Noradrenaline and Parkinson’s disease
- 1 UMR 5293, Institut des Maladies Neurodégénératives, Université de Bordeaux, Bordeaux, France
- 2 UMR 5293, Institut des Maladies Neurodégénératives, Centre National de la Recherche Scientifique, Bordeaux, France
- 3 Centre Hospitalier Universitaire, Bordeaux, France
Parkinson’s disease (PD) is characterized by the degeneration of dopamine (DA) neurons in the substantia nigra pars compacta, and motor symptoms including bradykinesia, rigidity, and tremor at rest. These symptoms are exhibited when striatal dopamine concentration has decreased by around 70%. In addition to motor deficits, PD is also characterized by the non-motor symptoms. However, depletion of DA alone in animal models has failed to simultaneously elicit both the motor and non-motor deficits of PD, possibly because the disease is a multi-system disorder that features a profound loss in other neurotransmitter systems. There is growing evidence that additional loss of noradrenaline (NA) neurons of the locus coeruleus, the principal source of NA in the brain, could be involved in the clinical expression of motor as well as in non-motor deficits. In the present review, we analyze the latest evidence for the implication of NA in the pathophysiology of PD obtained from animal models of parkinsonism and from parkinsonian patients. Recent studies have shown that NA depletion alone, or combined with DA depletion, results in motor as well as in non-motor dysfunctions. In addition, by using selective agonists and antagonists of noradrenaline alpha receptors we, and others, have shown that α2 receptors are implicated in the control of motor activity and that α2 receptor antagonists can improve PD motor symptoms as well as L-Dopa-induced dyskinesia. In this review we argue that the loss of NA neurons in PD has an impact on all PD symptoms and that the addition of NAergic agents to dopaminergic medication could be beneficial in the treatment of the disease.
Introduction
Parkinson’s disease (PD) is a degenerative disorder of the central nervous system that impairs motor skills, cognitive processes, and other functions. Motor symptoms are characterized by tremor, rigidity, bradykinesia, and postural instability. Among non-motor symptoms are autonomic dysfunction, sensory and sleep difficulties, cognitive, and neurobehavioral problems, including dementia and depression. For example, depression occurs in approximately 45% of all PD patients and reduces patient’s quality of life independently from motor symptoms (Lemke et al., 2004). These non-motor symptoms are common in the advanced stages of the disease.
Parkinson’s disease is a non-hereditary disease of unknown etiology that usually appears after the age of 50 and affects both sexes equally (Jankovic and Talosa, 1988). It is well-known to be characterized by a progressive degeneration of dopaminergic (DAergic) neurons (70–75%) in the substantia nigra pars compacta (SNc), which results in a dopamine (DA) depletion in the striatum (Ehringer and Hornykiewicz, 1960). However, it is misleading to reduce PD to a malady of the SNc. Indeed, it has been repeatedly shown over the last 50 years that noradrenergic (NAergic) cells from the locus coeruleus (LC) also degenerate in the disease (Greenfield and Bosanquet, 1953; Ehringer and Hornykiewicz, 1960; German et al., 1992; Bertrand et al., 1997; Tohgi et al., 1997; Ehringer and Hornykiewicz, 1998). The neuronal loss in the LC is greater (83%) than in the SNc (78%; Zarow et al., 2003). This is in agreement with the Braak’s theory (Braak and Del Tredici, 2008) that proposed a progressive caudo-rostral alteration of monoaminergic centers in the symptomatology of PD with a degeneration of LC NAergic neurons occurring before that of SNc DAergic neurons. Several studies revealed the existence of a correlation between the severity of DA and noradrenaline (NA) depletions with the severity of PD neurological symptoms (Marie et al., 1995; Selikhova et al., 2002). While the DAergic system is the main target of the pharmacological approaches to PD, corrections of the NA alterations inherent to the disease could improve the efficacy of current therapies. In this review we focus on the implication of the LC NA system in the pathophysiology of PD and on the different therapeutic approaches using NAergic agents.
Noradrenergic Systems in the Central Nervous System
The Noradrenergic System
Two main NAergic systems can be distinguished: one is composed of neurons belonging to the medulla oblongata (A1/C1 nucleus), whereas the other is more rostral (A2/C2 nucleus) and is located in the pons (Gaspar, 1994). The caudal NAergic system corresponds to diffuse groups of cells in the lower brainstem. The rostral NAergic system corresponds mainly to the locus coeruleus (LC, A6) located in the pons.
Noradrenaline release exerts potent neuromodulatory effects on synaptic transmission, changing the membrane potential, excitability of neurons and synaptic plasticity via adrenergic receptors (ARs). Two subtypes of ARs have been described: alpha ARs (α1 and α2) and beta ARs (β1, β2, and β3). These ARs are found throughout the brain including the striatum and substantia nigra. Different subtypes are coupled to different G proteins. In general, excitatory effects are mediated by α1 and β postsynaptic ARs (McCormick and Wang, 1991; McCormick et al., 1991; Arcos et al., 2003) and inhibitory effects by α2 presynaptic ARs (Belujon et al., 2007; Benarroch, 2009; Table 1).
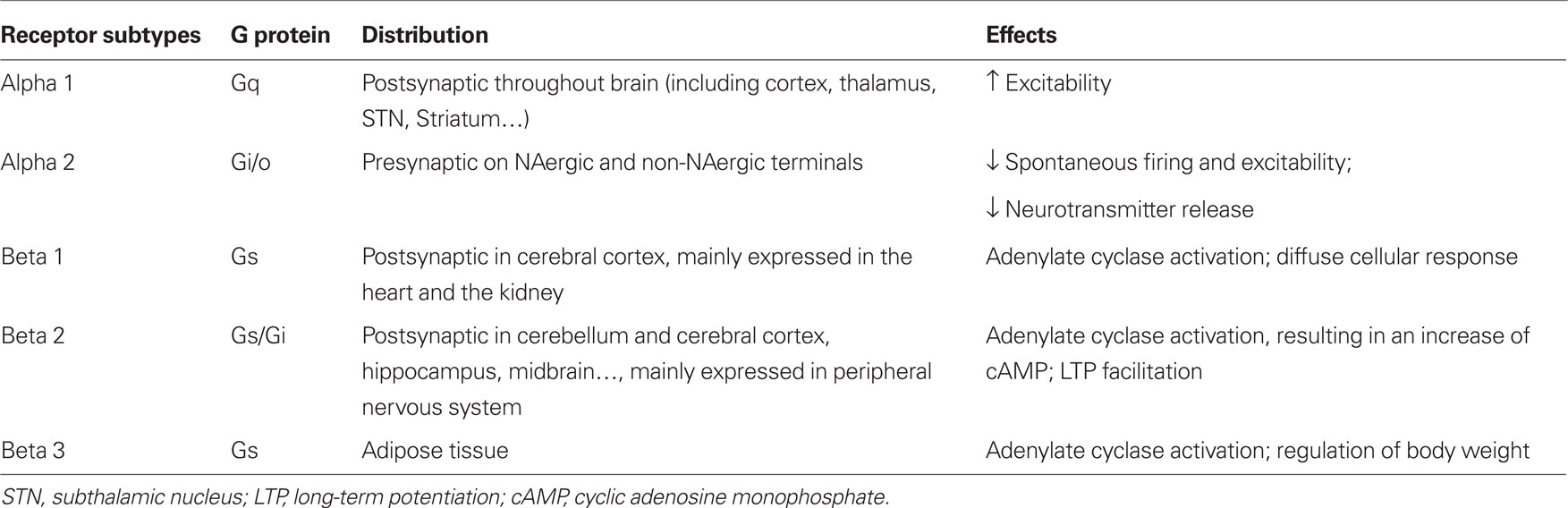
Table 1. Adrenergic receptor characteristics.
The Locus Coeruleus
The LC nucleus is present in all mammalian species and represents the main source of NA for the central nervous system (Mann, 1983; Aston-Jones, 2004). Synaptic inputs from several sources influence the activity of LC neurons. The LC NAergic system, via its widespread projections, modulates cortical, subcortical, and brainstem circuits. Due to the very small size of the LC, biochemical studies are unable to differentiate LC nucleus vs surrounding areas. A lot of retrograde and anterograde tract tracing studies over the years, that have been well recapitulated by Aston-Jones (2004) and Benarroch (2009), have demonstrated the numerous afferent and efferent connections of LC neurons. These reviews document that the orbitofrontal and anterior cingulate cortices provide the main source of glutamatergic afferent drive to the LC. LC modulates a variety of central functions through the release of NA into several brain areas including neocortex, hippocampus, thalamus, subthalamic nucleus, and substantia nigra. These projections are only sparse in striatum and spinal cord. Figure 1 summarizes the major afferent and efferent projections of the LC.
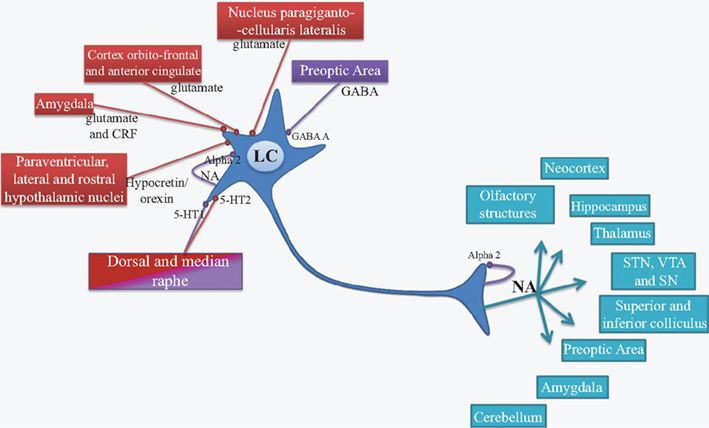
Figure 1. Afferent (red and purple) and efferent projections of LC neurons (blue). LC, locus coeruleus; NA, noradrenaline; Alpha2, Alpha2 receptor; GABA, γ-aminobutyrique acid; GABA A, GABA A receptor; 5-HT, serotonin; 5-HT1 and 5-HT2, serotonin receptor subtype 1 and 2; CRF, corticotropin releasing factor; STN, subthalamic nucleus; VTA, ventral tegmental area; SN, substantia nigra; NET, noradrenalinergic transporters; Red, excitatory input; Purple, inhibitory input.
