Memory, plasticity, and sleep – a role for calcium permeable AMPA receptors?
- Department of Brain and Cognitive Sciences, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, USA
Experience shapes and molds the brain throughout life.These changes in neuronal circuits are produced by a myriad of molecular and cellular processes. Simplistically, circuits are modified through changes in neurotransmitter release or through neurotransmitter detection at synapses. The predominant neurotransmitter receptor in excitatory transmission, the AMPA-type glutamate receptor (AMPAR), is exquisitely sensitive to changes in experience and synaptic activity. These ion channels are usually impermeable to calcium, a property conferred by the GluA2 subunit. However, GluA2-lacking AMPARs are permeable to calcium and have recently been shown to play a unique role in synaptic function. In this review, I will describe new findings on the role of calcium permeable AMPARs (CP-AMPARs) in experience-dependent and synaptic plasticity.These studies suggest that CP-AMPARs play a prominent role in maintaining circuits in a labile state where further plasticity can occur, thus promoting metaplasticity. Moreover, the abnormal expression of CP-AMPARs has been implicated in drug addiction and memory disorders and thus may be a novel therapeutic target.
Introduction
AMPA-type glutamate receptors (AMPARs) consist of heterotetrameric complexes composed of subunits GluA1–4 (Hollmann and Heinemann, 1994). Each subunit confers specific properties on the active receptor, including trafficking motifs, phosphorylation sites, and many other protein–protein interactions (Shepherd and Huganir, 2007). The GluA2 subunit is uniquely edited at the mRNA level, where a glutamine codon is switched to arginine (Lomeli et al., 1994), which confers channel resistance to calcium. Therefore, AMPAR complexes that lack GluA2 pass sodium and calcium ions resulting in higher conductance. In addition, calcium permeable AMPARs (CP-AMPARs) exhibit inward rectification in current–voltage plots that differs from the linear rectification of predominateAMPARs due to voltage-dependent block by intracellular polyamines (Burnashev et al., 1992; Donevan and Rogawski, 1995). GluA2 lacking AMPARs can be formed through GluA1 homomers or combinations of GluA1, 3, and 4. However, detection of these complexes in vivo in excitatory principle neurons has been somewhat controversial. Using single-cell analysis and conditional knock-out (KO) mice Nicoll and colleagues found that hippocampal excitatory neurons consist mostly of GluA1/2 or GluA2/3 receptors, with very few homomers detected (Lu et al., 2009). It is unclear if this is similar in other brain regions. In contrast, other studies have detected GluA2-lacking receptors, primarily through pharmacological use of external polyamine toxins such as Joro toxin and philanthotoxin (PhTx) or electrophysiological (by measuring rectification) methods in hippocampal neurons. In most of these studies, however, CP-AMPARs were only detected in young animals or after an epoch of plasticity-inducing neuronal activity (Isaac et al., 2007; Liu and Zukin, 2007). In contrast, aspiny, inhibitory interneurons throughout the CNS express GluA2-lacking, CP-AMPARs (Jonas et al., 1994; Goldberg et al., 2003). These receptors play an important role in interneuron synaptic plasticity including both Hebbian and anti-Hebbian forms (Alle et al., 2001; Lamsa et al., 2007).
Trafficking of AMPARs in and out of synapses is a major mechanism underlying synaptic plasticity (Shepherd and Huganir, 2007). Recent studies have shown that subunit switching of AMPAR complexes plays an important role in long-term forms of synaptic plasticity such long-term potentiation (LTP), long-term depression (LTD), and homeostatic plasticity (Man, 2011). This review concentrates on new findings that implicate CP-AMPARs in experience-dependent and synaptic plasticity (Figure 1). For brevity purposes, I focus only on the role of CP-AMPARs in excitatory neurons.
