The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution
 Nico P. Dantuma
Nico P. Dantuma Laura C. Bott
Laura C. Bott- 1Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden
- 2Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA
The ubiquitin-proteasome system (UPS) has been implicated in neurodegenerative diseases based on the presence of deposits consisting of ubiquitylated proteins in affected neurons. It has been postulated that aggregation-prone proteins associated with these disorders, such as α-synuclein, β-amyloid peptide, and polyglutamine proteins, compromise UPS function, and delay the degradation of other proteasome substrates. Many of these substrates play important regulatory roles in signaling, cell cycle progression, or apoptosis, and their inadvertent stabilization due to an overloaded and improperly functioning UPS may thus be responsible for cellular demise in neurodegeneration. Over the past decade, numerous studies have addressed the UPS dysfunction hypothesis using various model systems and techniques that differ in their readout and sensitivity. While an inhibitory effect of some disease proteins on the UPS has been demonstrated, increasing evidence attests that the UPS remains operative in many disease models, which opens new possibilities for treatment. In this review, we will discuss the paradigm shift that repositioned the UPS from being a prime suspect in the pathophysiology of neurodegeneration to an attractive therapeutic target that can be harnessed to accelerate the clearance of disease-linked proteins.
Introduction
Proteinopathies form a large group of pathologies that are characterized by the presence of abnormally folded proteins in affected cells (Carrell and Lomas, 1997). Among those diseases are hereditary neurodegenerative disorders, such as Huntington's disease (HD), spinal and bulbar muscular atrophy (SBMA), and several forms of autosomal dominant spinocerebellar ataxias (SCAs; types 1-3, 6, 7, 17), caused by polyglutamine (polyQ) repeat expansions in unrelated proteins (Orr and Zoghbi, 2007; La Spada and Taylor, 2010). The observation that the expanded polyQ repeat renders proteins prone to aggregation has raised the idea that members of this disease family may share a common pathogenic mechanism (Scherzinger et al., 1997). It has further been suggested that related pathogenic events may be an underlying cause in other neurodegenerative diseases characterized by the presence of protein aggregates, such as Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS) (Sherman and Goldberg, 2001). The technical advantage of working with monogenic polyQ diseases has brought together scientists from a broad variety of research disciplines who have studied the effects of the pathogenic proteins on a wide spectrum of cellular processes in cultured cells and animal models.
Since the discovery of repeat expansions as a genetic basis of hereditary neurodegenerative diseases more than two decades ago (La Spada et al., 1991), we have learned that polyQ proteins have an impact on diverse cellular processes such as transcription, transport, neuronal function, and viability. The long list of cellular functions affected by these proteins suggests that the disease-linked proteins disturb one or more systems central to these processes, causing many downstream pathways to collapse during the course of pathology. The ubiquitin-proteasome system (UPS) has received particular attention in the study of neurodegenerative disorders due to its role as a critical regulator of protein homeostasis in eukaryotic cells. It keeps the cellular environment free of misfolded, defective, and aggregation-prone proteins, which have been found to accumulate in neurodegenerative diseases (Ciechanover and Brundin, 2003). The protein quality control function of the UPS is, however, only one of the essential processes that engages this multitasking proteolytic system which governs also cell cycle progression and induction of apoptosis (Hershko and Ciechanover, 1998). It has been proposed that the vast amounts of aggregation-prone polyQ proteins may overwhelm the UPS and compromise other essential functions of the machinery required for maintaining cellular homeostasis (Mayer et al., 1989). According to this model, blockade of the UPS by the disease protein would result in global accumulation of proteasome substrates and thus provides an explanation for the broad and diverse effects on cellular homeostasis in affected cells.
Many laboratories have addressed the UPS dysfunction hypothesis over the last decade and it has become evident that UPS activity is preserved in the majority of neurodegenerative disorders. In the course of these studies, adaptive cellular responses have been identified that help to alleviate the burden of aggregation-prone proteins to keep ubiquitin-dependent proteolysis operative. Here we will discuss the transition of our view on the UPS from a dysfunctional system that catalyzes cytotoxicity in neurodegenerative diseases to a powerful proteolytic system that may be exploitable in therapeutic strategies aimed at clearing aggregation-prone proteins from cells.
The Ubiquitin-Proteasome System as Prime Suspect
The UPS, which is the principal pathway for the clearance of short-lived, damaged, and misfolded proteins in the nucleus and cytoplasm, consists of two separate, consecutive steps: ubiquitylation and proteasomal degradation (Hershko and Ciechanover, 1998; Kleiger and Mayor, 2014). An enzymatic cascade composed of ubiquitin activator, conjugase, and ligase catalyzes the covalent attachment of ubiquitin to a substrate protein. Ubiquitin is conjugated via its carboxy-terminal glycine to an internal lysine (Lys) residue or, less commonly, to the free amino (N) terminus of the substrate (Pickart, 2001). Multiple rounds of ubiquitylation lead to the formation of a polyubiquitin chain, which can function as a signal for degradation by the proteasome, a multi-protein complex consisting of a 20S core particle and 19S regulatory particles, at one or both ends. The active sites responsible for the chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasome are situated in the interior surface of the 20S core particle, thereby shielding their proteolytic activities from the rest of the cellular proteome (Bedford et al., 2010). The proteasome unfolds substrates and threads the polypeptide chains through the inner channel, where they are cleaved into short peptides (Bhattacharyya et al., 2014). Following their release from the barrel, peptides are rapidly processed into amino acids by cellular aminopeptidases and recycled (Reits et al., 2003).
Ubiquitylation has many other roles in cells besides proteasomal degradation (Figure 1). The destiny of a given substrate protein is determined by the type of ubiquitin assembly to which it is connected. This is possible because ubiquitin contains seven Lys residues in its amino acid sequence at positions 6, 11, 27, 29, 33, 48, and 63, which can serve as acceptors for additional ubiquitin monomers in the construction of polyubiquitin chains (Komander and Rape, 2012). As a result, many different chain topologies can be formed, which are recognized by specific ubiquitin-binding adaptors in the relevant cellular pathways. Chain topology and substrate specificity are determined by a large spectrum of ubiquitin-ligating and -modifying enzymes, whose expression levels and activities are tightly regulated in a tissue-, cell-, and compartment-specific manner. Several different types of polyubiquitin linkages target substrates to the proteasome, such as Lys11 and Lys29 in addition to the canonical Lys48-linked chains, as well as conjugation of a single ubiquitin molecule (Kravtsova-Ivantsiv and Ciechanover, 2012). Mono- and polyubiquitin chains also regulate non-proteolytic functions in cells, such as protein activity and localization (Seet et al., 2006). While most ubiquitin chains can target proteins for proteasomal degradation, it has been shown that Lys63-linked ubiquitin chains are the only modification that do not behave as a proteasome targeting signal in vivo (Nathan et al., 2013). Instead these chains play pivotal roles in signaling, endocytosis, and DNA repair. More recently, this chain topology has been implicated in macroautophagy (Kraft et al., 2010), a pathway that targets cytoplasmic proteins and organelles for degradation in lysosomes (Nakatogawa et al., 2009). Therefore, the UPS is highly interconnected with other proteolytic and non-proteolytic cellular processes at multiple levels, whereby it controls many diverse functions in cells.
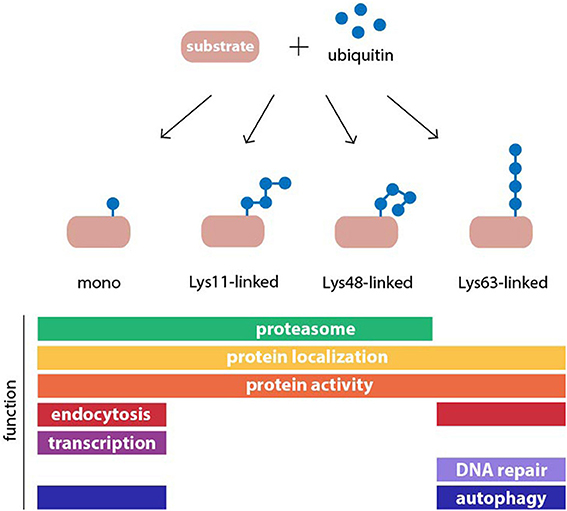
Figure 1. Structure and function of common ubiquitin modifications. Ubiquitin may be conjugated to protein substrates as either a monomer or a polymeric chain, in which one of seven internal lysine (Lys) residues of ubiquitin, or the N-terminal methionine, serves as an acceptor for additional ubiquitin moieties. The type of polyubiquitin linkage dictates the topology of the resulting chain. Ubiquitin modifications can regulate protein function or act as a signal in many cellular processes. Examples for functions of monoubiquitylation, and homogenous Lys11-, Lys48-, and Lys63-linked polyubiquitin chains are shown.
Given the vast amount of proteins that are involved in the UPS, it is perhaps not surprising that some of them have been genetically linked to neurodegenerative disorders (Ciechanover and Brundin, 2003). On first sight, the finding that mutations in genes encoding components of the UPS can cause or predispose for neurodegeneration supports the notion of inefficient ubiquitin-dependent proteolysis as a shared pathogenic mechanism. However, it should not be overlooked that the UPS and, in particular, the ubiquitin-targeting step are connected to many different processes besides proteasomal degradation. Several cases of neurodegeneration-linked mutations in UPS components are now known to affect ubiquitin-dependent processes that do not target proteins to the proteasome, while the UPS remains largely operative.
One of the best-studied examples of a UPS component that has been linked to neurodegeneration is the ubiquitin ligase Parkin, mutations in which cause an autosomal recessive juvenile-onset PD (Kitada et al., 1998). Early studies revealed that Parkin can target endoplasmic reticulum (ER)-derived proteins (Yang et al., 2003) and polyQ proteins for proteasomal degradation (Tsai et al., 2003) suggesting that a defect in the UPS-mediated protein quality control may be responsible for this pathology. However, during recent years it has become apparent that Parkin is also involved in the autophagy pathway that results in the degradation of dysfunctional mitochondria in lysosomes, a process known as mitophagy (Ashrafi and Schwarz, 2013). Parkin cooperates with the mitochondrial kinase PINK1, which has also been linked to PD, thus strengthening the genetic association between mitophagy and neurodegeneration (Clark et al., 2006; Park et al., 2006). Another theory suggests that Lys63-linked polyubiquitin chains generated by Parkin may be important for targeting aggregation-prone proteins to inclusions bodies (IBs) (Olzmann et al., 2007). Although the possibility remains that UPS dysfunction contributes to PD, recent studies point to autophagy-related processes as central to the pathology caused by Parkin mutations.
Another example is ubiquilin, a ubiquitin-binding shuttle factor that is involved in escorting polyubiquitylated proteins to the proteasome for degradation (Elsasser and Finley, 2005). Overexpression of ubiquilin has a neuroprotective effect in mice expressing a fragment of the polyQ protein causative for HD, huntingtin (El Ayadi et al., 2012). A single nucleotide polymorphism (SNP) that causes alternative splicing of the ubiquilin transcript predisposes for late-onset AD (Bertram et al., 2005). Given the function of ubiquilin as a shuttling substrate receptor in the UPS, it is tempting to speculate that the SNP in ubiquilin alters the ability of cells to recognize and destroy misfolded proteins. However, the AD-linked ubiquilin variants do not cause a general block of the UPS, but instead, were found to selectively cause accumulation of presenilin-1, which is involved in amyloid precursor protein (APP) processing (Viswanathan et al., 2011). Moreover, ubiquilin-1 functions as a molecular chaperone that regulates trafficking and processing of APP, which has been linked to its ability to stimulate polyubiquitylation of APP with non-proteolytic Lys63-linked polyubiquitin chains (El Ayadi et al., 2012). Together this suggests that while ubiquilin-1 may regulate the production of β-amyloid peptide at multiple levels in a ubiquitin-dependent fashion, the variants linked to AD do not seem to trigger a general failure of the UPS.
The ubiquitin-selective chaperone valosin-containing protein (VCP) has also attracted attention from researchers who study the link between the UPS and neurodegenerative disorders. Mutations in VCP cause multisystem proteinopathy (MSP), with among its spectrum of symptoms frontotemporal dementia (Watts et al., 2004), as well as the motor neuron disease amyotrophic lateral sclerosis (ALS) (Johnson et al., 2010). VCP is critical for proteasomal degradation of certain proteins and it is believed that it does so by means of its ATP-dependent chaperone activity that can unfold or segregate proteasome substrates from their environment (Stolz et al., 2011). However, ubiquitin-dependent functions of VCP are not limited to proteasomal degradation (Dantuma and Hoppe, 2012; Meyer et al., 2012) and include ubiquitin-selective autophagy (Ju et al., 2009; Tresse et al., 2010), the clearance of stress granules (Buchan et al., 2013), mitochondrial integrity (Bartolome et al., 2013), Parkin-dependent mitophagy (Kim et al., 2013), and the DNA damage response (Acs et al., 2011; Meerang et al., 2011). Overexpression of VCP mutants linked to MSP and ALS did not inhibit the UPS (Tresse et al., 2010), whereas some of the VCP-mediated events mentioned above were altered (Ju et al., 2009; Tresse et al., 2010; Bartolome et al., 2013; Fujita et al., 2013; Kim et al., 2013), suggesting that the pathology caused by mutant VCP likely involves essential ubiquitin-dependent processes that are different from proteasomal degradation.
