting the importance of the fine-tuning of the regulative function of Ca2+ in neurons. Several of these proteins have been grouped together and termed NCS proteins (Nef, 1996; Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001; Burgoyne, 2007). Fourteen NCS protein genes related to the ubiquitous Ca2+-sensor protein calmodulin exist in various species, and have been subdivided into five subfamilies. VILIP-1 (visinin-like protein 1, gene name VSNL1), VILIP-2 (visinin-like protein 2, gene name hippocalcin-like 4, HPCAL4), VILIP-3 (visinin-like protein 3, gene name HPCAL1), hippocalcin (gene name HPCA) and neurocalcin δ (gene name: NCALD) show amino acid identities between 67% and 94%, and form the subfamily of VSNLs (Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001; Spilker et al., 2002a; Burgoyne et al., 2004; Braunewell and Klein-Szanto, 2009). Branch 1 of the VSNL subfamily consists of VILIP-1 and VILIP-2, which are 89% homologous, and branch 2 consists of hippocalcin and VILIP-3, which share 94% identity, and both are 91% identical to neurocalcin δ (Spilker et al., 2002a). VILIP-1 was first cloned as visinin-like protein in chicken (Lenz et al., 1992), as neural visinin-like protein 1 in rat (NVP-1) (Kuno et al., 1992), as neurocalcin α from cow (Kato et al., 1998) and as VSNL1 in man (Polymeropoulos et al., 1995). VILIP-3 orthologs are rem-1 from chicken (Kraut et al., 1995), NVP-3 from rat (Kajimoto et al., 1993) and hHLP2 from man (Kobayashi et al., 1994). The protein sequences of VILIP-1 and VILIP-3 show 100% evolutionary conservation from chicken to man, indicating an important functional role of these Ca2+-sensors in the CNS of various species.
The Distribution of VILIP-1 and VILIP-3 in the CNS
VSNLs show a distinct but widespread expression pattern with high expression levels in nerve cells (Braunewell and Gundelfinger, 1999), but see Gierke et al. (2004) for an overview on peripheral distribution of the proteins at lower levels. In the periphery VILIP-1 has been implicated in cell migration and is a putative tumor migration suppressor gene in several forms of cancer (for review see Braunewell and Klein-Szanto, 2009). A comprehensive mRNA expression study of the VSNL subfamily describes the expression of VILIP-1, VILIP-2, VILIP-3 and hippocalcin in the rat brain (Paterlini et al., 2000). VILIP-1 mRNA shows a widespread distribution in most brain areas except the caudate-putamen. VILIP-3 exhibits strong expression in the cerebellum where it localizes to Purkinje and granule cells, and additional expression in the forebrain including neocortex, hippocampus and caudate-putamen (Paterlini et al., 2000). Comparative expression studies have been performed at the protein level, including studies of VILIP-1 and VILIP-3 in the rat cerebellum and hippocampus (Spilker et al., 2000), of neurocalcin isoforms α (VILIP-1) and δ in the rat cerebellum (Kato et al., 1998), and of VILIP-1 and VILIP-3 in the human brain (Bernstein et al., 1999). Immunohistochemical studies with VILIP-1-specific antibodies show expression in principal and non-principal neurons. Particularly strong expression levels are found in subpopulations of calbindin-D28K and calretinin-positive GABAergic interneurons in all hippocampal regions in the rat brain (Zhao and Braunewell, 2008). In hippocampal interneurons VILIP-1 co-localizes mainly with the so-called Ca2+-buffer proteins calbindin-D28K and calretinin (60–70%), but much less pronounced with parvalbumin (<10%) (Bernstein et al., 1999; Zhao and Braunewell, 2008). The rat expression profile differs from the profile in the human hippocampus. VILIP-1 immunoreactive neurons were found in the hippocampal CA1, CA4 and hilus regions, but were weak in the CA2 and CA3 areas in the human brain (Bernstein et al., 1999). Strong VILIP-3 protein and mRNA expression has been localized in the cerebellum, but expression in other brain regions including cortex and hippocampus has been observed (Spilker et al., 2000; Hamashima et al., 2001; Spilker and Braunewell, 2003). High expression levels of VILIP-3 exist in the dentate gyrus at the mRNA level (Spilker et al., 2000). VILIP-1 and VILIP-3 co-localize in hippocampal neurons in culture, showing a strong expression for VILIP-1 in many neurons and weaker expression of VILIP-3 in a subset of neurons (Spilker and Braunewell, 2003). To further understand the roles of these proteins in AD pathology, cellular and subcellular co-localization studies of VSNLs with their interaction partners need to be performed in AD brains and in AD animal models in the future.
The CA2+-myristoyl Switch, Target Interaction, and Neuronal CA2+-Signaling
VSNLs consist of 191–193 amino acid residues and harbor EF-hands as Ca2+-binding motif. EF-hands consist of several core amino acids involved in the coordinative binding of Ca2+ (D-X-D/N-X-D/N-X-Y-(X)4-E). All VSNLs possess 4 EF-hands, however, EF-hand 1 is dysfunctional due to changes in the core amino acid sequence (Braunewell, 2009; Braunewell and Klein-Szanto, 2009). At their N-terminus VSNLs bear a consensus sequence (M-G-(X)3-S) for N-terminal myristoylation, which leads to the co-translational attachment of a C14 myristic fatty acid. This modification enables all VSNLs to translocate to subcellular membrane compartments (Kobayashi et al., 1993; Ladant, 1995; Lenz et al., 1996; Spilker et al., 2002b) by a molecular mechanism termed Ca2+-myristoyl switch (Zozulya and Stryer, 1992). The molecular mechanism of the switch has been first analyzed in detail from tertiary structure data for the NCS protein recoverin. Binding of Ca2+ to recoverin induces a conformational change leading to surface exposure of hydrophobic protein parts and exposure of the myristoyl side chain, thereby making these structures available for interaction with cellular membranes and/or target proteins (Tanaka et al., 1995; Ames et al., 1996, 1997). In living cells, after increasing the intracellular Ca2+-concentration, VSNLs can translocate to subcellular membrane compartments (Ivings et al., 2002; Spilker et al., 2002b; O'Callaghan et al., 2002, 2003; Spilker and Braunewell, 2003). However, the Ca2+-dependent subcellular membrane localization of endogenously expressed VILIP-1 and VILIP-3 differed substantially in the same hippocampal neuron. VILIP-1 shows cell surface membrane association, including membranes of axons and dendrites, which is in line with the described function of VILIP-1 as modulator of cell surface associated proteins (Braunewell et al., 1997, 2001b; Lin et al., 2002a,b; Chaumont et al., 2008; Richler et al., 2011). In addition, VILIP-1 only affiliates with trans-Golgi membranes following a Ca2+-stimulus in hippocampal neurons (Spilker and Braunewell, 2003), while VILIP-3 showed a weak Ca2+-independent Golgi localization that was only gradually enhanced following stimulation of hippocampal neurons (Spilker et al., 2002b). Furthermore, VILIP-3 interacts with intracellular juxtanuclear membranes and granular structures in the whole cytosol (Spilker and Braunewell, 2003), which fits to a possible function as a modulator of MAP kinases (Spilker et al., 2002a), and the ER-localized plasma membrane redox system (PMRS) (Oikawa et al., 2004). Interestingly in this context, under conditions of disturbed Ca2+-homeostasis in AD an enhanced juxtanuclear membrane localization of VSNLs exists (Braunewell et al., 2001a; Blandini et al., 2004). We will need additional studies using markers for cellular organelles to understand their distinct subcellular distribution pattern and the re-distribution mechanisms following normal and pathological Ca2+-signals.
The reversible localization of Ca2+-sensors to distinct membrane compartments and signaling scaffolds in living neurons has been postulated to be a signal transduction mechanism for the selective activation of downstream signaling cascades, such as receptors, receptor signaling complexes and signal effector molecules (Spilker et al., 2002b; Spilker and Braunewell, 2003). Besides EF-hand 1 as a putative functional domain for VILIP-1 and VILIP-3, basic amino acids in the N-terminus have been postulated to be involved in interaction with phospholipids, particularly with their phosphatidylinositol phosphate (PIP) headgroups. Unmyristoylated VILIP-1 can bind to artificial phospholipid bilayers in the absence of Ca2+, and monolayer adsorption measurements showed a preference of binding to PI(4,5)P2 over PI(3,4,5)P3 (Braunewell et al., 2010; Wang et al., 2011). Furthermore, VILIP-1 and hippocalcin have been shown to interact with PI(4,5)P2 at the cell surface membrane in hippocampal neurons (O'Callaghan et al., 2005; Braunewell et al., 2010). The functional implications of the additional phospholipid interaction are not yet understood. EF-hand 1 forms the most variable part in the sequence of NCS proteins and, therefore, comprises a possible interaction site with target proteins (Lian et al., 2011). NCSs, such as VSNLs, serve as effectors to transduce cellular Ca2+-signals. Similar to the prototypical Ca2+-sensor calmodulin, the VSNLs appear to be modulators of multiple intracellular targets showing a “pleiotropy” of actions. VILIP-1 affects cAMP- and cGMP signaling and downstream signaling pathways including the rhoA/ROCK signaling pathway (Braunewell et al., 1997, 2001a; Mahloogi et al., 2003; Brackmann et al., 2005; Jheng et al., 2006; Chen et al., 2009). It interacts with several ligand-gated ion channels, such as glutamate receptors of the kainate subtype GluR6 (Coussen et al., 2005), the P2X2 ATP receptor (Chaumont et al., 2008), and the α4β2 nicotinic acetylcholine receptor (nAChR) (Lin et al., 2002a). Interestingly, VILIP-1 forms dimers, which appears to be important for the interaction with receptor dimers/multimers, such as α4β2 nAChR and the natriuretic peptide B receptor (NPR2, NPR-B) (Li et al., 2011; Wang et al., 2011). Functionally, VILIP-1 enhances surface expression of NPR-B, P2X2 ATP receptor and α4β2 nAChR (Brackmann et al., 2005; Chaumont et al., 2008; Gierke et al., 2008). In contrast to VILIP-1, comparatively little is known about signaling activities of VILIP-3. Although it forms dimers with VILIP-1 (Jheng et al., 2006), VILIP-3 does not affect the VILIP-1 target NPR-B (Spilker and Braunewell, 2003; Chen et al., 2009). In contrast, VILIP-3 directly or indirectly enhances activity of ERK1 and ERK2 (extracellular signal-regulated kinase 1 and 2) MAPK (mitogen-activated protein kinase) signaling (Spilker et al., 2002a), and interacts with cytochrome b5, with unknown functional impact (Oikawa et al., 2004).
