SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions
 Flavia Antonucci1
Flavia Antonucci1  Irene Corradini1,2
Irene Corradini1,2  Giuliana Fossati3
Giuliana Fossati3  Romana Tomasoni3
Romana Tomasoni3  Elisabetta Menna2,3
Elisabetta Menna2,3  Michela Matteoli2,3*
Michela Matteoli2,3*- 1Department of Medical Biotechnology and Translational Medicine, Università degli Studi di Milano, Milan, Italy
- 2Istituto di Neuroscienze, Centro Nazionale Ricerche, Milan, Italy
- 3Humanitas Clinical and Research Center, IRCCS Rozzano, Rozzano, Italy
A hallmark of synaptic specializations is their dependence on highly organized complexes of proteins that interact with each other. The loss or modification of key synaptic proteins directly affects the properties of such networks, ultimately impacting synaptic function. SNAP-25 is a component of the SNARE complex, which is central to synaptic vesicle exocytosis, and, by directly interacting with different calcium channels subunits, it negatively modulates neuronal voltage-gated calcium channels, thus regulating intracellular calcium dynamics. The SNAP-25 gene has been associated with distinct brain diseases, including Attention Deficit Hyperactivity Disorder (ADHD), schizophrenia and bipolar disorder, indicating that the protein may act as a shared biological substrate among different “synaptopathies”. The mechanisms by which alterations in SNAP-25 may concur to these psychiatric diseases are still undefined, although alterations in neurotransmitter release have been indicated as potential causative processes. This review summarizes recent work showing that SNAP-25 not only controls exo/endocytic processes at the presynaptic terminal, but also regulates postsynaptic receptor trafficking, spine morphogenesis, and plasticity, thus opening the possibility that SNAP-25 defects may contribute to psychiatric diseases by impacting not only presynaptic but also postsynaptic functions.
SNAP-25 is a component of the SNARE protein complex, which is involved in the exocytotic release of neurotransmitters during synaptic transmission. Through the coiled-coil assembly with syntaxin-1 and synaptobrevin, SNAP-25 mediates synaptic vesicle apposition to the presynaptic membrane permitting their Ca2+ triggered fusion. Consistently, the genetic ablation of this protein results in a complete block of synaptic transmission. SNAP-25 is present in two isoforms, a and b, resulting from alternative splicing of the exon 5 of the Snap-25 gene, which are differentially expressed during development. SNAP-25a is expressed at the embryonic stage, while SNAP-25b becomes the major isoform during postnatal life (Bark, 1993; Bark and Wilson, 1994; Bark et al., 1995), a developmental trend which has been confirmed in humans (Prescott and Chamberlain, 2011).
In line with its central role in neuronal function, the Snap-25 gene has been associated with several human neurological syndromes, including attention-deficit/hyperactivity disorder (ADHD), schizophrenia (Barr et al., 2000; Brophy et al., 2002; Kustanovich et al., 2003), and bipolar disorder (Etain et al., 2010). The protein appears therefore to represent a shared biological element among different psychiatric diseases.
Recently, several groups started to investigate the cellular and molecular mechanisms underpinning the SNAP-25 contribution to the onset of such pathologies, or, more likely, to the manifestations of specific traits typical of these diseases. A challenging scenario is now emerging, i.e., that some of the defects in diseases involving SNAP-25 might not exclusively result from the presynaptic role of the protein. Indeed, initially recognized as a presynaptic SNARE protein, the protein has been later shown to play additional non-SNARE roles and, very recently, even postsynaptic functions. The results of these lines of research are summarized in this review (see Table 1A).
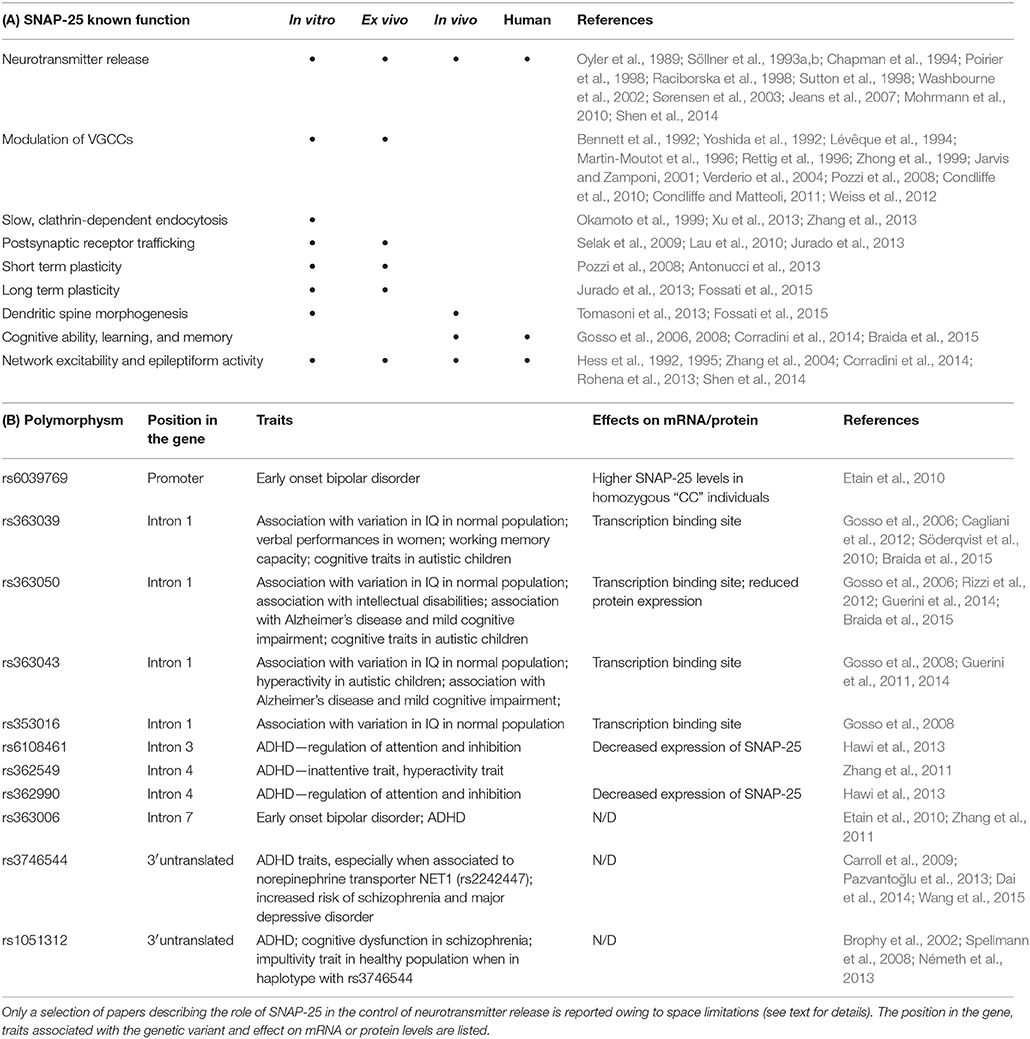
Table 1. (A) Functions of SNAP-25 protein, (B) Snap-25 polymorphisms discussed along the text.