A unique example of a protein responsible for a polyQ neurodegenerative disease that is involved in ubiquitin-dependent proteasomal degradation is ataxin-3. It functions as a deubiquitylation enzyme that reverses the ubiquitin mark on proteins by disassembling Lys48- and Lys63-linked polyubiquitin chains with a preference for the latter (Winborn et al., 2008). Expansion of the polyQ repeat that resides in ataxin-3 are the underlying cause for the most common form of autosomal dominant SCA, known as SCA-3 or Machado-Joseph disease. Notably, ataxin-3 physically interacts with VCP and regulates proteasomal degradation of ER-derived substrates (Wang et al., 2006; Zhong and Pittman, 2006). Overexpression of either wild-type or polyQ-expanded ataxin-3 compromises the functionality of the UPS resulting in increased levels of proteasome substrates (Burnett et al., 2003). Interestingly, non-expanded ataxin-3 is also a known suppressor of polyQ toxicity in models of SCA-3 and other polyQ diseases (Warrick et al., 2005). Although the reason for this protective effect is poorly understood, it may be attributed to the ability of ataxin-3 to stimulate sequestration of misfolded proteins (Burnett and Pittman, 2005). Finally, ataxin-3 regulates the ubiquitylation status of Parkin and stimulates its degradation by autophagy, linking also this protein to other ubiquitin-dependent systems (Durcan et al., 2011). This study, like the previous examples, suggests a complex relationship between the UPS and neurodegeneration and shows that the effects of disease-associated proteins on ubiquitin-dependent proteolysis are difficult to predict.
Two important lessons can be drawn from the afore-mentioned examples. First, each one of these examples underscores the prevalent notion that the UPS is tightly connected to neurodegenerative disorders. The fact that unrelated diseases share the presence of causative mutations in UPS components leaves little doubt that the UPS plays a central role in these pathologies. Second, they illustrate that care should be taken when extrapolating genetic data to disease mechanisms. Thus, the general view that a compromised UPS is responsible for the typical accumulation of misfolded proteins in neurodegenerative diseases is problematic due the complex and multilayered connection between the UPS and a variety of cellular functions. The fact that a large number of essential cellular functions other than protein quality control require ubiquitin conjugation or proteasomal degradation leaves the possibility open that the role of the UPS in these pathologies may be largely unrelated to ubiquitin-dependent protein quality control. A definite answer on whether, and if so, to what extent, impairment of the UPS contributes to the development of neurodegenerative diseases can only be obtained by empirically assessing the functional status of the UPS in each of these diseases.
Ubiquitin-Positive Inclusions
A hallmark of neurodegenerative diseases is the accumulation of abnormal proteins in insoluble deposists, or IBs. The idea that impaired clearance of misfolded proteins may be central to neurodegenerative diseases originates in part from the observation that IBs contain ubiquitin, proteasome subunits, and other UPS components (Cummings et al., 1998). Mori et al. who first described the presence of ubiquitin in IBs, already speculated that either the protease responsible for degrading ubiquitylated proteins (which was not known at that time to be the proteasome) was dysfunctional, or that ubiquitylated proteins residing in the inclusions resisted degradation (Mori et al., 1987).
For a long time, the nature of IBs has been under debate (Sisodia, 1998). While some researchers argued that they were innocent bystanders, others favored the view that IBs were directly implicated in the cellular pathology caused by aggregation-prone proteins (Sherman and Goldberg, 2001). One hypothesis that has appeared in different forms with various key players suggests that physical entrapment of proteins in IBs removes functional proteins from critical cellular processes. With respect to the UPS and chaperones, one can picture how sequestration may compromise their household function and elicit cellular pathology. According to yet another model, IBs are part of the cellular defense mechanism that protects cells against toxicity by functioning as a sink for misfolded proteins. A wealth of data in support for the latter option comes from cell biologists who found that ectopic overexpression of abnormal proteins triggers a sequence of reactions that results in the deposition of the misfolded proteins into specific perinuclear structures, which were coined aggresomes (Wojcik et al., 1996; Johnston et al., 1998; Garcia-Mata et al., 1999). These findings argue against the view that IBs are formed by passive aggregation, but instead require active, ATP-dependent processes in cells (Kopito, 2000). Moreover, related protein deposits have been documented in budding yeast, which suggests that this cellular response is conserved throughout evolution and serves cytoprotective functions in both unicellular and multicellular eukaryotic organisms (Kaganovich et al., 2008). In dividing cells, IBs are distributed asymmetrically to daughter cells during mitosis ensuring the generation of healthy progeny devoid of proteineous deposits (Rujano et al., 2006).
The most compelling evidence supporting a role of IBs as an adaptive response comes from live cell recordings, which show that IB formation correlates with increased survival in primary neurons expressing a mutant huntingtin fragment (Arrasate et al., 2004). This is consistent with the finding that reducing the load of IBs, achieved by genetically inhibiting ubiquitylation, enhances polyQ-mediated neurodegeneration in a SCA-1 mouse model (Cummings et al., 1999). Inclusions are reversible and highly dynamic structures, as switching off expression of a mutant huntingtin fragment (Yamamoto et al., 2000) or mutant ataxin-1 (Zu et al., 2004) in conditional mouse models results in the clearance of ubiquitin-positive IBs. When it comes to the UPS components and chaperones associated with IBs, it has been demonstrated that these factors do not appear to be physically trapped in IBs (Kim et al., 2002; Stenoien et al., 2002; Holmberg et al., 2004). A recent study even suggests that proteasomes play an active role in maintaining or dissolving these structures (Schipper-Krom et al., 2014). Furthermore, POH1/Rpn11, a proteasome subunit with deubiquitylation activity, stimulates the formation of IBs by generating free ubiquitin chains in proximity of the aggregated proteins and results in the recruitment of HDAC6 (Hao et al., 2013), which orchestrates ubiquitin-dependent transport of proteins to the aggresome (Kawaguchi et al., 2003). Altogether, these findings support a protective function of IBs in cells facing large amounts of misfolded proteins. Increasing evidence suggests that IBs may act as a hub for misfolded and aggregated proteins and redirect them to alternative destruction mechanisms, which will be discussed later.
Proteasome Activity in Neurodegenerative Diseases
One possibility to get a better insight into the functionality of the UPS is by assessing the individual enzymatic activities involved in ubiquitin-dependent proteasomal degradation (Lindsten and Dantuma, 2003). Ubiquitylation, on one hand, is the net result of a large family of enzymes that are involved in proteolytic and non-proteolytic processes and are therefore not straight-forward to address or to interpret. The proteasome, on the other hand, is the final destination of all ubiquitylated substrates to be degraded and creates a bottleneck in the UPS pathway. Its function is readily traceable to the individual proteolytic subunits whose activities can be measured by using specific fluorogenic substrates (Kisselev and Goldberg, 2005) or activity probes (Verdoes et al., 2006). It is therefore not surprising that the proteasome has received a lot of attention in studies that assess the functionality of the UPS in neurodegenerative diseases. However, correlation of proteasome activity measurements and UPS impairment in neurodegenerative diseases is complicated by the fact that it is presently unknown to what extent altered proteasome activity affects the overall flux of degradation of ubiquitylated substrates. Fibroblasts derived from mice with a heterozygous deletion of PSMC1/Rpt2, one of the ATPase subunits of the 19S regulatory particle, develop without obvious defects despite reduced proteasome function (Rezvani et al., 2012). Moreover, proteasome activity can be regulated through expression of individual subunits. For example, increasing the amount of active proteasomes, either through overexpression of the 20S core component PSMB5/β 5 responsible for the chymotrypsin-like activity or the proteasome assembly chaperone hUMP1/POMP, improves resistance to oxidative stress insults in human fibroblasts (Chondrogianni et al., 2005; Chondrogianni and Gonos, 2007). Likewise, levels of the PSMD11/Rpn6 proteasome subunit, which stabilizes the interaction between the regulatory and core particle and facilitates ubiquitin-dependent degradation (Pathare et al., 2012), can regulate proteasome activity (Vilchez et al., 2012a,b).
Translating how certain levels of proteasome inhibition will jeopardize the functional status of ubiquitin-dependent proteasomal degradation poses another unresolved problem in assessing the status of the UPS through proteasome activity measurements. Experiments with yeast strains expressing mutant proteasome subunits suggest that the proteolytic sites are non-redundant and vary in their contribution to overall protein degradation (Rubin et al., 1998). The chymotrypsin-like activity of the proteasome needs to be reduced by more than 80 percent in human cells before the clearance of ubiquitylated proteins becomes noticeably delayed (Dantuma et al., 2000; Bence et al., 2001). Even when this threshold is not reached, a lower degree of inhibition of the chymotrypsin-like activity does, however, impair the cells' ability to deal with an acute increase in the flux of ubiquitylated proteins during stress conditions (Dantuma et al., 2000). In mammalian cells, individual proteasome activities also differ in their contribution to overall protein degradation since the caspase-like activity can be chemically ablated without affecting the ability of cells to clear ubiquitylated proteins (Myung et al., 2001). Therefore, the differential contribution of individual proteolytic activities, the high level of redundant proteasome activity, and indirect effects of the stress status, which determine the load of ubiquitylated substrates, complicate the interpretation of proteasome activity measurements when it comes to the functional status of the UPS.
Initial studies have measured proteasome activity in postmortem human tissues and reported decreased activity in neurodegenerative disorders (Table 1). However, follow-up experiments with purified proteasomes, cell lines, and animal models have not always been consistent with those early reports. While in some studies a decrease in proteasome activity has been reported, others have found that the activity is unchanged or even increased (for references see Table 1). Increased proteasome activity could be an adaptive response to the augmented load of misfolded proteins in these diseases but there are also alternative explanations. In this respect, it is important to point out there is a set of alternative proteolytic subunits that can replace the constitutive proteases in the proteasome in response to interferon γ (Kniepert and Groettrup, 2014). The resulting immunoproteasome plays an important role in generating peptide fragments for antigen display by major histocompatibility complex class I proteins on the cell surface. Thus, changes in the activity of the proteasome may also be attributed to induction of immunoproteasomes in inflammatory response, which are commonly observed in neurodegenerative diseases (Czirr and Wyss-Coray, 2012). Indeed induction of immunoproteasome subunits have been reported in mouse models of polyQ disorders (Diaz-Hernandez et al., 2003) and other neurodegenerative diseases (Mishto et al., 2006; Cheroni et al., 2009; Orre et al., 2013).
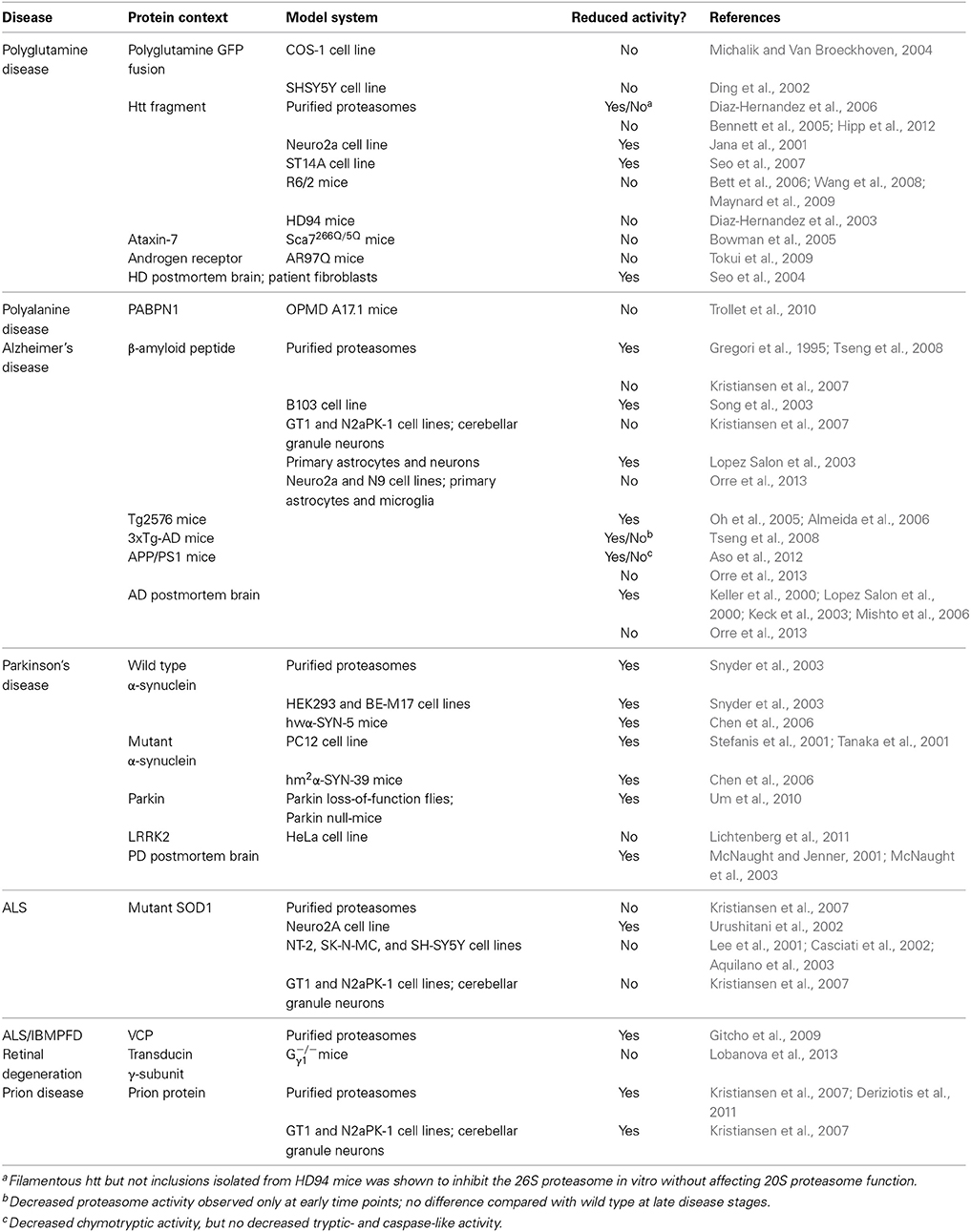
Table 1. Overview of measurements of the proteolytic activity of the proteasome in neurodegenerative disease models.