The biochemistry of NA neurons has relevance to PD. Because L-Dopa, the metabolic precursor of DA, given to patients to enhance DA synthesis, is also a metabolic precursor of NA. In the normal condition, tyrosine hydroxylase (TH) catalyzes the conversion of the amino acid tyrosine to L-Dopa. Both normally produced and administered L-Dopa are catalyzed to DA by L-aromatic amino acid decarboxylase (AADC). DA beta-hydroxylase (DBH) synthesizes NA from DA in the cytosol and either the vesicular monoamine transporter (VMAT) moves the NA into vesicles ready for exocytotic release or monoamine oxidase A degrades the NA. Once released into the extracellular space, reuptake of NA is performed by the plasmalemmal noradrenaline transporter (Rascol et al., 2001), while extracellular NA also limits its own release through the stimulation of auto-inhibitory α2 ARs (Figure 1). Of note, as reported in the cortex (Tanda et al., 1997), NET can also transport extracellular DA inside NAergic neurons.
The LC NAergic system contributes to long-term synaptic plasticity, pain modulation, energy homeostasis and control of local blood flow (McCormick, 1992). LC neurons fire in two distinct modes, tonic and phasic (Aston-Jones and Cohen, 2005a,b). Tonic activity is characterized by a sustained and highly regular pattern of discharge that is highest during wakefulness and decreases during slow-wave sleep. This tonic activity plays a central role in the sleep–waking cycle anticipating the fluctuations of electroencephalographic activity and promoting a state of vigilance. It is indeed well known that the stimulation of central NAergic receptors leads to changes in the state of vigilance. There is also a sustained increase in tonic discharge rate in response to environmental stimuli that elicit behavioral arousal and exploratory behavior. During focused attention and accurate task performance, LC neurons reduce their tonic firing to a moderate rate and respond phasically to task--relevant stimuli. The phasic bursts of LC activity are closely associated with highly accurate behavioral responses. Thus the LC nucleus has a major role in arousal, attention and stress response (Berridge and Waterhouse, 2003; Aston-Jones and Cohen, 2005a).
Alteration of the LC NAergic system has been implicated in many pathologies such as sleep and arousal disorders, attention deficit, hyperactivity disorder, post-traumatic stress disorder and psychiatric disorders, including anxiety, depression, schizophrenia, and mechanisms of opioid addiction (Benarroch, 2009). It constitutes a target for pharmacological treatment of these conditions (Aston-Jones, 2004; Rommelfanger and Weinshenker, 2007; Benarroch, 2009).
The Locus Coeruleus Noradrenergic System in PD
Locus coeruleus is often severely affected in neurodegenerative diseases (Saper et al., 1991) and a growing number of anatomical and biochemical data has stressed this point in PD. In line with the drastic decrease in NAergic cell bodies (Chan-Palay and Asan, 1989), significant depletions (>80%) of NA concentration have also been reported in the PD brain (Taquet et al., 1982; Jenner et al., 1983; Gaspar et al., 1991). Consistently, Positron emission tomography (Dailly et al., 2006) studies have reported that [11C]RTI-32, an in vivo marker of both DA transporter (DAT) and NET binding, was reduced in several regions including those poorly innervated by DAergic fibers (Marie et al., 1995; Remy et al., 2005). It has also been shown that NAergic synapses change size and shape of the pre- and postsynaptic components, polymorphism of the synaptic vesicles and marked morphological alterations of the mitochondria (Chan-Palay and Asan, 1989; Baloyannis et al., 2006). Cash et al. (1984, 1986) have reported an increase in the density of α1 and β1 ARs in synaptosomal and microsomal -fractions in the 6-hydroxydopamine (6-OHDA) rat model of PD (see below) as well as in the pre-frontal cortex of parkinsonian patients. The studies in animal models and PD patients suggest that the impairment of LC NAergic function could contribute to the motor and non-motor symptoms of PD.
The Locus Coeruleus Noradrenergic System in Animal Models of PD
A variety of models have been developed over the years with the increase in understanding of PD. Most, if not all, animal models have focused on DA depletion because PD symptoms typically occur in humans when approximately 70–80% of DA tissue content in the striatum has been lost (Bezard et al., 2001; McNamara and Durso, 2006). The impairment of the NAergic system, also typical of the human disease, has been neglected for several years to the point that most rat models of the disease, generated by administration of the catecholaminergic neurotoxin 6-OHDA, incorporate the concomitant protection of NAergic neurons with the NET blocker desipramine (see below). The reasons that have led to ignoring the role of NAergic neurons in the disease could be related to the demonstration that NAergic neurons are not directly responsible for the behavioral effects of L-Dopa in animal models. Also, the striatum has been the main target of the anti-parkinsonian treatments, a brain region that is poorly innervated by NAergic fibers. Nonetheless, NAergic mechanisms may participate in the therapeutic outcome of L-Dopa (Fox et al., 2001) and NA may act in brain regions that may profoundly impact on the control of motor behaviors, including the subthalamic nucleus (Belujon et al., 2007). The few data available regarding the various animal models of PD support a non-negligible role of NA in the aberrant motor output consequent to DA depletion.
Non-human primate models of the disease have been developed after the discovery of a specific neurotoxin 1-methyl-4-phenyl-l,2,3,6-tetrahydropyridine (MPTP) able to induce parkinsonism in humans (Langston and Ballard, 1983). MPTP has been shown to produce parkinsonian motor deficits in the non-human primate that are similar to PD (Langston and Ballard, 1984). This model has been essential in the understanding of the pathophysiology of PD (Fox and Brotchie, 2010) and the development of new therapeutic approaches, such as the lesion or high frequency stimulation of the subthalamic nucleus (Bergman et al., 1990; Benazzouz et al., 1993, 1996). MPTP-treated rhesus monkeys mimicked the DAergic cell degeneration in the SNc and profound striatal DA depletion. However, the inter-regional pattern of striatal DA loss typically encountered in the idiopathic PD, i.e., a significantly greater depletion of DA in the putamen compared to the caudate nucleus, is not reproduced in this model (Pifl et al., 1991). It was originally reported that MPTP did not cause LC cell death in non-human primates. However, it has become clear that MPTP also causes a distinct and often severe cell loss in the LC and/or NE depletion in terminal fields (Javitch et al., 1984; Mitchell et al., 1985; Di Paolo et al., 1986; Forno et al., 1986; Miyoshi et al., 1988; Pifl et al., 1991). MPTP-treated monkeys developed typical parkinsonian signs of bradykinesia, tremor, and hypokinesia. Behavioral symptoms of PD could involve the concomitant loss of DAergic and NAergic neurons, rather than DAergic neurons alone.
1-Methyl-4-phenyl-l,2,3,6-tetrahydropyridine has been also used in mice. MPTP-treated mice with 80% loss of DA do not exhibit profound motor impairments (German et al., 1992; Marien et al., 2004; Archer and Fredriksson, 2006). The fact that MPTP treatment in mice has little to no effect on LC neurons or NA tissue content may explain the lack of motor deficits. In support of a role of NA in motor symptoms in PD, DBH−/− mice display motor impairments associated with a hypodopaminergic state. These mice synthesize normal amounts of DA, but, in the absence of NA, the release of DA seems to be decreased (Rommelfanger and Weinshenker, 2007). This is consistent with data reporting the excitatory influence exerted by NA on DAergic neuron function (see below interaction between DA and NA). One important note is that DBH−/− mice are supplemented with NA agonists and NA itself during development as DBH is necessary for development (Thomas et al., 1995). Taken together, these data suggest that the behavioral symptoms of PD appear to be correlated with a concomitant loss of DA and NA.
The reserpinized rat model (Colpaert, 1987) and the VMAT KO mice (VMAT−/− mice; Taylor et al., 2009) also support an interaction between DAergic and NAergic systems in PD motor symptoms. Reserpine blocks the VMAT, leading to a rapid decrease in the release of monoamines. The cardinal signs of the disease including tremor, rigidity, and hypokinesia are induced by reserpine in rats. The order of occurrence and time course of these signs appear to resemble the natural history of the human disease (Colpaert, 1987; Taylor et al., 2009). Surprisingly, drugs that increase postsynaptic ARs activation or NA availability, but not DA modulation, are effective in reducing parkinsonian signs in reserpinized rats (Colpaert, 1987). These observations led to the proposal that adrenergic mechanisms in the brain may play an important role in modifying motor and non-motor signs. As VMAT is impaired in both the reserpinized rats and the VMAT−/− mice models, the involvement of serotonin (5-HT) in these mechanisms cannot be excluded because VMAT is also involved in the reuptake of serotonin. Reserpine treatments and VMAT−/− mice do not only result in SNc DAergic and LC NAergic systems depletions, they also induce an almost total depletion of DA and NA from the other catecholaminergic nuclei as well as 5-HT originating from the raphe nuclei. In PD, it is estimated that 40–60% of 5-HT neurons degenerate in some cases (Kish, 2003; Kish et al., 2008; Politis et al., 2010).
The 6-OHDA rat is probably the most popular model of PD. Different 6-OHDA models of PD have been developed in which the toxin has been injected into different parts of the nigrostriatal pathway to cause DAergic cell loss in the SNc, resulting in DA depletion in the striatum (Deumens et al., 2002). 6-OHDA can be injected directly into the SNc, the medial forebrain bundle (MFB) or the striatum. The first two modes of injection are used to develop severe lesions of DAergic neurons corresponding to an advanced stage of the disease, while striatal injection of 6-OHDA is used to produce partial DA cell loss corresponding to the early stage of the disease (Kirik et al., 1998; Yuan et al., 2005; Breit et al., 2007). Such models exhibit some motor disabilities related to sensorimotor degradation though freezing, hypokinesia and tremor are not present. In addition, the non-motor symptoms are not evident (Amalric et al., 1995; Olsson et al., 1995; Branchi et al., 2008). It is noteworthy that 6-OHDA lesion of DAergic neurons causes LC NAergic neurons to be overactive with an irregular pattern of discharge (Wang et al., 2009), suggesting that their influence is magnified in DA-lesioned rats.