Role of SNAP25 at the Presynapse: Synaptic Vesicles Exocytosis and Short Term Plasticity
SNAP-25 (synaptosomal-associated protein of 25 kDa) is a soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) protein that participates together with syntaxin-1 and synaptobrevin/VAMP (Jahn et al., 2003; Sudhof, 2004; Montecucco et al., 2005) in the regulation of synaptic vesicle exocytosis (Washbourne et al., 2002; reviewed in Milovanovic and Jahn, 2015). In the absence of SNAP-25, vesicle docking at the presynaptic active zones persists, but the pool of vesicles primed for release is empty, and fast calcium-triggered exocytosis is abolished (Sørensen et al., 2003). Furthermore, by calcium-dependent interaction with synaptotagmin, SNAP25 has a role in vesicle docking and priming as well as in triggering fast exocytosis (Mohrmann et al., 2010). Indeed the proteolytic cleavage of SNAP-25 by botulinum neurotoxins (BoNTs, serotypes A, C, and E) blocks exocytosis and neurotransmitter release (Schiavo et al., 2000; Ahnert-Hilger et al., 2013; Pantano and Montecucco, 2014), leading to the neuroparalysis characteristic of botulism (Aoki and Guyer, 2001).
Besides its well characterized role in exocytosis, SNAP-25 also modulates various voltage-gated calcium channels (VGCCs) (Atlas et al., 2001; Zamponi, 2003; Catterall and Few, 2008), by interacting with N-type (Sheng et al., 1996), P/Q-type (Martin-Moutot et al., 1996; Rettig et al., 1996), L-type (Wiser et al., 1999), and T-type channels (Weiss et al., 2012). SNAP-25 has been shown to negatively control neuronal calcium responsiveness to depolarization (Verderio et al., 2004) through voltage-gated calcium channel inhibition (Pozzi et al., 2008). Consistently, silencing endogenous SNAP-25 in glutamatergic neurons results in increased VGCC activity (Condliffe et al., 2010; Condliffe and Matteoli, 2011; see Figure 1).
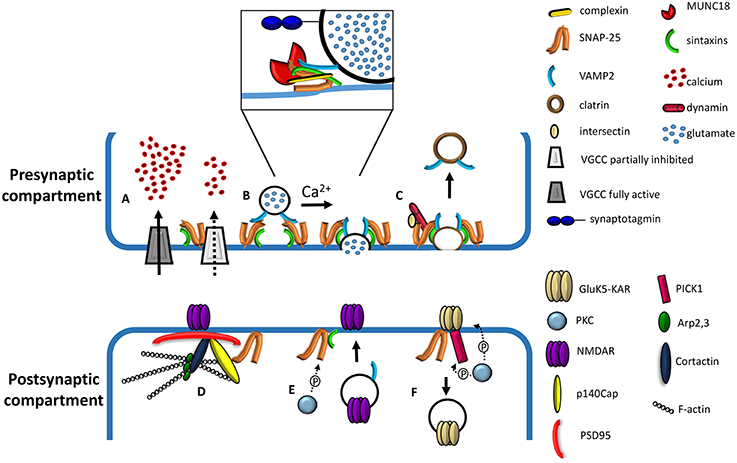
Figure 1. Cartoon depicting presynaptic and postsynaptic roles of SNAP-25. (A) Effect of presynaptic SNAP-25 on VGCCs. Calcium influx in the nerve terminal is negatively regulated by the complex formation between SNAP-25 and VGCCs; removal of the clamping role of SNAP-25, occurring upon reduction of the protein expression, results in elevated calcium influx through VGCCs (adapted from Kochlamazashvili and Haucke, 2013). (B) Involvement of SNAP-25 in the molecular machinery mediating Ca2+-triggered vesicle fusion. A docked synaptic vesicle is shown on the left. The core fusion machine is composed of synaptobrevin/VAMP2, syntaxin-1, and SNAP-25 (adapted from Kochlamazashvili and Haucke, 2013). The diagram in the box depicts a partially assembled SNARE complex including, besides synaptobrevin/VAMP2, syntaxin-1, and SNAP-25, complexins and MUNC18. The calcium sensor, synaptotagmin, is also depicted (adapted from Sudhof, 2012). (C) Role of SNAP-25 in slow clathrin-mediated synaptic vesicle endocytosis. SNAP-25 binds to the endocytic protein intersectin, while syntaxin binds dynamin, a GTPase mediating vesicle fission. The interactions between Synaptobrevin/VAMP2 and the ANTH domain of endocytic adaptors AP180 and CALM have been omitted for clarity. (D) Role of SNAP-25 in the organization of the postsynaptic density protein network. SNAP-25 interacts with p140Cap, which in turn forms a complex with PSD95, cortactin, Arp2,3, and F-actin (filamentous actin). NMDA receptors are depicted as interacting with PSD95 (adapted from Fossati et al., 2015). (E) Phosphorylation of SNAP-25 by PKC promotes the insertion of NMDA channels at the cell surface through the delivery of postsynaptic vesicles and their fusion with the plasma membrane, possibly via the formation of a SNARE complex (adapted from Lau et al., 2010). (F) Role of SNAP-25 in the removal of GluK5-contaning kainate receptors (KAR). KARs associate with SNAP-25 and the PKC-interacting protein PICK1. The PKC phosphorylation of the GluK5-C terminus may induce a conformational change facilitating the association with SNAP-25 and simultaneously decreasing GRIP binding affinity (adapted from Selak et al., 2009).
SNAP-25 also participates in slow, clathrin-dependent endocytosis at hippocampal synapses, possibly contributing to the coupling between exocytosis and endocytosis (Zhang et al., 2013). Given that SNARE proteins mediate exocytosis at all nerve terminals, their dual role in exo- and endocytosis is likely a general principle. Although how exactly SNARE proteins are involved in endocytosis remains unclear, the following binding studies provide some indications. Synaptobrevin/VAMP2 binds to the AP180 N-terminal homology (ANTH) domain of endocytic adaptors AP180 and Clathrin Assembly Lymphoid Myeloid leukemia (CALM) protein (Koo et al., 2011; Miller et al., 2011); also, stonin 2, facilitates clathrin/AP-2-dependent internalization of synaptotagmin and targets it to a recycling vesicle pool in living neurons (Diril et al., 2006).
SNAP-25 binds to the endocytic protein intersectin (Okamoto et al., 1999); syntaxin binds to dynamin (Galas et al., 2000). Based on these evidence it was proposed that the exocytosis machinery, including SNARE proteins (synaptobrevin, SNAP-25, and syntaxin), is needed in the initiating step of endocytosis and likely controls the amount of endocytosis (for a review see Wu et al., 2014).
