Nitric oxide–soluble guanylyl cyclase–cyclic GMP signaling in the striatum: new targets for the treatment of Parkinson’s disease?
- 1 Department of Neuroscience, The Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
- 2 Department of Cellular and Molecular Pharmacology, The Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
Striatal nitric oxide (NO)-producing interneurons play an important role in the regulation of corticostriatal synaptic transmission and motor behavior. Striatal NO synthesis is driven by concurrent activation of NMDA and dopamine (DA) D1 receptors. NO diffuses into the dendrites of medium-sized spiny neurons which contain high levels of NO receptors called soluble guanylyl cyclases (sGC). NO-mediated activation of sGC leads to the synthesis of the second messenger cGMP. In the intact striatum, transient elevations in intracellular cGMP primarily act to increase neuronal excitability and to facilitate glutamatergic corticostriatal transmission. NO–cGMP signaling also functionally opposes the inhibitory effects of DA D2 receptor activation on corticostriatal transmission. Not surprisingly, abnormal striatal NO–sGC–cGMP signaling becomes apparent following striatal DA depletion, an alteration thought to contribute to pathophysiological changes observed in basal ganglia circuits in Parkinson’s disease (PD). Here, we discuss recent developments in the field which have shed light on the role of NO–sGC–cGMP signaling pathways in basal ganglia dysfunction and motor symptoms associated with PD and L-DOPA-induced dyskinesias.
No Signaling in the Striatum
Nitric oxide (NO) is a gaseous neuromodulator and is implicated in the regulation of numerous physiological and pathophysiological processes in both the peripheral and central nervous system (Boehning and Snyder, 2003; Bredt, 2003; Garthwaite, 2008). Since its discovery in 1987 as the “endothelial derived relaxation factor” (EDRF) in peripheral blood vessels (Palmer et al., 1987), three distinct isoforms of the NO-producing enzyme nitric oxide synthase (NOS; brain/neuronal NOS, inducible NOS, endothelial NOS) have been described (Alderton et al., 2001; Garthwaite, 2008). Of particular interest is the neuronal NOS (nNOS), which is ubiquitously distributed throughout the brain and relatively abundant in the dorsal striatum and nucleus accumbens (Bredt et al., 1990; Vincent, 1994). Striatal NO is synthesized primarily in nNOS-containing interneurons, which are readily revealed using NADPH-diaphorase histochemical staining (Figures 1A,B) as well as nNOS immunohistochemical labeling (Hope et al., 1991; Kawaguchi, 1993; Gracy and Pickel, 1997). The remainder of this review will focus in detail on how NO synthesis is regulated by DA D1- and D2-like receptors and their interactions with glutamate inputs/receptors, and how nitrergic signaling then affects striatal output in normal and parkinsonian animals.
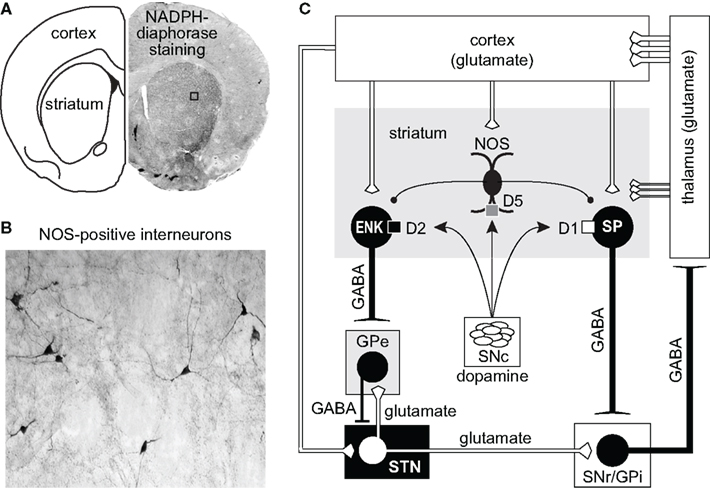
Figure 1. (A,B) Coronal section of the forebrain revealing the presence of NADPH-diaphorase staining and NOS-positive interneurons in cortex and striatum. (C) Diagram illustrating the major connections of the basal ganglia. The cerebral cortex provides the major inputs to the striatum. Both direct (D1/substance P -SP-) and indirect (D2/enkephalin -ENK-) striatal output neurons receive modulatory input arising from local NOS interneurons. Direct projecting neurons provide axon collaterals to the output nuclei of the basal ganglia: internal segment of the globus pallidus (GPi) and to the substantia nigra pars reticulata (SNr). Indirect striatal output neurons are indirectly connected to the GPi and SNr through connections that involve the external segment of the globus pallidus (GPe) and the subthalamic nucleus (STN). Feedback pathways to the cortex arise from GABAergic output neurons in the GPi and the SNr via the thalamo-cortical circuit.