Neuronal CA2+-Signaling and AD
According to the amyloid β (Aβ) hypothesis, excessive accumulation of Aβ assemblies in the brain is involved in the etiology of AD (Figure 1). Gradual accumulation of aggregated Aβ initiates a complex, multistep cascade that includes inflammatory changes, gliosis, neuritic/synaptic changes, transmitter loss, and formation of neurofibrillary tangles (NFT), leading to cognitive impairments and ultimately to extensive cell death in both sporadic (late-onset AD, LOAD: ApoE4 and other risk factors) and familial or genetically linked AD (FAD with the genetic risk factors APP—amyloid precursor protein, PS1 and PS2—presenilins 1 and 2) (Hardy and Selkoe, 2002; Holtzman et al., 2011). The molecular mechanisms involved are not completely understood. However, any AD hypothesis is challenged to explain that the changes in synaptic physiology and the onset of cognitive impairments long precede the massive cell death that characterizes the later stages of AD. An extension of the Aβ hypothesis to account for this fact, is the Ca2+-hypothesis of AD. The hypothesis aims to explain how abnormal Aβ metabolism induces a change in Ca2+-homeostasis, which then initiates both, the early decline in memory and the later, more massive changes in Ca2+-levels and the following increase in neuronal cell death (for review see Foster, 2007; Bezprozvanny and Mattson, 2008; Berridge, 2010; Supnet and Bezprozvanny, 2010; Chakroborty and Stutzmann, 2011). Aβ induces a massive disturbance of Ca2+-homeostasis by enhancing both, the entry of external Ca2+ as well as the sensitivity of the InsP3 and ryanodine receptors, that release Ca2+ from internal stores. Aβ oligomers have been reported to increase Ca2+-influx by forming Ca2+-pores in the plasma membrane and by regulating existing plasma membrane Ca2+-channels. However, increased intracellular Ca2+-levels are not only functionally linked to Aβ, but also to presenilin mutations and ApoE4 expression. The initial localized up-regulation of Ca2+-levels will then lead to re-modeling of Ca2+-signaling pathways, such as for instance NCS signaling (Figure 1), which leads to subtle effects on neurotransmission and synaptic plasticity underlying cognition (Chakroborty and Stutzmann, 2011), and later to changes affecting mitochondrial (Supnet and Bezprozvanny, 2010) and endoplasmatic reticulum Ca2+-pathways (Bezprozvanny and Mattson, 2008), causing massive neuronal cell death. A current focus of the Ca2+-hypothesis is to understand how the initial subtle dys-regulation of Ca2+-signaling affects neuroplasticity and brings about the early loss of memory. It also has been postulated that since the defect in cognition occurs before there is any sign of massive cell death, the development of drugs to normalize the subtle changes in Ca2+-signaling may arrest the slow progression of AD (Berridge, 2010).
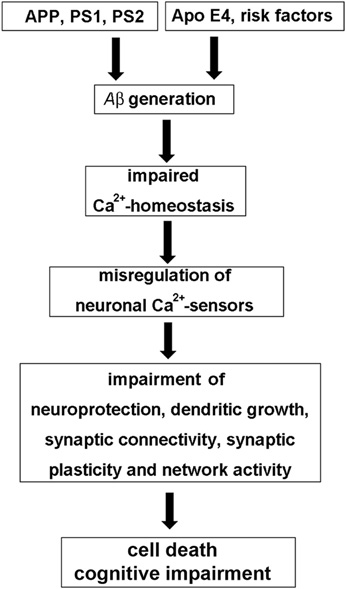
Figure 1. The “neuronal Ca2+-sensor hypothesis” of Alzheimer's disease. In the Aβ hypothesis of AD genetic risk factors, such as mutations in the amyloid precursor protein (APP) and the presenilins PS1 and PS2, but also risks factors such as the apolipoprotein gene ApoeE4, age, nutrition or exercise in sporadic AD lead to the excessive generation of toxic forms of the Aβ peptide, which is derived from APP. Aβ then causes an impairment of neuronal Ca2+-homeostasis resulting in the misregulation of neuronal Ca2+-sensor (NCS) proteins. Pathological expression and function of NCS proteins affects various neuronal signaling pathways involved in neuroprotection, neuronal connectivity, and synaptic plasticity, which, among other mechanisms, contribute to cognitive impairment and cell death observed in AD.
The Role of NCS Proteins in AD: Neuroprotection Versus Neurotoxicity
NCS proteins have been implicated in cognitive processes and in the pathology of AD (for review see Blandini et al., 2004; Buxbaum, 2004; Braunewell, 2005; Braunewell and Bernstein, 2009; Craig-Schapiro et al., 2009). There is less protein expression of VILIP-1, and the numbers of VILIP-1- and, to a lesser degree, VILIP-3-immunoreactive neurons are reduced in the temporal cortex of AD patients, (Bernstein et al., 1999). These data point to a disease-related loss of VSNLs. In AD brains, extracellularly located VSNLs are in close association with the pathologic lesions, such as dystrophic nerve cell processes, amorphous and neuritic plaques, and extracellular NFTs, indicating that they may be involved in the pathophysiology of altered Ca2+-homeostasis in AD (Braunewell et al., 2001a). In PC12 pheochromocytoma cells, VILIP-1 over-expression enhances hyper-phosphorylation of tau protein, which destabilizes microtubules, and, in transfected PC12 cells, it increases Ca2+-mediated cell death (Schnurra et al., 2001). Co-expression of the Ca2+-buffer protein, calbindin-D28K, which is neuroprotective and down-regulated in AD (Iacopino and Christakos, 1992; McMahon et al., 1998), attenuates the effect of VILIP-1 on cell death induced by ionomycin. In this context, VILIP-1 has a widespread distribution in the brain, including in GABAergic neurons (Bernstein et al., 1999). In addition to being expressed in most pyramidal neurons of human and rat hippocampi, it largely co-localizes with calbindin-D28K and calretinin in GABAergic interneurons (Zhao and Braunewell, 2008), but less with parvalbumin-positive interneurons. In interneurons of transgenic AD mouse models and in AD brains, the Ca2+-buffer proteins, calbindin-D28K and calretinin, are down-regulated (Kaufmann et al., 1998; Palop et al., 2003, 2007; Popović et al., 2008; Baglietto-Vargas et al., 2010; Takahashi et al., 2010). These findings suggest that the ratio of expression of Ca2+-sensor to Ca2+-buffer proteins may define subpopulations of neurons particularly vulnerable to Aβ-induced and Ca2+-mediated neurotoxicity (Figure 2). Since interneurons are essential for the generation of synchronous rhythmic activity in the hippocampus, which is underlying cognitive processing/memory encoding, the early alterations of hippocampal inhibitory functionality in AD may result in the cognitive impairments seen in the initial stages of the disease (Palop and Mucke, 2010). On the other hand, for the related VSNL hippocalcin a neuroprotective role has been postulated in age-related neurodegeneration (Masuo et al., 2007). The only other NCS protein that has been implicated in AD (Figure 2) and which is not belonging to the VILIP subfamily, is Calsenilin/DREAM/KChIP3. It was independently identified as a NCS protein that interacts with presenilins (PS1 and 2), serves as a transcription repressor, and binds to A-type potassium channels (Buxbaum et al., 1998; Carrión et al., 1999; An et al., 2000). Calsenilin levels are elevated in the cortex region of AD brains and in the neocortex and the hippocampus of brains of Swedish mutant beta-amyloid precursor protein (sweAPP) transgenic mice. When cultured cortical and hippocampal neurons are exposed to Aβ this induces both calsenilin protein and mRNA expression, and cell death, whereas calsenilin expression blockade protects against Aβ toxicity (Jo et al., 2004). In contrast, in Xenopus oocytes calsenilin reversed the pathogenic effects of mutant PS1 on Ins(1,4,5)P3-mediated Ca2+-signaling. Presenilin mutations perturb intracellular Ca2+-signaling pathways contributing to the key features of AD, such as increased Aβ production, tau hyper-phosphorylation, and enhanced vulnerability to cell death. Calsenilin expression reversed the mutant PS-1-enhanced amplitudes and altered kinetics of Ca2+-signals in oocytes (Leissring et al., 2000). Rivas et al. found recently, using the yeast two-hybrid assay, that DREAM interacts with peroxiredoxin 3 (Prdx3), an antioxidant enzyme found in mitochondria (Rivas et al., 2011). The peroxiredoxin system is a cellular defense system against oxidative stress, and the decreased protein levels of Prdx3 in AD has been discussed as the results of mitochondrial damage, which may reduce cellular protection against oxidative damage (Kim et al., 2001). The Prdx3-DREAM interaction modulates the DREAM redox state and in turn modulates transcriptional repression by DREAM. Since transient DREAM knockdown in PC12 cells sensitizes these cells to H(2)O(2)-induced oxidative stress, this would suggest a protective role for DREAM against oxidative damage (Rivas et al., 2011). In the future the careful re-evaluation of the potential neurotoxic versus neuroprotective roles of NCS proteins in AD-related cellular models and in AD animal models is necessary to define whether different NCS proteins show neurotoxic and/or neuroprotective properties under the specific conditions of disturbed Ca2+-homeostasis in AD brains.