Therefore, SNAP-25 represents a multifunctional protein involved in the control of secretion by multiple interactions. In line with the multiple roles of the protein, different neuronal processes are affected, in an unexpected way, in conditions characterized by SNAP-25 reduction. Indeed halved SNAP-25 levels in 13–14 DIV neuronal cultures not only failed to impair synaptic transmission, as expected by the SNARE role of SNAP-25, but instead enhanced evoked glutamatergic neurotransmission (Antonucci et al., 2013). This effect was dependent on presynaptic voltage-gated calcium channel activity and was not accompanied by changes in spontaneous quantal events or in the pool of readily releasable synaptic vesicles (Antonucci et al., 2013). Notably, synapses of 13–14 DIV neurons with reduced SNAP-25 expression showed paired-pulse depression as opposed to paired-pulse facilitation occurring in their wild-type counterparts (Antonucci et al., 2013). These data suggest that the more sensitive phenotype for reduced SNAP-25 levels may be the regulation of calcium channels, not the role of SNAP-25 in transmitter release. Based on these results, a dual role of SNAP-25 not only as a carrier but also as a “guardian of synaptic transmission” was proposed: in particular, reduced SNAP-25 expression, although sufficient to sustain SNARE-mediated synaptic vesicle fusion, partially releases VGCCs from SNAP-25-mediated inhibition, thus resulting in elevated calcium influx and facilitated neurotransmission (Kochlamazashvili and Haucke, 2013).
An Unexpected Role of SNAP-25 at the Post-Synapse: Spine Morphogenesis and Plasticity
In the last years, different evidence indicated an unexpected postsynaptic role for SNAP-25 (see Figure 1). The protein was indeed shown to control NMDA and kainate-type receptors trafficking (Selak et al., 2009; Lau et al., 2010). In particular the interaction of SNAP-25 with the GluK5 subunit of KARs and PICK1 reduces the GluK5 stability on the membrane, thus favoring KAR internalization (Selak et al., 2009), whereas the PKC-mediated phosphorylation of SNAP-25 on serine 187, promotes NMDAR delivery to the cell surface via SNARE-dependent exocytosis (Lau et al., 2010). In the latter study the authors elegantly demonstrated that introduction of the constitutively active form of PKC via the recording pipette to neurons rapidly potentiated NMDA currents in cells treated with inactive BoNT/A whereas treatment of neurons with active BoNT/A abolished PKC potentiation of NMDA currents without altering basal NMDA currents, thus unveiling SNAP-25 involvement in the potentiation of the synapse. Given that LTP-inducing protocols can induce SNAP-25 phosphorylation (Genoud et al., 1999), high frequency stimulation protocols may act via phosphorylation of SNAP-25 to promote insertion of NMDARs and elicit LTP. Indeed acute SNAP-25 downregulation resulted in LTP impairment (Jurado et al., 2013). These data opened the possibility that, besides a presynaptic impact, reductions of SNAP-25 levels may affect the structure, and/or the function of the postsynaptic compartment, which would provide a logical frame for the protein involvement in psychiatric diseases, such as schizophrenia or intellectual disability, which are known to be also characterized by defects at the postsynaptic compartment (Fernández et al., 2009; Penzes et al., 2011).
Despite the evidence pointing to a postsynaptic role of SNAP-25, a clear demonstration of whether SNAP-25 localizes in the dendritic spines of the postsynaptic neuron is still lacking. Some recent studies attempted to locate SNAP-25 in the postsynaptic terminal either by immunofluorescence (Selak et al., 2009), or ground state depletion (GSD) microscopy, which allows protein localization with a precision up to 20 nm (Tomasoni et al., 2013). Also by coimmunoprecipitation, bimolecular fluorescence complementation (BiFC) and biochemical fractionation, a molecular complex of SNAP-25 with postsynaptic proteins was detected (Selak et al., 2009; Tomasoni et al., 2013; Fossati et al., 2015). Nevertheless this is still a controversial topic, since other studies showed an exclusively presynaptic location of SNAP-25 through immunogold labeling of synaptic boutons (Holderith et al., 2012; Kerti et al., 2012). Certainly, the SNAP-25 expression levels in the postsynaptic compartment are quantitatively much lower than at the presynaptic one (Tao-Cheng et al., 2000; Sharma et al., 2012) and this could account for its difficult detection in dendritic spines.
In recent years, the postsynaptic role of SNAP-25 has been supported by evidence showing a structural modification of the postsynaptic compartment upon SNAP-25 reduction. In particular, acute reduction of SNAP-25 expression in primary hippocampal cultures led to an immature phenotype of dendritic spines, while overexpression of the protein resulted in an increase in the density of mature, PSD-95-positive spines (Tomasoni et al., 2013). The effect was shown to be truly postsynaptic, and not secondary to altered presynaptic function as demonstrated by co-culturing of SNAP25 heterozygous and GFP-expressing wild type neurons. SNAP-25 reductions were also shown to affect the localization of PSD95, with acute downregulation of SNAP-25 resulting in a significant reduction of PSD95-positive puncta (Fossati et al., 2015). Correspondingly, acute down-regulation of SNAP-25 in CA1 hippocampal region by lentiviral expression reduced spine density and resulted in immature spine morphology, thus recapitulating in vivo the spine abnormalities observed in cultures upon acute SNAP-25 silencing (Fossati et al., 2015).
Which could be the mechanism by which SNAP-25 controls dendritic spine morphology and PSD95 mobility? The cleavage of SNAP-25 by BoNT/E, which prevents the protein to enter the fusion complex, did not reduce spine density or PSD95 size, thus excluding that SNAP-25 controls PSD95 recruitment through its SNARE function and suggesting instead a protein scaffolding role at the spine level (Fossati et al., 2015). This hypothesis was supported by the finding that p140Cap, a scaffold protein located into dendritic spines with a crucial role in regulating actin cytoskeleton, spine formation (Jaworski et al., 2009), and learning processes (Repetto et al., 2014), is a key member of the molecular complex which includes SNAP-25 and PSD95 (Tomasoni et al., 2013; Fossati et al., 2015).
The correct formation of this molecular complex preserves the proper organization of the dendritic spine. Maintaining spine integrity could further facilitate the formation of the protein complexes which contain also SNAP-25 and that regulate receptor trafficking (Selak et al., 2009; Lau et al., 2010). Based on these results, it is conceivable that postsynaptic SNAP-25 may be important for orchestrating a dynamic equilibrium among the glutamate receptors at a given synapse, thereby regulating synapse efficacy also at the postsynaptic side.
SNAP-25, A Shared Biological Pathway Among Different Psychiatric Diseases
The defective formation of the SNARE complex for vesicle fusion and the aberrant regulation of voltage-gated calcium channels are the processes generally taken into account to explain the involvement of the protein in those psychiatric diseases which have been linked to the Snap-25 gene. However, the recent data indicating a postsynaptic role for the protein raise the possibility that SNAP-25 defects may contribute, in these disorders, also through alterations of postsynaptic receptors trafficking or spine morphogenesis.