Noradrenaline depletion in the rodent by N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) is an approach commonly used to model human neuropsychiatric disorders in rodents. Even though the efficacy of DSP-4 in destroying NA fibers is region- and species-dependent (Dailly et al., 2006), convincing evidence indicates that DSP-4 is a neurotoxin that lesions NAergic terminals arising selectively from the LC (Fritschy and Grzanna, 1989; Grzanna et al., 1989; Fritschy and Grzanna, 1991). Concomitant to the data obtained in non-human primates and genetic mice models of PD (Pifl et al., 1991; Rommelfanger and Weinshenker, 2007), we have recently found that NA depletion alone induces severe motor deficits similar to those reported after 6-OHDA lesion (Delaville et al., 2010). In contrast to a 6-OHDA lesion (Deumens et al., 2002), the motor impairments consequent to NA depletion were not related to DAergic cell loss. Furthermore, the motor deficits reported in 6-OHDA-lesioned rats were not aggravated by the additional depletion of NA (Delaville et al., 2010). Another study has reported that denervation of LC NAergic terminals potentiated the 6-OHDA-induced partial DAergic neurodegeneration and akinesia only in rats treated with a D2 receptor antagonist, haloperidol (Srinivasan and Schmidt, 2003). Only a few studies have specifically addressed the non-motor symptoms of PD in animal models. Our recent data (Delaville et al., 2010) show that additional NA depletion in 6-OHDA-lesioned rats induced anxiety and fear behavior in the elevated plus maze, both being non-motor symptoms of PD. Recently, it has been reported that additional NA depletion impaired working memory in a manner similar to that observed after bilateral inactivation of LC suggesting that the NAergic system of the LC may play an important role in acquisition of spatial memory (Khakpour-Taleghani et al., 2009). Nevertheless, it has been reported that the deficits of spatial information processing in PD are solely a consequence of the striatal DAergic dysfunction (De Leonibus et al., 2007).
In conclusion, most animal models have focused on the motor symptoms related to the nigrostriatal DAergic system. A thorough examination of the data suggests that NAergic neuron dysfunction plays a non-negligible role in the motor and non-motor symptoms. The link between non-motor symptoms and the impairment of NAergic system has been also noted in humans.
Involvement of NA in PD Symptoms
Although it is relatively common to describe a variety of significant biochemical alterations in PD patients, it is more difficult to demonstrate for each neurotransmitter a specific role in the pathophysiology of the disease. This point could be addressed by careful post-mortem studies which should correlate a specific biochemical pattern observed in a given patient with his detailed clinical history. PET imaging studies demonstrate that it is possible to assess in vivo the neocortical monoamine terminal loss (including NA), and to elucidate its potential role in the complex cognitive and affective impairment in PD (Marie et al., 1995). This part of the review is focused on the most relevant studies investigating the role of NA depletion in specific motor and non-motor symptoms related to PD in human patients and associated with the above-mentioned animal models (Table 2).
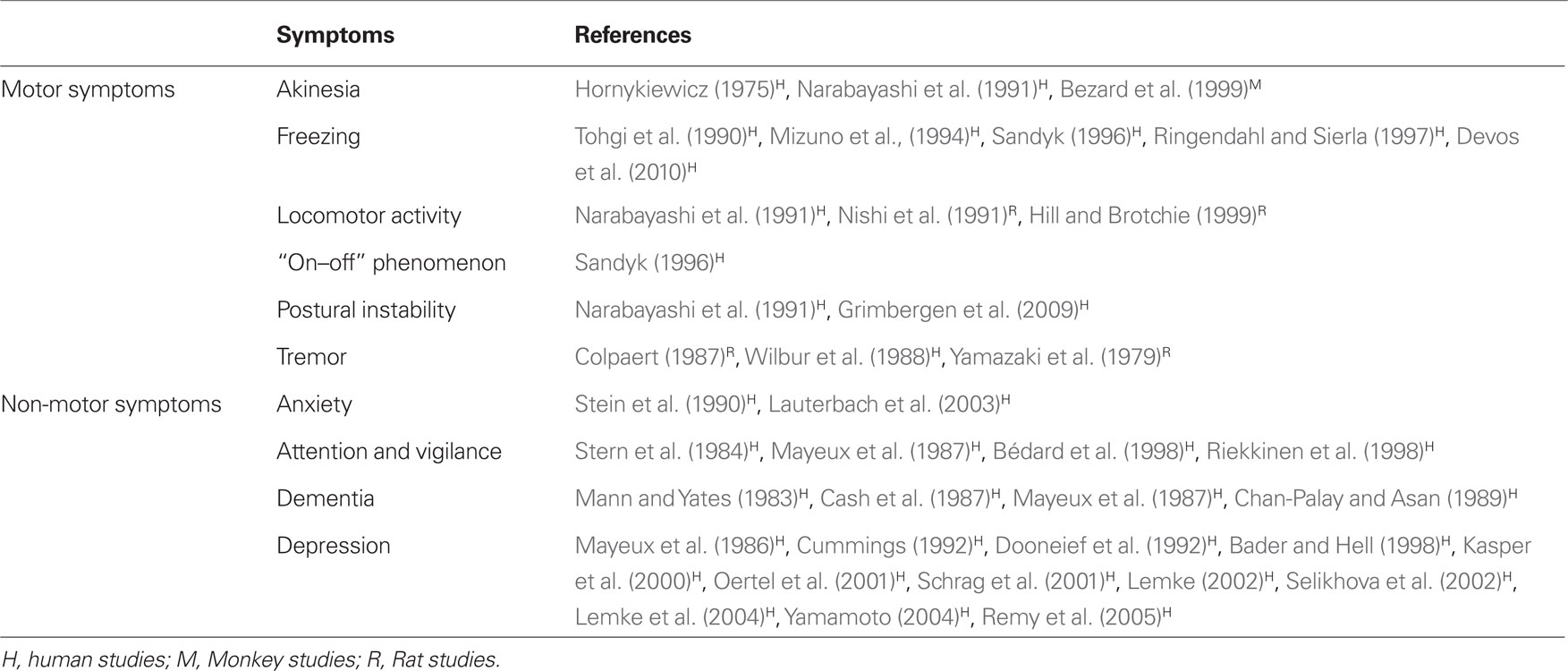
Table 2. Involvement of the LC in specific motor and non-motor symptoms of PD and their reference studies.
(a) Motor symptoms
Parkinson’s disease presents a wider spectrum of motor symptoms when both DA and NA deficiency are combined (Narabayashi et al., 1991). α2ARs agonism potentiates the anti-parkinsonian action of L-Dopa treatment (Hill and Brotchie, 1999). It is also suggested that alpha-adrenergic mechanisms and in particular α2ARs, may be involved in reserpine-induced tremor and rigidity (Colpaert, 1987). In human, akathisia and pseudo-parkinsonian tremor are modulated by a NAergic pathway that projects from the LC to the limbic system (Wilbur et al., 1988); these symptoms are improved by an α2ARs agonist or a βARs antagonist (Wilbur et al., 1988). Several studies have also reported the involvement of the LC NA pathway in freezing (Mizuno et al., 1994; Sandyk, 1996; Ringendahl and Sierla, 1997; Devos et al., 2010), the “on–off” phenomenon (Sandyk, 1996), postural instability (Narabayashi et al., 1991), and akinesia (Hornykiewicz, 1975; Narabayashi et al., 1991).
(b) Non-motor symptoms
Improvements in medication to treat the motor symptoms have highlighted the non-motor symptoms, which represent one of the main concerns of preclinical and clinical studies (Chaudhuri and Odin, 2010). The non-motor symptoms may appear many years before the onset of the motor complications in PD (Lemke et al., 2004). Whilst we focus on potential DAergic sources of mental dysfunction in PD, the potential role of NAergic systems should be kept in mind as well. Indeed, specific loss of DAergic and NAergic innervation of the limbic system is associated with cognitive and neurobehavioral problems, including dementia (Cash et al., 1987), depression (Cummings, 1992; Remy et al., 2005), anxiety (Stein et al., 1990; Lauterbach et al., 2003), and attention deficits (Riekkinen et al., 1998).
In a rat model of early stage PD, striatal DAergic degeneration and NAergic alterations in prefrontal cortex may have caused emotional reactivity and anxiety (Tadaiesky et al., 2008). These -findings are consistent with data showing that, in naive rats, NAergic function regulates the responsiveness to environmental cues and anxiety behavior (Lapiz et al., 2001). In humans, it is also thought that the enhanced anxiety found in elderly people may be related to the loss of DAergic and NAergic innervations (Gareri et al., 2002).
One of the most devastating non-motor symptoms in PD is depression (Cummings, 1992; Bader and Hell, 1998; Oertel et al., 2001; Schrag et al., 2001; Lemke, 2002; Yamamoto, 2004; Remy et al., 2005). The incidence of depression is currently high in PD, reaching a prevalence of 40% in some studies (Mayeux et al., 1986; Dooneief et al., 1992). The reason for the high frequency of depression in PD is poorly understood but, even if in some patients a dopamine agonist (pramipexole) can improve this symptom (Bxarone et al., 2010), the combined depletion of the three monoaminergic systems may be an explanation. In the unilateral rat model of PD, we have found that the DA depletion combined with NA depletion induced by DSP-4 and 5-HT depletion induced by the tryptophane hydroxylase inhibitor parachlorophenylalanine reduced the sucrose consumption and enhanced the time of immobility in the Porsolt test (Delaville et al., 2010). Importantly, only the combined depletions of all three monoamine systems resulted in a depressive-like behavior. It has been reported in humans that the binding of [11C]RTI-32 in the LC and in several regions of the limbic system including the anterior cingulate cortex, the amygdala, and the ventral striatum was inversely correlated with the degree of apathy and the intensity of depression in the patients (Remy et al., 2005). [11C]RTI-32 is a ligand displaying similar affinities to DAT and NET but with far lower affinity for the 5-HTergic transporter (Carroll et al., 1995). Thus, it seems that the DAergic and NAergic innervations are involved in PD depression and might specifically play a role in apathy, which is a major feature of depression. A histological study has shown that LC neuron morphology is more severely altered in PD with depression than in PD without depression (Chan-Palay and Asan, 1989).