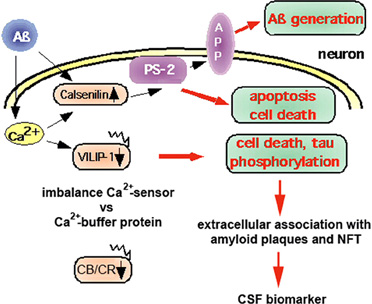
Figure 2. Neuroprotective versus neurotoxic role of Ca2+-binding proteins in the pathology of AD. Aβ deposition is believed to cause changes in the Ca2+-homeostasis that lead to pathological expression and activation of NCS proteins, such as calsenilin and VILIP-1. Calsenilin, which is up-regulated in AD, promotes apoptotic cell death in a manner dependent on presenilin-2 (PS2), but has been found to be neuroprotective in PC12 cells. In contrast, VILIP-1 is down-regulated in AD. Over-expression of VILIP-1 leads to tau phosphorylation and cell death in PC12 neurons, which is attenuated by expression of the Ca2+-buffer protein calbindin-D28K. Thus, in AD brains, an imbalance in the ratio of Ca2+-sensor (Calsenilin, VILIP-1) versus Ca2+-buffer proteins (CB/CR: calbindin-D28K, calretinin) in subpopulations of neurons may lead to their selective vulnerability and early loss, and, in turn, to the appearance of extracellular VSNLs, which are associated with the pathologic hallmarks amyloid plaques and neurofibrillary tangles (NFT). VILIP-1 is detected in the cerebrospinal fluid (CSF) of AD patients, and serves as a CSF biomarker. Increased levels correlate with CSF t-tau, p-tau, the ApoE ε4/ε4 genotype, and with mini mental state examination (MMSE) scores of AD patients.
VSNLs as Biomarkers for AD—Old Wine in New Bottles
Association of VSNLs with the pathologic hallmarks of AD have been published more than a decade ago. More recent support for a functional role of VSNLs in AD comes from genome-wide association studies (GWAS). An SNP for VILIP-1 associates (rs4038131, p = 5.9 × 10−7) with AD and subsequent psychosis (Hollingworth et al., 2011). For VILIP-3, there was an association of 2 SNPs (rs1019785, rs10197851, p = 3.67 × 10−6, p = 7.13 × 10−6) with LOAD; this was replicated in the NIH LOAD data set (Lee et al., 2011). VILIP-1 seems to have additional roles in the cognitive impairments associated with AD. The early findings of reduced protein expression of VSNLs (Bernstein et al., 1999; Schnurra et al., 2001) have been confirmed for brain areas other than the temporal cortex, and extended to changes in mRNA levels. For example, in postmortem brains of AD patients, there is lower expression of VILIP-1 mRNA in the amygdala, cingulate cortex, hippocampus, and cerebellum (Loring et al., 2001; Youn et al., 2007). Whole-genome expression profiling of RNA obtained from the frontal cortex identified genes associated with cognitive decline, and expression of VILIP-1 mRNA correlated with NFT content and with the MMSE scores of AD patients (Wilmot et al., 2008). Similarly, two independent re-analyses were performed on a microarray dataset corresponding to hippocampus gene expression for AD subjects with varying degrees of severity (contributed by Blalock et al., 2004), found down-regulation of VILIP-1 mRNA, association with NFT content in the hippocampus, and again an association with the MMSE score (Miller et al., 2008; Gómez Ravetti et al., 2010). These data indicate that VILIP-1 expression is lost not only due to loss of VILIP-1-expressing neurons (Bernstein et al., 1999; Schnurra et al., 2001), but also to pathological down-regulation of VILIP-1 mRNA levels. The lowered VILIP-1 mRNA expression correlated with the severity of cognitive decline, as measured by MMSE scores for AD subjects. Similarly, an observation by Lee et al., 2008 ties in with these results on VILIP-1-MMSE correlation in microarray studies (Miller et al., 2008; Gómez Ravetti et al., 2010). In this study, CSF samples were analyzed by ELISA to measure concentrations of Aβ1–42, t-tau, p-tau, and VILIP-1. However, in contrast to VILIP-1 mRNA signals, in the CSF of AD subjects, VILIP-1 protein is increased, relative to controls. There was also strong correlation of increased CSF-VILIP-1 with CSF t-tau, p-tau, the ApoE ε4/ε4 genotype, and lowered MMSE scores of AD patients (Lee et al., 2008). The new findings of the appearance of the intracellular protein VILIP-1 in the CSF is consistent with the older view that VILIP-1 is released from neurons during neurotoxic insults, and that extracellular VILIP-1 then associates with the pathologic characteristics of AD (Schnurra et al., 2001; Braunewell et al., 2001a). Thus, although VILIP-1 mRNA appears to be actively down-regulated in AD and the down-regulation correlates with reduced MMSE scores, at the same time neurons expressing VILIP-1 seem to be particularly vulnerable against Aβ-induced disturbances of Ca2+-homeostasis, and these neurons appear to die early on in the disease. This would explain how the intracellular protein is released from neurons and can then be found associated with amyloid plaques and NFT, and finally makes its way into the CSF. Thus, there appears to be significant correlation of reduced VILIP-1 mRNA as well as enhanced CSF protein levels with reduced MMSE scores and early cognitive decline in AD.
VILIP-1 and Cognitive Impairment in AD?
Since VILIP-1 concentrations in the CSF correlate with MMSE scores, CSF-VILIP-1 has been proposed as a marker for declined cognition and disease severity (Verbeek and Olde Rikkert, 2008; Craig-Schapiro et al., 2009). This assumption from the Lee et al., 2008 study prompted a larger study in 300 subjects, where VILIP-1 was confirmed as a CSF biomarker for early AD (Tarawneh et al., 2011). In this study VILIP-1 also showed elevated plasma levels. Importantly, the Holtzman group also describes a 2–3 year follow-up study in cognitively healthy control subjects, in which CSF levels of VILIP-1 in still healthy individuals had predictive value for future cognitive decline (Tarawneh et al., 2011). Thus, the correlation of CSF VILIP-1 with MMSE scores suggests that VILIP-1 in the CSF is a valid biomarker, and is a prognostic marker for cognitive decline in early AD (Craig-Schapiro et al., 2009; Tarawneh et al., 2011). Notably, in non-AD dementias no increased VILIP-1 CSF levels were detected, pointing to the possibility that VILIP-1 may be linked to disease-specific mechanisms or alterations in signaling pathways (Tarawneh et al., 2011). These novel observations should raise a variety of new research questions. More support that VILIP-1 is directly associated with cognitive capabilities comes from a study showing that VSNL1 SNPs are associated with performance in the Wisconsin Card Sorting Test, an assessment of frontal cortical function in schizophrenia patients with cognitive impairments (Braunewell et al., 2011). These results also raise the question of whether VILIP-1, in addition to its role in Aβ-induced and Ca2+-mediated neuronal death, might be involved in neuronal signaling pathways and mechanisms of impaired synaptic plasticity and cognition in AD. Other related NCS proteins, including Calsenilin/DREAM/KChIP3 and NCS-1, affect synaptic plasticity (Sippy et al., 2003; Fontán-Lozano et al., 2009; Saab et al., 2009; Wu et al., 2010). VSNLs are specialized mediators of Ca2+-signals in neuronal signaling processes known to affect cognition. VILIP-2 was shown to slow inactivation of Ca(V)2.1 channels in a myristoylation dependent manner (Few et al., 2005). Ca(V)2.1 channels conduct P/Q-type Ca2+-currents, and initiate synaptic transmission at most synapses in the CNS. The VILIP-2-dependent facilitation and inactivation of these channels contributes to short-term synaptic plasticity (Nanou et al., 2012). Hippocalcin acts as Ca2+-sensor for hippocampal long-term depression (Palmer et al., 2005), and hippocalcin knockout animals display impaired spatial and associative memory (Kobayashi et al., 2005). Moreover, it was shown show that expression of hippocalcin, but not of a hippocalcin myristoylation mutant, leads to an enhanced slow afterhyperpolarization current I(sAHP) in cultured hippocampal neurons. A train of action potentials activates potassium channels in a Ca2+-dependent manner to produce the sAHP current, which in turns dampens neuronal excitability. This was strongly reduced in hippocalcin knockout animals (Tzingounis et al., 2007). The modulation of sAHP currents is believed to regulate neuronal excitability, synaptic efficacy, and the threshold for tetanus-induced synaptic plasticity. Hippocalcin and neurocalcin δ, but not VILIP-2, can also act as a Ca2+-sensor for the sAHP current in the cerebral cortex, indicating that VSNLs can gate sAHPs and thus neuronal excitability in various brain regions (Villalobos and Andrade, 2010). It is likely that VILIP-1 and -3 have similar functions in regulating neuronal excitability and synaptic plasticity, particularly since VILIP-1 up-regulation has been linked to mGluR-dependent long-term potentiation (Braunewell et al., 2003; Brackmann et al., 2004).