Several reports have shown the presence of polymorphisms in the Snap-25 gene, which have been associated with ADHD (Barr et al., 2000; Faraone et al., 2005; Zhang et al., 2011; Hawi et al., 2013; Pazvantoğlu et al., 2013), schizophrenia (Thompson et al., 2003), and early-onset bipolar disorders (Etain et al., 2010; see Table 1B). Notably, some of these polymorphisms were found to control not only specific traits of the disease, but even behavioral tracts in healthy individuals. As an example, several single nucleotide polymorphisms (i.e., rs363043, rs353016, rs363039, rs363050) of the Snap-25 gene have been associated with Intelligence Quotient (IQ) phenotypes in healthy individuals (Gosso et al., 2006, 2008). Also, although autism spectrum disorder (ASD) has not been directly linked to the Snap-25 gene, polymorphisms analyzed in a cohort of children affected by ASD revealed a significant association between Snap-25 SNP rs363043 and hyperactivity traits (Guerini et al., 2011), while rs363050 and rs363039 polymorphisms were shown to correlate with cognitive deficits in ASD children (Braida et al., 2015). Notably, a first analysis of transcriptional activity through luciferase reporter gene revealed that SNP rs363050, which is localized in the intron 1 of the Snap-25 gene, leads to reduced protein expression (Braida et al., 2015). Therefore, the possibility that reduced SNAP-25 levels may contribute to specific behavioral traits, such as hyperactivity or cognitive performances in healthy individuals or in different psychiatric diseases, including those to which the gene has not been directly associated, like in the case of ASD, remains a challenging possibility to be tested in the future.
Notably, even in schizophrenia, where the SNAP-25 levels are significantly lower in the hippocampus (Young et al., 1998; Fatemi et al., 2001; Thompson et al., 2003) and in the frontal lobe Broadman's area 10 (Thompson et al., 1998), an association between the rs1051312 polymorphism of the Snap-25 gene and cognitive dysfunctions was reported (Spellmann et al., 2008). Furthermore, and consistent with the observations already reported in SNAP-25 heterozigous mice (Antonucci et al., 2013), even in schizophrenic patients the reduction of SNAP-25 levels does not seem to correlate with an impairment in the SNARE complex formation (Ramos-Miguel et al., 2015). Of interest, and in line with the possible relevance of SNAP-25 expression levels in different psychiatric diseases, a SNAP-25 variant located in the promoter region (rs6039769) and associated with early-onset bipolar disorder was found to correlate with a significantly higher SNAP-25b expression in prefrontal cortex (Etain et al., 2010). Higher levels of the SNARE in dorsolateral prefrontal cortex of patients affected by bipolar disorder were already reported by Scarr et al. (2006).
As a support to the functional impact of the protein levels in cognitive or motor functions, genetic mice models showed that the chronic reduction of SNAP-25 affects mouse behavior. The coloboma mouse model, characterized by halved SNAP-25 levels (Hess et al., 1992), displays indeed a hyperactive phenotype (Hess et al., 1992), associated with abnormal thalamic spike-wave discharges (Hess et al., 1995; Zhang et al., 2004; Faraone et al., 2005; Russell, 2007). Similarly, juvenile SNAP-25 heterozygous mice displays a moderate hyperactivity, which disappears in the adult animals, and impaired associative learning and memory, which persist instead in the adults. Electroencephalographic recordings revealed the occurrence of frequent spikes, suggesting a diffuse network hyperexcitability, accompanied by a higher susceptibility to kainate-induced seizures, and degeneration of hilar neurons. Notably, both EEG alterations and cognitive defects were improved by antiepileptic drugs, in particular valproic acid (Corradini et al., 2014; Braida et al., 2015). A defective negative control of voltage gated calcium channels resulting from the reduced SNAP-25 levels could be at the origin of the network hyperexcitability (Corradini et al., 2014). Although, the demonstration of a direct causal link between altered SNAP-25 expression and psychiatric diseases is still lacking, evidences obtained in the coloboma mouse suggest that reduction of SNAP-25 expression may be directly involved in some psychiatric traits, rather than simply represent an epiphenomenon; indeed, when a transgene expressing SNAP-25 was bred into the coloboma strain in order to complement Snap-25 depletion, the hyperactivity displayed by the mutant mice was rescued (Hess et al., 1995 J Neurosci).
Recently a de novo variant was identified in the Snap-25 exon 4 (Phe48Val), in a 15-years-old girl with intractable epilepsy and severe encephalopathy, but no neuromuscular symptoms (Rohena et al., 2013). Later on, exome sequencing identified a de novo dominant mutation of a conserved residue in exon 5 of Snap-25b in an 11-years-old patient displaying congenital myasthenia, cortical hyperexcitability, cerebellar ataxia, and intellectual disability (Shen et al., 2014). The Ile67Asn variant was reported to be pathogenic because, by disrupting the hydrophobic alpha-helical coiled-coil structure of the SNARE complex, it inhibits synaptic vesicle exocytosis (Shen et al., 2014). Of note, a heterozygous Ile67Thr missense mutation in Snap-25b gene was observed in the so-called blind-drunk (1/Bdr) mouse, which shows a mild ataxic gait around age 4 weeks, impaired sensorimotor gating and increased anxiety (Jeans et al., 2007; Oliver and Davies, 2009). This mutation is located in a highly conserved codon and parallels Ile67Asn mutation observed in the 11 years-old patient (Shen et al., 2014). In the case of the Ile67Asn mutation, Shen and colleagues propose that the substitution of a hydrophobic residue with a hydrophilic one destabilizes the coiled-coil SNARE complex structure, thus hindering vesicle fusion (Shen et al., 2014); however, it is also possible that the Ile67Asn mutation causes a distortion of the coiled coil structure in such a way as to affect the interaction of the SNARE complex with its protein partners. This appears to be the case in the blind-drunk mutation which results in the enhancement of the affinity of SNAP-25 for its binding partners and is therefore likely to cause an increase in association of the SNARE complex (Jeans et al., 2007). No information about the impact of Val48Phe variant on SNAP-25 structure and function is still available.
Additional genetic mouse models underlined the role of Snap-25 mutations in specific traits of psychiatric diseases. Single nucleotide substitution resulting in a missense Ser187Ala mutation at the site of phosphorylation of SNAP-25 by PKC has been associated with increased anxiety, decreased dopamine and serotonine release (Kataoka et al., 2011), impaired PPI of the startle response, a typical parameter of schizophrenia, deficits in working memory, immature features of dentate granule cells (Ohira et al., 2013), and epileptic seizures (Watanabe et al., 2015). Interestingly, Ser187 phosphorylation of SNAP-25 is development- and activity-dependent both in vitro and in vivo (Kataoka et al., 2006; Pozzi et al., 2008); it is associated with synaptic vesicles availability (Nagy et al., 2002; Houeland et al., 2007) and it is necessary for the negative control of voltage-gated calcium channels (Pozzi et al., 2008).