An exceptional case, in which the presence of an aggregation-protein has been mechanistically linked to inhibition of the proteasome, is the prion protein, which causes when misfolded the fatal, transmissible neurodegenerative disorder Creutzfeld-Jacob disease. Wild-type prion proteins residing in the cytosolic compartment of cells are efficiently degraded in a ubiquitin-dependent fashion (Yedidia et al., 2001) whereas cytosolic mutant prion proteins form aggresomes, which is accompanied by signs of apoptotic cell death (Kristiansen et al., 2005). It has been shown that oligomers of mutant prion protein effectively inhibit the activity of the proteasome in vitro and in vivo (Kristiansen et al., 2007) through direct binding of oligomers to the 20S proteasome core particle, which results in stabilization of a closed confirmation of the proteasome (Deriziotis et al., 2011). As a consequence, prion protein inhibits proteolysis by preventing substrate access to the proteasome core.
The main constituent of the Lewy bodies in PD, α-synuclein, has also been shown to bind to proteasomes (Snyder et al., 2003) and inhibit UPS function in vitro and in vivo (Stefanis et al., 2001; Snyder et al., 2003; Chen et al., 2006). Cytoplasmic amyloid β peptide can also interact with the proteasome (Gregori et al., 1995), however, its effect on proteasome activity is debated (for references see Table 1). Moreover, it is not clear if proteasome inhibition by α-synuclein or β-amyloid is mechanistically related to that of prion proteins.
For polyQ proteins, it has been shown that ubiquitylated filamentous aggregates of mutant huntingtin isolated from mouse or human brain samples can selectively inhibit the proteolytic activity of the proteasome (Diaz-Hernandez et al., 2006). We will discuss below that several studies suggest that this inhibitory activity of huntingtin aggregates does not compromise the UPS in vivo (Bett et al., 2009; Maynard et al., 2009; Ortega et al., 2010). Moreover, mutant huntingtin fragments have not been found to inhibit degradation of ubiquitylated substrates by the 26S proteasome in vitro (Hipp et al., 2012). Whether or not polyQ proteins can be degraded by the mammalian proteasome has been the subject of a considerable debate (Venkatraman et al., 2004; Pratt and Rechsteiner, 2008; Juenemann et al., 2013). Puromycin-sensitive aminopeptidase has been identified as the main cytosolic protease to efficiently clear expanded polyQ peptides generated by the proteasome in vitro (Bhutani et al., 2007). It has been hypothesized that proteasome-derived polyQ fragments may enhance aggregate formation (Venkatraman et al., 2004; Raspe et al., 2009), but it is unclear whether these species are actually generated in vivo. In cultured cells, aggregation of mutant huntingtin fragment is exacerbated following treatment with the proteasome inhibitor lactacystin (Waelter et al., 2001). Moreover, enhancing UPS-mediated clearance of polyQ proteins reduces their levels and toxicity, suggesting that the net outcome of accelerated proteasomal degradation is beneficial in polyQ diseases (Verhoef et al., 2002; Michalik and Van Broeckhoven, 2004).
Functional Status of the UPS in Neurodegenerative Diseases
UPS functionality, which describes the relative rate at which cells ubiquitylate and degrade proteins at a given time, can be addressed in cells and tissues in several ways. These approaches are based on the assumption that cells that cannot maintain a constant flux through the UPS will gradually build up ubiquitylated proteasome substrates irrespective of their nature. As explained earlier, the relationship between UPS activity and levels of substrates is often complex. Reduced proteasome activity does not necessarily lead to functional impairment of the UPS as long as the activity is sufficient to process ubiquitylated proteins targeted for destruction. Vice versa, ubiquitin-dependent proteolysis in cells can be severely impaired despite the presence of a normal proteasome activity profile. Furthermore, cells may mobilize compensatory mechanisms to assist a suboptimal but functioning UPS.
A straight-forward approach to assess UPS activity in cells or tissues measures the abundance or quality of ubiquitylated proteins. However, ubiquitylation is involved in many cellular processes other than proteasomal degradation (Komander and Rape, 2012) and it is therefore difficult to extrapolate whether a possible build up in ubiquitylated proteins is a consequence of UPS impairment or reflects other changes in the ubiquitin homeostasis (Groothuis et al., 2006). Substrate flux through the UPS can be assessed by determining the levels or half-lives of cellular proteins. An important caveat with this approach is that the degradation of endogenous proteasome substrates is often tightly regulated and likely to change depending on internal or external cues. Particularly in the context of neurodegenerative diseases, altered degradation rates of endogenous substrates likely reflect functional differences in protein stabilization rather than changes in overall UPS activity. This problem can be circumvented by the use of reporter substrates, which have been developed for monitoring protein degradation by the UPS. These reporters are typically based on fluorescent proteins fused to degradation signals that target the fusion protein for constitutive turnover via the UPS (Neefjes and Dantuma, 2004). They include the ubiquitin-dependent reporters UbG76V-green fluorescent protein (GFP) and GFPu that are targeted via an N-terminal ubiquitin or a short CL1 peptide motif, respectively (Dantuma et al., 2000; Bence et al., 2001). Another commonly used reporter, based on the degron derived from mouse ornithine decarboxylase (ODC), is degraded in a ubiquitin-independent fashion (Murakami et al., 1992). UPS reporter substrates are in principle devoid of intrinsic regulatory elements to eliminate the impact of variables such as posttranslational modifications, which can affect the half-life of endogenous protein substrates. Although all these reporter proteins are subject to degradation by the 26S proteasome, they are recognized by distinct targeting pathways and therefore differ in sensitivity and signal-to-background ratio. It has been shown that levels of the reporters inversely correlate with UPS activity (Dantuma et al., 2000; Bence et al., 2001). Accumulation of reporter protein relative to baseline levels is typically interpreted as reduced UPS function, however, reporter protein levels may be affected by changes in expression (Bowman et al., 2005; Tokui et al., 2009) or protein synthesis, to which they are very sensitive as a consequence of their short half-life (Li et al., 1998). These reporter substrates are important tools for studying UPS functionality in cell and mouse models of neurodegenerative diseases (Table 2).
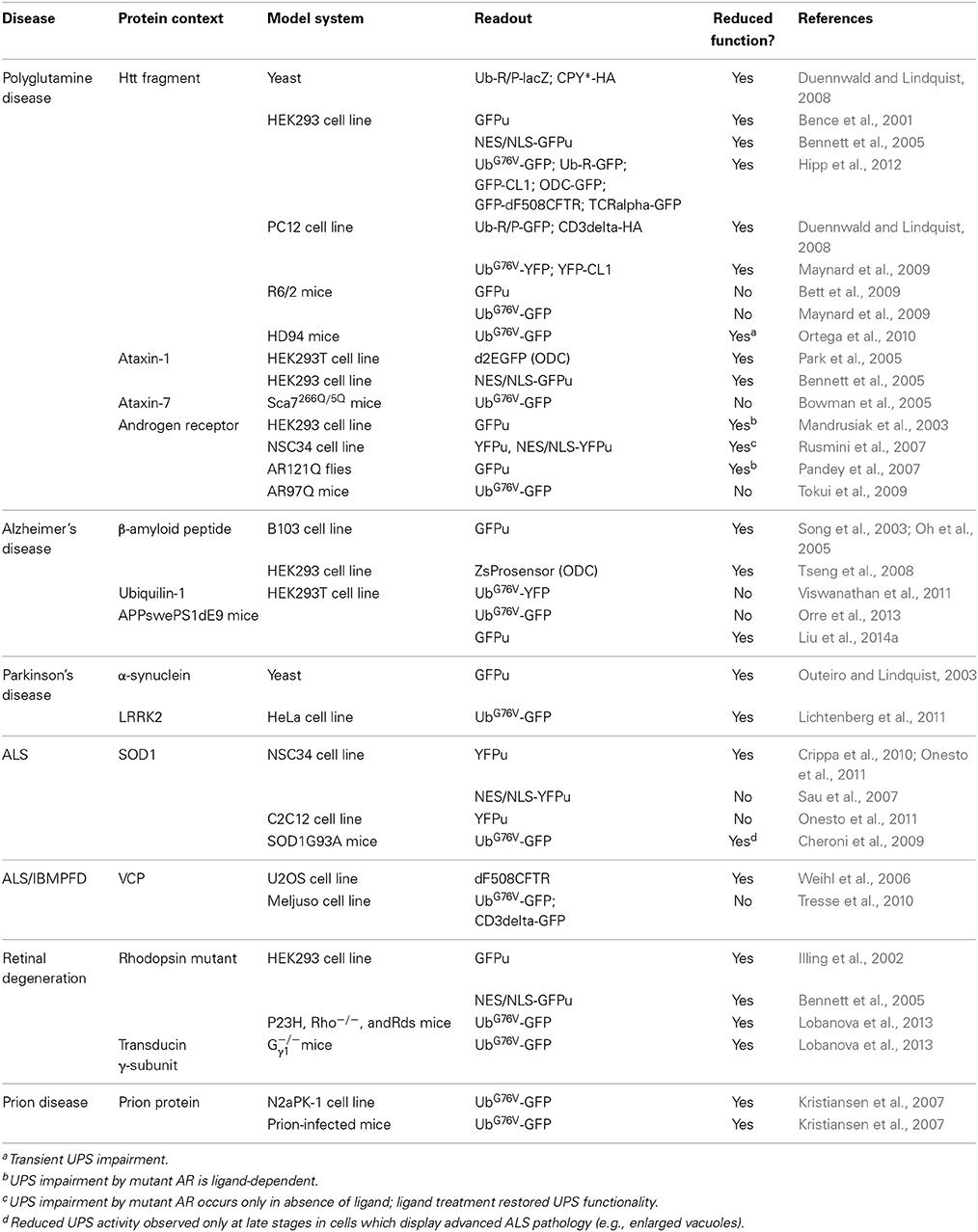
Table 2. Overview of measurements of the functionality of the ubiquitin/proteasome system in neurodegenerative disease models.
In cell culture experiments, an N-terminal fragment of polyQ-expanded huntingtin has been shown to impair the degradation of the endogenous substrate p53 (Jana et al., 2001) and GFPu (Bence et al., 2001). UPS inhibition appears to be a consequence of protein aggregation and not a unique feature of polyQ proteins since the unrelated aggregation-prone protein mutant cystic fibrosis membrane conductance regulator (CFTR) has a similar effect on the UPS (Bence et al., 2001). GFPu reporter accumulation correlates with induction of apoptosis, supporting a model in which UPS impairment induced by protein aggregation proteins is responsible for cell death although it cannot exclude that other adverse effects are responsible for the cytotoxicity (Bence et al., 2001). UPS impairment as a result of polyQ proteins affects both the nuclear and cytosolic UPS and does not seem to be confined to the compartment that accumulates the aggregation-prone proteins, arguing for general interference with UPS function (Bennett et al., 2005). A study utilizing a version of the GFPu reporter targeted to the synapse of neurons suggested that mutant huntingtin primarily affects UPS function in the synaptic compartment (Wang et al., 2008). It is noteworthy that later studies found the artificial CL1 degradation signal in GFPu to render proteins aggregation-prone (Menendez-Benito et al., 2005; Link et al., 2006), which is not surprising if one considers that this artificial degradation signal mimics targeting signals present in ER-derived proteasome substrates (Gilon et al., 2000). Subsequent studies have shown that the polyQ huntingtin fragment also affects the degradation of other UPS reporters, such as UbG76V-GFP, signifying that this effect is not limited to aggregation-prone proteins (Maynard et al., 2009; Mitra et al., 2009; Hipp et al., 2012). Analysis in a cellular model demonstrates that aggregates of N-terminal huntingtin do not directly impair the proteasome, instead, the increase in ubiquitylated proteins likely reflects a general disturbance of the cellular proteostasis network (Hipp et al., 2012).