VILIP-1, Dendritic Connectivity and Cognitive Impairment in AD
A major question is how VILIP-1 and VILIP-3 may influence cognition. One possibility for VILIP-1 is that it activates cyclic AMP- and cyclic GMP-signaling by enhancing surface expression of membrane-localized adenylyl and guanylyl cyclases (Brackmann et al., 2005; Braunewell et al., 2011). Thereby, VILIP-1 might influence cAMP and cGMP-dependent neuronal processes, including neuronal differentiation, neurite outgrowth, different forms of synaptic plasticity and learning and memory (Schuman and Madison, 1991; Telegdy, 1994; Monfort et al., 2002). The regulation of cAMP-levels by VILIP-1 has been initially detected in stably transfected rat C6 glioma cells (Braunewell et al., 1997). The myristoylation-deficient mutant of VILIP-1, which lacks the myristoylation consensus motif and, therefore, does not exhibit the Ca2+-myristoyl switch, showed a dominant-negative effect on cAMP-levels in C6 cells. Already basic cAMP levels appeared to be elevated in VILIP-1-transfected C6 cells, which is the cause for induction of differentiation of those glioma cells (Braunewell and Gundelfinger, 1997). VILIP-1 appears to influence adenylyl cyclase activity in selected cell types including human embryonic kidney cells (Lin et al., 2002b), the pancreatic β cell line MIN6 (Dai et al., 2006), and various skin tumor cell lines, where it also affects rhoA signaling (Mahloogi et al., 2003; Schönrath et al., 2011). No direct interaction of VILIP-1 with adenylyl cyclase isoforms has been detected, but VILIP-1 expression was shown to enhance surface expression of different adenylyl cyclase isoforms in skin tumor cell lines leading to enhanced tumor cell migration (Schönrath et al., 2011).
VILIP-1 enhances neurite outgrowth in SH-SY5Y neuroblastoma via effects on cAMP-signaling (Braunewell et al., 2011). The cAMP signaling pathway overcomes Aβ-induced inhibition of neurite formation in SH-SY5Y neurons and in the hippocampus in transgenic APP/PS1 mice, carrying human AD mutations (Smith et al., 2009). In SH-SY5Y cells, Aβ reduces cAMP levels and increases levels of the GTP-bound (active) form of rhoA, which is eliminated by the rho-associated protein kinase (ROCK) inhibitor, Y-27632 (Petratos et al., 2008). Manipulation of the cAMP-rhoA/ROCK signaling pathway using the ROCK inhibitor leads to the extension of long neurites in SH-SY5Y cells. Consistent with the effect of VILIP-1 on cAMP signaling in SH-SY5Y neurons, the reduction of its expression through exposure of cells to siRNA results in loss of dendritic arborisation as measured as reduced number of dendrites in a Sholl analysis in hippocampal neurons. This effect is likely due to the observed reduced VILIP-1-dependent formation of cAMP in hippocampal neurons. As expected over-expression of VILIP-1 led to an increase in the number of dendrites in these neurons (Braunewell et al., 2011). It is noteworthy that calbindin-D28K, the neuroprotective Ca2+-buffer protein which is down-regulated in AD and co-localizes with VILIP-1 in hippocampal interneurons (Gierke et al., 2008; Zhao and Braunewell, 2008), promotes neuronal differentiation and neurite outgrowth of hippocampal precursor cells and dopaminergic neurons (Choi et al., 2001; Kim et al., 2006). Similarly, hippocalcin enhances basic fibroblast growth factor-induced neurite outgrowth in a hippocampal cell line (Oh et al., 2008). Interestingly in another study, VILIP-1 and neurocalcin δ were found to be developmentally up-regulated in axon tracts in the olfactory system. Counter-intuitively, their over-expression led to reduced axon outgrowth, but left dendrite length unaffected in hippocampal neurons in the same study (Yamatani et al., 2010). Whereas neurocalcin δ also showed reduced branch point and dendrite numbers, VILIP-1 appeared to only slightly increase dendrite numbers, although not significantly (Yamatani et al., 2010). It is highly likely that different VSNLs may affect different neuronal compartments, with VILIP-1 reducing axonal (Yamatani et al., 2010), but enhancing dendritic differentiation (Braunewell et al., 2011). However, comparative studies on axonal versus dendritic differentiation will have to be performed in the same experimental set up to further substantiate this hypothesis. Particularly the effect of VILIP-1 and, possibly, VILIP-3 on dendritic arborization and connectivity may underlie altered synaptic function and hippocampal network connectivity, and thus may contribute to the cognitive decline in early phases of AD. The loss of distinct Ca2+-buffer and Ca2+-sensor proteins in subpopulations of hippocampal interneurons, may render these neurons particularly vulnerable against Aβ-induced morphological disturbances, such as reduced dendritic spinogenesis in AD, which are significantly contributing to cognitive decline (Smith et al., 2009; Wei et al., 2010).
VILIP-1, Nicotinergic Signaling and Cognitive Impairment in AD
One other possibility how VILIP-1 may affect cognition lies in the fact that VILIP-1 interacts with the α4β2 nAChR (Lin et al., 2002a). Reduced levels of nAChRs and cholinergic neurotransmission are involved in the etiology of AD, and acetylcholinesterase inhibitors are used for the treatment of AD (Buckingham et al., 2009). The high-affinity α4β2 nAChR is the main nicotine binding site in the brain, and appears to mediate nicotine-dependent improvements in attention, learning, and working memory (Rezvani and Levin, 2001; Levin et al., 2006). These facts have sparked interest in the development of novel treatments for cognitive dysfunction in CNS disorders, based on modulation of nAChR activity. In clinical trials, agonists and antagonists of the major α7- and α4-containing nAChRs are beneficial (Buckingham et al., 2009; Bacher et al., 2009; Fedorov et al., 2009). Since VILIP-1 is an endogenous modulator of α4β2 nAChR, it is conceivable that the down-regulation of VILIP-1 mRNA at early stages of AD leads to deficits in α4β2 nAChR activity in the hippocampus. In a yeast two-hybrid screen for protein-protein interactions, VILIP-1 bound to a 30-amino acid region in the large intracellular loop of the α4 -subunit of the α4β2 nAChR. Co-expression of VILIP-1 with recombinant α4β2 nAChR up-regulated the surface expression levels by twofold and increased the agonist-sensitivity to acetylcholine by threefold. The VILIP-1 myristoylation mutant or mutants not able to bind Ca2+ are found to attenuate the modulation of α4β2 nAChR (Lin et al., 2002a). Similarly, in hippocampal neurons, co-expression of VILIP-1 with recombinant α4β2 nAChR up-regulated its surface expression and increased the agonist sensitivity to acetylcholine, suggesting that VILIP-1 is a modulator of α4β2 nAChR, and leading to functional up-regulation of the receptor (Zhao et al., 2009a). VILIP-1 and α4β2 nAChR were found in a complex with the trans-Golgi SNARE syntaxin 6, involved in Golgi to surface membrane trafficking and constitutive exocytosis. Moreover, the nicotine-induced, and α7 nAChR-mediated Ca2+-myristoyl switch of VILIP-1 in hippocampal neurons provides a crosstalk mechanism for the interaction of α7 with α4β2 nAChRs, in that the α7 nAChR mediated Ca2+-influx in neurons can activate α4β2 nAChRs via VILIP-1 (Zhao et al., 2009b Figure 3). In rat and human hippocampi, VILIP-1 and α4β2 nAChR are co-localized in a subpopulation of interneurons (Gierke et al., 2008; Zhao and Braunewell, 2008). In view of the prominent expression of VILIP-1 in calbindin-positive interneurons and in disinhibitory, calretinin-positive interneurons in the hippocampal formation, which are also partly positive for α4β2 nAChR (Zhao and Braunewell, 2008), VILIP-1 is well positioned to regulate hippocampal network activity. The interaction of VILIP-1 with α4β2 nAChR enhances GABAergic signaling in interneurons, and the frequency of IPSCs (inhibitory postsynaptic currents) in pyramidal neurons (Gierke et al., 2008), thereby likely changing the activity of the hippocampal neuronal network, and thus synaptic plasticity and cognition (Rezvani and Levin, 2001; Levin et al., 2006). Interneurons are involved in the generation of synchronous rhythmic activity in the hippocampus essential for cognitive processing/memory encoding. Therefore, early alterations in hippocampal interneurons in AD may result in the cognitive impairments seen in the initial stages of the disease (Palop et al., 2003, 2007; Baglietto-Vargas et al., 2010). In AD the loss of VILIP-1 expression may thus lead to a loss of surface expression and functional activity of α4β2 nAChRs in interneurons, and in turn reduction of GABAergic interneuron activity (Figure 3). The effect of VILIP-1 on nicotinergic signaling, in hippocampal interneurons, for instance, may explain the correlation of CSF VILIP-1 levels and VILIP-1 mRNA levels with MMSE scores and cognition. Thus, down-regulation of VILIP-1 affecting α4β2 nAChR expression and activity in interneurons may contribute to cognitive impairments in AD.
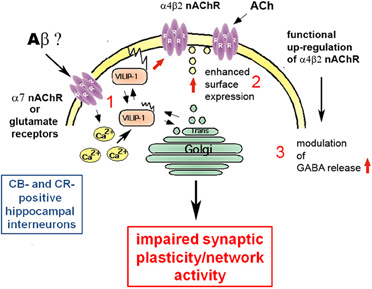
Figure 3. Molecular mechanisms of the involvement of VILIP-1 in pathological network activity and cognition. (1) The amyloid β peptide binds to and activates receptors, such as the α7 nAChR and NMDA-type glutamate receptors, leading to changes in the cellular Ca2+-homeostasis. The increase in Ca2+ concentrations shuttles VILIP-1 to the cell surface and Golgi membranes. (2) VILIP-1 enhances surface expression and sensitivity of α4β2 nAChRs in interneurons. (3) In hippocampal interneurons VILIP-1 enhances up-regulation of α4β2 nAChRs and thereby modulates GABA release. This leads to a VILIP-1-dependent enhancement of ACh-evoked IPSCs in hippocampal pyramidal cells. The pathological down-regulation of VILIP-1 in AD may thus negatively affect hippocampal network activity and synaptic plasticity, and lead to cognitive disturbances in the AD brain.