Conclusions
The recent discovery of SNAP-25 role in the control of receptor trafficking and spine morphogenesis, which points to the protein role as a postsynaptic structural hub, opens new avenues for the comprehension of the physiological role of the protein at the synapse and offers new mechanistic insights as to SNAP-25 involvement in synaptopathies that go beyond the protein's established roles in presynaptic function. The finding that the activity-driven spine remodeling is defective in neuronal networks constitutively developing in the presence of reduced levels of SNAP-25, makes a provocative link to human pathologies, such as schizophrenia, where both a reduction of SNAP-25 expression and a decrease in dendritic spine density have been described.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in our lab is supported by PRIN (2010JFYFY2-008), Ministero della Salute (HEALTH-F2-2009-241498), Telethon GGP12115, Cariplo 2015-0594, Fondazione Veronesi, and Fondazione Vollaro to MM; Ministero dell'Istruzione, dell'Università e della Ricerca (FIRB-RBFR10ZBYZ) to FA; Progetto Bandiera Interomics to EM and Cariplo 2015-0952 to RT. We thank Prof. Cesare Montecucco (University of Padova) for constructive discussions and for critical reading of the manuscript.
References
Ahnert-Hilger, G., Munster-Wandowski, A., and Höltje, M. (2013). Synaptic vesicle proteins: targets and routes for botulinum neurotoxins. Curr. Top. Microbiol. Immunol. 364, 159–177. doi: 10.1007/978-3-662-45790-0_8
Antonucci, F., Corradini, I., Morini, R., Fossati, G., Menna, E., Pozzi, D., et al. (2013). Reduced SNAP-25 alters short-term plasticity at developing glutamatergic synapses. EMBO Rep. 14, 645–651. doi: 10.1038/embor.2013.75
Aoki, K. R., and Guyer, B. (2001). Botulinum toxin type A and other botulinum toxin serotypes: a comparative review of biochemical and pharmacological actions. Eur. J. Neurol. 8(Suppl. 5), 21–29. doi: 10.1046/j.1468-1331.2001.00035.x
Atlas, D., Wiser, O., and Trus, M. (2001). The voltage-gated Ca2+ channel is the Ca2+ sensor of fast neurotransmitter release. Cell. Mol. Neurobiol. 21, 717–731. doi: 10.1023/A:1015104105262
Bark, I. C. (1993). Structure of the chicken gene for SNAP-25 reveals duplicated exon encoding distinct isoforms of the protein. J. Mol. Biol. 233, 67–76. doi: 10.1006/jmbi.1993.1485
Bark, I. C., Hahn, K. M., Ryabinin, A. E., and Wilson, M. C. (1995). Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc. Natl. Acad. Sci. U.S.A. 92, 1510–1514. doi: 10.1073/pnas.92.5.1510
Bark, I. C., and Wilson, M. C. (1994). Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene 139, 291–292. doi: 10.1016/0378-1119(94)90773-0
Barr, C. L., Feng, Y., Wigg, K., Bloom, S., Roberts, W., Malone, M., et al. (2000). Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol. Psychiatry 5, 405–409. doi: 10.1038/sj.mp.4000733
Bennett, M. K., Calakos, N., and Scheller, R. H. (1992). Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 257, 255–259. doi: 10.1126/science.1321498
Braida, D., Guerini, F. R., Ponzoni, L., Corradini, I., De Astis, S., Pattini, L., et al. (2015). Association between SNAP-25 gene polymorphisms and cognition in autism: functional consequences and potential therapeutic strategies. Transl. Psychiatry 5:e500. doi: 10.1038/tp.2014.136
Brophy, K., Hawi, Z., Kirley, A., Fitzgerald, M., and Gill, M. (2002). Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): evidence of linkage and association in the Irish population. Mol. Psychiatry 7, 913–917. doi: 10.1038/sj.mp.4001092
Cagliani, R., Riva, S., Marino, C., Fumagalli, M., D'Angelo, M. G., Riva, V., et al. (2012). Variants in SNAP25 are targets of natural selection and influence verbal performances in women. Cell. Mol. Life Sci. 69, 1705–1715. doi: 10.1007/s00018-011-0896-y
Carroll, L. S., Kendall, K., O'Donovan, M. C., Owen, M. J., and Williams, N. M. (2009). Evidence that putative ADHD low risk alleles at SNAP25 may increase the risk of schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 893–899. doi: 10.1002/ajmg.b.30915
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Chapman, E. R., An, S., Barton, N., and Jahn, R. (1994). SNAP-25, a t-SNARE which binds to both syntaxin and synaptobrevin via domains that may form coiled coils. J. Biol. Chem. 269, 27427–27432.
Condliffe, S. B., Corradini, I., Pozzi, D., Verderio, C., and Matteoli, M. (2010). Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J. Biol. Chem. 285, 24968–24976. doi: 10.1074/jbc.M110.145813
Condliffe, S. B., and Matteoli, M. (2011). Inactivation kinetics of voltage-gated calcium channels in glutamatergic neurons are influenced by SNAP-25. Channels 5, 304–307. doi: 10.4161/chan.5.4.16228
Corradini, I., Donzelli, A., Antonucci, F., Welzl, H., Loos, M., Martucci, R., et al. (2014). Epileptiform activity and cognitive deficits in SNAP-25+/- mice are normalized by antiepileptic drugs. Cereb. Cortex 24, 364–376. doi: 10.1093/cercor/bhs316
Dai, D., Wang, Y., Yuan, J., Zhou, X., Jiang, D., Li, J., et al. (2014). Meta-analyses of 10 polymorphisms associated with the risk of schizophrenia. Biomed. Rep. 2, 729–736. doi: 10.3892/br.2014.308
Diril, M. K., Wienisch, M., Jung, N., Klingauf, J., and Haucke, V. (2006). Stonin 2 is an AP-2-dependent endocytic sorting adaptor for synaptotagmin internalization and recycling. Dev. Cell 10, 233–244. doi: 10.1016/j.devcel.2005.12.011
Etain, B., Dumaine, A., Mathieu, F., Chevalier, F., Henry, C., Kahn, J. P., et al. (2010). A SNAP25 promoter variant is associated with early-onset bipolar disorder and a high expression level in brain. Mol. Psychiatry 15, 748–755. doi: 10.1038/mp.2008.148
Faraone, S. V., Perlis, R. H., Doyle, A. E., Smoller, J. W., Goralnick, J. J., Holmgren, M. A., et al. (2005). Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1313–1323. doi: 10.1016/j.biopsych.2004.11.024