Two transgenic mouse models expressing GFP-based reporter substrates for the UPS, UbG76V-GFP (Lindsten et al., 2003) and GFPu (Bove et al., 2006), have been instrumental in addressing UPS functionality in neurodegeneration in vivo. Using these mice, it has been demonstrated that UPS function is preserved in animals expressing polyQ-expanded proteins such as the N-terminal huntingtin fragment, R6/2 (Bett et al., 2009; Maynard et al., 2009; Ortega et al., 2010), androgen receptor (AR) responsible for SBMA (Tokui et al., 2009), and ataxin-7 that causes SCA-7 (Bowman et al., 2005). The lack of UPS impairment in the R6/2 mouse model has been particularly puzzling since the evidence for a globally dysfunctional UPS in cultured cells has largely been based on ectopic expression of the very same huntingtin fragment that is expressed in these mice (Bence et al., 2001; Jana et al., 2001; Bennett et al., 2007; Wang et al., 2008).
The development of quantitative mass spectrometry combined with efficient purification of ubiquitylated proteins or signature peptides from biological samples has been invaluable in undertaking detailed analysis of the ubiquitylated proteome in human diseases (Kessler, 2013). This method not only allows the determination of total ubiquitin levels, but also enables discrimination of different types of ubiquitin chains. Quantitative mass spectrometry has been successfully applied to investigate the composition of ubiquitin conjugates in R6/2 mice and HD patient brain and revealed accumulation of Lys11-, Lys48-, and Lys63-linked ubiquitin chains (Bennett et al., 2007). In theory, the increase in Lys11- and Lys48-linked ubiquitin chains would be consistent with a functional blockade of the UPS as both chain topologies can target proteins to the proteasome (Komander and Rape, 2012). However, Lys63-linked polyubiquitin chains also accumulate in HD and are not normally associated with proteasomal degradation (Nathan et al., 2013), suggesting that the effect of polyQ-expanded huntingtin on ubiquitin homeostasis is more complex than simple blockade of the UPS. In support of this view, two independent studies have not detected stabilization of UbG76V-GFP (Maynard et al., 2009) or GFPu (Bett et al., 2006) in R6/2 mice. Ubiquitin conjugates observed in R6/2 brains have also been shown to differ qualitatively from those observed upon proteasome inhibition (Maynard et al., 2009), indicating that the observed increase in ubiquitylation in HD may be due to changes in ubiquitin-dependent processes other than proteasomal degradation.
While data obtained with mouse models for polyQ diseases have unequivocally supported the presence of a preserved UPS, UPS impairment has been detected in models of other neurodegenerative diseases. For example studies with the UbG76V-GFP reporter mice have shown that UPS impairment occurs during progression in an ALS mouse model expressing mutant SOD1 (Cheroni et al., 2009). Likewise, UPS impairment has been demonstrated in a mouse model of prion pathology, which is consistent with a role for direct UPS impairment by the prion protein (Kristiansen et al., 2007). Opposing findings have been reported in the well-characterized APPswe/PS1dE9 mouse model for AD. A recent study has demonstrated accumulation of GFPu reporter as well as the endogenous proteasome substrate p53 in this model (Liu et al., 2014a), whereas the same mouse model did not accumulate UbG76V-GFP according to another report (Orre et al., 2013). Although documented for some disorders, global inhibition of ubiquitin-dependent proteolysis does not appear to be a universal feature of neurodegenerative diseases.
Compensatory Mechanisms
The absence of functional impairment of the UPS in mouse models of polyQ diseases does not imply that the UPS is unaffected by the presence of aggregation-prone proteins. In cellular models, polyQ proteins have been shown to elicit UPS inhibition, indicating that they can have a general negative impact on intracellular proteolysis. Transgene expression levels are an important difference between the cell lines and mouse models. In cells, proteins are typically transiently overexpressed in an acute manner whereas proteins are expressed chronically at modest levels in transgenic mice, more similar to the situation in human patients. The effect of acute induction of the N-terminal mutant huntingtin fragment has been addressed in vivo (Ortega et al., 2010) using a mouse model in which huntingtin expression can be regulated by administration of doxycyclin (Yamamoto et al., 2000). Interestingly, acute overexpression of the mutant huntingtin fragment in these mice is accompanied by increased levels of the UbG76V-GFP reporter substrate in neuronal and non-neuronal cells. Importantly, accumulation of the UPS reporter is of a transient nature, suggesting that cells activate adaptive responses to restore UPS activity (Ortega et al., 2010). Restoration of UPS function coincides with IB formation in neurons (Mitra et al., 2009; Ortega et al., 2010), which is consistent with a protective role of inclusions through sequestration of toxic oligomeric species. In support of this model, the UbG76V-GFP reporter has been shown to accumulate also in R6/2 mice following treatment with chemical inhibitors of protein aggregation, which block the formation of IBs (Heiser et al., 2002; Ortega et al., 2010).
The autophagy pathway is tightly connected with the UPS and can play a compensatory role in maintaining intracellular protein degradation under conditions of reduced UPS activity. Autophagy counteracts the toxicity of mutant huntingtin, possibly through promoting the removal of aggregated, oligomeric species (Ravikumar et al., 2004). Moreover, autophagy induction reversed UPS impairment in a Drosophila model of SBMA (Pandey et al., 2007). Chemicals that inhibit lysosomal degradation also compromise the UPS, suggesting that autophagy is required for proper functioning of ubiquitin-dependent proteasomal degradation (Korolchuk et al., 2009). Autophagy likely assists the UPS in the removal of a pool of problematic proteasome substrates, such as aggregated or damaged proteins, which would otherwise impede UPS activity. It has been suggested that, analogous to UPS dysfunction in neurodegeneration, autophagy may be compromised in polyQ diseases (Ravikumar et al., 2004). Interestingly, experiments in mice have shown that autophagy dysfunction results in a neurodegeneration phenotype associated with ubiquitin-positive inclusions, indicating that an impaired autophagolysosomal system can recapitulate neurodegeneration (Hara et al., 2006; Komatsu et al., 2006).
Ubiquitin, which serves a wide range of cellular functions besides proteasomal degradation, is closely involved in these compensatory mechanisms (Figure 2). It has been shown to target proteins to aggresomes by a mechanism that involves HDAC6, a cytosolic deacteylation enzyme that binds unanchored ubiquitin chains and facilitates the sequestration of aggregated proteins in IBs (Kawaguchi et al., 2003). The ability of specific adaptor proteins, such as p62 and NBR1, to simultaneously bind polyubiquitylated cargo and the autophagosome marker LC3 also supports a central role for ubiquitin in selective autophagy (Kraft et al., 2010). However, the role of ubiquitin as a decisive signal in autophagy has been questioned and instead it has been argued that protein oligomerization and not ubiquitylation is the primary signal (Riley et al., 2010). Both the formation of aggresomes (Olzmann et al., 2007) and ubiquitin-selective autophagy (Tan et al., 2008) are typically associated with Lys63-linked ubiquitin chains, although the involvement of other chain topologies is less clear. Notably, monoubiquitin can suffice as a signal to direct proteins to the autophagosome (Kim et al., 2008). More experiments are needed to dissect the functions of ubiquitin and the relative contribution of individual chain topologies in cellular processes.
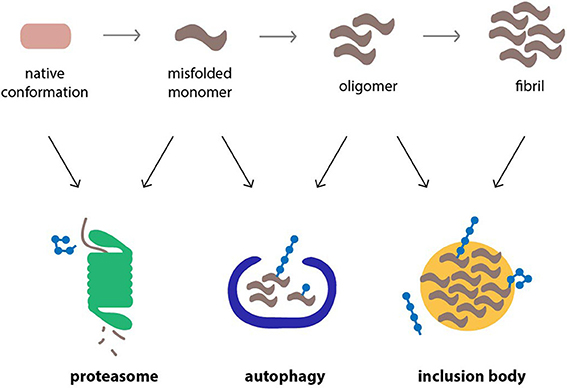
Figure 2. Cellular pathways that counteract protein aggregation are ubiquitin-dependent processes. Proteins linked to neurodegenerative diseases, such as α-synuclein, β-amyloid peptide and polyQ proteins, are prone to misfolding and aggregation in the cellular environment. The proteasome, autophagy, and inclusion bodies form a network of quality control systems which reduces levels of misfolded proteins and counteracts aggregation. All three pathways are regulated by ubiquitylation.
The fact that ubiquitin is shared between targeting mechanisms that direct substrates to the proteolytic machinery and adaptive responses, which counteract toxic proteins, suggests a functional significance in directing the joint efforts of these pathways in eliminating harmful proteins (Groothuis et al., 2006). It is possible that the accumulation of polyubiquitin conjugates in polyQ diseases may be due to upregulation of ubiquitin-dependent adaptive responses, such as the formation of inclusions or activation of ubiquitin-selective autophagy, which through their actions may preserve UPS activity. Thus, rather than being a consequence of a functional impaired UPS, the accumulation of ubiquitin conjugates may be part of the reason why the UPS remains operative even in the challenging intracellular environment of polyQ disease.
Reducing Levels of Neurodegeneration-Associated Proteins
In monogenic, dominant neurodegenerative disorders in which the identity of the disease-causing mutant proteins is known, as in the case of the polyQ disorders, an obvious but technically less trivial therapeutic approach would be to reduce the levels of the mutant protein. However, some of the gene products fulfill important functions in cells and may even be essential for viability, as is the case for huntingtin (Dragatsis et al., 2000) and ataxin-7 (Helmlinger et al., 2004). This problem may be circumvented by designing strategies to selectively target the mutated protein without affecting the wild-type form. Additionally, experiments in inducible mouse models for HD (Yamamoto et al., 2000) and SCA-1 (Zu et al., 2004) have shown that switching off expression of polyQ proteins reverses disease symptoms and neuronal pathology. A recent study has demonstrated that decreasing expression of polyQ-expanded ataxin-7 by 50 percent results in a full phenotypic rescue in a conditional SCA-7 mouse model (Furrer et al., 2013), which suggests that modest reduction of polyQ proteins may suffice for therapeutic intervention in humans. Targeting expression of mutant transcripts using RNA interference or antisense oligonucleotide technology is a promising approach to achieve this goal (Bonini and La Spada, 2005).
An alternative strategy for reducing levels of disease proteins is to accelerate their turnover by the UPS, whose function is preserved even at late stages of pathology. However, the question remains whether or not the proteasome can efficiently degrade disease-linked proteins. Some studies have suggested that the proteasome cannot degrade the polyQ proteins (Dyer and McMurray, 2001; Jana et al., 2001; Holmberg et al., 2004; Venkatraman et al., 2004) whereas others other reports show that they can be efficiently degraded by the proteasome as long as they remain in a soluble state (Verhoef et al., 2002; Kaytor et al., 2004; Michalik and Van Broeckhoven, 2004; Juenemann et al., 2013; Tsvetkov et al., 2013). Also, α-synuclein, expression levels of which can predispose individuals to PD (Singleton et al., 2003), can be degraded by the proteasome (Bennett et al., 1999), suggesting that other aggregation-prone proteins can be targeted by the UPS.
Several studies show that stimulating ubiquitylation and degradation of disease proteins through the UPS can rescue pathology in cell and animal models. For example, increasing the pool of free ubiquitin through genetic overexpression suppresses α-synuclein-induced neurodegeneration in Drosophila (Lee et al., 2009). The phenotypic rescue depends on the ability of the transgenic ubiquitin to form Lys48-linked polyubiquitin chains that typically target proteins for proteasomal degradation. Overexpression of certain ubiquitin ligases, which determine substrate specificity in the UPS pathway, also can confer protection in neurodegeneration models. Increasing levels of the ubiquitin ligase CHIP (C terminus of Hsc-70 interacting protein) delays the disease phenotype in SCA-1 (Al-Ramahi et al., 2006) and SBMA animal models (Adachi et al., 2007) through enhanced ubiquitylation and subsequent clearance of polyQ-expanded proteins. Moreover, Parkin has been shown to ubiquitylate ataxin-3 and reduces polyQ toxicity in cells (Tsai et al., 2003; Morishima et al., 2008). These findings show that the UPS is a plastic and versatile system that can be harnessed to accelerate the clearance of disease-linked proteins.
Leveraging Proteasomal Degradation
Augmenting UPS activity or targeting its activity toward disease-associated proteins may be achieved through small molecules and, though challenging, opens up the possibility of counteracting protein accumulation in neurodegeneration (Figure 3). One such strategy takes advantage of the fact that aggregation-prone proteins are better substrates for the UPS in their monomeric, soluble state compared to oligomeric assemblies. Compounds, which stimulate expression or activity of heat shock proteins, effectively counteract aggregation and increase the clearance of misfolded proteins through the UPS. The compound arimoclomol induces chaperone expression and has been shown to ameliorate disease in SBMA (Malik et al., 2013) and ALS mice (Kalmar et al., 2012). YM-1, a small molecule that increases the ability of the molecular chaperone Hsp70 to bind unfolded substrates, increases degradation of polyQ-expanded AR in cell culture and rescues toxicity in SBMA flies (Wang et al., 2013).