It has also been shown that LC cell loss correlates significantly with performance on reaction time tasks and continuous performance tasks that measure attention and vigilance (Stern et al., 1984; Mayeux et al., 1987; Bedard et al., 1998; Riekkinen et al., 1998; McNamara and Durso, 2006). LC damage results in severe loss of cortical and limbic NAergic innervation with a 40–78% decrease in NA, its metabolites and related enzymes in PD. These changes are more marked in patients with dementia than in those without dementia (Cash et al., 1987; Mayeux et al., 1987).
Evidence for the Interaction between NA and DA in PD
The concomitant loss of NA and DA could therefore promote aberrant motor and non-motor symptoms, suggesting that the two systems could interact in the brain. Only a few studies demonstrate the role of DA in NAergic modulation in PD. In addition to the increase in firing rate of LC NA neurons (Wang et al., 2009), an increase in the density of β ARs in the cerebral cortex, the forebrain, thalamic nuclei, the midbrain, the hippocampus, and the cerebellum has been reported in 6-OHDA rats with a selective DAergic neuron lesion (Johnson et al., 1989). In general, a decrease in DAergic neuron function seems to enhance NAergic system activity.
Although anatomical evidence for innervation of DA containing brain regions by NAergic fibers in the rat is scarce, functional studies suggest the existence of NAergic inputs that facilitate DAergic transmission (Jenner et al., 1983). Indeed, Ponzio et al. (1981) demonstrated that NAergic nerve terminals originating from the LC may be involved in regulating the functional activity of the DAergic nerve terminals both in the cerebral cortex and the striatum. This regulation appears to be excitatory in nature and is present early in development. These data are confirmed by pharmacological studies showing that αlARs antagonism may reduce the sensitivity of the mesolimbic DAergic system to pharmacological or environmental challenge (Davis et al., 1985; Snoddy and Tessel, 1985; Auclair et al., 2002). Since most antipsychotic drugs exhibit both D2 receptor and α1ARs antagonist properties, they may alleviate psychosis not only through blockade of postsynaptic DAergic receptors, but also presynaptically on the mesolimbic DAergic system, through their α1ARs antagonistic action (Mathe et al., 1996). Conversely, stimulation of α2ARs and β1ARs elevates DA extracellular levels in the striatum by acting at sites downstream of the DAergic neurons themselves (Chopin et al., 1999; Hudson et al., 1999). More specifically, β1ARs and α2ARs regulate the phosphorylation of DARPP-32, a phosphoprotein regulated by DAergic transmission, in neostriatal neurons. Gi protein activation by α2ARs antagonizes Gs/PKA signaling mediated by D1 receptors and α2ARs in striatonigral and striatopallidal neurons respectively, thereby enhancing D2 receptor/Gi signaling in striatopallidal neurons (Hara et al., 2010). Reches and Meiner (1992) did not report any change in striatal DA synthesis and release after lesions of the dorsal NA bundle (DNEB). It is therefore suggested that any possible effect of the LC on DA transmission in the striatum is not mediated by the DNEB.
In parallel, the LC NAergic system is thought to exert a neuroprotective influence on SNc DAergic neurons. The first hints that NA promotes DAergic neuron survival came from MPTP studies in non-human primates and mice (Mavridis et al., 1991; Fornai et al., 1996). These studies reported that the damage to nigrostriatal DAergic neurons induced by MPTP was potentiated by pre-treatment with DSP4. Rommelfanger et al. (2004) subsequently showed that either pharmacological or genetic blockade of NET, which increases extracellular NA, also protects DAergic neurons from MPTP damage in mice. The mechanisms underlying the neuroprotective influence of NA are not clearly determined (Rommelfanger and Weinshenker, 2007).
Therapeutic Approaches Targeting the Naergic System
Effects of L-Threo-Dops on Motor and Non-Motor Symptoms
Treatment of PD has focused on supplementing DA levels indirectly through L-Dopa administration or on stimulating directly DAergic receptors with DA agonists. Despite the success of these therapies, certain features of PD fail to respond to treatment and even worsen with DA replacement therapy. Among these incapacitating refractory symptoms are apathy, depression, memory loss, akathisia, postural instability, and sudden transient freezing. Clinical and experimental evidence suggests that NA deficiency may be responsible for some of these PD symptoms (see above).
The DA replacement therapy with L-Dopa probably alters the activity of NAergic neurons. Indeed, L-Dopa may enter NAergic neurons to be decarboxylated to DA. Nishi et al. (1991) but not Hollister et al. (1979), have suggested that central NAergic terminals play a significant role in the increase of locomotor activity induced by L-Dopa administration in rats with striatal DA deficiency. Nevertheless, NAergic neurons do not release DA synthesized from L-Dopa as it has been reported that the total destruction of 5-HTergic neurons abolished the releasing effect of L-Dopa (Tanaka et al., 1999), even in brain regions receiving substantial amount of NAergic fibers (Navailles et al., 2010a,b). On the other hand, NAergic neurons could participate in the clearance of extracellular DA. Indeed, because NET is able to transport DA, NAergic fibers could modify the pattern of L-Dopa-induced DA release depending on the relative innervation of brain regions by NAergic neurons. Thus, in the striatum, it has been reported that desipramine enhanced L-Dopa-induced DA release (Arai et al., 2008), suggesting that the clearance could be higher in brain regions more densely innervated. Finally, one could expect an increase in NA tissue content and/or an increase in NA release induced by L-Dopa. The finding that the α2AR antagonist idazoxan is efficient in reducing the dyskinesia induced by L-Dopa but not by the DA agonist apomorphine (Fox et al., 2001) in MPTP-treated monkeys supports a change in NAergic neuron function induced by L-Dopa.
L-Threo-dihydroxyphenylserine (L-threo-DOPS), an non-natural NAergic precursor, has been shown to be effective in relieving motor and non-motor symptoms of PD (Kuno, 1997). Indeed, in the rat, the duration of harmaline induced tremor was significantly reduced by intraventricular administration of L-thero-DOPS (Yamazaki et al., 1979). In PD patients, Ogawa et al. (1984, 1985) have shown that 10 days treatment with L-threo-DOPS substantially improved patient’s freezing episodes, rigidity, and dysarthria. Narabayashi et al. (1991) have reported that treatment with L-threo-DOPS, in combination with a peripheral AADC inhibitor, may have profound effects on freezing phenomena in patients with PD (Tohgi et al., 1990; Tohgi et al., 1993). Other aspects of akinesia also improved considerably in a majority of these patients (Yamamoto et al., 1986), although rigidity and tremor were barely affected (Jenner et al., 1983). When L-threo-DOPS treatment was stopped, the freezing returned to almost the former level, even though the patients continued to receive L-Dopa and benserazide. This could be explained by the fact that L-Dopa measurably affects NA content in the cerebrospinal fluid compared to L-threo-DOPS treatment (Maruyama et al., 1996). In addition to the improvement of motor symptoms, psychological symptoms have also been improved by L-threo-DOPS (Suzuki et al., 1984; Azuma et al., 1991).
Effects of Naergic Receptor Modulation on Motor and Non-Motor Symptoms
α2ARs are distributed widely within the basal ganglia, including the substantia nigra (Schapira, 2005). Mavridis et al. (1991) have suggested that the activation of α1ARs, which results in an increase in NAergic tone, facilitates locomotor activity, and inversely, α2ARs activation, by decreasing NAergic tone, inhibits locomotor activity. In PD, hypoactivation of NAergic tone may be involved in the manifestation of tremor and rigidity. In the reserpine rat, yohimbine, an α2ARs antagonist blocked tremor and improved rigidity but not hypokinesia (Colpaert, 1987).
In the 6-OHDA rat and MPTP monkey models of PD, blockade of α2ARs by idazoxan improved motor disabilities (Bezard et al., 1999; Belujon et al., 2007) in a manner comparable to that induced by a minimal dose of L-Dopa (Bezard et al., 1999). Although these findings provide support for the therapeutic potential of α2ARs in the treatment of PD, idazoxan as a monotherapy in PD patients did not display anti-parkinsonian actions (Henry et al., 1999; Rascol et al., 2001; Colosimo and Craus, 2003). However, co-administration of idazoxan with L-Dopa can provide an anti-parkinsonian action lasting more than twice the duration obtained with L-Dopa alone. Interestingly, the α2AR agonist clonidine and βARs blockers like propranolol are effective in treating akathisia and tardive dyskinesia (Wilbur et al., 1988). However, clonidine is more often used to treat attention deficit in PD. Attention accuracy was not affected by withdrawal of DAergic drugs in mild or severe PD patients. Clonidine retarded accuracy of performance in a difficult attention test in PD patients (Riekkinen et al., 1998). It seems that the NAergic system via α2AR may act differentially on the manifestation of motor and non-motor symptoms in PD. α2AR antagonism would lead to motor amelioration whereas α2ARs agonism would have non-motor benefits.
Non-motor symptoms are also improved by the use of selective α1AR agonists. For example, naphtoxazine may improve performance in some cognitive tests of “frontal functions,” including the Stroop and the odd-man-out tests, which have been previously found to be affected in PD (Bedard et al., 1998). This drug reduced the percentage of errors and restored the lateralization of N100 during the shifting reaction time task, suggesting that it may act on the processes underlying the shifting deficit in these patients (Bedard et al., 1998). NAergic compounds could also be efficient in depression in PD. Reboxetine, a specific NET inhibitor, significantly improved depression scores (Lemke, 2002). This agent did not significantly change parkinsonian motor symptoms or dosage of L-Dopa. Reboxetine was effective and well tolerated in PD patients receiving 4 weeks of treatment of moderate-to-severe depression. Recently, however, the other NET inhibitor atomoxetine failed to reduce depression in PD patients (Weintraub et al., 2010). The use of selective NET inhibitors may be critically dependent on the status of NA neurons which, as mentioned above, can be severely damaged.
There are good theoretical and clinical reasons, including pharmacological specificity of effects and low incidence of side effects, to consider reboxetine, idazoxan, clonidine, or naphtoxazine for the treatment of different symptoms in different cases of PD. The efficacy of some of these drugs including the selective NET inhibitors would, however, be critically dependent on the status of LC NAergic fibers. Finally, the wide spectrum of pathophysiological conditions found in the human disease is still not directly addressed in animal models. The predictive efficacy of NAergic compounds in the treatment of non-motor symptoms in PD may require the development of better behavioral tests in animal models.