Fatemi, S. H., Earle, J. A., Stary, J. M., Lee, S., and Sedgewick, J. (2001). Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport 12, 3257–3262. doi: 10.1097/00001756-200110290-00023
Fernández, E., Collins, M. O., Uren, R. T., Kopanitsa, M. V., Komiyama, N. H., Croning, M. D., et al. (2009). Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 5, 269. doi: 10.1038/msb.2009.27
Fossati, G., Morini, R., Corradini, I., Antonucci, F., Trepte, P., Edry, E., et al. (2015). Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 22, 1425–1436. doi: 10.1038/cdd.2014.227
Galas, M. C., Chasserot-Golaz, S., Dirrig-Grosch, S., and Bader, M. F. (2000). Presence of dynamin–syntaxin complexes associated with secretory granules in adrenal chromaffin cells. J. Neurochem. 75, 1511–1519. doi: 10.1046/j.1471-4159.2000.0751511.x
Genoud, S., Pralong, W., Riederer, B. M., Eder, L., Catsicas, S., and Muller, D. (1999). Activity-dependent phosphorylation of SNAP-25 in hippocampal organotypic cultures. J. Neurochem. 72, 1699–1706. doi: 10.1046/j.1471-4159.1999.721699.x
Gosso, M. F., de Geus, E. J., Polderman, T. J., Boomsma, D. I., Heutink, P., and Posthuma, D. (2008). Common variants underlying cognitive ability: further evidence for association between the SNAP-25 gene and cognition using a family-based study in two independent Dutch cohorts. Genes Brain Behav. 7, 355–364. doi: 10.1111/j.1601-183X.2007.00359.x
Gosso, M. F., de Geus, E. J., van Belzen, M. J., Polderman, T. J., Heutink, P., Boomsma, D. I., et al. (2006). The SNAP-25 gene is associated with cognitive ability: evidence from a family-based study in two independent Dutch cohorts. Mol. Psychiatry 11, 878–886. doi: 10.1038/sj.mp.4001868
Guerini, F. R., Agliardi, C., Sironi, M., Arosio, B., Calabrese, E., Zanzottera, M., et al. (2014). Possible association between SNAP-25 single nucleotide polymorphisms and alterations of categorical fluency and functional MRI parameters in Alzheimer's disease. J. Alzheimers Dis. 42, 1015–1028. doi: 10.3233/JAD-140057
Guerini, F. R., Bolognesi, E., Chiappedi, M., Manca, S., Ghezzo, A., Agliardi, C., et al. (2011). SNAP-25 single nucleotide polymorphisms are associated with hyperactivity in autism spectrum disorders. Pharmacol. Res. 64, 283–288. doi: 10.1016/j.phrs.2011.03.015
Hawi, Z., Matthews, N., Wagner, J., Wallace, R. H., Butler, T. J., Vance, A., et al. (2013). DNA variation in the SNAP25 gene confers risk to ADHD and is associated with reduced expression in prefrontal cortex. PLoS ONE 8:e60274. doi: 10.1371/journal.pone.0060274
Hess, E. J., Jinnah, H. A., Kozak, C. A., and Wilson, M. C. (1992). Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on chromosome 2. J. Neurosci. 12, 2865–2874.
Hess, E. J., Rogan, P. K., Domoto, M., Tinker, D. E., Ladda, R. L., and Ramer, J. C. (1995). Absence of linkage of apparently single gene mediated ADHD with the human syntenic region of the mouse mutant Coloboma. Am. J. Med. Genet. 60, 573–579. doi: 10.1002/ajmg.1320600619
Holderith, N., Lorincz, A., Katona, G., Rózsa, B., Kulik, A., Watanabe, M., et al. (2012). Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat. Neurosci. 15, 988–997. doi: 10.1038/nn.3137
Houeland, G., Nakhost, A., Sossin, W. S., and Castellucci, V. F. (2007). PKC modulation of transmitter release by SNAP-25 at sensory-to-motor synapses in Aplysia. J. Neurophysiol. 97, 134–143. doi: 10.1152/jn.00122.2006
Jahn, R., Lang, T., and Sudhof, T. C. (2003). Membrane fusion. Cell 112, 519–533. doi: 10.1016/S0092-8674(03)00112-0
Jarvis, S. E., and Zamponi, G. W. (2001). Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. J. Neurosci. 21, 2939–2948.
Jaworski, J., Kapitein, L. C., Gouveia, S. M., Dortland, B. R., Wulf, P. S., Grigoriev, I., et al. (2009). Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. doi: 10.1016/j.neuron.2008.11.013
Jeans, A. F., Oliver, P. L., Johnson, R., Capogna, M., Vikman, J., Molnár, Z., et al. (2007). A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc. Natl. Acad. Sci. U.S.A. 104, 2431–2436. doi: 10.1073/pnas.0610222104
Jurado, S., Goswami, D., Zhang, Y., Molina, A. J., Sudhof, T. C., and Malenka, R. C. (2013). LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77, 542–558. doi: 10.1016/j.neuron.2012.11.029
Kataoka, M., Kuwahara, R., Matsuo, R., Sekiguchi, M., Inokuchi, K., and Takahashi, M. (2006). Development- and activity-dependent regulation of SNAP-25 phosphorylation in rat brain. Neurosci. Lett. 407, 258–262. doi: 10.1016/j.neulet.2006.08.055
Kataoka, M., Yamamori, S., Suzuki, E., Watanabe, S., Sato, T., Miyaoka, H., et al. (2011). A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS ONE 6:e25158. doi: 10.1371/journal.pone.0025158
Kerti, K., Lorincz, A., and Nusser, Z. (2012). Unique somato-dendritic distribution pattern of Kv4.2 channels on hippocampal CA1 pyramidal cells. Eur. J. Neurosci. 35, 66–75. doi: 10.1111/j.1460-9568.2011.07907.x
Kochlamazashvili, G., and Haucke, V. (2013). A dual role of SNAP-25 as carrier and guardian of synaptic transmission. EMBO Rep. 14, 579–580. doi: 10.1038/embor.2013.74
Koo, S. J., Markovic, S., Puchkov, D., Mahrenholz, C. C., Beceren-Braun, F., Maritzen, T., et al. (2011). SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. U.S.A. 108, 13540–13545. doi: 10.1073/pnas.1107067108
Kustanovich, V., Merriman, B., McGough, J., McCracken, J. T., Smalley, S. L., and Nelson, S. F. (2003). Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol. Psychiatry 8, 309–315. doi: 10.1038/sj.mp.4001247
Lau, C. G., Takayasu, Y., Rodenas-Ruano, A., Paternain, A. V., Lerma, J., Bennett, M. V., et al. (2010). SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 30, 242–254. doi: 10.1523/JNEUROSCI.4933-08.2010
Lévêque, C., el Far, O., Martin-Moutot, N., Sato, K., Kato, R., Takahashi, M., et al. (1994). Purification of the N-type calcium channel associated with syntaxin and synaptotagmin. A complex implicated in synaptic vesicle exocytosis. J. Biol. Chem. 269, 6306–6312.