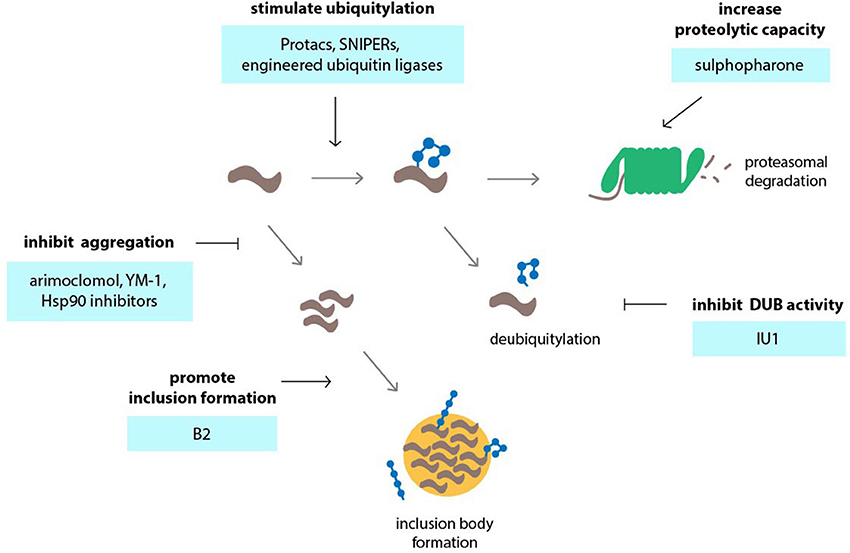
Figure 3. Targeting the ubiquitin-proteasome system (UPS) in neurodegenerative disorders using small molecules or engineering approaches. Various events in the UPS can be targeted by compounds in order to stimulate UPS activity. Among those events are accelerating of ubiquitylation by compounds or engineered ubiquitin ligases, inhibition of deubiquitylation, inhibition of protein aggregation so that the proteins remain in a state that is permissible to proteasomal degradation and stimulation of the formation of inclusion bodies which may reduce the load of aggregation-prone proteins and preserve UPS activity.
Stimulation of UPS activity is another means by which degradation of disease protein can be achieved. An important question is whether such a complex system involving a large number of proteins can be effectively activated by small molecules in a therapeutic setting. Recent studies have revealed that the capacity of the UPS can be regulated via two related transcription factors, Nrf1 and Nrf2. Nrf1 is synthesized as an ER-anchored protein and only becomes transcriptionally active in the nucleus when increased UPS function is required (Radhakrishnan et al., 2010; Grimberg et al., 2011). Nrf2 regulates the antioxidant response in cells and can stimulate expression of proteasome subunits, which likely increases degradation of oxidized proteins during stress (Pickering et al., 2012). Nrf2 can be induced by the small molecule sulforaphane, which increases proteasome levels and activity (Kwak et al., 2003, 2007), and enhances UPS function in vivo (Liu et al., 2014b). Sulforaphane has been shown to reduce mutant huntingtin protein and mitigate polyQ toxicity in neuronal cells (Liu et al., 2014b).
Ubiquitin-dependent proteasomal degradation of aggregation-prone proteins can also be stimulated through direct modulation of proteasome function (Lee et al., 2010). The regulatory particle of the proteasome harbors two deubiquitylation enzymes, namely UCH-L5 and USP14/Ubp6, that counteract proteasomal degradation by trimming ubiquitin chains of recruited substrates (Finley, 2009). It is has been proposed that chain trimming by proteasome-associated deubiquitylation enzymes may function as a molecular timer that determines the time window during which the proteasome can initiate degradation, thereby rescuing poorly ubiquitylated proteins from degradation (Lam et al., 1997). IU1, a selective small molecule inhibitor of USP14, has been shown to accelerate proteasomal degradation of aggregation-prone proteins, including proteins associated with neurodegenerative diseases such as tau, TDP43 and the polyQ protein ataxin-3, as well as oxidized proteins (Lee et al., 2010). However, it remains to be seen whether IU1 is effective in mitigating disease manifestations in animal models of neurodegenerative disorders.
Selective Proteasomal Targeting of Disease Proteins
Increasing overall degradation by stimulating UPS activity may be accompanied by unwanted side effects due to their general nature. Selectively targeting the disease-causing proteins for proteasomal is therefore expected to have many advantages over general stimulation of UPS activity. The specificity of the UPS action towards disease-linked proteins can be increased by taking advantage of the natural regulatory systems that dictate the degradation rates of these proteins. While this method has the potential of reducing side-effects of increasing overall UPS activity, it requires detailed insights in the biological function and regulation of the disease-causing protein.
Among the proteins implicated in polyQ diseases, AR is probably the one whose functions are best understood. AR is a nuclear hormone receptor that is linked to various types of cancers, most notoriously prostate cancer (Matsumoto et al., 2013). The idea of using small molecule inhibitors of heat shock protein 90 (Hsp90) as therapeutic tools in SBMA originates from cancer studies which revealed that AR is an Hsp90 client and requires heat shock proteins for proper functioning (Prescott and Coetzee, 2006). It has been shown that geldanamycin analogs 17-AAG and 17-DMAG accelerate degradation of AR and other Hsp90 client proteins and reduce polyQ-expanded AR toxicity in cell and mouse models (Waza et al., 2005; Tokui et al., 2009). Interestingly, geldanamycin is also effective in preventing aggregation of mutant huntingtin, which has been attributed to the ability of Hsp90 inhibitors to induce a general heat shock response (Sittler et al., 2001). Activation of the heat shock response may also contribute to lowering of AR half-life by geldanamycin analogs in SBMA models, as overexpression of heat shock proteins has been shown to also promote AR degradation (Bailey et al., 2002). Small molecules that promote the deposition of aggregation-prone proteins into IBs may also be beneficial in neurodegenerative diseases. For example, the compound B2 has been shown to increase inclusion formation of the mutant huntingtin fragment, polyQ-expanded AR, and α-synuclein, and thereby suppresses toxicity in cell and fly models (Bodner et al., 2006; Palazzolo et al., 2010).
Protein stability can be influenced by posttranslational modifications. One example for this is again AR, whose levels and subcellular localization is controlled by the PI3K-Akt signaling pathway. It has been shown that phosphorylation of mutant AR by the Akt kinase, which can be induced by insulin-like growth factor-1 (IGF-1), accelerates clearance of the receptor by the proteasome (Palazzolo et al., 2007). Importantly, IGF-1 reduces mutant AR aggregation and toxicity in SBMA models in vitro and in vivo (Palazzolo et al., 2009; Rinaldi et al., 2012). More recently, it has been shown that the turnover of ataxin-1 is regulated by phosphorylation by MSK1 and, accordingly, the levels of polyQ-expanded ataxin-1 could be reduced by depletion of MSK1 (Park et al., 2013). The phosphorylation event is controlled by components in the MAPK-Ras pathway, and pharmacological curtailing of the MAPK pathway with small compound inhibitors of MEK1/2 or Raf1 similarly reduced levels and toxicity of mutant ataxin-1 (Park et al., 2013).
In recent years, scientists have begun to explore the possibility to mobilize branches of the UPS that are not normally involved in handling aggregation-prone proteins. Earlier studies have shown that engineered chimeric ubiquitin ligases can be used to redirect the ubiquitylation machinery to proteins of interest (Zhou et al., 2000). Engineered ubiquitin ligases typically contain two domains: a recognition domain specific to the protein of interest, and a ubiquitylation domain. Examples for the latter are RING, HECT, and U-box domains, which directly catalyze the addition of ubiquitin to the substrate. Ubiquitylation domains may also guide the substrate protein to the enzymatic activity of a ubiquitin ligase, such as the SCF complex via the F box domain (Zhang and Zhou, 2005). An example of engineered ubiquitin ligases in the context of neurodegenerative diseases is a chimeric Dorfin-CHIP fusion that combines the substrate binding domain of the ubiquitin ligase Dorfin with the U-box of CHIP. The resulting fusion protein was found to ubiquitylate mutant SOD1 more efficiently than the wild-type Dorfin or CHIP ubiquitin ligases (Ishigaki et al., 2007). Off-target effects due to residual activity toward their native substrates, or unwanted regulation in the cellular microenvironment are a major caveat with current chimeras based on domains derived from endogenous proteins. However, an important advantage of the chimeras is that they can be designed with high specificity to certain sub-populations of the protein-of-interest. For instance, a recent study reported the generation of an SCF-βTrCP ubiquitin ligase that selectively targets only activated ErbB receptor tyrosine kinases for degradation (Kong et al., 2014).
Nanobody-based fusions are variations on protein chimeras based on single chain antibodies, which, when expressed in the intracellular environment, bind to target proteins with high specificity (Muyldermans, 2013). A fusion consisting of an F-box domain and a nanobody directed to GFP has been successfully used to deplete cytoplasmic, nuclear, and transmembrane GFP-fusion proteins in cultured cells and in vivo via the SCF complex (Caussinus et al., 2012). In theory, this approach may also be used to engineer intrabody-based ubiquitin ligases specific to disease-associated proteins, or even to pathogenic protein conformations. Although engineered ubiquitin ligases can be of great value in experimental settings and provide us with a proof-of-principle that ubiquitylation can be tailored to accelerated degradation of desired proteins, there are currently many practical limitations that limit their adaptation to therapeutic settings, such as safety and delivery to target tissues.
A number of studies have explored whether small molecules can be used to redirect endogenous ubiquitylation enzymes to defined target substrates. Most interesting in this respect is the work on molecules known as Protacs (Proteolysis-targeting chimeric molecules) and SNIPERs (Specific and non-genetic IAP-dependent Protein Erasers) (Buckley and Crews, 2014). Both types of molecules combine a ubiquitin ligase interaction peptide with a small molecule ligand specific for the target protein (Sakamoto et al., 2001). Protacs have mainly been developed to target cancer-relevant gene products but may be applicable as well to misfolded proteins in neurological disorders. One example is a chimeric molecule that unites a peptide motif from HIF1α with the AR ligand dihydrotestosterone (DHT) and can be used to accelerate degradation of AR in cultured cells (Rodriguez-Gonzalez et al., 2008). Although it would be interesting to test whether this molecule can also accelerate the degradation of mutant AR in SBMA or whether derivatives can be generated for other polyQ proteins, a limiting factor in the development of Protacs is poor permeability across cell membranes and potential antigenicity due to their peptidic nature. However, in the cases where the ubiquitin ligase can be recruited by small synthetic molecules instead of polypeptides, it may be possible to obtain compounds with potential therapeutic value. A functional cell-permeable Protac combines two active molecules, the AR ligand DHT and the Mdm2-interacting compound Nutlin, to increase ubiquitylation of AR by Mdm2 (Schneekloth et al., 2008). In SNIPERs, an IAP1-interacting bestatin ester replaces the peptide domain found in Protacs that links the chimeric molecule to ubiquitin ligase activity. SNIPERs have been used successfully in the context of steroid receptors, including AR, and efficiently reduce receptor protein levels at lower molar concentrations than Protacs (Okuhira et al., 2013). Small molecules for targeted degradation of disease-associated proteins by the UPS have so far mainly been explored in the cancer field, but the successful application in malignant cells may serve as a proof-of-principle to warrant their investigation in neurodegenerative disorders.
Concluding Remarks
Increasing evidence suggests that ubiquitin-dependent proteolysis is largely operative in many neurodegenerative diseases, and hence a causal relationship does not exist between aggregation-prone proteins and global UPS impairment. While perturbations in UPS function due to disease proteins cannot be excluded, adaptive responses such as IB formation and autophagy likely contribute to the restoration of cellular protein homeostasis. This knowledge justifies further exploration of the protein degradation machinery for treating or preventing debilitating neurodegenerative disorders. However, in order to fully appreciate the potential of the UPS as a therapeutic target, we need to continue to decipher how the UPS and compensatory pathways coordinate the detoxification and clearance of misfolded proteins.
While the current data do not support a scenario in which global impairment of the protein quality control function of the UPS lies at the basis of proteinopathies, our present understanding strongly supports an involvement of ubiquitin-dependent processes in the development or progression of neurodegenerative diseases. Dissecting the molecular mechanisms and key players responsible for changes in ubiquitylation may help to identify suitable therapeutic targets. Because it appears to be unlikely that this inhibitory activity is a shared intrinsic feature of aggregation-prone proteins, it will be important to study the interplay between the UPS and the individual disease-associated proteins in the context of their native functions.
This change of perspective from the UPS as a potential cause for neurodegeneration to it being a preserved proteolytic pathway that may be exploited in therapeutic approaches has been an important contribution of the large number of studies that have probed into the role of the UPS in polyQ disorders. Inspiring examples can be found in the identification of compounds which stimulate UPS activity and the development of small molecules that target desired proteins for proteasomal degradation. Although these studies provide us with proofs-of-principle for redirecting proteins to endogenous ubiquitin ligases, there is a clear need for compounds with more favorable drug-like properties in order to accomplish this aim in a physiological setting. If successful, such small molecules could be used on their own or in combination with other polyQ protein-reducing therapies, such as RNA interference, to reduce the levels of the toxic proteins in patients. Despite the fact that much needs to be done before we can start to evaluate the applicability of such approaches, the recent insight that at least the UPS is still on our side in fighting the toxic effects of the these rogue proteins opens other opportunities in the pursuit for therapeutics for neurodegenerative diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Florian Salomons and Emily Foran for critical reading of the manuscript. The Dantuma lab is supported by the Swedish Research Council, the Swedish Cancer Society and the Karolinska Institute.