Effects of Naergic Agents on L-Dopa Induced Dyskinesia
The side effects associated with long-term L-Dopa treatment today constitute an important cause of functional disability. Side effects such as abnormal involuntary movements and psychiatric disorders remain difficult to manage without causing an increase in parkinsonian immobility. Moreover, management of the phenomenon of the “off” period is limited by these same side effects. The causes of L-Dopa-induced dyskinesia are unclear. It probably involves non-physiological pulsatile stimulation of DAergic receptors or non-physiological release of DA (e.g., from 5-HTergic nerve terminals) in the striatum (Tanaka et al., 1999; Carta et al., 2007; Navailles et al., 2010b). Treatments targeting non-DAergic neurotransmitter systems including glutamate, GABA, NA, acetylcholine, 5-HT, adenosine, and cholecystokinin have been studied (Colosimo and Craus, 2003). It has been recently proposed that lesion of the LC is correlated with the onset of L-Dopa-induced dyskinesia (Fornai et al., 2007). In the 6-OHDA rat model, lesioned rats showed a sensitization–desensitization turning response, whereas in 6-OHDA rats with an additional NAergic degeneration induced by DSP-4, the turning activity was maximal throughout the test. Double-lesioned rats exhibited a lower percentage of dose failure episodes during treatment. NAergic denervation appears to be associated with prolonged long-term DAergic sensitization.
This type of response appears to be comparable to that reported in the clinical setting with intermittent L-Dopa administration where no desensitization occurs once the abnormal response is established (Ruckert et al., 1997; Perez et al., 2009a,b). Fulceri et al. (2007) also demonstrated that unilateral damage to the NAergic system precedes the onset of dyskinesia and worsens the severity of L-Dopa-induced contralateral abnormal involuntary movements in hemi-parkinsonian rats. Furthermore, increases in locomotor activity after L-Dopa administration were markedly suppressed in DA and NA deficient groups. This may suggest that additional NAergic denervation selectively decreases the motor response to L-Dopa treatment (Nishi et al., 1991; Mizuno et al., 1994; Perez et al., 2007).
In view of these results, NAergic modulation has been considered to decrease L-Dopa side effects. In 6-OHDA rats, the α2AR antagonist atipamezole and the α2AR agonist dexmedetomidine increased and decreased contralateral circling evoked by L-Dopa respectively. Atipamezole also prolonged the duration of action of L-Dopa. The α1AR antagonist prazosin partially -antagonized the effect of L-Dopa and had a strong inhibitory effect on the -atipamezole-induced potentiation of the L-Dopa response (Haapalinna et al., 2003; Yavich et al., 2003). This suggests that atipamezole can modulate motor function indirectly, by stimulating the release of NA and directly, by blocking postsynaptic α2ARs in neurons other than NAergic ones.
In MPTP-treated monkeys, blockade of α2ARs by idazoxan in combination with L-Dopa did not impair the anti-parkinsonian response but significantly reduced dyskinesias and delayed their onset, so that the “on” state without dyskinesias was prolonged. The antidyskinetic effect of idazoxan was maintained when repeatedly administered for 10 days and the locomotor response to L-Dopa was significantly increased by chronic administration of idazoxan (Henry et al., 1999; Grondin et al., 2000; Fox et al., 2001). The same antidyskinetic effect has been shown with the α2ARs antagonists yohimbine, rauwolscine, and fipamezole (Gomez-Mancilla and Bedard, 1993; Henry et al., 1999; Grondin et al., 2000; Savola et al., 2003; Fox and Brotchie, 2010). In the case of idazoxan and fipamezole, an extension of the duration of action of levodopa was also observed. All these antagonists had no effect on their own on DA overflow (Yavich et al., 2003). On the other hand, the α2ARs agonist clonidine and the βARs antagonist propanolol markedly reduced the dyskinetic movements induced by L-Dopa at the cost of a return to parkinsonism (Gomez-Mancilla and Bedard, 1993). Another study in the MPTP-lesioned primate has shown that idazoxan significantly reduced L-Dopa-induced dyskinesia, suggesting that α2ARs stimulation may be involved in the generation of L-Dopa-induced dyskinesia (Fox et al., 2001; Bara-Jimenez et al., 2004).
In humans, however, neither idazoxan nor fipamezole had any effect on duration of on time. These drugs have shown variable benefits in clinical trials (Rascol et al., 1994). The poor efficacy could be explained by the dose used in the different studies. In the MPTP primate model, a significant effect of idazoxan on dyskinesia was observed over a range of doses from 1 to 10 mg/kg whereas much lower doses were used in human studies (approximately 0.25–1.0 mg/kg). Similarly, the maximal efficacy of fipamezole in reducing L-Dopa-induced dyskinesia in humans was low compared to that reported in the monkey. It is not clear whether the doses and routes employed of either drug might provide equivalent plasma or brain drug levels across the two species. Side effects with α2ARs antagonists used in human studies included hypertension, facial flushing, and headache which could be inherent to α2ARs blockade. These side effects were dose-dependent and doses higher than 20 mg were not well tolerated (Fox and Brotchie, 2010). However, other α2ARs antagonists could be developed. For example, it has been shown in rats that atipamezole had no effect on blood pressure (Haapalinna et al., 2003). Further studies are warranted to confirm the potential benefit of α2ARs antagonists in L-Dopa-induced dyskinesia.
Concluding Remarks
Studies in a variety of models and species indicate that NAergic cell loss in the LC could be a conditioning factor for the natural history of PD. LC NA exerts an excitatory drive on the nigrostriatal DAergic system. The use of therapeutic strategies leading to an increase in NA in the brain of PD patients may have a specific place in the treatment of the disease. Pharmacological modulation of the NAergic system, specifically with α2-AR antagonists, perhaps by inhibiting autoreceptor function, could be important in the treatment of different symptoms of PD and of L-Dopa-induced dyskinesia.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Claire Delaville was supported by a fellowship from the Ministère de l’Education Nationale, de la Recherche et de la Technologie (MENRT). The University Bordeaux Segalen and the Centre National de la Recherche Scientifique (CNRS) funded this study. We wish to thank Dr. Martin Guthrie for English reading of the manuscript.
References
Amalric, M., Moukhles, H., Nieoullon, A., and Daszuta, A. (1995). Complex deficits on reaction time performance following bilateral intrastriatal 6-OHDA infusion in the rat. Eur. J. Neurosci. 7, 972–980.
Arai, A., Tomiyama, M., Kannari, K., Kimura, T., Suzuki, C., Watanabe, M., Kawarabayashi, T., Shen, H., and Shoji, M. (2008). Reuptake of l-DOPA-derived extracellular DA in the striatum of a rodent model of Parkinson’s disease via norepinephrine transporter. Synapse 62, 632–635.
Archer, T., and Fredriksson, A. (2006). Influence of noradrenaline denervation on MPTP-induced deficits in mice. J. Neural Transm. 113, 1119–1129.
Arcos, D., Sierra, A., Nunez, A., Flores, G., Aceves, J., and Arias-Montano, J. A. (2003). Noradrenaline increases the firing rate of a subpopulation of rat subthalamic neurones through the activation of alpha 1-adrenoceptors. Neuropharmacology 45, 1070–1079.
Aston-Jones, G. (2004). “Locus coeruleus, A5 and A7 noradrenergic cell groups,” in The Rat Nervous System, 3rd Edn, ed G. Paxinos (New York: Elsevier), 259–284.
Aston-Jones, G., and Cohen, J. D. (2005a). Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J. Comp. Neurol. 493, 99–110.
Aston-Jones, G., and Cohen, J. D. (2005b). An integrative theory of locus -coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 28, 403–450.
Auclair, A., Cotecchia, S., Glowinski, J., and Tassin, J. P. (2002). D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha 1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J. Neurosci. 22, 9150–9154.
Azuma, T., Suzuki, T., Sakoda, S., Mizuno, R., Tsujino, S., Kobayashi, T., Kishimoto, S., Hiraga, T., Matsubara, T., Yoshida, S., and Tone, K. (1991). Effect of long-term l-threo-3, 4--dihydroxyphenylserine administration on alpha 2-adrenergic receptors in platelet membranes in neurologic disorders. Acta Neurol. Scand. 84, 46–50.
Bader, J. P., and Hell, D. (1998). [Parkinson syndrome and depression]. Fortschr. Neurol. Psychiatr. 66, 303–312.
Baloyannis, S. J., Costa, V., and Baloyannis, I. S. (2006). Morphological alterations of the synapses in the locus coeruleus in Parkinson’s disease. J. Neurol. Sci. 248, 35–41.
Bara-Jimenez, W., Dimitrova, T., Sherzai, A., Favit, A., Mouradian, M. M., and Chase, T. N. (2004). Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson’s disease. Mov. Disord. 19, 1183–1186.
Bedard, M. A., el Massioui, F., Malapani, C., Dubois, B., Pillon, B., Renault, B., and Agid, Y. (1998). Attentional deficits in Parkinson’s disease: partial reversibility with naphtoxazine (SDZ NVI-085), a selective noradrenergic alpha 1 agonist. Clin. Neuropharmacol. 21, 108–117.
Belujon, P., Bezard, E., Taupignon, A., Bioulac, B., and Benazzouz, A. (2007). Noradrenergic modulation of subthalamic nucleus activity: behavioral and electrophysiological evidence in intact and 6-hydroxydopamine-lesioned rats. J. Neurosci. 27, 9595–9606.
Benarroch, E. E. (2009). The locus ceruleus norepinephrine system: functional organization and potential clinical significance. Neurology 73, 1699–1704.
Benazzouz, A., Boraud, T., Feger, J., Burbaud, P., Bioulac, B., and Gross, C. (1996). Alleviation of experimental hemiparkinsonism by high-frequency stimulation of the subthalamic nucleus in primates: a comparison with l-Dopa treatment. Mov. Disord. 11, 627–632.
Benazzouz, A., Gross, C., Feger, J., Boraud, T., and Bioulac, B. (1993). Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur. J. Neurosci. 5, 382–389.
Bergman, H., Wichmann, T., and DeLong, M. R. (1990). Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249, 1436–1438.
Berridge, C. W., and Waterhouse, B. D. (2003). The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev. 42, 33–84.
Bertrand, E., Lechowicz, W., Szpak, G. M., and Dymecki, J. (1997). Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson’s disease. Folia Neuropathol. 35, 80–86.