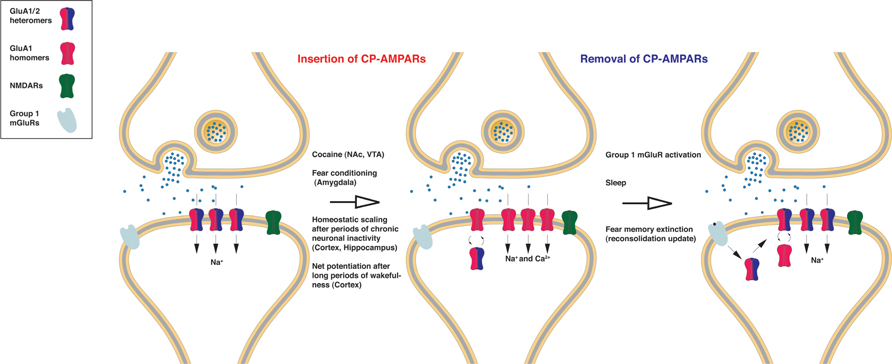
FIGURE 1. The role of CP-AMPARs in neuronal plasticity. GluA2-lacking receptors are relatively rare at baseline conditions in most excitatory neurons. However, GluA1/2 heteromers are replaced with GluA1 homomers after the induction of numerous plasticity paradigms. Since GluA1 homomers conduct Na+ and Ca2+ ions, synaptic transmission is usually potentiated even if the absolute numbers of receptors are the same. Precisely how this AMPAR subunit switch or replacement takes place is unclear. In some cases homomers may be directly inserted without replacing heteromers, which would also cause net potentiation of synaptic transmission. Although the time-course of induction may differ, cocaine injection; fear conditioning; homeostatic scaling; and long periods of wakefulness all result in an increase in synaptic/surface CP-AMPARs, which usually results in potentiation of synaptic transmission. In many cases CP-AMPAR surface/synaptic expression is transient but removal of CP-AMPARs can be facilitated by the activation of group 1 mGluR activation, perhaps by synthesizing GluA2 subunits locally at synapses, which also results in mGluR-dependent synaptic depression.
Sensory-dependent Plasticity
Sensory information from the environment can have long-lasting changes on the brain. Many, now classic, experiments have used the sensory system to probe mechanisms of experience-dependent plasticity. These studies have been especially useful in defining the basic properties of cortical networks in vivo (Feldman, 2009).
Rodents use their whiskers to explore their surroundings and the barrel cortex exhibits very stereotypic anatomy and plasticity, making it an excellent model to probe mechanisms of experience-dependent plasticity (Feldman and Brecht, 2005). Single whisker stimulation in the absence of adjacent whiskers results in potentiation of spared whisker responses in the cortex. This potentiation occurs through specific strengthening of excitatory transmission at layer 4-2/3 pyramidal neurons in the somatosensory cortex, only in neurons that respond to the intact whisker. This potentiation was accompanied by a change in AMPAR rectification properties suggesting a role of CP-AMPARs (Clem and Barth, 2006). More recently, the same group surprisingly found that CP-AMPARs were only transiently involved in this form of potentiation as 4-2/3 synapses older than P13 underwent potentiation but there was no evidence of CP-AMPARs. Moreover, CP-AMPARs do not underlie potentiation of layer 2/3-2/3 synapses, even when these synapses display robust single whisker-induced potentiation (Wen and Barth, 2011). This data suggests that the trafficking of CP-AMPARs is synapse specific, perhaps requiring distinct pools of AMPARs within the same post-synaptic cell (Kielland et al., 2009) and tightly regulated during development. Precisely why these synapse-specific differences in AMPAR subunit trafficking occur remains a major question to be addressed.