Modulation of Striatal NOS Activity by Dopamine D1 and D2 Receptor Activation
Remarkably consistent outcomes have been reported in studies examining the impact of DA D1- and D2-like receptor agonists/antagonists on striatal NOS activity and cGMP production (Altar et al., 1990; Morris et al., 1997; Di Stefano et al., 2005; Sammut et al., 2006, 2007b; Siuciak et al., 2006; Park and West, 2009; Hoque et al., 2010). Early studies by Altar et al. (1990) were the first to show that D1-like receptor activation induced by SKF 38393 increases striatal tissue levels of cGMP, whereas antagonism of this receptor with SCH 23390 decreases similar measures of cGMP. In the same study these authors reported that the D2 receptor antagonists haloperidol and sulpiride robustly elevated striatal tissue levels of cGMP. These findings have been confirmed and extended in more recent studies (Di Stefano et al., 2005; Siuciak et al., 2006). Thus, consistent with the above work, D2-like receptor agonism was shown to decrease striatal tissue levels of cGMP (Di Stefano et al., 2005). Moreover, work by Schmidt and colleagues showed that the facilitatory effects of D1-like receptor agonist and D2 receptor antagonist on striatal tissue levels of cGMP are abolished in nNOS−/− (i.e., knockout) mice (Siuciak et al., 2006). Together, the above studies confirm that both D1- and D2-like receptor activation strongly regulates striatal nNOS activity, albeit in opposing manners (i.e, D1 receptor activation is facilitatory, D2 receptor activation is inhibitory), and identify a critical role of nNOS–NO signaling in the generation of striatal cGMP.
Consistent with the above studies of striatal cGMP synthesis, our laboratory has reported that electrical and chemical stimulation of the substantia nigra and systemic administration of the D1 receptor agonist SKF 81297 all robustly increase amperometric measures of striatal NO efflux via nNOS and D1-like receptor-dependent mechanisms (Sammut et al., 2006, 2007a; Park and West, 2009). Interestingly, the facilitatory effects of nigrostriatal DA cell activation and SKF 81297 on striatal NO efflux were both attenuated by systemic administration of the D2-like receptor agonist quinpirole, whereas administration of the D2-like receptor antagonist eticlopride augmented evoked NO efflux (Sammut et al., 2007a).
Other work has examined the impact of D1 and D2 receptor interactions using histochemical measures of striatal NOS activity (NADPH-d staining). Reports from leading laboratories have demonstrated that the catalytic activity of the nNOS enzyme is responsible for producing NADPH-d staining (Dawson et al., 1991; Hope et al., 1991) and that measurements of staining in striatal interneurons accurately reflect enzyme activity (Morris et al., 1997; Sancesario et al., 2004). Moreover, quantification of NADPH-d staining using optical density is a valuable index of striatal nNOS activity which can be observed in identified interneurons across striatal subregions (Kuo et al., 1994; Morris et al., 1997; Sanceserio et al., 2004; Hoque et al., 2010). Studies using NADPH-diaphorase staining as an indirect measure of striatal nNOS activity have also shown that administration of D1-like receptor antagonist decreases enzyme activity, whereas D2-like receptor antagonists have the opposite effect (Morris et al., 1997; Hoque et al., 2010). Similar to our above studies using NO microsensor recordings, pretreatment with the D2 receptor agonist quinpirole abolished the facilitatory effect of SKF 81297 on NADPH-diaphorase staining/nNOS activity (Hoque et al., 2010). These studies indicate that D1- and D2-like receptor activation has opposing effects on striatal nNOS activity (reviewed in West, 2010).
Impact of Dopamine–Glutamate Interactions on Neuronal NOS Activity
Reciprocal functional interactions between D1 and NMDA receptors are believed to occur in a variety of neurons in the CNS via direct physical interactions and following activation of second messengers (Cepeda and Levine, 2006). In the striatum, DA–glutamate interactions involved in the regulation of nNOS activity are likely to be complex as these transmitter systems converge both at the level of the NOS interneurons (Fujiyama and Masuko, 1996; Hidaka and Totterdell, 2001) and the principle medium-sized spiny neurons (MSNs; Morello et al., 1997; Sancesario et al., 2000; Hidaka and Totterdell, 2001). We have recently begun to study the interaction between DA and glutamate as it pertains to striatal NOS activity using NO microsensor recordings combined with local reverse microdialysis for intrastriatal drug delivery (Park and West, 2009). These studies found that local infusion of D1 agonist potentiates nNOS activity elicited via electrical stimulation of cortical afferents (Park and West, 2009). Interestingly, both the effects of electrical stimulation and D1 agonist were blocked by local D1 antagonist infusion, suggesting that D1 receptor co-activation is required for NOS stimulation by cortical inputs (Park and West, 2009). Further studies revealed that the increase in striatal NO efflux elicited by systemic administration of D1 agonist is blocked by intrastriatal infusion of the selective nNOS inhibitor 7-nitroindazole, the DA D1 receptor antagonist SCH 23390, and NMDA receptor antagonists (CPP and kynurenic acid), indicating that D1 receptor-mediated NO efflux is dependent on concurrent D1 and NMDA receptor activation (see Figure 2). Our studies using NADPH-diaphorase staining as a complementary measure of striatal nNOS activity have also shown that systemic administration of the NMDA receptor antagonist CPP attenuated staining in the dorsal striatum (Hoque et al., 2010). As expected NOS activity stimulated by systemic administration of D1 receptor agonist or D2 receptor antagonist was attenuated by D1 antagonism and D2 agonism, respectively. Moreover, pretreatment with an NMDA receptor antagonist blocked the facilitatory effects of D1 receptor agonist and D2 receptor antagonist on NOS activity. Importantly, in all studies statistical interactions were observed between drug pretreatments and D1 agonist/D2 antagonist treatments, indicating that the drug pretreatments were acting to block the effect of the DA modulation, and not by simply lowering overall basal levels of NOS activity in a manner independent of a DA receptor-mediated mechanism (Hoque et al., 2010).