VILIP-3 and the Plasma Membrane Redox System in AD
Comparatively little is known about functional activities of VILIP-3. VILIP-3 affects ERK1/2-phosphorylation in PC12 cells (Spilker et al., 2002a), has been discussed to enhance cAMP levels in the prostatic epithelial cell line NbE-1 (Tang et al., 2012), and VILIP-3 as well as hippocalcin, interact with the microsomal enzyme cytochrome b5, which in turn interacts with cytochrome b5 reductase located in the endoplasmic reticulum-perinuclear region in microsomal membranes (Oikawa et al., 2004). Cytochrome b5 belongs to the PMRS (Hyun et al., 2006). Membrane-associated oxidative stress has been implicated in the synaptic dysfunction and neuronal degeneration that occurs in AD, but the underlying mechanisms are unknown. The PMRS provides electrons for energy metabolism and recycling of antioxidants, and is impaired in AD (Hyun et al., 2010). The activities of several PMRS enzymes are decreased in plasma membranes from the hippocampus and cerebral cortex of 3xTgAD mice, an animal model of AD. Neurons over-expressing the PMRS enzymes (NQO1 or cytochrome b5 reductase) exhibit increased resistance to Aβ (Hyun et al., 2010). Under conditions of disturbed Ca2+-homeostasis in AD, there is an enhanced pathological, juxtanuclear localization of VILIP-3 (Braunewell et al., 2001a). The Ca2+-dependent translocation of VILIP-3 to the endoplasmic reticulum (ER)-rich perinuclear region, may indicate that VILIP-3 impacts the microsomal monooxygenase complex composed of cytchrome b5-reductase, cytochrome P450, and other reductases of the ER. Although the functional implications of the interaction are not yet known, reduced expression of VILIP-3 may decrease PMSR activity and lead to enhanced oxidative stress. Thus, increased levels of VILIP-3 may be neuroprotective against Aβ-induced oxidative stress. In this context, hippocalcin also appears to be neuroprotective (Mercer et al., 2000; Lindholm et al., 2002). Hippocalcin−/− mice are more sensitive to thapsigargin-induced cell death and to excitotoxicity caused by kainic acid and quinolinic acid (Korhonen et al., 2005; Masuo et al., 2007). Moreover, these mice display increased caspase-12 activation and an age-dependent increase in neurodegeneration (Masuo et al., 2007). Interestingly, DREAM, which is upregulated in AD (Jo et al., 2004), was found to interact with the mitochondrial antioxidant enzyme Prdx3 (Rivas et al., 2011). Prdx3 expression protected against pesticide-induced mitochondrial damage, it improved cognition and decreased Aβ levels in APP transgenic mice (Chen et al., 2012). DREAM knockdown sensitizes H(2)O(2)-induced oxidative stress in PC12 cells, thus DREAM up-regulation has been discussed to be neuroprotective against oxidative stress (Rivas et al., 2011). It will be interesting to investigate whether there is actually co-localization of DREAM with Prdx3 in mitochondria or in the cytosol, and whether this interaction leads to neuroprotection under pathological conditions in an AD model. Taken together, these results indicate that plasma membrane, ER and mitochondrial redox systems play a crucial role in both oxidative stress-induced cell death and in cognitive impairment in AD, and it will be important to clarify whether the observed interactions of NCS proteins with different components of redox systems may affect the functionality of antioxidant systems in AD, and vice versa. These interactions may underlie some of the neurotoxic/neuroprotective and cognitive functions of NCS proteins in AD.
Future Perspectives
There are many open questions concerning the role of Ca2+-signaling and particularly the role of Ca2+-sensors, such as Calsenilin and VSNLs, in the etiology of AD. The complex pattern of up- and down-regulation of NCSs, and of Ca2+-buffers, such as calbindin-D28K and calretinin, may have multiple additive effects on neuronal pathways and systems at various levels and intensities, and thereby significantly contribute to cognitive impairments in AD and to neuronal death at the later stages of AD. In order to determine the effects of VSNLs, in conjunction with their respective interaction partners and associated signaling pathways, and to determine whether they are therapeutic targets for treatment of cognitive impairments in AD, several important questions need to be answered in the future (Working hypothesis Figure 4). First, (1) what are the mechanisms of VILIP down-regulation in AD. One possibility is a direct down-regulation by Aβ, or alternatively via Aβ-supression of neurotrophin signaling. Next, (2) what is the role VSNLs play in Aβ-induced Ca2+-overload, leading to reduced activity of the PMRS and to impaired neuroprotection in vulnerable subpopulations of neurons. (3) What are the signaling pathways involved in the effect of VILIP-1 on tau-phosphorylation and (4) on dendritic growth and spinogenesis in neurons? Finally, whether there is changed expression of VSNLs, particularly in interneurons, that affects network activity and synaptic plasticity in transgenic AD animal models, and most importantly (5) whether for instance VILIP-1, via its modulatory effect on cAMP and nicotinergic signaling plays a role for impaired synaptic plasticity and cognition in animal models. Such an effect could contribute to some of the early cognitive impairments observed in AD.
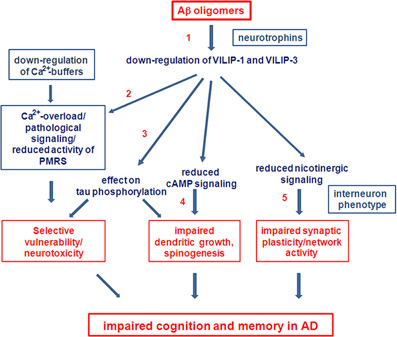
Figure 4. Future perspectives. Working hypothesis and open questions on the involvement of VSNLs and associated signaling pathways as mediators of Aβ oligomer-induced cell death and detrimental effects on cognition in AD.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in Karl Heinz Braunewell's laboratories has been supported by grants from DFG (Br1579/8–1 and Br1579/9–1/2, Priority Program of the German Research Foundation SPP1226), Alzheimer's Drug Discovery foundation (ADDF), and Southern Research Institute (SRI) startup funding. The comprehensive and authoritative peer-review of this article is greatly appreciated.
References
Ames, J. B., Ishima, R., Tanaka, T., Gordon, J. I., Stryer, L., and Ikura, M. (1997). Molecular mechanics of calcium-myristoyl switches. Nature 389, 198–202.
Ames, J. B., Tanaka, T., Stryer, L., and Ikura, M. (1996). Portrait of a myristoyl switch protein. Curr. Opin. Struct. Biol. 6, 432–438.
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., Hinson, J. W., Mattsson, K. I., Strassle, B. W., Trimmer, J. S., and Rhodes, K. J. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556.
Bacher, I., Wu, B., Shytle, D. R., and George, T. P. (2009). Mecamylamine-a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin. Pharmacother. 10, 2709–2721.
Baglietto-Vargas, D., Moreno-Gonzalez, I., Sanchez-Varo, R., Jimenez, S., Trujillo-Estrada, L., Sanchez-Mejias, E., Torres, M., Romero-Acebal, M., Ruano, D., Vizuete, M., Vitorica, J., and Gutierrez, A. (2010). Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/AbetaPP Alzheimer mice hippocampus. J. Alzheimers Dis. 21, 119–132.
Bernstein, H-G., Baumann, B., Danos, P., Diekmann, S., Bogerts, B., Gundelfinger, E. D., and Braunewell, K-H. (1999). Regional and cellular distribution of neural visinin-like protein immunoreactivities (VILIP-1 and VILIP-3) in human brain. J. Neurocytol. 28, 655–662.
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463.
Blalock, E. M., Geddes, J. W., Chen, K. C., Porter, N. M., Markesbery, W. R., and Landfield, P. W. (2004). Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 101, 2173–2178.
Blandini, F., Braunewell, K-H., Manahan-Vaughan, D., Orzi, F., and Sarti, P. (2004). Neurodegeneration and energy metabolism: from chemistry and clinics. Cell Death Differ. 11, 479–484.
Brackmann, M., Schuchmann, S., Anand, R., and Braunewell, K-H. (2005). Neuronal Ca2+ sensor protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J. Cell. Sci. 118, 2495–2505.
Brackmann, M., Zhao, C., Kuhl, D., Manahan-Vaughan, D., and Braunewell, K. H. (2004). MGluRs regulate the expression of neuronal calcium sensor proteins NCS-1 and VILIP-1 and the immediate early gene arg3.1/arc in the hippocampus in vivo. Biochem. Biophys. Res. Commun. 322, 1073–1079.
Braunewell, K. H. (2005). The dark side of Ca2+ signalling by neuronal Ca2+- sensor proteins: from Alzheimer's disease to cancer. Trends Pharmacol. Sci. 26, 345–351.
Braunewell, K. H., and Bernstein, H. G. (2009). Notes on visinin-like proteins and Alzheimer's disease. Clin. Chem. 55, 1041–1043.
Braunewell, K. H., Brackmann, M., and Manahan-Vaughan, D. (2003). Group I mGluRs regulate the expression of the intracellular neuronal calcium sensor protein VILIP-1 in vitro and in vivo: possible implications for mGluR-dependent hippocampal plasticity? Neuropharmacology 44, 707–715.
Braunewell, K. H., and Gundelfinger, E. D. (1997). Low level expression of calcium-sensor protein VILIP induces cAMP- dependent differentiation in rat C6 glioma cells. Neurosci. Lett. 234, 139–142.
Braunewell, K. H., and Gundelfinger, E. D. (1999). Intracellular neuronal calcium sensor proteins: a family of EF-hand calcium-binding proteins in search of a function. Cell Tissue Res. 299, 1–12.