Addiction
Drugs of addiction have long-lasting effects on brain circuitry and often can induce changes rapidly with a single exposure. The expression mechanisms of drug-evoked plasticity have been explored, implicating AMPAR trafficking and plasticity as important molecular substrates (Luscher and Malenka, 2011). In the ventral tegmental area (VTA) of the basal ganglia, dopamine neurons are thought to be a prime mediator of drug-induced reward responses. One of the first studies implicating a role for CP-AMPARs in addiction found that a single injection of cocaine significantly increased the ratio of AMPAR/NMDAR responses (as measured by AMPA/NMDA EPSCs from midbrain slices) for approximately a week following the injection of cocaine (Ungless et al., 2001). Prolonged self-administration of cocaine results in an increase in the AMPA/NMDA ratio that lasts several months. Electrophysiological measures suggested that the change in the ratio was caused partly by an increase in AMPAR-EPSCs. This increase was accompanied by a change in rectification properties (Bellone and Luscher, 2006), suggesting the presence of GluA2-lacking AMPARs. CP-AMPARs were confirmed by the sensitivity of the EPSCs to Joro toxin. However, thetotal numbers of AMPARs seemed to be the same, suggesting that a subunit switch had occurred rather than direct insertion of CP-AMPARs. What are the consequences of this subunit switch? Further studies revealed that induction protocols that would normally elicit LTP in DA neurons did not induce LTP in cocaine-treated animals and inverted the rules for the induction of synaptic plasticity (Ungless et al., 2001; Mameli et al., 2011) as described below. Normally depolarization induces potentiation of transmission because NMDARs become unblocked but cocaine administration reduced NMDAR currents, which would account for the lack LTP induction. Conversely, a slight hyperpolarization of DA neurons during afferent stimulation induced a strengthening of AMPAR transmission only in slices from mice that had received cocaine. This is probably due to the fact that CP-AMPARs are blocked under depolarizing conditions but pass more current when hyperpolarized. Thus, cocaine administration induces changes in the basal properties of excitatory synapses as well as in the rules that govern the induction of activity-dependent synaptic plasticity.
An intriguing idea that results from these findings is that reversing the AMPAR subunit switch may be a method to treat drug addiction. Activation of group 1 metabotropic glutamate receptors (mGluR1/5) in slices from cocaine-treated mice quickly removes CP-AMPARs and replaces them with GluA2-containing ones, leading to LTD (Bellone and Luscher, 2005, 2006). This form of LTD requires rapid synthesis of GluA2 subunits via local translation from mRNA present in dendrites of DA neurons (Mameli et al., 2007). Interfering with mGluR-LTD, via expression of a dominant-negative peptide that disrupts the mGluR1/5–Homer interaction selectively in the VTA, significantly prolongs the persistence of cocaine-evoked plasticity (Mameli et al., 2009). Another recent study found AMPAR potentiation due to an increase in CP-AMPARs in the nucleus accumbens (NAc), another midbrain structure implicated in drug addiction (Conrad et al., 2008). The CP-AMPAR antagonist 1-naphthylacetyl spermine (NASPM) was injected into the accumbens of cocaine-exposed rats before tests for cue-induced cocaine-seeking resulted in decreased cue-induced cocaine-seeking after cocaine withdrawal. The authors suggest that this potentiation mediates the incubation of cocaine craving due to increased reactivity of accumbens neurons to cocaine-related cues. As follow-up studies show, an mGluR1/5 agonist could reverse the abnormal accumulation of CP-AMPARs in the NAc (McCutcheon et al., 2011) and depotentiation of cortical NAc inputs by optogenetic stimulation in vivo restored normal transmission and abolished cocaine-induced locomotor sensitization (Pascoli et al., 2012). Taken together, these results suggest that mGluR1/5 activation in VTA DA neurons may be a novel therapeutic target for addiction by decreasing the abnormal levels of CP-AMPARs.