References
Acs, K., Luijsterburg, M. S., Ackermann, L., Salomons, F. A., Hoppe, T., and Dantuma, N. P. (2011). The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18, 1345–1350. doi: 10.1038/nsmb.2188
Adachi, H., Waza, M., Tokui, K., Katsuno, M., Minamiyama, M., Tanaka, F., et al. (2007). CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J. Neurosci. 27, 5115–5126. doi: 10.1523/JNEUROSCI.1242-07.2007
Almeida, C. G., Takahashi, R. H., and Gouras, G. K. (2006). Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 26, 4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006
Al-Ramahi, I., Lam, Y. C., Chen, H. K., De Gouyon, B., Zhang, M., Perez, A. M., et al. (2006). CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 281, 26714–26724. doi: 10.1074/jbc.M601603200
Aquilano, K., Rotilio, G., and Ciriolo, M. R. (2003). Proteasome activation and nNOS down-regulation in neuroblastoma cells expressing a Cu,Zn superoxide dismutase mutant involved in familial ALS. J. Neurochem. 85, 1324–1335. doi: 10.1046/j.1471-4159.2003.01783.x
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810. doi: 10.1038/nature02998
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Aso, E., Lomoio, S., Lopez-Gonzalez, I., Joda, L., Carmona, M., Fernandez-Yague, N., et al. (2012). Amyloid generation and dysfunctional immunoproteasome activation with disease progression in animal model of familial Alzheimer's disease. Brain Pathol. 22, 636–653. doi: 10.1111/j.1750-3639.2011.00560.x
Bailey, C. K., Andriola, I. F., Kampinga, H. H., and Merry, D. E. (2002). Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 11, 515–523. doi: 10.1093/hmg/11.5.515
Bartolome, F., Wu, H. C., Burchell, V. S., Preza, E., Wray, S., Mahoney, C. J., et al. (2013). Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64. doi: 10.1016/j.neuron.2013.02.028
Bedford, L., Paine, S., Sheppard, P. W., Mayer, R. J., and Roelofs, J. (2010). Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 20, 391–401. doi: 10.1016/j.tcb.2010.03.007
Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. doi: 10.1126/science.292.5521.1552
Bennett, E. J., Bence, N. F., Jayakumar, R., and Kopito, R. R. (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351–365. doi: 10.1016/j.molcel.2004.12.021
Bennett, E. J., Shaler, T. A., Woodman, B., Ryu, K. Y., Zaitseva, T. S., Becker, C. H., et al. (2007). Global changes to the ubiquitin system in Huntington's disease. Nature 448, 704–708. doi: 10.1038/nature06022
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Bertram, L., Hiltunen, M., Parkinson, M., Ingelsson, M., Lange, C., Ramasamy, K., et al. (2005). Family-based association between Alzheimer's disease and variants in UBQLN1. N. Engl. J. Med. 352, 884–894. doi: 10.1056/NEJMoa042765
Bett, J. S., Cook, C., Petrucelli, L., and Bates, G. P. (2009). The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS ONE 4:e5128. doi: 10.1371/journal.pone.0005128
Bett, J. S., Goellner, G. M., Woodman, B., Pratt, G., Rechsteiner, M., and Bates, G. P. (2006). Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington's disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum. Mol. Genet. 15, 33–44. doi: 10.1093/hmg/ddi423
Bhattacharyya, S., Yu, H., Mim, C., and Matouschek, A. (2014). Regulated protein turnover: snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 15, 122–133. doi: 10.1038/nrm3741
Bhutani, N., Venkatraman, P., and Goldberg, A. L. (2007). Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J. 26, 1385–1396. doi: 10.1038/sj.emboj.7601592
Bodner, R. A., Outeiro, T. F., Altmann, S., Maxwell, M. M., Cho, S. H., Hyman, B. T., et al. (2006). Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc. Natl. Acad. Sci. U.S.A. 103, 4246–4251. doi: 10.1073/pnas.0511256103
Bonini, N. M., and La Spada, A. R. (2005). Silencing polyglutamine degeneration with RNAi. Neuron 48, 715–718. doi: 10.1016/j.neuron.2005.11.008
Bove, J., Zhou, C., Jackson-Lewis, V., Taylor, J., Chu, Y., Rideout, H. J., et al. (2006). Proteasome inhibition and Parkinson's disease modeling. Ann. Neurol. 60, 260–264. doi: 10.1002/ana.20937
Bowman, A. B., Yoo, S. Y., Dantuma, N. P., and Zoghbi, H. Y. (2005). Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 20,20. doi: 10.1093/hmg/ddi064
Buchan, J. R., Kolaitis, R. M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
Buckley, D. L., and Crews, C. M. (2014). Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew. Chem. Int. Ed Engl. 53, 2312–2330. doi: 10.1002/anie.201307761
Burnett, B. G., and Pittman, R. N. (2005). The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. U.S.A. 102, 4330–4335. doi: 10.1073/pnas.0407252102
Burnett, B., Li, F., and Pittman, R. N. (2003). The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 12, 3195–3205. doi: 10.1093/hmg/ddg344
Carrell, R. W., and Lomas, D. A. (1997). Conformational disease. Lancet 350, 134–138. doi: 10.1016/S0140-6736(97)02073-4
Casciati, A., Ferri, A., Cozzolino, M., Celsi, F., Nencini, M., Rotilio, G., et al. (2002). Oxidative modulation of nuclear factor-kappaB in human cells expressing mutant fALS-typical superoxide dismutases. J. Neurochem. 83, 1019–1029. doi: 10.1046/j.1471-4159.2002.01232.x
Caussinus, E., Kanca, O., and Affolter, M. (2012). Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 19, 117–121. doi: 10.1038/nsmb.2180
Chen, L., Thiruchelvam, M. J., Madura, K., and Richfield, E. K. (2006). Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol. Dis. 23, 120–126. doi: 10.1016/j.nbd.2006.02.004
Cheroni, C., Marino, M., Tortarolo, M., Veglianese, P., De Biasi, S., Fontana, E., et al. (2009). Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 82–96. doi: 10.1093/hmg/ddn319
Chondrogianni, N., and Gonos, E. S. (2007). Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp. Gerontol. 42, 899–903. doi: 10.1016/j.exger.2007.01.012
Chondrogianni, N., Tzavelas, C., Pemberton, A. J., Nezis, I. P., Rivett, A. J., and Gonos, E. S. (2005). Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 280, 11840–11850. doi: 10.1074/jbc.M413007200
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neuordegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. doi: 10.1016/S0896-6273(03)00606-8
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Crippa, V., Sau, D., Rusmini, P., Boncoraglio, A., Onesto, E., Bolzoni, E., et al. (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. doi: 10.1093/hmg/ddq257
Cummings, C. J., Mancini, M. A., Antalffy, B., Defranco, D. B., Orr, H. T., and Zoghbi, H. Y. (1998). Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19, 148–154. doi: 10.1038/502
Cummings, C. J., Reinstein, E., Sun, Y., Antalffy, B., Jiang, Y., Ciechanover, A., et al. (1999). Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24, 879–892. doi: 10.1016/S0896-6273(00)81035-1
Czirr, E., and Wyss-Coray, T. (2012). The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163. doi: 10.1172/JCI58656
Dantuma, N. P., and Hoppe, T. (2012). Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin-dependent extraction from chromatin. Trends Cell Biol. 22, 483–491. doi: 10.1016/j.tcb.2012.06.003
Dantuma, N. P., Lindsten, K., Glas, R., Jellne, M., and Masucci, M. G. (2000). Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543. doi: 10.1038/75406
Deriziotis, P., Andre, R., Smith, D. M., Goold, R., Kinghorn, K. J., Kristiansen, M., et al. (2011). Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 30, 3065–3077. doi: 10.1038/emboj.2011.224
Diaz-Hernandez, M., Hernandez, F., Martin-Aparicio, E., Gomez-Ramos, P., Moran, M. A., Castano, J. G., et al. (2003). Neuronal induction of the immunoproteasome in Huntington's disease. J. Neurosci. 23, 11653–11661. Available online at: http://www.jneurosci.org/content/23/37/11653.abstract
Diaz-Hernandez, M., Valera, A. G., Moran, M. A., Gomez-Ramos, P., Alvarez-Castelao, B., Castano, J. G., et al. (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 98, 1585–1596. doi: 10.1111/j.1471-4159.2006.03968.x
Ding, Q., Lewis, J. J., Strum, K. M., Dimayuga, E., Bruce-Keller, A. J., Dunn, J. C., et al. (2002). Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. J. Biol. Chem. 277, 13935–13942. doi: 10.1074/jbc.M107706200
Dragatsis, I., Levine, M. S., and Zeitlin, S. (2000). Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 26, 300–306. doi: 10.1038/81593
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
Durcan, T. M., Kontogiannea, M., Thorarinsdottir, T., Fallon, L., Williams, A. J., Djarmati, A., et al. (2011). The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 20, 141–154. doi: 10.1093/hmg/ddq452
Dyer, R. B., and McMurray, C. T. (2001). Mutant protein in Huntington disease is resistant to proteolysis in affected brain. Nat. Genet. 29, 270–278. doi: 10.1038/ng745
El Ayadi, A., Stieren, E. S., Barral, J. M., and Boehning, D. (2012). Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc. Natl. Acad. Sci. U.S.A. 109, 13416–13421. doi: 10.1073/pnas.1206786109
Elsasser, S., and Finley, D. (2005). Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 7, 742–749. doi: 10.1038/ncb0805-742
Finley, D. (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513. doi: 10.1146/annurev.biochem.78.081507.101607
Fujita, K., Nakamura, Y., Oka, T., Ito, H., Tamura, T., Tagawa, K., et al. (2013). A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat. Commun. 4, 1816. doi: 10.1038/ncomms2828
Furrer, S. A., Waldherr, S. M., Mohanachandran, M. S., Baughn, T. D., Nguyen, K. T., Sopher, B. L., et al. (2013). Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum. Mol. Genet. 22, 890–903. doi: 10.1093/hmg/dds495
Garcia-Mata, R., Bebok, Z., Sorscher, E. J., and Sztul, E. S. (1999). Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 146, 1239–1254. doi: 10.1083/jcb.146.6.1239
Gilon, T., Chomsky, O., and Kulka, R. G. (2000). Degradation signals recognized by the Ubc6p-Ubc7p ubiquitin-conjugating enzyme pair. Mol. Cell. Biol. 20, 7214–7219. doi: 10.1128/MCB.20.19.7214-7219.2000
Gitcho, M. A., Strider, J., Carter, D., Taylor-Reinwald, L., Forman, M. S., Goate, A. M., et al. (2009). VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284, 12384–12398. doi: 10.1074/jbc.M900992200
Gregori, L., Fuchs, C., Figueiredo-Pereira, M. E., Van Nostrand, W. E., and Goldgaber, D. (1995). Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J. Biol. Chem. 270, 19702–19708. doi: 10.1074/jbc.270.34.19702
Grimberg, K. B., Beskow, A., Lundin, D., Davis, M. M., and Young, P. (2011). Basic leucine zipper protein Cnc-C is a substrate and transcriptional regulator of the Drosophila 26S proteasome. Mol. Cell. Biol. 31, 897–909. doi: 10.1128/MCB.00799-10
Groothuis, T. A., Dantuma, N. P., Neefjes, J., and Salomons, F. A. (2006). Ubiquitin crosstalk connecting cellular processes. Cell Div. 1:21. doi: 10.1186/1747-1028-1-21
Hao, R., Nanduri, P., Rao, Y., Panichelli, R. S., Ito, A., Yoshida, M., et al. (2013). Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828. doi: 10.1016/j.molcel.2013.08.016
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Heiser, V., Engemann, S., Brocker, W., Dunkel, I., Boeddrich, A., Waelter, S., et al. (2002). Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc. Natl. Acad. Sci. U.S.A. 99(Suppl. 4), 16400–16406. doi: 10.1073/pnas.182426599
Helmlinger, D., Hardy, S., Sasorith, S., Klein, F., Robert, F., Weber, C., et al. (2004). Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 13, 1257–1265. doi: 10.1093/hmg/ddh139
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hipp, M. S., Patel, C. N., Bersuker, K., Riley, B. E., Kaiser, S. E., Shaler, T. A., et al. (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J. Cell Biol. 196, 573–587. doi: 10.1083/jcb.201110093
Holmberg, C. I., Staniszewski, K. E., Mensah, K. N., Matouschek, A., and Morimoto, R. I. (2004). Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23, 4307–4318. doi: 10.1038/sj.emboj.7600426
Illing, M. E., Rajan, R. S., Bence, N. F., and Kopito, R. R. (2002). A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 277, 34150–34160. doi: 10.1074/jbc.M204955200
Ishigaki, S., Niwa, J., Yamada, S., Takahashi, M., Ito, T., Sone, J., et al. (2007). Dorfin-CHIP chimeric proteins potently ubiquitylate and degrade familial ALS-related mutant SOD1 proteins and reduce their cellular toxicity. Neurobiol. Dis. 25, 331–341. doi: 10.1016/j.nbd.2006.09.017
Jana, N. R., Zemskov, E. A., Wang, G., and Nukina, N. (2001). Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10, 1049–1059. doi: 10.1093/hmg/10.10.1049
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. doi: 10.1083/jcb.143.7.1883