Bezard, E., Brefel, C., Tison, F., Peyro-Saint-Paul, H., Ladure, P., Rascol, O., and Gross, C. E. (1999). Effect of the alpha 2 adrenoreceptor antagonist, idazoxan, on motor disabilities in MPTP-treated monkey. Prog. Neuropsychopharmacol. Biol. Psychiatry 23, 1237–1246.
Bezard, E., Dovero, S., Prunier, C., Ravenscroft, P., Chalon, S., Guilloteau, D., Crossman, A. R., Bioulac, B., Brotchie, J. M., and Gross, C. E. (2001). Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J. Neurosci. 21, 6853–6861.
Braak, H., and Del Tredici, K. (2008). Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 70, 1916–1925.
Branchi, I., D’Andrea, I., Armida, M., Cassano, T., Pezzola, A., Potenza, R. L., Morgese, M. G., Popoli, P., and Alleva, E. (2008). Nonmotor symptoms in Parkinson’s disease: investigating early-phase onset of behavioral dysfunction in the 6-hydroxydopamine-lesioned rat model. J. Neurosci. Res. 86, 2050–2061.
Breit, S., Bouali-Benazzouz, R., Popa, R. C., Gasser, T., Benabid, A. L., and Benazzouz, A. (2007). Effects of 6-hydroxydopamine-induced severe or partial lesion of the nigrostriatal pathway on the neuronal activity of pallido-subthalamic network in the rat. Exp. Neurol. 205, 36–47.
Bxarone, P., Poewe, W., Albrecht, S., Debieuvre, C., Massey, D., Rascol, O., Tolosa, E., and Weintraub, D. (2010). Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9, 573–580.
Carroll, F. I., Scheffel, U., Dannals, R. F., Boja, J. W., and Kuhar, M. J. (1995). Development of imaging agents for the dopamine transporter. Med. Res. Rev. 15, 419–444.
Carta, M., Carlsson, T., Kirik, D., and Bjorklund, A. (2007). Dopamine released from 5-HT terminals is the cause of l-DOPA-induced dyskinesia in parkinsonian rats. Brain 130, 1819–1833.
Cash, R., Dennis, T., L’Heureux, R., Raisman, R., Javoy-Agid, F., and Scatton, B. (1987). Parkinson’s disease and dementia: norepinephrine and dopamine in locus ceruleus. Neurology 37, 42–46.
Cash, R., Raisman, R., Lanfumey, L., Ploska, A., and Agid, Y. (1986). Cellular localization of adrenergic receptors in rat and human brain. Brain Res. 370, 127–135.
Cash, R., Ruberg, M., Raisman, R., and Agid, Y. (1984). Adrenergic receptors in Parkinson’s disease. Brain Res. 322, 269–275.
Chan-Palay, V., and Asan, E. (1989). Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol. 287, 373–392.
Chaudhuri, K. R., and Odin, P. (2010). The challenge of non-motor symptoms in Parkinson’s disease. Prog. Brain Res. 184, 325–341.
Chopin, P., Colpaert, F. C., and Marien, M. (1999). Effects of alpha-2 adrenoceptor agonists and antagonists on circling behavior in rats with unilateral 6-hydroxydopamine lesions of the nigrostriatal pathway. J. Pharmacol. Exp. Ther. 288, 798–804.
Colosimo, C., and Craus, A. (2003). Noradrenergic drugs for levodopa-induced dyskinesia. Clin. Neuropharmacol. 26, 299–305.
Colpaert, F. C. (1987). Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology 26, 1431–1440.
Cummings, J. L. (1992). Depression and Parkinson’s disease: a review. Am. J. Psychiatry 149, 443–454.
Dailly, E., Chenu, F., Petit-Demouliere, B., and Bourin, M. (2006). Specificity and efficacy of noradrenaline, serotonin depletion in discrete brain areas of Swiss mice by neurotoxins. J. Neurosci. Methods 150, 111–115.
Davis, M., Kehne, J. H., and Commissaris, R. L. (1985). Antagonism of -apomorphine-enhanced startle by alpha 1-adrenergic antagonists. Eur. J. Pharmacol. 108, 233–241.
Delaville, C., Chetrit, J., Abdallah, K., Morin, S., Cardoit, L., De Deurwaerdère, P., and Benazzouz, A. (2010). Involvement of monoamine deficiency in motor and nonmotor disabilities of Parkinson’s disease: behavioural, biochemical and electrophysiological studies (Int. Basal Ganglia Soc. Abstr.).
De Leonibus, E., Pascucci, T., Lopez, S., Oliverio, A., Amalric, M., and Mele, A. (2007). Spatial deficits in a mouse model of Parkinson disease. Psychopharmacology (Berl.) 194, 517–525.
Deumens, R., Blokland, A., and Prickaerts, J. (2002). Modeling Parkinson’s disease in rats: an evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 175, 303–317.
Devos, D., Defebvre, L., and Bordet, R. (2010). Dopaminergic and non-dopaminergic pharmacological hypotheses for gait disorders in Parkinson’s disease. Fundam. Clin. Pharmacol. 24, 407–421.
Di Paolo, T., Bedard, P., Daigle, M., and Boucher, R. (1986). Long-term effects of MPTP on central and peripheral catecholamine and indoleamine concentrations in monkeys. Brain Res. 379, 286–293.
Dooneief, G., Mirabello, E., Bell, K., Marder, K., Stern, Y., and Mayeux, R. (1992). An estimate of the incidence of depression in idiopathic Parkinson’s disease. Arch. Neurol. 49, 305–307.
Ehringer, H., and Hornykiewicz, O. (1960). [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system]. Klin. Wochenschr. 38, 1236–1239.
Ehringer, H., and Hornykiewicz, O. (1998). Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Parkinsonism Relat. Disord. 4, 53–57.
Fornai, F., di Poggio, A. B., Pellegrini, A., Ruggieri, S., and Paparelli, A. (2007). Noradrenaline in Parkinson’s disease: from disease progression to current therapeutics. Curr. Med. Chem. 14, 2330–2334.
Fornai, F., Vaglini, F., Maggio, R., Bonuccelli, U., and Corsini, G. U. (1996). Excitatory amino acids and MPTP toxicity. Adv. Neurol. 69, 167–176.
Forno, L. S., Langston, J. W., DeLanney, L. E., Irwin, I., and Ricaurte, G. A. (1986). Locus ceruleus lesions and eosinophilic inclusions in MPTP-treated monkeys. Ann. Neurol. 20, 449–455.
Fox, S. H., and Brotchie, J. M. (2010). The MPTP-lesioned non-human primate models of Parkinson’s disease. Past, present, and future. Prog. Brain Res. 184, 133–157.
Fox, S. H., Henry, B., Hill, M. P., Peggs, D., Crossman, A. R., and Brotchie, J. M. (2001). Neural mechanisms underlying peak-dose dyskinesia induced by levodopa and apomorphine are distinct: evidence from the effects of the alpha(2) adrenoceptor antagonist idazoxan. Mov. Disord. 16, 642–650.
Fritschy, J. M., and Grzanna, R. (1989). Immunohistochemical analysis of the neurotoxic effects of DSP-4 identifies two populations of noradrenergic axon terminals. Neuroscience 30, 181–197.
Fritschy, J. M., and Grzanna, R. (1991). Selective effects of DSP-4 on locus coeruleus axons: are there pharmacologically different types of noradrenergic axons in the central nervous system? Prog. Brain Res. 88, 257–268.
Fulceri, F., Biagioni, F., Ferrucci, M., Lazzeri, G., Bartalucci, A., Galli, V., Ruggieri, S., Paparelli, A., and Fornai, F. (2007). Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res. 1135, 219–229.
Gareri, P., De Fazio, P., and De Sarro, G. (2002). Neuropharmacology of depression in aging and age-related diseases. Ageing Res. Rev. 1, 113–134.
Gaspar, P. (1994). Anatomy of the Noradrenergic Pathways in the Primate Brain Alteration in Parkinson’s Disease. Boca Raton: CRC Press, 73–88.
Gaspar, P., Duyckaerts, C., Alvarez, C., Javoy-Agid, F., and Berger, B. (1991). Alterations of dopaminergic and noradrenergic innervations in motor cortex in Parkinson’s disease. Ann. Neurol. 30, 365–374.
German, D. C., Manaye, K. F., White, C. L. III, Woodward, D. J., McIntire, D. D., Smith, W. K., Kalaria, R. N., and Mann, D. M. (1992). Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 32, 667–676.
Gomez-Mancilla, B., and Bedard, P. J. (1993). Effect of nondopaminergic drugs on l-dopa-induced dyskinesias in MPTP-treated monkeys. Clin. Neuropharmacol. 16, 418–427.
Greenfield, J. G., and Bosanquet, F. D. (1953). The brain-stem lesions in Parkinsonism. J. Neurol. Neurosurg. Psychiatr. 16, 213–226.
Grimbergen, Y. A., Langston, J. W., Roos, R. A., and Bloem, B. R. (2009). Postural instability in Parkinson’s disease: the adrenergic hypothesis and the locus coeruleus. Expert Rev. Neurother. 9, 279–290.
Grondin, R., Hadj Tahar, A., Doan, V. D., Ladure, P., and Bedard, P. J. (2000). Noradrenoceptor antagonism with idazoxan improves l-dopa-induced dyskinesias in MPTP monkeys. Naunyn Schmiedebergs Arch. Pharmacol. 361, 181–186.
Grzanna, R., Berger, U., Fritschy, J. M., and Geffard, M. (1989). Acute action of DSP-4 on central norepinephrine axons: biochemical and immunohistochemical evidence for differential effects. J. Histochem. Cytochem. 37, 1435–1442.
Haapalinna, A., Leino, T., and Heinonen, E. (2003). The alpha 2-adrenoceptor antagonist atipamezole potentiates anti-Parkinsonian effects and can reduce the adverse cardiovascular effects of dopaminergic drugs in rats. Naunyn Schmiedebergs Arch. Pharmacol. 368, 342–351.
Hara, M., Fukui, R., Hieda, E., Kuroiwa, M., Bateup, H. S., Kano, T., Greengard, P., and Nishi, A. (2010). Role of adrenoceptors in the regulation of dopamine/DARPP-32 signaling in neostriatal neurons. J. Neurochem. 113, 1046–1059.