CP-AMPAR Induced Metaplasticity in Memory Consolidation
Fear memory has been extensively studied in rodents, with much of the circuitry now well defined (Rodrigues et al., 2004). A standard protocol to induce fear memory involves pairing a tone [a neutral cue/conditioned stimulus (CS)] with a foot shock [an unconditioned stimulus (US)]. Cue-dependent fear conditioning is mediated by the potentiation of glutamatergic synaptic transmission in the lateral amygdala (LA) (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). In a recent study by Clem and Huganir (2010) an enhancement of AMPAR-EPSCs at thalamic afferents to LA neurons occurs after auditory fear conditioning in mice. This enhancement was observed as both an increase in the AMPA/NMDA ratio as well as larger AMPAR miniature EPSC amplitudes and these changes lasted for days. Potentiation of AMPAR transmission was accompanied by a slowly developing enhancement of rectification that was sensitive to NASPM. These changes peaked at 24 h after conditioning but intriguingly were not evident 1 week later even though potentiation remained. This data suggests a transient role for CP-AMPARs in the expression of fear conditioning but not a role in the maintenance of potentiation or memory. This exquisite and specific stability of potentiation in the face of significant AMPAR subunit changes is remarkable considering the differences in channel properties of CP-AMPARs. Based on the cocaine studies discussed above, the authors sought out an LTD protocol that would remove CP-AMPARs and found that LTD (mediated through NMDA and mGluR1 receptors) was enhanced after fear conditioning in LA neurons. The authors then reasoned that removing CP-AMPARs could underlie protocols of fear memory extinction. Standard extinction protocols present the CS without a reinforcing US, resulting in diminished responses to the CS. However, the memory is not fully erased, as spontaneous recovery of the memory can occur by specific cues (Herry et al., 2010). In contrast, if the fear memory is retrieved or reactivated by a single presentation of the tone without the shock 1 hour before the extinction session, the fear responses are permanently removed (Monfils et al., 2009). Previous studies have shown that recall of a memory renders that memory trace labile again, where it becomes sensitive to disruption in similar manner to when the original memory was being consolidated. Over a certain time period the memory is reconsolidated. Clem and Huganir (2010) find that reconsolidation is blocked by mGluR1 antagonists and results in a decrease in AMPAR transmission that was previously potentiated by fear conditioning. This reversal in potentiation is due to the selective removal of CP-AMPARs, and the LTD induction was greatly reduced in mice receiving the retrieval–extinction protocol, compared with those receiving extinction alone. These findings suggest that the presence of CP-AMPARs renders the memory trace labile and allows full memory erasure or modification but only in a constrained time window. It will be of some interest to determine how CP-AMPARs mediate this phenomenon but the results are consistent with an emerging theme that CP-AMPARs allow metaplasticity (i.e., plasticity changes in response to the activity historyof the neuron) to occur (Abraham and Bear, 1996).
Homeostatic AMPAR Scaling
Neuronal output is normally very stable and chronic changes in activity/input levels result in compensatory homeostatic changes that maintain a constant output (Turrigiano, 2011). One particular mechanism of homeostatic plasticity involves the synaptic scaling of post-synaptic AMPARs (Turrigiano et al., 1998). Levels of synaptic AMPARs scale up in conditions of low activity and down in conditions of high activity are typically evident in bidirectional changes in mini EPSC amplitude. The molecular mechanisms of AMPAR scaling are beginning to emerge and many studies have shown that CP-AMPARs play a role. AMPAR currents show inward rectification and become sensitive to CP-AMPAR antagonists after neuronal activity blockade (Thiagarajan et al., 2005; Sutton et al., 2006) and scaling is blocked in neurons chronically incubated with PhTx (Hou et al., 2008). Moreover, molecules that can induce synaptic up-scaling of AMPARs such as retinoic acid and the cytokine tumor necrosis factor-alpha cause an increase in surface CP-AMPARs (Stellwagen and Malenka, 2006; Aoto et al., 2008). Another molecular player involved in AMPAR scaling, Arc/arg3.1, may also preferentially regulate GluA1 during scaling as Arc KO neurons exhibit a significant increase in surface GluA1 but not GluA2 expression and synaptic scaling of AMPARs in the visual cortex is abolished both in vitro and in vivo (Shepherd et al., 2006; Gao et al., 2010; McCurry et al., 2010). GluA2-lacking receptors are also inserted after prolonged inactivity at individual synapses, a form of very local homeostatic plasticity that is also Arc-dependent (Beique et al., 2011).