Ju, J. S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Juenemann, K., Schipper-Krom, S., Wiemhoefer, A., Kloss, A., Sanz Sanz, A., and Reits, E. A. (2013). Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 288, 27068–27084. doi: 10.1074/jbc.M113.486076
Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. doi: 10.1038/nature07195
Kalmar, B., Edet-Amana, E., and Greensmith, L. (2012). Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 378–392. doi: 10.3109/17482968.2012.660953
Kawaguchi, Y., Kovacs, J. J., McLaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738. doi: 10.1016/S0092-8674(03)00939-5
Kaytor, M. D., Wilkinson, K. D., and Warren, S. T. (2004). Modulating huntingtin half-life alters polyglutamine-dependent aggregate formation and cell toxicity. J. Neurochem. 89, 962–973. doi: 10.1111/j.1471-4159.2004.02376.x
Keck, S., Nitsch, R., Grune, T., and Ullrich, O. (2003). Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J. Neurochem. 85, 115–122. doi: 10.1046/j.1471-4159.2003.01642.x
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer's disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Kessler, B. M. (2013). Ubiquitin—omics reveals novel networks and associations with human disease. Curr. Opin. Chem. Biol. 17, 59–65. doi: 10.1016/j.cbpa.2012.12.024
Kim, N. C., Tresse, E., Kolaitis, R. M., Molliex, A., Thomas, R. E., Alami, N. H., et al. (2013). VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80. doi: 10.1016/j.neuron.2013.02.029
Kim, P. K., Hailey, D. W., Mullen, R. T., and Lippincott-Schwartz, J. (2008). Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. U.S.A. 105, 20567–20574. doi: 10.1073/pnas.0810611105
Kim, S., Nollen, E. A., Kitagawa, K., Bindokas, V. P., and Morimoto, R. I. (2002). Polyglutamine protein aggregates are dynamic. Nat. Cell Biol. 4, 826–831. doi: 10.1038/ncb863
Kisselev, A. F., and Goldberg, A. L. (2005). Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Meth. Enzymol. 398, 364–378. doi: 10.1016/S0076-6879(05)98030-0
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kleiger, G., and Mayor, T. (2014). Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. 24, 352–359. doi: 10.1016/j.tcb.2013.12.003
Kniepert, A., and Groettrup, M. (2014). The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 39, 17–24. doi: 10.1016/j.tibs.2013.10.004
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi: 10.1146/annurev-biochem-060310-170328
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kong, F., Zhang, J., Li, Y., Hao, X., Ren, X., Li, H., et al. (2014). Engineering a single ubiquitin ligase for the selective degradation of all activated ErbB receptor tyrosine kinases. Oncogene 33, 986–995. doi: 10.1038/onc.2013.33
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/S0962-8924(00)01852-3
Korolchuk, V. I., Mansilla, A., Menzies, F. M., and Rubinsztein, D. C. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527. doi: 10.1016/j.molcel.2009.01.021
Kraft, C., Peter, M., and Hofmann, K. (2010). Selective autophagy: ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12, 836–841. doi: 10.1038/ncb0910-836
Kravtsova-Ivantsiv, Y., and Ciechanover, A. (2012). Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 125, 539–548. doi: 10.1242/jcs.093567
Kristiansen, M., Deriziotis, P., Dimcheff, D. E., Jackson, G. S., Ovaa, H., Naumann, H., et al. (2007). Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 26, 175–188. doi: 10.1016/j.molcel.2007.04.001
Kristiansen, M., Messenger, M. J., Klohn, P. C., Brandner, S., Wadsworth, J. D., Collinge, J., et al. (2005). Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J. Biol. Chem. 280, 38851–38861. doi: 10.1074/jbc.M506600200
Kwak, M. K., Cho, J. M., Huang, B., Shin, S., and Kensler, T. W. (2007). Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 43, 809–817. doi: 10.1016/j.freeradbiomed.2007.05.029
Kwak, M. K., Wakabayashi, N., Greenlaw, J. L., Yamamoto, M., and Kensler, T. W. (2003). Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 23, 8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003
Lam, Y. A., Xu, W., Demartino, G. N., and Cohen, R. E. (1997). Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737–740. doi: 10.1038/385737a0
La Spada, A. R., and Taylor, J. P. (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247–258. doi: 10.1038/nrg2748
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Lee, B. H., Lee, M. J., Park, S., Oh, D. C., Elsasser, S., Chen, P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. doi: 10.1038/nature09299
Lee, F. K., Wong, A. K., Lee, Y. W., Wan, O. W., Chan, H. Y., and Chung, K. K. (2009). The role of ubiquitin linkages on alpha-synuclein induced-toxicity in a Drosophila model of Parkinson's disease. J. Neurochem. 110, 208–219. doi: 10.1111/j.1471-4159.2009.06124.x
Lee, M., Hyun, D. H., Marshall, K. A., Ellerby, L. M., Bredesen, D. E., Jenner, P., et al. (2001). Effect of overexpression of BCL-2 on cellular oxidative damage, nitric oxide production, antioxidant defenses, and the proteasome. Free Radic. Biol. Med. 31, 1550–1559. doi: 10.1016/S0891-5849(01)00633-5
Li, X., Zhao, X., Fang, Y., Jiang, X., Duong, T., Fan, C., et al. (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975. doi: 10.1074/jbc.273.52.34970
Lichtenberg, M., Mansilla, A., Zecchini, V. R., Fleming, A., and Rubinsztein, D. C. (2011). The Parkinson's disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis. 2:e196. doi: 10.1038/cddis.2011.81
Lindsten, K., and Dantuma, N. P. (2003). Monitoring the ubiquitin/proteasome system in conformational diseases. Ageing Res. Rev. 2, 433–449. doi: 10.1016/S1568-1637(03)00031-X
Lindsten, K., Menendez-Benito, V., Masucci, M. G., and Dantuma, N. P. (2003). A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 21, 897–902. doi: 10.1038/nbt851
Link, C. D., Fonte, V., Hiester, B., Yerg, J., Ferguson, J., Csontos, S., et al. (2006). Conversion of green fluorescent protein into a toxic, aggregation-prone protein by C-terminal addition of a short peptide. J. Biol. Chem. 281, 1808–1816. doi: 10.1074/jbc.M505581200
Liu, Y., Hettinger, C. L., Zhang, D., Rezvani, K., Wang, X., and Wang, H. (2014a). The proteasome function reporter GFPu accumulates in young brains of the APPswe/PS1dE9 Alzheimer's disease mouse model. Cell. Mol. Neurobiol. 34, 315–322. doi: 10.1007/s10571-013-0022-9
Liu, Y., Hettinger, C. L., Zhang, D., Rezvani, K., Wang, X., and Wang, H. (2014b). Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington's disease. J. Neurochem. 129, 539–547. doi: 10.1111/jnc.12647
Lobanova, E. S., Finkelstein, S., Skiba, N. P., and Arshavsky, V. Y. (2013). Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 9986–9991. doi: 10.1073/pnas.1305521110
Lopez Salon, M., Morelli, L., Castano, E. M., Soto, E. F., and Pasquini, J. M. (2000). Defective ubiquitination of cerebral proteins in Alzheimer's disease. J. Neurosci. Res. 62, 302–310. doi: 10.1002/1097-4547(20001015)62:2<302::AID-JNR15>3.0.CO;2-L
Lopez Salon, M., Pasquini, L., Besio Moreno, M., Pasquini, J. M., and Soto, E. (2003). Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 180, 131–143. doi: 10.1016/S0014-4886(02)00060-2
Malik, B., Nirmalananthan, N., Gray, A. L., La Spada, A. R., Hanna, M. G., and Greensmith, L. (2013). Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy. Brain 136, 926–943. doi: 10.1093/brain/aws343
Mandrusiak, L. M., Beitel, L. K., Wang, X., Scanlon, T. C., Chevalier-Larsen, E., Merry, D. E., et al. (2003). Transglutaminase potentiates ligand-dependent proteasome dysfunction induced by polyglutamine-expanded androgen receptor. Hum. Mol. Genet. 12, 1497–1506. doi: 10.1093/hmg/ddg161
Matsumoto, T., Sakari, M., Okada, M., Yokoyama, A., Takahashi, S., Kouzmenko, A., et al. (2013). The androgen receptor in health and disease. Annu. Rev. Physiol. 75, 201–224. doi: 10.1146/annurev-physiol-030212-183656
Mayer, R. J., Lowe, J., Lennox, G., Doherty, F., and Landon, M. (1989). Intermediate filaments and ubiquitin: a new thread in the understanding of chronic neurodegenerative diseases. Prog. Clin. Biol. Res. 317, 809–818.
Maynard, C. J., Bottcher, C., Ortega, Z., Smith, R., Florea, B. I., Diaz-Hernandez, M., et al. (2009). Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc. Natl. Acad. Sci. U.S.A. 106, 13986–13991. doi: 10.1073/pnas.0906463106
McNaught, K. S., Belizaire, R., Isacson, O., Jenner, P., and Olanow, C. W. (2003). Altered proteasomal function in sporadic Parkinson's disease. Exp. Neurol. 179, 38–46. doi: 10.1006/exnr.2002.8050
McNaught, K. S., and Jenner, P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci. Lett. 297, 191–194. doi: 10.1016/S0304-3940(00)01701-8
Meerang, M., Ritz, D., Paliwal, S., Garajova, Z., Bosshard, M., Mailand, N., et al. (2011). The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 13, 1376–1382. doi: 10.1038/ncb2367
Menendez-Benito, V., Verhoef, L. G., Masucci, M. G., and Dantuma, N. P. (2005). Endoplasmic reticulum stress compromises the ubiquitin-proteasome system. Hum. Mol. Genet. 14, 2787–2799. doi: 10.1093/hmg/ddi312
Meyer, H., Bug, M., and Bremer, S. (2012). Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123. doi: 10.1038/ncb2407
Michalik, A., and Van Broeckhoven, C. (2004). Proteasome degrades soluble expanded polyglutamine completely and efficiently. Neurobiol. Dis. 16, 202–211. doi: 10.1016/j.nbd.2003.12.020
Mishto, M., Bellavista, E., Santoro, A., Stolzing, A., Ligorio, C., Nacmias, B., et al. (2006). Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol. Aging 27, 54–66. doi: 10.1016/j.neurobiolaging.2004.12.004
Mitra, S., Tsvetkov, A. S., and Finkbeiner, S. (2009). Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 284, 4398–4403. doi: 10.1074/jbc.M806269200
Mori, H., Kondo, J., and Ihara, Y. (1987). Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science 235, 1641–1644. doi: 10.1126/science.3029875
Morishima, Y., Wang, A. M., Yu, Z., Pratt, W. B., Osawa, Y., and Lieberman, A. P. (2008). CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum. Mol. Genet. 17, 3942–3952. doi: 10.1093/hmg/ddn296
Murakami, Y., Matsufuji, S., Kameji, T., Hayashi, S., Igarashi, K., Tamura, T., et al. (1992). Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360, 597–599. doi: 10.1038/360597a0
Muyldermans, S. (2013). Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797. doi: 10.1146/annurev-biochem-063011-092449
Myung, J., Kim, K. B., Lindsten, K., Dantuma, N. P., and Crews, C. M. (2001). Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol. Cell 7, 411–420. doi: 10.1016/S1097-2765(01)00188-5
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi: 10.1038/nrm2708
Nathan, J. A., Kim, H. T., Ting, L., Gygi, S. P., and Goldberg, A. L. (2013). Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 32, 552–565. doi: 10.1038/emboj.2012.354
Neefjes, J., and Dantuma, N. P. (2004). Fluorescent probes for proteolysis: tools for drug discovery. Nat. Rev. Drug Discov. 3, 58–69. doi: 10.1038/nrd1282
Oh, S., Hong, H. S., Hwang, E., Sim, H. J., Lee, W., Shin, S. J., et al. (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 126, 1292–1299. doi: 10.1016/j.mad.2005.07.006
Okuhira, K., Demizu, Y., Hattori, T., Ohoka, N., Shibata, N., Nishimaki-Mogami, T., et al. (2013). Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 104, 1492–1498. doi: 10.1111/cas.12272
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. doi: 10.1083/jcb.200611128
Onesto, E., Rusmini, P., Crippa, V., Ferri, N., Zito, A., Galbiati, M., et al. (2011). Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 118, 266–280. doi: 10.1111/j.1471-4159.2011.07298.x
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. doi: 10.1146/annurev.neuro.29.051605.113042
Orre, M., Kamphuis, W., Dooves, S., Kooijman, L., Chan, E. T., Kirk, C. J., et al. (2013). Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain 136, 1415–1431. doi: 10.1093/brain/awt083
Ortega, Z., Diaz-Hernandez, M., Maynard, C. J., Hernandez, F., Dantuma, N. P., and Lucas, J. J. (2010). Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J. Neurosci. 30, 3675–3688. doi: 10.1523/JNEUROSCI.5673-09.2010
Outeiro, T. F., and Lindquist, S. (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. doi: 10.1126/science.1090439
Palazzolo, I., Burnett, B. G., Young, J. E., Brenne, P. L., La Spada, A. R., Fischbeck, K. H., et al. (2007). Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 16, 1593–1603. doi: 10.1093/hmg/ddm109
Palazzolo, I., Nedelsky, N. B., Askew, C. E., Harmison, G. G., Kasantsev, A. G., Taylor, J. P., et al. (2010). B2 attenuates polyglutamine-expanded androgen receptor toxicity in cell and fly models of spinal and bulbar muscular atrophy. J. Neurosci. Res. 88, 2207–2216. doi: 10.1002/jnr.22389