Braunewell, K. H., Dwary, A. D., Richter, F., Trappe, K., Zhao, C., Giegling, I., Schönrath, K., and Rujescu, D. (2011). Association of VSNL1 with schizophrenia, frontal cortical function, and biological significance for its gene product as a modulator of cAMP levels and neuronal morphology. Transl Psychol 1, e22.
Braunewell, K. H., and Klein-Szanto, A. J. P. (2009). Visinin-like proteins (VSNLs): interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell Tissue Res. 335, 301–316.
Braunewell, K. H., Paul, B., Altarche-Xifro, W., Noack, C., Lange, K., and Hofmann, A. (2010). Interactions of visinin-like proteins with phospho-inositides. Aust. J. Chem. 63, 350.
Braunewell, K. H., Riederer, P., Spilker, C., Gundelfinger, E. D., Bogerts, B., and Bernstein, H. G. (2001a). Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (VILIPs)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement. Geriatr. Cogn. Disord. 2, 110–115.
Braunewell, K. H., Brackmann, M., Schaupp, M., Spilker, C., Anand, R., and Gundelfinger, E. D. (2001b). Intracellular neuronal calcium sensor (NCS) protein VILIP-1 modulates cGMP signalling pathways in transfected neural cells and cerebellar granule neurones. J. Neurochem. 78, 1277–1286.
Braunewell, K. H., Spilker, C., Behnisch, T., and Gundelfinger, E. D. (1997). The neuronal calcium-sensor protein VILIP modulates cyclic AMP accumulation in stably transfected C6 glioma cells: amino-terminal myristoylation determines functional activity. J. Neurochem. 68, 2129–2139.
Buckingham, S. D., Jones, A. K., Brown, L. A., and Sattelle, D. B. (2009). Nicotinic acetylcholine receptor signalling: roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol. Rev. 61, 39–61.
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193.
Burgoyne, R. D., O'Callaghan, D. W., Hasdemir, B., Haynes, L. P., and Tepikin, A. V. (2004). Neuronal Ca2+-sensor proteins: multitalented regulators of neuronal function. Trends Neurosci. 27, 203–209.
Burgoyne, R. D., and Weiss, J. L. (2001). The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 353, 1–12.
Buxbaum, J. D. (2004). A role for calsenilin and related proteins in multiple aspects of neuronal function. Biochem. Biophys. Res. Commun. 322, 1140–1144.
Buxbaum, J. D., Choi, E. K., Luo, Y., Lilliehook, C., Crowley, A. C., Merriam, D. E., and Wasco, W. (1998). Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 4, 1177–1181.
Carrión, A. M., Link, W. A., Ledo, F., Mellström, B., and Naranjo, J. R. (1999). DREAM is a Ca2+-regulated transcriptional repressor. Nature 398, 80–84.
Chaumont, S., Compan, V., Toulme, E., Richler, E., Housley, G. D., Rassendren, F., and Khakh, B. S. (2008). Regulation of P2X2 receptors by the neuronal calcium sensor VILIP1. Sci. Signal. 1, ra8.
Chakroborty, S., and Stutzmann, G. E. (2011). Early calcium dysregulation in Alzheimer's disease: setting the stage for synaptic dysfunction. Sci. China Life Sci. 54, 752–762.
Chen, L., Yoo, S. E., Na, R., Liu, Y., and Ran, Q. (2012). Cognitive impairment and increased Aβ levels induced by paraquat exposure are attenuated by enhanced removal of mitochondrial H(2)O(2). Neurobiol. Aging 33, 432.e15–26.
Chen, K. C., Wang, L. K., and Chang, L. S. (2009). Regulatory elements and functional implication for the formation of dimeric visinin-like protein-1. J. Pept. Sci. 15, 89–94.
Choi, W. S., Chun, S. Y., Markelonis, G. J., Oh, T. H., and Oh, Y. J. (2001). Overexpression of calbindin-D28K induces neurite outgrowth in dopaminergic neuronal cells via activation of p38 MAPK. Biochem. Biophys. Res. Commun. 287, 656–661.
Coussen, F., Perrais, D., Jaskolski, F., Sachidhanandam, S., Normand, E., Bockaert, J., Marin, P., and Mulle, C. (2005). Co-assembly of two GluR6 kainate receptor splice variants within a functional protein complex. Neuron 47, 555–566.
Craig-Schapiro, R., Fagan, A. M., and Holtzman, D. M. (2009). Biomarkers of Alzheimer's disease. Neurobiol. Dis. 35, 128–140.
Dai, F. F., Zhang, Y., Kang, Y., Wang, Q., Gaisano, H. Y., Braunewell, K. H., Chan, C. B., and Wheeler, M. B. (2006). The neuronal Ca2+ sensor protein visinin-like protein-1 is expressed in pancreatic islets and regulates insulin secretion. J. Biol. Chem. 281, 21942–22153.
Fedorov, N. B., Benson, L. C., Graef, J., Lippiello, P. M., and Bencherif, M. (2009). Differential pharmacologies of mecamylamine enantiomers: positive allosteric modulation and noncompetitive inhibition. J. Pharmacol. Exp. Ther. 328, 525–532.
Few, A. P., Lautermilch, N. J., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2005). Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J. Neurosci. 25, 7071–7080.
Fontán-Lozano, A., Romero-Granados, R., del-Pozo-Martín, Y., Suárez-Pereira, I., Delgado-García, J. M., Penninger, J. M., and Carrión, A. M. (2009). Lack of DREAM protein enhances learning and memory and slows brain aging. Curr. Biol. 19, 54–60.
Foster, T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6, 319–325.
Gierke, P., Zhao, C., Bernstein, H. G., Noack, C., Anand, R., Heinemann, U., and Braunewell, K. H. (2008). Implication of neuronal Ca(2+)-sensor protein VILIP-1 in the glutamate hypothesis of schizophrenia. Neurobiol. Dis. 32, 162–175.
Gierke, P., Zhao, C., Brackmann, M., Linke, B., Heinemann, U., and Braunewell, K. H. (2004). Expression analysis of members of the neuronal calcium sensor protein family: combining bioinformatics and Western blot analysis. Biochem. Biophys. Res. Commun. 323, 38–43.
Gómez Ravetti, M., Rosso, O. A., Berretta, R., and Moscato, P. (2010). Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus' gene expression profiles in Alzheimer's disease. PLoS One 5:e10153 doi: 10.1371/journal.pone.0010153
Hamashima, H., Tamaru, T., Noguchi, H., Kobayashi, M., and Takamatsu, K. (2001). Immunochemical assessment of neural visinin-like calcium-binding protein 3 expression in rat brain. Neurosci. Res. 39, 133–143.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Hollingworth, P., Sweet, R., Sims, R., Harold, D., Russo, G., Abraham, R., Stretton, A., Jones, N., Gerrish, A., Chapman, J., Ivanov, D., Moskvina, V., Lovestone, S., Priotsi, P., Lupton, M., Brayne, C., Gill, M., Lawlor, B., Lynch, A., Craig, D., McGuinness, B., Johnston, J., Holmes, C., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., the GERAD Consortium; the National Institute on Aging Late-Onset Alzheimer's Disease Family Study Group Holmans, P., Jones, L., Devlin, B., Klei, L., Barmada, M. M., Demirci, F. Y., Dekosky, S. T., Lopez, O. L., Passmore, P., Owen, M. J., O'Donovan, M. C., Mayeux, R., Kamboh, M. I., and Williams, J. (2011). Genome-wide association study of Alzheimer's disease with psychotic symptoms. Mol. Psychiatry [Epub ahead of print].
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer's disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr1.
Hyun, D. H., Emerson, S. S., Jo, D. G., Mattson, M. P., and de Cabo, R. (2006). Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc. Natl. Acad. Sci. U.S.A. 103, 19908–19912.
Hyun, D. H., Mughal, M. R., Yang, H., Lee, J. H., Ko, E. J., Hunt, N. D., de Cabo, R., and Mattson, M. P. (2010). The plasma membrane redox system is impaired by amyloid β-peptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp. Neurol. 225, 423–429.
Iacopino, A. M., and Christakos, S. (1992). Specific reduction of calcium-binding protein (28-kilodalton calbindin-D) gene expression in aging and neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 87, 4078–4082.
Ivings, L., Pennington, S. R., Jenkins, R., Weiss, J. L., and Burgoyne, R. D. (2002). Identification of Ca2+-dependent binding partners for the neuronal calcium sensor protein neurocalcin delta: interaction with actin, clathrin and tubulin. Biochem. J. 363, 599–608.
Jheng, F. F., Wang, L., Lee, L., and Chang, L. S. (2006). Functional contribution of Ca2+ and Mg2+ to the intermolecular interaction of visinin-like proteins. Protein J. 25, 250–256.
Jo, D. G., Lee, J. Y., Hong, Y. M., Song, S., Mook-Jung, I., Koh, J. Y., and Jung, Y. K. (2004). Induction of pro-apoptotic calsenilin/DREAM/KChIP3 in Alzheimer's disease and cultured neurons after amyloid-beta exposure. J. Neurochem. 88, 604–611.
Kajimoto, Y., Shirai, Y., Mukai, H., Kuno, T., and Tanaka, C. (1993). Molecular cloning of two additional members of the neural visinin-like Ca(2+)-binding protein gene family. J. Neurochem. 61, 1091–1096.
Kato, M., Watanabe, Y., Iino, S., Takaoka, Y., Kobayashi, S., Haga, T., and Hidaka, H. (1998). Cloning and expression of a cDNA encoding a new neurocalcin isoform (neurocalcin alpha) from bovine brain. Biochem. J. 331, 871–876.