The role of GluA1 in scaling, however, does not seem to be universal. Tetrodotoxin (TTX) treatment, which blocks action potentials, induced synaptic scaling in cultured visual cortical neurons through the insertion of GluA2-containing AMPARs at synapses, which was blocked by a GluA2 C-tail peptide but not by a GluA1 C-tail peptide (Gainey et al., 2009). Similarly, reducing GluA2 levels with a short hairpin RNA (shRNA) blocks synaptic scaling. These data are consistent with reports from cortical, hippocampal, and spinal cultures where scaling was induced by TTX-dependent activity blockade (O’Brien et al., 1998; Wierenga et al., 2005; Sutton et al., 2006). In contrast, several other studies have shown that scaling induced by chronic action potential and NMDAR blockade (using the drug APV) together is mediated by CP-AMPARs (Ju et al., 2004; Thiagarajan et al., 2005; Sutton et al., 2006). The discrepancy in these findings seems to result from the methods used to modulate neuronal activity, i.e., blockade using TTX, or APV or TTX + APV. The blocking of NMDARs seems to evoke local protein synthesis of GluA1 protein (Sutton et al., 2006). It remains unclear why scaling would require CP-AMPARs or whether this type of scaling is qualitatively different from scaling mediated by GluA2.
CP-AMPARs are Modulated by Sleep/Wake Cycles
Sleep remains one of the biggest enigmas in science. Why do we sleep and what why is it useful? Emerging evidence suggests that sleep is integral for normal brain functioning and memory consolidation (Diekelmann and Born, 2010). A leading theory suggests that sleep sub-serves a homeostatic role in maintaining optimal levels of synaptic transmission (Vyazovskiy et al., 2008). At a simplistic level the theory proposes that during wakefulness net synaptic transmission slowly potentiates, perhaps due to LTP-like processes during learning. If synaptic transmission continues to potentiate learning may be obstructed due to saturation of net synaptic weights. The theory therefore contends that during sleep this potentiation is reversed due to LTD-like processes, which allows memory to consolidate. One study found increases in GluA1-containing AMPAR levels in synaptoneurosomes during wakefulness and decreases during sleep in both cortex and hippocampus. Other molecular changes in AMPAR, CaMKII, and GSK3β phosphorylation were also consistent with synaptic potentiation during wakefulness and with depression during sleep (Vyazovskiy et al., 2008). Another study found that AMPAR mEPSCs, in slices of frontal cortex from mice and rats, were increased in frequency and amplitude after wakefulness and decreased after sleep (Liu et al., 2010). Consistent with these results, the slope and amplitude of field potentials increase during wakefulness and decrease during sleep in freely moving rats (Vyazovskiy et al., 2008).
A recent study found that the trafficking of CP-AMPARs is modulated during sleep/wake cycles, perhaps via CP-AMPAR- dependent homeostatic AMPAR scaling (Lante et al., 2011). CP-AMPARs accounted for ~25% of total EPSP size in layer 5 cortical pyramidal neurons from somatosensory cortex at the end of the wakefulness/dark period as assessed using PhTx and rectification properties. In contrast EPSPs were PhTx insensitive with linear current–voltage characteristics at the end of the wake/light period. The CP-AMPAR removal during the wake period occurred gradually over time and was significant after 4 h. Similarly, CP-AMPARs were evident 4 h after light offset. The significance of these findings are unclear, but are very intriguing. Further investigation into the role of CP-AMPARs in sleep may shed light on the role of sleep in memory consolidation.
Conclusion
Research on CP-AMPARs is in its infancy, but the role of CP-AMPARs in synaptic and experience-dependent plasticity is beginning to be elucidated. It seems clear that there are specific trafficking rules that govern when and where these particular AMPARs are expressed. A general theme that emerges from the studies above is that CP-AMPARs are transiently expressed after plasticity-inducing stimuli in many contexts. This transient expression is usually accompanied by potentiation of synaptic transmission. However, CP-AMPARs generally do not mediate maintenance of plasticity but instead seem to play a role in maintaining neural circuits in a “plasticity-ready” state (i.e., they act as a switch to turn on metaplasticity) that can then keep memory traces labile, but only in a time-delimited manner. Indeed, long-term expression of CP-AMPARs has been associated with neuronal pathology such as ischemia and therefore long-term expression of CP-AMPARs may have a detrimental effect on neuronal function (Liu and Zukin, 2007).