Martin-Moutot, N., Charvin, N., Leveque, C., Sato, K., Nishiki, T., Kozaki, S., et al. (1996). Interaction of SNARE complexes with P/Q-type calcium channels in rat cerebellar synaptosomes. J. Biol. Chem. 271, 6567–6570. doi: 10.1074/jbc.271.12.6567
Miller, S. E., Sahlender, D. A., Graham, S. C., Höning, S., Robinson, M. S., Peden, A. A., et al. (2011). The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 147, 1118–1131. doi: 10.1016/j.cell.2011.10.038
Milovanovic, D., and Jahn, R. (2015). Organization and dynamics of SNARE proteins in the presynaptic membrane. Front. Physiol. 6:89. doi: 10.3389/fphys.2015.00089
Mohrmann, R., de Wit, H., Verhage, M., Neher, E., and Sørensen, J. B. (2010). Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330, 502–505. doi: 10.1126/science.1193134
Montecucco, C., Schiavo, G., and Pantano, S. (2005). SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem. Sci. 30, 367–372. doi: 10.1016/j.tibs.2005.05.002
Nagy, G., Matti, U., Nehring, R. B., Binz, T., Rettig, J., Neher, E., et al. (2002). Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser(187) potentiates vesicle recruitment. J. Neurosci. 22, 9278–9286.
Németh, N., Kovács-Nagy, R., Székely, A., Sasvári-Székely, M., and Rónai, Z. (2013). Association of impulsivity and polymorphic microRNA-641 target sites in the SNAP-25 gene. PLoS ONE 8:e84207. doi: 10.1371/journal.pone.0084207
Ohira, K., Kobayashi, K., Toyama, K., Nakamura, H. K., Shoji, H., Takao, K., et al. (2013). Synaptosomal-associated protein 25 mutation induces immaturity of the dentate granule cells of adult mice. Mol. Brain 6:12. doi: 10.1186/1756-6606-6-12
Okamoto, M., Schoch, S., and Sudhof, T. C. (1999). EHSH1/intersectin, a protein that contains EH and SH3 domains and binds to dynamin and SNAP-25. A protein connection between exocytosis and endocytosis? J. Biol. Chem. 274, 18446–18454. doi: 10.1074/jbc.274.26.18446
Oliver, P. L., and Davies, K. E. (2009). Interaction between environmental and genetic factors modulates schizophrenic endophenotypes in the Snap-25 mouse mutant blind-drunk. Hum. Mol. Genet. 18, 4576–4589. doi: 10.1093/hmg/ddp425
Oyler, G. A., Higgins, G. A., Hart, R. A., Battenberg, E., Billingsley, M., Bloom, F. E., et al. (1989). The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J. Cell Biol. 109, 3039–3052. doi: 10.1083/jcb.109.6.3039
Pantano, S., and Montecucco, C. (2014). The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 71, 793–811. doi: 10.1007/s00018-013-1380-7
Pazvantoğlu, O., Güneş, S., Karabekiroğlu, K., Yeğin, Z., Erenkuş, Z., Akbaş, S., et al. (2013). The relationship between the presence of ADHD and certain candidate gene polymorphisms in a Turkish sample. Gene 528, 320–327. doi: 10.1016/j.gene.2013.07.004
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Poirier, M. A., Hao, J. C., Malkus, P. N., Chan, C., Moore, M. F., King, D. S., et al. (1998). Protease resistance of syntaxin.SNAP-25.VAMP complexes. Implications for assembly and structure. J. Biol. Chem. 273, 11370–11377. doi: 10.1074/jbc.273.18.11370
Pozzi, D., Condliffe, S., Bozzi, Y., Chikhladze, M., Grumelli, C., Proux-Gillardeaux, V., et al. (2008). Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. U.S.A. 105, 323–328. doi: 10.1073/pnas.0706211105
Prescott, G. R., and Chamberlain, L. H. (2011). Regional and developmental brain expression patterns of SNAP25 splice variants. BMC Neurosci. 12:35. doi: 10.1186/1471-2202-12-35
Raciborska, D. A., Trimble, W. S., and Charlton, M. P. (1998). Presynaptic protein interactions in vivo: evidence from botulinum A, C, D and E action at frog neuromuscular junction. Eur. J. Neurosci. 10, 2617–2628. doi: 10.1046/j.1460-9568.1998.00270.x
Ramos-Miguel, A., Beasley, C. L., Dwork, A. J., Mann, J. J., Rosoklija, G., Barr, A. M., et al. (2015). Increased SNARE protein-protein interactions in orbitofrontal and anterior cingulate cortices in Schizophrenia. Biol. Psychiatry 78, 361–373. doi: 10.1016/j.biopsych.2014.12.012
Repetto, D., Camera, P., Melani, R., Morello, N., Russo, I., Calcagno, E., et al. (2014). p140Cap regulates memory and synaptic plasticity through Src-mediated and citron-N-mediated actin reorganization. J. Neurosci. 34, 1542–1553. doi: 10.1523/JNEUROSCI.2341-13.2014
Rettig, J., Sheng, Z. H., Kim, D. K., Hodson, C. D., Snutch, T. P., and Catterall, W. A. (1996). Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc. Natl. Acad. Sci. U.S.A. 93, 7363–7368. doi: 10.1073/pnas.93.14.7363
Rizzi, T. S., Beunders, G., Rizzu, P., Sistermans, E., Twisk, J. W., van Mechelen, W., et al. (2012). Supporting the generalist genes hypothesis for intellectual ability/disability: the case of SNAP25. Genes Brain Behav. 11, 767–771. doi: 10.1111/j.1601-183X.2012.00819.x
Rohena, L., Neidich, J., Truitt Cho, M., Gonzalez, K. D., Tang, S., Devinsky, O., et al. (2013). Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis. 1:e26314. doi: 10.4161/rdis.26314
Russell, V. A. (2007). Neurobiology of animal models of attention-deficit hyperactivity disorder. J. Neurosci. Methods 161, 185–198. doi: 10.1016/j.jneumeth.2006.12.005
Scarr, E., Gray, L., Keriakous, D., Robinson, P. J., and Dean, B. (2006). Increased levels of SNAP-25 and synaptophysin in the dorsolateral prefrontal cortex in bipolar I disorder. Bipolar Disord. 8, 133–143. doi: 10.1111/j.1399-5618.2006.00300.x
Schiavo, G., Matteoli, M., and Montecucco, C. (2000). Neurotoxins affecting neuroexocytosis. Physiol. Rev. 80, 717–766.