Henry, B., Fox, S. H., Peggs, D., Crossman, A. R., and Brotchie, J. M. (1999). The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of l-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. 14, 744–753.
Hill, M. P., and Brotchie, J. M. (1999). The adrenergic receptor agonist, clonidine, potentiates the anti-parkinsonian action of the selective kappa-opioid receptor agonist, enadoline, in the monoamine-depleted rat. Br. J. Pharmacol. 128, 1577–1585.
Hollister, A. S., Breese, G. R., and Mueller, R. A. (1979). Role of monoamine neural systems in l-dihydroxyphenylalanine-stimulated activity. J. Pharmacol. Exp. Ther. 208, 37–43.
Hornykiewicz, O. (1975). Brain monoamines and parkinsonism. Natl. Inst. Drug Abuse Res. Monogr. Ser. 11, 13–21.
Hudson, A. L., Robinson, E. S., Lalies, M. D., Tyacke, R. J., Jackson, H. C., and Nutt, D. J. (1999). In vitro and in vivo approaches to the characterization of the alpha2-adrenoceptor. J. Auton. Pharmacol. 19, 311–320.
Jankovic, J., and Talosa, E. (1988). Parkinson’s Disease and Movement Disorders, 5th Edn. Baltimore: Lippincott William and Wilkins.
Javitch, J. A., Uhl, G. R., and Snyder, S. H. (1984). Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: characterization and localization of receptor binding sites in rat and human brain. Proc. Natl. Acad. Sci. U.S.A. 81, 4591–4595.
Jenner, P., Sheehy, M., and Marsden, C. D. (1983). Noradrenaline and 5-hydroxytryptamine modulation of brain dopamine function: implications for the treatment of Parkinson’s disease. Br. J. Clin. Pharmacol. 15(Suppl. 2), 277S–289S.
Johnson, E. W., Wolfe, B. B., and Molinoff, P. B. (1989). Regulation of subtypes of beta-adrenergic receptors in rat brain following treatment with 6-hydroxydopamine. J. Neurosci. 9, 2297–2305.
Kasper, S., el Giamal, N., and Hilger, E. (2000). Reboxetine: the first selective noradrenaline re-uptake inhibitor. Expert Opin. Pharmacother. 1, 771–782.
Khakpour-Taleghani, B., Lashgari, R., Motamedi, F., and Naghdi, N. (2009). Effect of reversible inactivation of locus ceruleus on spatial reference and working memory. Neuroscience 158, 1284–1291.
Kirik, D., Rosenblad, C., and Bjorklund, A. (1998). Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp. Neurol. 152, 259–277.
Kish, S. J. (2003). Biochemistry of Parkinson’s disease: is a brain serotonergic deficiency a characteristic of idiopathic Parkinson’s disease? Adv. Neurol. 91, 39–49.
Kish, S. J., Tong, J., Hornykiewicz, O., Rajput, A., Chang, L. J., Guttman, M., and Furukawa, Y. (2008). Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain 131, 120–131.
Langston, J. W., and Ballard, P. (1984). Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson’s disease. Can. J. Neurol. Sci. 11, 160–165.
Langston, J. W., and Ballard, P. A. Jr. (1983). Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 309, 310.
Lapiz, M. D., Mateo, Y., Durkin, S., Parker, T., and Marsden, C. A. (2001). Effects of central noradrenaline depletion by the selective neurotoxin DSP-4 on the behaviour of the isolated rat in the elevated plus maze and water maze. Psychopharmacology (Berl.) 155, 251–259.
Lauterbach, E. C., Freeman, A., and Vogel, R. L. (2003). Correlates of generalized anxiety and panic attacks in dystonia and Parkinson disease. Cogn. Behav. Neurol. 16, 225–233.
Lemke, M. R. (2002). Effect of reboxetine on depression in Parkinson’s disease patients. J. Clin. Psychiatry 63, 300–304.
Lemke, M. R., Fuchs, G., Gemende, I., Herting, B., Oehlwein, C., Reichmann, H., Rieke, J., and Volkmann, J. (2004). Depression and Parkinson’s disease. J. Neurol. 251 (Suppl. 6), 24–27.
Mann, D. M. (1983). The locus coeruleus and its possible role in ageing and degenerative disease of the human central nervous system. Mech. Ageing Dev. 23, 73–94.
Mann, D. M., and Yates, P. O. (1983). Pathological basis for neurotransmitter changes in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 9, 3–19.
Marie, R. M., Barre, L., Rioux, P., Allain, P., Lechevalier, B., and Baron, J. C. (1995). PET imaging of neocortical monoaminergic terminals in Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 9, 55–71.
Marien, M. R., Colpaert, F. C., and Rosenquist, A. C. (2004). Noradrenergic mechanisms in -neurodegenerative diseases: a theory. Brain Res. Brain Res. Rev. 45, 38–78.
Maruyama, W., Naoi, M., and Narabayashi, H. (1996). The metabolism of l-DOPA and l-threo-3,4-dihydroxyphenylserine and their effects on monoamines in the human brain: analysis of the intraventricular fluid from parkinsonian patients. J. Neurol. Sci. 139, 141–148.
Mathe, J. M., Nomikos, G. G., Hildebrand, B. E., Hertel, P., and Svensson, T. H. (1996). Prazosin inhibits MK-801-induced hyperlocomotion and dopamine release in the nucleus accumbens. Eur. J. Pharmacol. 309, 1–11.
Mavridis, M., Degryse, A. D., Lategan, A. J., Marien, M. R., and Colpaert, F. C. (1991). Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 41, 507–523.
Mayeux, R., Stern, Y., Sano, M., Cote, L., and Williams, J. B. (1987). Clinical and biochemical correlates of bradyphrenia in Parkinson’s disease. Neurology 37, 1130–1134.
Mayeux, R., Stern, Y., Williams, J. B., Cote, L., Frantz, A., and Dyrenfurth, I. (1986). Clinical and biochemical features of depression in Parkinson’s disease. Am. J. Psychiatry 143, 756–759.
McCormick, D. A. (1992). Neurotransmitter actions in the thalamus and cerebral cortex. J. Clin. Neurophysiol. 9, 212–223.
McCormick, D. A., Pape, H. C., and Williamson, A. (1991). Actions of norepinephrine in the cerebral cortex and thalamus: implications for function of the central noradrenergic system. Prog. Brain Res. 88, 293–305.
McCormick, D. A., and Wang, Z. (1991). Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 442, 235–255.
McNamara, P., and Durso, R. (2006). Neuropharmacological treatment of mental dysfunction in Parkinson’s disease. Behav. Neurol. 17, 43–51.
Mitchell, I. J., Cross, A. J., Sambrook, M. A., and Crossman, A. R. (1985). Sites of the neurotoxic action of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the macaque monkey include the ventral tegmental area and the locus coeruleus. Neurosci. Lett. 61, 195–200.
Miyoshi, R., Kito, S., Ishida, H., and Katayama, S. (1988). Alterations of the central noradrenergic system in MPTP-induced monkey parkinsonism. Res. Commun. Chem. Pathol. Pharmacol. 62, 93–102.
Mizuno, Y., Kondo, T., and Mori, H. (1994). Various aspects of motor fluctuations and their management in Parkinson’s disease. Neurology 44, S29–S34.
Narabayashi, H., Yokochi, F., Ogawa, T., and Igakura, T. (1991). [Analysis of l-threo-3, 4-dihydroxyphenylserine effect on motor and psychological symptoms in Parkinson’s disease]. No To Shinkei 43, 263–268.
Navailles, S., Bioulac, B., Gross, C., and De Deurwaerdere, P. (2010a). Chronic l-DOPA therapy alters central serotonergic function and l-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 41, 585–590.
Navailles, S., Bioulac, B., Gross, C., and De Deurwaerdere, P. (2010b). Serotonergic neurons mediate ectopic release of dopamine induced by l-DOPA in a rat model of Parkinson’s disease. Neurobiol. Dis. 38, 136–143.
Nishi, K., Kondo, T., and Narabayashi, H. (1991). Destruction of norepinephrine terminals in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice reduces locomotor activity induced by l-dopa. Neurosci. Lett. 123, 244–247.
Oertel, W. H., Hoglinger, G. U., Caraceni, T., Girotti, F., Eichhorn, T., Spottke, A. E., Krieg, J. C., and Poewe, W. (2001). Depression in Parkinson’s disease. An update. Adv. Neurol. 86, 373–383.
Ogawa, N., Kuroda, H., Yamamoto, M., Nukina, I., and Ota, Z. (1984). Improvement in freezing phenomenon of Parkinson’s disease after D l-threo-3, 4-dihydroxyphenylserine. Acta Med. Okayama 38, 301–304.
Ogawa, N., Yamamoto, M., and Takayama, H. (1985). L-threo-3, 4-dihydroxyphenylserine treatment of Parkinson’s disease. J. Med. 16, 525–534.
Olsson, M., Nikkhah, G., Bentlage, C., and Bjorklund, A. (1995). Forelimb akinesia in the rat Parkinson model: differential effects of dopamine agonists and nigral transplants as assessed by a new stepping test. J. Neurosci. 15, 3863–3875.
Perez, V., Marin, C., Rubio, A., Aguilar, E., Barbanoj, M., and Kulisevsky, J. (2009a). Effect of the additional noradrenergic neurodegeneration to 6-OHDA-lesioned rats in levodopa-induced dyskinesias and in cognitive disturbances. J. Neural Transm. 116, 1257–1266.
Perez, V., Sosti, V., Rubio, A., Barbanoj, M., Gich, I., Rodriguez-Alvarez, J., and Kulisevsky, J. (2009b). Noradrenergic modulation of the motor response induced by long-term levodopa administration in Parkinsonian rats. J. Neural Transm. 116, 867–874.
Perez, V., Sosti, V., Rubio, A., Barbanoj, M., Rodriguez-Alvarez, J., and Kulisevsky, J. (2007). Modulation of the motor response to dopaminergic drugs in a parkinsonian model of combined dopaminergic and noradrenergic degeneration. Eur. J. Pharmacol. 576, 83–90.
Pifl, C., Schingnitz, G., and Hornykiewicz, O. (1991). Effect of 1-methyl-4--phenyl-1,2,3,6-tetrahydropyridine on the regional distribution of brain monoamines in the rhesus monkey. Neuroscience 44, 591–605.