Many questions remain and very little, at the mechanistic level, is known about how the expression of CP-AMPARs affects neuronal properties. Are there specific second messenger systems controlled through calcium flux via CP-AMPARs? What are the trafficking rules that govern activity-dependent CP-AMPAR expression? How are CP-AMPARs specifically removed and replaced by GluA2-containing AMPARs? Not only are these questions important for understanding basic brain function but, as seen in the studies mentioned in this article, CP-AMPARs may be an exciting new target for therapeutics in drug addiction and memory disorders.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Dr. Asha Bhakar for helpful comments on the manuscript. Jason D. Shepherd is funded by the NIH (1-K99-NS076364-01).
References
Abraham, W. C., and Bear, M. F. (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130.
Alle, H., Jonas, P., and Geiger, J. R. (2001). PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc. Natl. Acad. Sci. U.S.A. 98, 14708–14713.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320.
Beique, J. C., Na, Y., Kuhl, D., Worley, P. F., and Huganir, R. L. (2011). Arc-dependent synapse-specific homeostatic plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 816–821.
Bellone, C., and Luscher, C. (2005). mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. Eur. J. Neurosci. 21, 1280–1288.
Bellone, C., and Luscher, C. (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9, 636–641.
Burnashev, N., Monyer, H., Seeburg, P. H., and Sakmann, B. (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8, 189–198.
Clem, R. L., and Barth, A. (2006). Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49, 663–670.
Clem, R. L., and Huganir, R. L. (2010). Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330, 1108–1112.
Conrad, K. L., Tseng, K. Y., Uejima, J. L., Reimers, J. M., Heng, L. J., Shaham, Y., Marinelli, M., and Wolf, M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121.
Donevan, S. D., and Rogawski, M. A. (1995). Intracellular polyamines mediate inward rectification of Ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropio nic acid receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 9298–9302.
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55.
Feldman, D. E., and Brecht, M. (2005). Map plasticity in somatosensory cortex. Science 310, 810–815.
Gainey, M. A., Hurvitz-Wolff, J. R., Lambo, M. E., and Turrigiano, G. G. (2009). Synaptic scaling requires the GluR2 subunit of theAMPA receptor. J. Neurosci. 29, 6479–6489.
Gao, M., Sossa, K., Song, L., Errington, L., Cummings, L., Hwang, H., Kuhl, D., Worley, P., and Lee, H. K. (2010). A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci. 30, 7168–7178.
Goldberg, J. H., Tamas, G., Aronov, D., and Yuste, R. (2003). Calcium microdomains in aspiny dendrites. Neuron 40, 807–821.
Herry, C., Ferraguti, F., Singewald, N., Letzkus, J. J., Ehrlich, I., and Luthi, A. (2010). Neuronal circuits of fear extinction. Eur. J. Neurosci. 31, 599–612.
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108.
Hou, Q., Zhang, D., Jarzylo, L., Huganir, R. L., and Man, H. Y. (2008). Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 105, 775–780.
Isaac, J. T., Ashby, M. C., and Mcbain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Jonas, P., Racca, C., Sakmann, B., Seeburg, P. H., and Monyer, H. (1994). Differences in Ca2+ permeability of AMPA-typeglutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron 12, 1281–1289.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C. C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253.
Kielland, A., Bochorishvili, G., Corson, J., Zhang, L., Rosin, D. L., Heggelund, P., and Zhu, J. J. (2009). Activity patterns govern synapse-specific AMPA receptor trafficking between deliverable and synaptic pools. Neuron 62, 84–101.
Lamsa, K. P., Heeroma, J. H., Somogyi, P., Rusakov, D. A., and Kullmann, D. M. (2007). Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science 315, 1262–1266.