Kaufmann, W. A., Barnas, U., Humpel, C., Nowakowski, K., DeCol, C., Gurka, P., Ransmayr, G., Hinterhuber, H., Winkler, H., and Marksteiner, J. (1998). Synaptic loss reflected by secretoneurin-like immunoreactivity in the human hippocampus in Alzheimer's disease. Eur. J. Neurosci. 10, 1084–1094.
Kim, J. H., Lee, J. A., Song, Y. M., Park, C. H., Hwang, S. J., Kim, Y. S., Kaang, B. K., and Son, H. (2006). Overexpression of calbindin-D28K in hippocampal progenitor cells increases neuronal differentiation and neurite outgrowth. FASEB J. 20, 109–111.
Kim, S. H., Fountoulakis, M., Cairns, N., and Lubec, G. (2001). Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer's disease and Down syndrome. J. Neural Transm. Suppl. 61, 223–235.
Kobayashi, M., Masaki, T., Hori, K., Masuo, Y., Miyamoto, M., Tsubokawa, H., Noguchi, H., Nomura, M., and Takamatsu, K. (2005). Hippocalcin-deficient mice display a defect in cAMP response element-binding protein activation associated with impaired spatial and associative memory. Neuroscience 133, 471–484.
Kobayashi, M., Takamatsu, K., Fujishiro, M., Saitoh, S., and Noguchi, T. (1994). Molecular cloning of a novel calcium-binding protein structurally related to hippocalcin from human brain and chromosomal mapping of its gene. Biochim. Biophys. Acta 1222, 515–518.
Kobayashi, M., Takamatsu, K., Saitoh, S., and Nogushi, T. (1993). Myristoylation of hippocalcin is linked to its membrane association properties. J. Biol. Chem. 268, 18898–18904.
Korhonen, L., Hansson, I., Kukkonen, J. P., Brännvall, K., Kobayashi, M., Takamatsu, K., and Lindholm, D. (2005). Hippocalcin protects against caspase-12-induced and age-dependent neuronal degeneration. Mol. Cell. Neurosci. 28, 85–95.
Kraut, N., Frampton, J., and Graf, T. (1995). Rem-1, a putative direct target gene of the Myb-Ets fusion oncoprotein in haematopoietic progenitors, is a member of the recoverin family. Oncogene 10, 1027–1036.
Kuno, T., Kajimoto, Y., Hashimoto, T., Mukai, H., Shirai, Y., Saheki, S., and Tanaka, C. (1992). cDNA cloning of a neural visinin-like Ca(2+)-binding protein. Biochem. Biophys. Res. Commun. 184, 1219–1225.
Ladant, D. (1995). Calcium and membrane binding properties of bovine neurocalcin delta expressed in Escherichia coli. J. Biol. Chem. 270, 3179–3185.
Lee, J. M., Blennow, K., Andreasen, N., Laterza, O., Modur, V., Olander, J., Gao, F., Ohlendorf, M., and Ladenson, J. H. (2008). The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin. Chem. 54, 1617–1623.
Lee, J. H., Cheng, R., Barral, S., Reitz, C., Medrano, M., Lantigua, R., Jiménez-Velazquez, I. Z., Rogaeva, E., StGeorge-Hyslop, P. H., and Mayeux, R. (2011). Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol. 68, 320–328.
Leissring, M. A., Yamasaki, T. R., Wasco, W., Buxbaum, J. D., Parker, I., and LaFerla, F. M. (2000). Calsenilin reverses presenilin-mediated enhancement of calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 8590–8593.
Lenz, S. E., Braunewell, K. H., Weise, C., Nedlina-Chittka, A., and Gundelfinger, E. D. (1996). The neuronal EF-hand Ca(2+)-binding protein VILIP: interaction with cell membrane and actin-based cytoskeleton. Biochem. Biophys. Res. Commun. 225, 1078–1083.
Lenz, S. E., Henschel, Y., Zopf, D., Voss, B., and Gundelfinger, E. D. (1992). VILIP, a cognate protein of the retinal calcium binding proteins visinin and recoverin, is expressed in the developing chicken brain. Brain Res. Mol. Brain Res. 15, 133–140.
Levin, E. D., McClernon, F. J., and Rezvani, A. H. (2006). Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl.) 184, 523–539.
Li, C., Pan, W., Braunewell, K. H., and Ames, J. B. (2011). Structural analysis of Mg2+ and Ca2+ binding, myristoylation, and dimerization of the neuronal calcium sensor and visinin-like protein 1 (VILIP-1). J. Biol. Chem. 286, 6354–6366.
Lian, L. Y., Pandalaneni, S. R., Patel, P., McCue, H. V., Haynes, L. P., and Burgoyne, R. D. (2011). Characterisation of the interaction of the C-terminus of the dopamine D2 receptor with neuronal calcium sensor-1. PLoS One 6:e27779. doi: 10.1371/journal.pone.0027779
Lin, L., Jeanclos, E. M., Treuil, M., Braunewell, K. H., Gundelfinger, E. D., and Anand, R. (2002a). The calcium sensor protein visinin-like protein-1 modulates the surface expression and agonist-sensitivity of the a4β2 nicotinic acetylcholine receptor. J. Biol. Chem. 277, 41872–41878.
Lin, L., Braunewell, K. H., Gundelfinger, E. D., and Anand, R. (2002b). Functional analysis of calcium-binding EF-hand motifs of visinin-like protein-1. Biochem. Biophys. Res. Commun. 296, 827–832.
Lindholm, D., Mercer, E. A., Yu, L. Y., Chen, Y., Kukkonen, J., Korhonen, L., and Arumäe, U. (2002). Neuronal apoptosis inhibitory protein: structural requirements for hippocalcin binding and effects on survival of NGF-dependent sympathetic neurons. Biochim. Biophys. Acta 1600, 138–147.
Loring, J. F., Porter, J. G., Seilhammer, J., Kaser, M. R., and Wesselschmidt, R. (2001). A gene expression profile of Alzheimer's disease. DNA Cell Biol. 20, 683–695.
Mahloogi, H., Gonzalez-Guerrico, A. M., De Cicco, R. L., Bassi, D. E., Goodrow, T., Braunewell, K. H., and Klein-Szanto, A. J. P. (2003). Graduate decrease of VILIP-1 Expression during mouse skin tumor progression and its role in regulating tumor cell invasive behavior. Cancer Res. 63, 4997–5004.
Masuo, Y., Ogura, A., Kobayashi, M., Masaki, T., Furuta, Y., Ono, T., and Takamatsu, K. (2007). Hippocalcin protects hippocampal neurons against excitotoxin damage by enhancing calcium extrusion. Neuroscience 145, 495–504.
McMahon, A., Wong, B. S., Iacopino, A. M., Ng, M. C., Chi, S., and German, D. C. (1998). Calbindin-D28k buffers intracellular calcium and promotes resistance to degeneration in PC12 cells. Brain Res. Mol. Brain Res. 54, 56–63.
Mercer, E. A., Korhonen, L., Skoglösa, Y., Olsson, P. A., Kukkonen, J. P., and Lindholm, D. (2000). NAIP interacts with hippocalcin and protects neurons against calcium-induced cell death through caspase-3-dependent and -independent pathways. EMBO J. 19, 3597–3607.
Miller, J. A., Oldham, M. C., and Geschwind, D. H. (2008). A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J. Neurosci. 28, 1410–1420.
Monfort, P., Munoz, M. D., Kosenko, E., and Felipo, V. (2002). Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J. Neurosci. 22, 10116–10124.
Nanou, E., Martinez, G. Q., Scheuer, T., and Catterall, W. A. (2012). Molecular determinants of modulation of CaV2.1 channels by visinin-like protein 2. J. Biol. Chem. 287, 504–513.
Nef, P. (1996). “Neuron-specific calcium sensors (the NCS subfamily),” in: Guidebook to the Calcium-Binding Proteins, ed M. R. Celio (New York: Oxford University Press), 94–98.
O'Callaghan, D. W., Haynes, L. P., and Burgoyne, R. D. (2005). High-affinity interaction of the N-terminal myristoylation motif of the neuronal calcium sensor protein hippocalcin with phosphatidylinositol 4,5-bisphosphate. Biochem. J. 391, 231–238.
O'Callaghan, D. W., Ivings, L., Weiss, J. L., Ashby, M. C., Tepikin, A. V., and Burgoyne, R. D. (2002). Differential use of myristoyl groups on neuronal calcium sensor proteins as a determinant of spatio-temporal aspects of Ca2+ signal transduction. J. Biol. Chem. 277, 14227–14237.
O'Callaghan, D. W., Tepikin, A. V., and Burgoyne, R. D. (2003). Dynamics and calcium sensitivity of the Ca2+-myristoyl switch protein hippocalcin in living cells. J. Cell Biol. 163, 715–721.
Oh, D. Y., Cho, J. H., Park, S. Y., Kim, Y. S., Yoon, Y. J., Yoon, S. H., Chung, K. C., Lee, K. S., and Han, J. S. (2008). A novel role of hippocalcin in bFGF-induced neurite outgrowth of H19–7 cells. J. Neurosci. Res. 86, 1557–1565.
Oikawa, K., Kimura, S., Aoki, N., Atsuta, Y., Takiyama, Y., Nagato, T., Yanai, M., Kobayashi, H., Sato, K., Sasajima, T., and Tateno, M. (2004). Neuronal calcium sensor protein visinin-like protein-3 interacts with microsomal cytochrome b5 in a Ca2+-dependent manner. J. Biol. Chem. 279, 15142–11552.
Palmer, C. L., Lim, W., Hastie, P. G., Toward, M., Korolchuk, V. I., Burbidge, S. A., Banting, G., Collingridge, G. L., Isaac, J. T., and Henley, J. M. (2005). Hippocalcin functions as a calcium sensor in hippocampal LTD. Neuron 47, 487–494.