Politis, M., Wu, K., Loane, C., Kiferle, L., Molloy, S., Brooks, D. J., and Piccini, P. (2010). Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol. Dis. 40, 216–221.
Ponzio, F., Hallman, H., and Jonsson, G. (1981). Noradrenaline and dopamine interaction in rat brain during development. Med. Biol. 59, 161–169.
Rascol, O., Arnulf, I., Peyro-Saint Paul, H., Brefel-Courbon, C., Vidailhet, M., Thalamas, C., Bonnet, A. M., Descombes, S., Bejjani, B., Fabre, N., Montastruc, J. L., and Agid, Y. (2001). Idazoxan, an alpha-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson’s disease. Mov. Disord. 16, 708–713.
Rascol, O., Fabre, N., Blin, O., Poulik, J., Sabatini, U., Senard, J. M., Ane, M., Montastruc, J. L., and Rascol, A. (1994). Naltrexone, an opiate antagonist, fails to modify motor symptoms in patients with Parkinson’s disease. Mov. Disord. 9, 437–440.
Reches, A., and Meiner, Z. (1992). The locus coeruleus and dopaminergic function in rat brain: implications to parkinsonism. Brain Res. Bull. 28, 663–666.
Remy, P., Doder, M., Lees, A., Turjanski, N., and Brooks, D. (2005). Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 128, 1314–1322.
Riekkinen, M., Kejonen, K., Jakala, P., Soininen, H., and Riekkinen, P. Jr. (1998). Reduction of noradrenaline impairs attention and dopamine depletion slows responses in Parkinson’s disease. Eur. J. Neurosci. 10, 1429–1435.
Ringendahl, H., and Sierla, T. (1997). [Freezing phenomenon in Parkinson disease]. Fortschr. Neurol. Psychiatr. 65, 435–445.
Rommelfanger, K. S., and Weinshenker, D. (2007). Norepinephrine: the redheaded stepchild of Parkinson’s disease. Biochem. Pharmacol. 74, 177–190.
Rommelfanger, K. S., Weinshenker, D., and Miller, G. W. (2004). Reduced MPTP toxicity in noradrenaline transporter knockout mice. J. Neurochem. 91, 1116–1124.
Ruckert, N., Bubser, M., and Schmidt, W. J. (1997). 6-Hydroxydopamine lesion of locus coeruleus and the antiparkinsonian potential of NMDA-receptor antagonists in rats. J. Neural Transm. 104, 363–377.
Sandyk, R. (1996). Freezing of gait in Parkinson’s disease is improved by treatment with weak electromagnetic fields. Int. J. Neurosci. 85, 111–124.
Saper, C. B., Sorrentino, D. M., German, D. C., and de Lacalle, S. (1991). Medullary catecholaminergic neurons in the normal human brain and in Parkinson’s disease. Ann. Neurol. 29, 577–584.
Savola, J. M., Hill, M., Engstrom, M., Merivuori, H., Wurster, S., McGuire, S. G., Fox, S. H., Crossman, A. R., and Brotchie, J. M. (2003). Fipamezole (JP-1730) is a potent alpha2 adrenergic receptor antagonist that reduces -levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. 18, 872–883.
Schapira, A. H. (2005). Present and future drug treatment for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. 76, 1472–1478.
Schrag, A., Jahanshahi, M., and Quinn, N. P. (2001). What contributes to depression in Parkinson’s disease? Psychol. Med. 31, 65–73.
Selikhova, M. V., Kogan, B. M., Serkin, G. V., and Gusev, E. I. (2002). [Catecholamine metabolism in different forms of Parkinson’s disease]. Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova. 102, 37–40.
Snoddy, A. M., and Tessel, R. E. (1985). Prazosin: effect on psychomotor-stimulant cues and locomotor activity in mice. Eur. J. Pharmacol. 116, 221–228.
Srinivasan, J., and Schmidt, W. J. (2003). Potentiation of parkinsonian symptoms by depletion of locus coeruleus noradrenaline in 6-hydroxydopamine-induced partial degeneration of substantia nigra in rats. Eur. J. Neurosci. 17, 2586–2592.
Stein, M. B., Heuser, I. J., Juncos, J. L., and Uhde, T. W. (1990). Anxiety disorders in patients with Parkinson’s disease. Am. J. Psychiatry 147, 217–220.
Stern, Y., Mayeux, R., and Cote, L. (1984). Reaction time and vigilance in Parkinson’s disease. Possible role of altered norepinephrine metabolism. Arch Neurol. 41, 1086–1089.
Suzuki, T., Sakoda, S., Ueji, M., Kishimoto, S., Hayashi, A., Kondo, T., and Narabayashi, H. (1984). Treatment of parkinsonism with L-threo-3,4-dihydroxyphenylserine: a pharmacokinetic study. Neurology 34, 1446–1450.
Tadaiesky, M. T., Dombrowski, P. A., Figueiredo, C. P., Cargnin-Ferreira, E., Da Cunha, C., and Takahashi, R. N. (2008). Emotional, cognitive and neurochemical alterations in a -premotor stage model of Parkinson’s disease. Neuroscience 156, 830–840.
Tanaka, H., Kannari, K., Maeda, T., Tomiyama, M., Suda, T., and Matsunaga, M. (1999). Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 10, 631–634.
Tanda, G., Pontieri, F. E., Frau, R., and Di Chiara, G. (1997). Contribution of blockade of the noradrenaline carrier to the increase of extracellular dopamine in the rat prefrontal cortex by amphetamine and cocaine. Eur. J. Neurosci. 9, 2077–2085.
Taquet, H., Javoy-Agid, F., Cesselin, F., Hamon, M., Legrand, J. C., and Agid, Y. (1982). Microtopography of methionine-enkephalin, dopamine and noradrenaline in the ventral mesencephalon of human control and Parkinsonian brains. Brain Res. 235, 303–314.
Taylor, T. N., Caudle, W. M., Shepherd, K. R., Noorian, A., Jackson, C. R., Iuvone, P. M., Weinshenker, D., Greene, J. G., and Miller, G. W. (2009). Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 29, 8103–8113.
Thomas, S. A., Matsumoto, A. M., and Palmiter, R. D. (1995). Noradrenaline is essential for mouse fetal development. Nature 374, 643–646.
Tohgi, H., Abe, T., Saheki, M., Yamazaki, K., and Murata, T. (1997). Concentration of catecholamines and indoleamines in the cerebrospinal fluid of patients with vascular parkinsonism compared to Parkinson’s disease patients. J. Neural Transm. 104, 441–449.
Tohgi, H., Abe, T., and Takahashi, S. (1993). The effects of L-threo-3,4-dihydroxyphenylserine on the total norepinephrine and dopamine concentrations in the cerebrospinal fluid and freezing gait in parkinsonian patients. J. Neural Transm. Park. Dis. Dement. Sect. 5, 27–34.
Tohgi, H., Abe, T., Takahashi, S., Takahashi, J., Ueno, M., and Nozaki, Y. (1990). Effect of a synthetic norepinephrine precursor, L-threo-3,4- dihydroxyphenylserine on the total norepinephrine concentration in the cerebrospinal fluid of parkinsonian patients. Neurosci. Lett. 116, 194–197.
Wang, T., Zhang, Q. J., Liu, J., Wu, Z. H., and Wang, S. (2009). Firing activity of locus coeruleus noradrenergic neurons increases in a rodent model of Parkinsonism. Neurosci. Bull. 25, 15–20.
Weintraub, D., Mavandadi, S., Mamikonyan, E., Siderowf, A. D., Duda, J. E., Hurtig, H. I., Colcher, A., Horn, S. S., Nazem, S., Ten Have, T. R., and Stern, M. B. (2010). Atomoxetine for depression and other neuropsychiatric symptoms in Parkinson disease. Neurology 75, 448–455.
Wilbur, R., Kulik, F. A., and Kulik, A. V. (1988). Noradrenergic effects in tardive dyskinesia, akathisia and pseudoparkinsonism via the limbic system and basal ganglia. Prog. Neuropsychopharmacol. Biol. Psychiatry 12, 849–864.
Yamamoto, M., Ogawa, N., and Ujike, H. (1986). Effect of L-threo-3,4-dihydroxyphenylserine chronic administration on cerebrospinal fluid and plasma free 3-methoxy-4-hydroxy-phenylglycol concentration in patients with Parkinson’s disease. J. Neurol. Sci. 73, 39–44.
Yamazaki, M., Tanaka, C., and Takaori, S. (1979). Significance of central noradrenergic system on harmaline induced tremor. Pharmacol. Biochem. Behav. 10, 421–427.
Yavich, L., Sirvio, J., Haapalinna, A., Ylinen, A., and Mannisto, P. T. (2003). Atipamezole, an alpha2-adrenoceptor antagonist, augments the effects of L-DOPA on evoked dopamine release in rat striatum. Eur. J. Pharmacol. 462, 83–89.
Yuan, H., Sarre, S., Ebinger, G., and Michotte, Y. (2005). Histological, behavioural and neurochemical evaluation of medial forebrain bundle and striatal 6-OHDA lesions as rat models of Parkinson’s disease. J. Neurosci. Methods 144, 35–45.
Keywords: Parkinson’s disease, motor and non-motor symptoms, noradrenaline, dopamine, locus coeruleus
Citation: Delaville C, De Deurwaerdère P and Benazzouz A (2011) Noradrenaline and Parkinson’s disease. Front. Syst. Neurosci. 5:31. doi: 10.3389/fnsys.2011.00031
Received: 02 February 2011;
Paper pending published: 27 March 2011;
Accepted: 04 May 2011;
Published online: 18 May 2011.
Edited by:
Elizabeth Abercrombie, Rutgers-Newark: The State University of New Jersey, USAReviewed by:
Christine E. Collins, Vanderbilt University, USAAlessandro Stefani, University of Rome, Italy
Copyright: © 2011 Delaville, De Deurwaerdère and Benazzouz. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Abdelhamid Benazzouz, Institut des Maladies Neurodégénératives, Université Bordeaux Segalen, 146, rue Léo-Saignat, 33076 Bordeaux Cedex, France. e-mail: abdelhamid.benazzouz@u-bordeaux2.fr