Palazzolo, I., Stack, C., Kong, L., Musaro, A., Adachi, H., Katsuno, M., et al. (2009). Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63, 316–328. doi: 10.1016/j.neuron.2009.07.019
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863. doi: 10.1038/nature05853
Park, J., Al-Ramahi, I., Tan, Q., Mollema, N., Diaz-Garcia, J. R., Gallego-Flores, T., et al. (2013). RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature 498, 325–331. doi: 10.1038/nature12204
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Park, Y., Hong, S., Kim, S. J., and Kang, S. (2005). Proteasome function is inhibited by polyglutamine-expanded ataxin-1, the SCA1 gene product. Mol. Cells 19, 23–30. Available online at: http://www.molcells.org/journal/view.html?year=2005&volume=19&number=1&spage=23
Pathare, G. R., Nagy, I., Bohn, S., Unverdorben, P., Hubert, A., Korner, R., et al. (2012). The proteasomal subunit Rpn6 is a molecular clamp holding the core and regulatory subcomplexes together. Proc. Natl. Acad. Sci. U.S.A. 109, 149–154. doi: 10.1073/pnas.1117648108
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pickering, A. M., Linder, R. A., Zhang, H., Forman, H. J., and Davies, K. J. (2012). Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J. Biol. Chem. 287, 10021–10031. doi: 10.1074/jbc.M111.277145
Pratt, G., and Rechsteiner, M. (2008). Proteasomes cleave at multiple sites within polyglutamine tracts: activation by PA28gamma(K188E). J. Biol. Chem. 283, 12919–12925. doi: 10.1074/jbc.M709347200
Prescott, J., and Coetzee, G. A. (2006). Molecular chaperones throughout the life cycle of the androgen receptor. Cancer Lett. 231, 12–19. doi: 10.1016/j.canlet.2004.12.037
Radhakrishnan, S. K., Lee, C. S., Young, P., Beskow, A., Chan, J. Y., and Deshaies, R. J. (2010). Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17–28. doi: 10.1016/j.molcel.2010.02.029
Raspe, M., Gillis, J., Krol, H., Krom, S., Bosch, K., Van Veen, H., et al. (2009). Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci. 122, 3262–3271. doi: 10.1242/jcs.045567
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Reits, E., Griekspoor, A., Neijssen, J., Groothuis, T., Jalink, K., Van Veelen, P., et al. (2003). Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 18, 97–108. doi: 10.1016/S1074-7613(02)00511-3
Rezvani, N., Elkharaz, J., Lawler, K., Mee, M., Mayer, R. J., and Bedford, L. (2012). Heterozygosity for the proteasomal Psmc1 ATPase is insufficient to cause neuropathology in mouse brain, but causes cell cycle defects in mouse embryonic fibroblasts. Neurosci. Lett. 521, 130–135. doi: 10.1016/j.neulet.2012.05.070
Riley, B. E., Kaiser, S. E., Shaler, T. A., Ng, A. C., Hara, T., Hipp, M. S., et al. (2010). Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 191, 537–552. doi: 10.1083/jcb.201005012
Rinaldi, C., Bott, L. C., Chen, K. L., Harmison, G. G., Katsuno, M., Sobue, G., et al. (2012). Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol. Med. 18, 1261–1268. doi: 10.2119/molmed.2012.00271
Rodriguez-Gonzalez, A., Cyrus, K., Salcius, M., Kim, K., Crews, C. M., Deshaies, R. J., et al. (2008). Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 27, 7201–7211. doi: 10.1038/onc.2008.320
Rubin, D. M., Glickman, M. H., Larsen, C. N., Dhruvakumar, S., and Finley, D. (1998). Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 17, 4909–4919. doi: 10.1093/emboj/17.17.4909
Rujano, M. A., Bosveld, F., Salomons, F. A., Dijk, F., Van Waarde, M. A., Van Der Want, J. J., et al. (2006). Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 4:e417. doi: 10.1371/journal.pbio.0040417
Rusmini, P., Sau, D., Crippa, V., Palazzolo, I., Simonini, F., Onesto, E., et al. (2007). Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 28, 1099–1111. doi: 10.1016/j.neurobiolaging.2006.05.015
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559. doi: 10.1073/pnas.141230798
Sau, D., De Biasi, S., Vitellaro-Zuccarello, L., Riso, P., Guarnieri, S., Porrini, M., et al. (2007). Mutation of SOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 16, 1604–1618. doi: 10.1093/hmg/ddm110
Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., et al. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558. doi: 10.1016/S0092-8674(00)80514-0
Schipper-Krom, S., Juenemann, K., Jansen, A. H., Wiemhoefer, A., Van Den Nieuwendijk, R., Smith, D. L., et al. (2014). Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 588, 151–159. doi: 10.1016/j.febslet.2013.11.023
Schneekloth, A. R., Pucheault, M., Tae, H. S., and Crews, C. M. (2008). Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908. doi: 10.1016/j.bmcl.2008.07.114
Seet, B. T., Dikic, I., Zhou, M. M., and Pawson, T. (2006). Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 7, 473–483. doi: 10.1038/nrm1960
Seo, H., Sonntag, K. C., and Isacson, O. (2004). Generalized brain and skin proteasome inhibition in Huntington's disease. Ann. Neurol. 56, 319–328. doi: 10.1002/ana.20207
Seo, H., Sonntag, K. C., Kim, W., Cattaneo, E., and Isacson, O. (2007). Proteasome activator enhances survival of Huntington's disease neuronal model cells. PLoS ONE 2:e238. doi: 10.1371/journal.pone.0000238
Sherman, M. Y., and Goldberg, A. L. (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32. doi: 10.1016/S0896-6273(01)00177-5
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. doi: 10.1126/science.1090278
Sisodia, S. S. (1998). Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 95, 1–4. doi: 10.1016/S0092-8674(00)81743-2
Sittler, A., Lurz, R., Lueder, G., Priller, J., Lehrach, H., Hayer-Hartl, M. K., et al. (2001). Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum. Mol. Genet. 10, 1307–1315. doi: 10.1093/hmg/10.12.1307
Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A., and Wolozin, B. (2003). Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. doi: 10.1074/jbc.M208641200
Song, S., Kim, S. Y., Hong, Y. M., Jo, D. G., Lee, J. Y., Shim, S. M., et al. (2003). Essential role of E2-25K/Hip-2 in mediating amyloid-beta neurotoxicity. Mol. Cell 12, 553–563. doi: 10.1016/j.molcel.2003.08.005
Stefanis, L., Larsen, K. E., Rideout, H. J., Sulzer, D., and Greene, L. A. (2001). Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 21, 9549–9560. Available online at: http://www.jneurosci.org/content/21/24/9549.abstract
Stenoien, D. L., Mielke, M., and Mancini, M. A. (2002). Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat. Cell Biol. 4, 806–810. doi: 10.1038/ncb859
Stolz, A., Hilt, W., Buchberger, A., and Wolf, D. H. (2011). Cdc48: a power machine in protein degradation. Trends Biochem. Sci. 36, 515–523. doi: 10.1016/j.tibs.2011.06.001
Tan, J. M., Wong, E. S., Kirkpatrick, D. S., Pletnikova, O., Ko, H. S., Tay, S. P., et al. (2008). Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439. doi: 10.1093/hmg/ddm320
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E., et al. (2001). Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10, 919–926. doi: 10.1093/hmg/10.9.919
Tokui, K., Adachi, H., Waza, M., Katsuno, M., Minamiyama, M., Doi, H., et al. (2009). 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum. Mol. Genet. 18, 898–910. doi: 10.1093/hmg/ddn419
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T. P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi: 10.4161/auto.6.2.11014
Trollet, C., Anvar, S. Y., Venema, A., Hargreaves, I. P., Foster, K., Vignaud, A., et al. (2010). Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 19, 2191–2207. doi: 10.1093/hmg/ddq098
Tsai, Y. C., Fishman, P. S., Thakor, N. V., and Oyler, G. A. (2003). Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 278, 22044–22055. doi: 10.1074/jbc.M212235200
Tseng, B. P., Green, K. N., Chan, J. L., Blurton-Jones, M., and Laferla, F. M. (2008). Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29, 1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014
Tsvetkov, A. S., Arrasate, M., Barmada, S., Ando, D. M., Sharma, P., Shaby, B. A., et al. (2013). Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 9, 586–592. doi: 10.1038/nchembio.1308
Um, J. W., Im, E., Lee, H. J., Min, B., Yoo, L., Yoo, J., et al. (2010). Parkin directly modulates 26S proteasome activity. J. Neurosci. 30, 11805–11814. doi: 10.1523/JNEUROSCI.2862-09.2010
Urushitani, M., Kurisu, J., Tsukita, K., and Takahashi, R. (2002). Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 83, 1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104. doi: 10.1016/S1097-2765(04)00151-0
Verdoes, M., Florea, B. I., Menendez-Benito, V., Maynard, C. J., Witte, M. D., Van Der Linden, W. A., et al. (2006). A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem. Biol. 13, 1217–1226. doi: 10.1016/j.chembiol.2006.09.013
Verhoef, L. G., Lindsten, K., Masucci, M. G., and Dantuma, N. P. (2002). Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700. doi: 10.1093/hmg/11.22.2689
Vilchez, D., Boyer, L., Morantte, I., Lutz, M., Merkwirth, C., Joyce, D., et al. (2012a). Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308. doi: 10.1038/nature11468
Vilchez, D., Morantte, I., Liu, Z., Douglas, P. M., Merkwirth, C., Rodrigues, A. P., et al. (2012b). RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature 489, 263–268. doi: 10.1038/nature11315
Viswanathan, J., Haapasalo, A., Bottcher, C., Miettinen, R., Kurkinen, K. M., Lu, A., et al. (2011). Alzheimer's disease-associated ubiquilin-1 regulates presenilin-1 accumulation and aggresome formation. Traffic 12, 330–348. doi: 10.1111/j.1600-0854.2010.01149.x
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H., et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407. doi: 10.1091/mbc.12.5.1393
Wang, A. M., Miyata, Y., Klinedinst, S., Peng, H. M., Chua, J. P., Komiyama, T., et al. (2013). Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 9, 112–118. doi: 10.1038/nchembio.1140
Wang, J., Wang, C. E., Orr, A., Tydlacka, S., Li, S. H., and Li, X. J. (2008). Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J. Cell Biol. 180, 1177–1189. doi: 10.1083/jcb.200709080
Wang, Q., Li, L., and Ye, Y. (2006). Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 174, 963–971. doi: 10.1083/jcb.200605100
Warrick, J. M., Morabito, L. M., Bilen, J., Gordesky-Gold, B., Faust, L. Z., Paulson, H. L., et al. (2005). Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell 18, 37–48. doi: 10.1016/j.molcel.2005.02.030
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Waza, M., Adachi, H., Katsuno, M., Minamiyama, M., Sang, C., Tanaka, F., et al. (2005). 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 11, 1088–1095. doi: 10.1038/nm1298
Weihl, C. C., Dalal, S., Pestronk, A., and Hanson, P. I. (2006). Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15, 189–199. doi: 10.1093/hmg/ddi426
Winborn, B. J., Travis, S. M., Todi, S. V., Scaglione, K. M., Xu, P., Williams, A. J., et al. (2008). The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 283, 26436–26443. doi: 10.1074/jbc.M803692200
Wojcik, C., Schroeter, D., Wilk, S., Lamprecht, J., and Paweletz, N. (1996). Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur. J. Cell Biol. 71, 311–318.
Yamamoto, A., Lucas, J. J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101, 57–66. doi: 10.1016/S0092-8674(00)80623-6
Yang, Y., Nishimura, I., Imai, Y., Takahashi, R., and Lu, B. (2003). Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by pael-R in Drosophila. Neuron 37, 911–924. doi: 10.1016/S0896-6273(03)00143-0
Yedidia, Y., Horonchik, L., Tzaban, S., Yanai, A., and Taraboulos, A. (2001). Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 20, 5383–5391. doi: 10.1093/emboj/20.19.5383
Zhang, J., and Zhou, P. (2005). Ectopic targeting of substrates to the ubiquitin pathway. Meth. Enzymol. 399, 823–833. doi: 10.1016/S0076-6879(05)99053-8
Zhong, X., and Pittman, R. N. (2006). Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 15, 2409–2420. doi: 10.1093/hmg/ddl164
Zhou, P., Bogacki, R., McReynolds, L., and Howley, P. M. (2000). Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 6, 751–756. doi: 10.1016/S1097-2765(00)00074-5
Keywords: neurodegeneration, ubiquitin, proteasome, proteolysis, protein quality control
Citation: Dantuma NP and Bott LC (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 7:70. doi: 10.3389/fnmol.2014.00070
Received: 11 May 2014; Accepted: 09 July 2014;
Published online: 31 July 2014.
Edited by:
Fred Van Leeuwen, Maastricht University, NetherlandsReviewed by:
A. Kimberley McAllister, University of California, USAElly M. Hol, University Medical Center Utrecht, Netherlands
Copyright © 2014 Dantuma and Bott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nico P. Dantuma, Department of Cell and Molecular Biology, Karolinska Institutet, von Eulers väg 3, S-17177 Stockholm, Sweden e-mail: nico.dantuma@ki.se