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., Yoo, J., Ho, K. O., Yu, G. Q., Kreitzer, A., Finkbeiner, S., Noebels, J. L., and Mucke, L. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711.
Palop, J. J., Jones, B., Kekonius, L., Chin, J., Yu, G. Q., Raber, J., Masliah, E., and Mucke, L. (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 100, 9572–9577.
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818.
Paterlini, M., Revilla, V., Grant, A. L., and Wisden, W. (2000). Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience 99, 205–216.
Petratos, S., Li, Q. X., George, A. J., Hou, X., Kerr, M. L., Unabia, S. E., Hatzinisiriou, I., Maksel, D., Aguilar, M. I., and Small, D. H. (2008). The beta-amyloid protein of Alzheimer's disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 131, 90–108.
Polymeropoulos, M. H., Ide, S., Soares, M. B., and Lennon, G. G. (1995). Sequence characterization and genetic mapping of the human VSNL1 gene, a homolo gue of the rat visinin-like peptide RNVP1. Genomics 29, 273–275.
Popoviæ, M., Caballero-Bleda, M., Kadish, I., and Van Groen, T. (2008). Subfield and layer-specific depletion in calbindin-D28K, calretinin and parvalbumin immunoreactivity in the dentate gyrus of amyloid precursor protein/presenilin 1 transgenic mice. Neuroscience 155, 182–191.
Rezvani, A. H., and Levin, E. D. (2001). Cognitive effects of nicotine. Biol. Psychiatry 49, 258–267.
Richler, E., Shigetomi, E., and Khakh, B. S. (2011). Neuronal P2X2 receptors are mobile ATP sensors that explore the plasma membrane when activated. J. Neurosci. 31, 16716–16730.
Rivas, M., Aurrekoetxea, K., Mellström, B., and Naranjo, J. R. (2011). Redox signaling regulates transcriptional activity of the Ca2+-dependent repressor DREAM. Antioxid. Redox Signal. 14, 1237–1243.
Saab, B. J., Georgiou, J., Nath, A., Lee, F. J., Wang, M., Michalon, A., Liu, F., Mansuy, I. M., and Roder, J. C. (2009). NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 63, 643–656.
Schnurra, I., Riederer, P., Bernstein, H. G., and Braunewell, K. H. (2001). The neuronal calcium sensor (NCS) protein VILIP-1 is associated with amyloid plaques and extracellular tangles and promotes cell death and tau-phosphorylation in vitro: a link between calcium sensors and Alzheimer's disease? Neurobiol. Dis. 8, 900–909.
Schönrath, K., Pan, W., Klein-Szanto, A. J., and Braunewell, K. H. (2011). Involvement of VILIP-1 (visinin-like protein) and opposite roles of cyclic AMP and GMP signalling in in vitro cell migration of murine skin squamous cell carcinoma. Mol. Carcinog. 50, 319–333.
Schuman, E. M., and Madison, D. V. (1991). A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506.
Sippy, T., Cruz-Martín, A., Jeromin, A., and Schweizer, F. E. (2003). Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat. Neurosci. 6, 1031–1038.
Smith, D. L., Pozueta, J., Gong, B., Arancio, O., and Shelanski, M. (2009). Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. U.S.A. 106, 16877–16882.
Spilker, C., and Braunewell, K-H. (2003). The calcium-myristoyl switch of neuronal calcium sensor (NCS) proteins: same biochemical principle but different calcium-dependent localization of VILIP-3 and -1 in hippocampal neurons. Mol. Cell. Neurosci. 24, 766–778.
Spilker, C., Gundelfinger, E. D., and Braunewell, K-H. (2002a). Different properties of the intracellular neuronal Calcium sensor proteins VILIP-1 and VILIP-3: from subcellular localization to cellular function. Biochim. Biophys. Acta proteins and proteomics 1600, 118–127.
Spilker, C., Dresbach, T., and Braunewell, K-H. (2002b). Reversible translocation and activity-dependent localization of the calcium-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J. Neurosci. 22, 7331–7339.
Spilker, C., Richter, K., Smalla, K-H., Manahan-Vaughan, D., Gundelfinger, E. D., and Braunewell, K-H. (2000). The neuronal EF-hand calcium-binding protein VILIP-3 is expressed in cerebellar Purkinje cells and shows a calcium-dependent membrane association. Neuroscience 96, 121–129.
Supnet, C., and Bezprozvanny, I. (2010). The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47, 183–189.
Takahashi, H., Brasnjevic, I., Rutten, B. P., Van Der Kolk, N., Perl, D. P., Bouras, C., Steinbusch, H. W., Schmitz, C., Hof, P. R., and Dickstein, D. L. (2010). Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer's disease. Brain Struct. Funct. 214, 145–160.
Tanaka, T., Ames, J. B., Harvey, T. S., Stryer, L., and Ikura, M. (1995). Sequestration of the membrane-targeting myristoyl group of recoverin in the calcium-free state. Nature 376, 444–447.
Tang, W. Y., Morey, L. M., Cheung, Y. Y., Birch, L., Prins, G. S., and Ho, S. M. (2012). Neonatal exposure to estradiol/bisphenol A alters promoter methylation and expression of Nsbp1 and Hpcal1 genes and transcriptional programs of Dnmt3a/b and Mbd2/4 in the rat prostate gland throughout life. Endocrinology 153, 42–55.
Tarawneh, R., D'Angelo, G., Macy, E., Xiong, C., Carter, D., Cairns, N. J., Fagan, A. M., Head, D., Mintun, M. A., Ladenson, J. H., Lee, J. M., Morris, J. C., and Holtzman, D. M. (2011). Visinin-like protein-1:diagnostic and prognostic biomarker in Alzheimer disease. Ann. Neurol. 70, 274–285.
Telegdy, G. (1994). The action of ANP, BNP and related peptides on motivated behavior in rats. Rev. Neurosci. 5, 309–315.
Tzingounis, A. V., Kobayashi, M., Takamatsu, K., and Nicoll, R. A. (2007). Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron 53, 487–493.
Verbeek, M. M., and Olde Rikkert, M. G. (2008). Cerebrospinal fluid biomarkers in the evaluation of Alzheimer disease. Clin. Chem. 54, 1589–1591.
Villalobos, C., and Andrade, R. (2010). Visinin-like neuronal calcium sensor proteins regulate the slow calcium-activated afterhyperpolarizing current in the rat cerebral cortex. J. Neurosci. 30, 14361–14365.
Wang, C. K., Simon, A., Jessen, C. M., Oliveira, C. L., Mack, L., Braunewell, K. H., Ames, J. B., Pedersen, J. S., and Hofmann, A. (2011). Divalent cations and redox conditions regulate the molecular structure and function of Visinin-like Protein-1. PLoS One 6:e26793. doi: 10.1371/journal.pone.0026793
Wei, W., Nguyen, L. N., Kessels, H. W., Hagiwara, H., Sisodia, S., and Malinow, R. (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196.
Wilmot, B., McWeeney, S. K., Nixon, R. R., Montine, T. J., Laut, J., Harrington, C. A., Kaye, J. A., and Kramer, P. L. (2008). Translational gene mapping of cognitive decline. Neurobiol. Aging 29, 524–541.
Wu, L. J., Mellström, B., Wang, H., Ren, M., Domingo, S., Kim, S. S., Li, X. Y., Chen, T., Naranjo, J. R., and Zhuo, M. (2010). DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Mol. Brain 3, 3
Yamatani, H., Kawasaki, T., Mita, S., Inagaki, N., and Hirata, T. (2010). Proteomics analysis of the temporal changes in axonal proteins during maturation. Dev. Neurobiol. 70, 523–537.
Youn, H., Jeoung, M., Koo, Y., Ji, H., Markesbery, W. R., Ji, I., and Ji, T. H. (2007). Kalirin is under-expressed in Alzheimer's disease hippocampus. J. Alzheimers Dis. 11, 385–397.
Zhao, C., and Braunewell, K-H. (2008). Expression of the neuronal calcium sensor VILIP-1 in the rat hippocampus. Neuroscience 153, 1202–1212.
Zhao, C. J., Noack, C., Brackmann, M., Gloveli, T., Maelicke, A., Heinemann, U., Anand, R., and Braunewell, K. H. (2009a). Neuronal Ca2+ sensor VILIP-1 leads to the upregulation of functional alpha4beta2 nicotinic acetylcholine receptors in hippocampal neurons. Mol. Cell. Neurosci. 40, 280–292.
Zhao, C., Anand, R., and Braunewell, K. H. (2009b). Nicotine-induced Ca2+-myristoyl switch of neuronal Ca2+ sensor VILIP-1 in hippocampal neurons: a possible crosstalk mechanism for nicotinic receptors. Cell. Mol. Neurobiol. 29, 273–286.
Keywords: cAMP/cGMP signaling, cognition, MAPK pathways, neurite outgrowth, neuroprotection, neuronal Ca2+-sensors, nicotinic acetylcholine receptors, plasma membrane redox system
Citation: Braunewell KH (2012) The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer's disease—old wine in new bottles. Front. Mol. Neurosci. 5:20. doi: 10.3389/fnmol.2012.00020
Received: 13 January 2012; Paper pending published: 19 January 2012;
Accepted: 09 February 2012; Published online: 23 February 2012.
Edited by:
Jose R. Naranjo, Centro Nacional de Biotecnología, SpainReviewed by:
Laura Mateos, Karolinska Institutet, SwedenRobert Burgoyne, University of Liverpool, UK
Copyright: © 2012 Braunewell. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Karl H. Braunewell, Molecular and Cellular Neuroscience Laboratory, Department Biochemistry and Molecular Biology, Southern, Research Institute, 2000 Ninth Avenue South, Birmingham, AL 35205, USA. e-mail: braunewell@sri.org