Selak, S., Paternain, A. V., Aller, M. I., Picó, E., Rivera, R., and Lerma, J. (2009). A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron 63, 357–371. doi: 10.1016/j.neuron.2009.07.017
Sharma, M., Burré, J., Bronk, P., Zhang, Y., Xu, W., and Südhof, T. C. (2012). CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841. doi: 10.1038/emboj.2011.467
Shen, X. M., Selcen, D., Brengman, J., and Engel, A. G. (2014). Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 83, 2247–2255. doi: 10.1212/WNL.0000000000001079
Sheng, Z. H., Rettig, J., Cook, T., and Catterall, W. A. (1996). Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature 379, 451–454. doi: 10.1038/379451a0
Söderqvist, S., McNab, F., Peyrard-Janvid, M., Matsson, H., Humphreys, K., Kere, J., et al. (2010). The SNAP25 gene is linked to working memory capacity and maturation of the posterior cingulate cortex during childhood. Biol. Psychiatry 68, 1120–1125. doi: 10.1016/j.biopsych.2010.07.036
Söllner, T., Bennett, M. K., Whiteheart, S. W., Scheller, R. H., and Rothman, J. E. (1993a). A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418. doi: 10.1016/0092-8674(93)90376-2
Söllner, T., Whiteheart, S. W., Brunner, M., Erdjument-Bromage, H., Geromanos, S., Tempst, P., et al. (1993b). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. doi: 10.1038/362318a0
Sørensen, J. B., Nagy, G., Varoqueaux, F., Nehring, R. B., Brose, N., Wilson, M. C., et al. (2003). Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114, 75–86. doi: 10.1016/S0092-8674(03)00477-X
Spellmann, I., Müller, N., Musil, R., Zill, P., Douhet, A., Dehning, S., et al. (2008). Associations of SNAP-25 polymorphisms with cognitive dysfunctions in Caucasian patients with schizophrenia during a brief trail of treatment with atypical antipsychotics. Eur. Arch. Psychiatry Clin. Neurosci. 258, 335–344. doi: 10.1007/s00406-007-0800-9
Sudhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Sudhof, T. C. (2012). Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 4:a011353. doi: 10.1101/cshperspect.a011353
Sutton, R. B., Fasshauer, D., Jahn, R., and Brunger, A. T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353. doi: 10.1038/26412
Tao-Cheng, J. H., Du, J., and McBain, C. J. (2000). Snap-25 is polarized to axons and abundant along the axolemma: an immunogold study of intact neurons. J. Neurocytol. 29, 67–77. doi: 10.1023/A:1007168231323
Thompson, P. M., Egbufoama, S., and Vawter, M. P. (2003). SNAP-25 reduction in the hippocampus of patients with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 411–417. doi: 10.1016/S0278-5846(03)00027-7
Thompson, P. M., Sower, A. C., and Perrone-Bizzozero, N. I. (1998). Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia. Biol. Psychiatry 43, 239–243. doi: 10.1016/S0006-3223(97)00204-7
Tomasoni, R., Repetto, D., Morini, R., Elia, C., Gardoni, F., Di Luca, M., et al. (2013). SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat. Commun. 4, 2136. doi: 10.1038/ncomms3136
Verderio, C., Pozzi, D., Pravettoni, E., Inverardi, F., Schenk, U., Coco, S., et al. (2004). SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 41, 599–610. doi: 10.1016/S0896-6273(04)00077-7
Wang, Q., Wang, Y., Ji, W., Zhou, G., He, K., Li, Z., et al. (2015). SNAP25 is associated with schizophrenia and major depressive disorder in the Han Chinese population. J. Clin. Psychiatry 76, e76–82. doi: 10.4088/JCP.13m08962
Washbourne, P., Thompson, P. M., Carta, M., Costa, E. T., Mathews, J. R., Lopez-Bendito, G., et al. (2002). Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat. Neurosci. 5, 19–26. doi: 10.1038/nn783
Watanabe, S., Yamamori, S., Otsuka, S., Saito, M., Suzuki, E., Kataoka, M., et al. (2015). Epileptogenesis and epileptic maturation in phosphorylation site-specific SNAP-25 mutant mice. Epilepsy Res. 115, 30–44. doi: 10.1016/j.eplepsyres.2015.05.004
Weiss, N., Hameed, S., Fernández-Fernández, J. M., Fablet, K., Karmazinova, M., Poillot, C., et al. (2012). A Ca(v)3.2/Syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J. Biol. Chem. 287, 2810–2818. doi: 10.1074/jbc.M111.290882
Wiser, O., Trus, M., Hernández, A., Renstrom, E., Barg, S., Rorsman, P., et al. (1999). The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc. Natl. Acad. Sci. U.S.A. 96, 248–253. doi: 10.1073/pnas.96.1.248
Wu, L. G., Hamid, E., Shin, W., and Chiang, H. C. (2014). Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu. Rev. Physiol. 76, 301–331. doi: 10.1146/annurev-physiol-021113-170305
Xu, J., Luo, F., Zhang, Z., Xue, L., Wu, X. S., Chiang, H. C., et al. (2013). SNARE proteins synaptobrevin, SNAP-25, and syntaxin are involved in rapid and slow endocytosis at synapses. Cell Rep. 3, 1414–1421. doi: 10.1016/j.celrep.2013.03.010
Yoshida, A., Oho, C., Omori, A., Kuwahara, R., Ito, T., and Takahashi, M. (1992). HPC-1 is associated with synaptotagmin and omega-conotoxin receptor. J. Biol. Chem. 267, 24925–24928.
Young, C. E., Arima, K., Xie, J., Hu, L., Beach, T. G., Falkai, P., et al. (1998). SNAP-25 deficit and hippocampal connectivity in schizophrenia. Cereb. Cortex 8, 261–268. doi: 10.1093/cercor/8.3.261
Zamponi, G. W. (2003). Regulation of presynaptic calcium channels by synaptic proteins. J. Pharmacol. Sci. 92, 79–83. doi: 10.1254/jphs.92.79
Zhang, H., Zhu, S., Zhu, Y., Chen, J., Zhang, G., and Chang, H. (2011). An association study between SNAP-25 gene and attention-deficit hyperactivity disorder. Eur. J. Paediatr. Neurol. 15, 48–52. doi: 10.1016/j.ejpn.2010.06.001
Zhang, Y., Vilaythong, A. P., Yoshor, D., and Noebels, J. L. (2004). Elevated thalamic low-voltage-activated currents precede the onset of absence epilepsy in the SNAP25-deficient mouse mutant coloboma. J. Neurosci. 24, 5239–5248. doi: 10.1523/JNEUROSCI.0992-04.2004
Zhang, Z., Wang, D., Sun, T., Xu, J., Chiang, H. C., Shin, W., et al. (2013). The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J. Neurosci. 33, 9169–9175. doi: 10.1523/JNEUROSCI.0301-13.2013
Keywords: SNAP-25, synaptopathies, presynaptic role, postsynaptic role, brain diseases
Citation: Antonucci F, Corradini I, Fossati G, Tomasoni R, Menna E and Matteoli M (2016) SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions. Front. Synaptic Neurosci. 8:7. doi: 10.3389/fnsyn.2016.00007
Received: 25 January 2016; Accepted: 07 March 2016;
Published: 24 March 2016.
Edited by:
Lucia Tabares, University of Seville, SpainReviewed by:
Gerald W. Zamponi, University of Calgary, CanadaWayne S. Sossin, McGill University, Canada
Copyright © 2016 Antonucci, Corradini, Fossati, Tomasoni, Menna and Matteoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michela Matteoli, m.matteoli@in.cnr.it