Lante, F., Toledo-Salas, J. C., Ondrejcak, T., Rowan, M. J., and Ulrich, D. (2011). Removal of synaptic Ca2+-permeable AMPA receptors during sleep. J. Neurosci. 31, 3953–3961.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Liu, Z. W., Faraguna, U., Cirelli, C., Tononi, G., and Gao, X. B. (2010). Direct evidence for wake-related increases and sleep-related decreases in synaptic strength in rodent cortex. J. Neurosci. 30, 8671–8675.
Lomeli, H., Mosbacher, J., Melcher, T., Hoger, T., Geiger, J. R., Kuner, T., Monyer, H., Higuchi, M., Bach, A., and Seeburg, P. H. (1994). Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266, 1709–1713.
Lu, W., Shi, Y., Jackson, A. C., Bjorgan, K., During, M. J., Sprengel, R., Seeburg, P. H., and Nicoll, R. A. (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268.
Luscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663.
Mameli, M., Balland, B., Lujan, R., and Luscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Mameli, M., Bellone, C., Brown, M. T., and Luscher, C. (2011). Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat. Neurosci. 14, 414–416.
Mameli, M., Halbout, B., Creton, C., Engblom, D., Parkitna, J. R., Spanagel, R., and Luscher, C. (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 12, 1036–1041.
Man, H. Y. (2011). GluA2-lacking, calcium-permeable AMPA receptors – inducers of plasticity? Curr. Opin. Neurobiol. 21, 291–298.
McCurry, C. L., Shepherd, J. D., Tropea, D., Wang, K. H., Bear, M. F., and Sur, M. (2010). Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat. Neurosci. 13, 450–457.
McCutcheon, J. E., Loweth, J. A., Ford, K. A., Marinelli, M., Wolf, M. E., and Tseng, K. Y. (2011). Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J. Neurosci. 31, 14536–14541.
McKernan, M. G., and Shinnick-Gallagher, P. (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611.
Monfils, M. H., Cowansage, K. K., Klann, E., and Ledoux, J. E. (2009). Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 324, 951–955.
O’Brien, R. J., Kamboj, S., Ehlers, M. D., Rosen, K. R., Fischbach, G. D., and Huganir, R. L. (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078.
Pascoli, V., Turiault, M., and Luscher, C. (2012). Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481, 71–75.
Rodrigues, S. M., Schafe, G. E., and Ledoux, J. E. (2004). Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron 44, 75–91.
Rogan, M. T., Staubli, U. V., and Ledoux, J. E. (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607.
Shepherd, J. D., and Huganir, R. L. (2007). The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643.
Shepherd, J. D., Rumbaugh, G., Wu, J., Chowdhury, S., Plath, N., Kuhl, D., Huganir, R. L., and Worley, P. F. (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484.
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Thiagarajan, T. C., Lindskog, M., and Tsien, R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737.
Turrigiano, G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Ungless, M. A., Whistler, J. L., Malenka, R. C., and Bonci, A. (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411, 583–587.
Vyazovskiy, V. V., Cirelli, C., Pfister-Genskow, M., Faraguna, U., and Tononi, G. (2008). Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat. Neurosci. 11, 200–208.
Wen, J. A., and Barth, A. L. (2011). Input-specific critical periods for experience-dependent plasticity in layer 2/3 pyramidal neurons. J. Neurosci. 31, 4456–4465.
Keywords: calcium permeable AMPA receptors, sleep, memory, homeostatic plasticity, synaptic plasticity, drug addiction, metaplasticity, fear conditioning
Citation: Shepherd JD (2012) Memory, plasticity, and sleep – a role for calcium permeable AMPA receptors? Front. Mol. Neurosci. 5:49. doi: 10.3389/fnmol. 2012.00049
Received: 23 August 2011; Accepted: 26 March 2012;
Published online: 11 April 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USACopyright: © 2012 Shepherd. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jason D. Shepherd, Department of Brain and Cognitive Sciences, The Picower Institute for Learning and Memory, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Building 46, Room 3301, Cambridge, MA 02139, USA. e-mail: jshephe@mit.edu