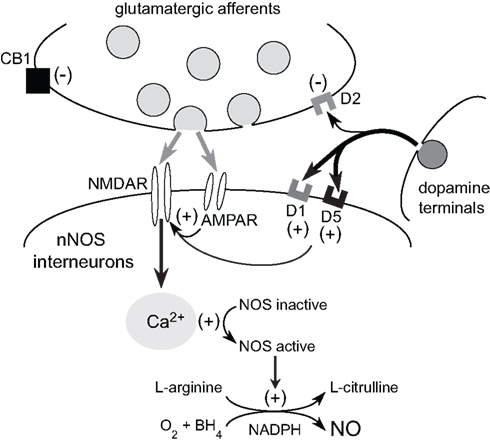
Figure 2. Glutamatergic modulation of striatal NOS interneurons by cortical inputs. Corticostriatal activation exerts direct excitatory effects on NOS interneurons via AMPA and NMDA receptor activation. NMDA receptor-dependent calcium influx activates nNOS and facilitates the conversion of L-arginine into L-citrulline and NO production. Dopamine release from the nigrostriatal pathway facilitates nNOS activity via D1/5 receptor stimulation. Furthermore, striatal NMDA receptor activation plays a critical role in the facilitatory effects of D1/5 receptor activation on striatal NO synthesis. Given that D1/5 receptor tone is also necessary for NMDA receptor activation of NOS, it is likely that reciprocal DA and glutamate interactions are crucial for the activation of striatal nNOS and NO transmission.
Taken together, these findings are consistent with the multitude of studies reporting that D1 receptor activation potentiates NMDA-induced responses in cortical and striatal neurons (Cepeda and Levine, 1998). Moreover, these novel observations indicate that reciprocal D1–NMDA and D2–NMDA receptor interactions play critical and opposing roles in regulating striatal NOS activity. Future studies will need to determine which specific modes of DA transmission favor D1–NMDA facilitation and which lead to D2–NMDA suppression of NOS activity. However, based on the above discussion of the effects of D1 and D2 antagonism on nNOS activity, we predict that tonic levels of DA act to suppress NMDA receptor-mediated nNOS stimulation by activation of D2-like receptors. During robust phasic activation of DA transmission occurring during burst firing (mimicked by stimulation of the substantia nigra and D1 receptor agonist treatment), D1–NMDA receptor interactions would facilitate nNOS activity and NO transmission. In the DA-depleted parkinsonian striatum or following D1 receptor blockade, we predict that loss of D1 tone would prohibit activation of nNOS and suppress NO signaling (Park and West, 2009). As discussed below in our summary of work performed in animal models of Parkinson’s disease (PD), currently there is evidence in support of and against this model.
Role of NOS Interneurons in the Regulation of Corticostriatal Transmission
Corticostriatal afferents target two functionally distinct groups of MSNs that form the “direct” (striatonigral MSNs) and the “indirect” (striatopallidal MSNs) pathways (Albin et al., 1989; Alexander and Crutcher, 1990; Parent, 1990; DeLong and Wichmann, 2007; Figure 1C). Corticostriatal projections also provide excitatory input to striatal interneurons (Kawaguchi, 1993) which are involved in the feed-forward regulation of MSNs by GABA (Tepper et al., 2004; Mallet et al., 2005) and NO (Sammut et al., 2007a, 2010; Ondracek et al., 2008). While GABA binds to receptors on the surface of the plasma membrane, newly synthesized NO diffuses past the plasma membrane into the dendrites of striatal MSNs, which contain high levels of NO receptors called soluble guanylyl cyclases (sGC; Figure 3; Ariano, 1983; Ding et al., 2004). In fact, sGC expression and activity are reportedly higher in the striatum than in any other brain region (Hofmann et al., 1977; Matsuoka et al., 1992). Once generated, NO has been reported to induce or modulate various forms of short and long-term corticostriatal synaptic plasticity (Calabresi et al., 1999, 2000; Doreulee et al., 2003; West and Grace, 2004; Ondracek et al., 2008; Sammut et al., 2010), and alter synchrony within neuronal networks (O’Donnell and Grace, 1997; Sammut et al., 2007a; Chepkova et al., 2009). Most of these studies indicate that this NO signal is derived from nNOS localized to striatal interneurons. However, evidence that LTP of corticostriatal transmission is blocked in slices following administration of a non-selective NOS inhibitor and in mutant mice lacking the endothelial NOS gene (Doreulee et al., 2003) indicates that NO derived from the vasculature is also capable of modulating corticostriatal transmission.
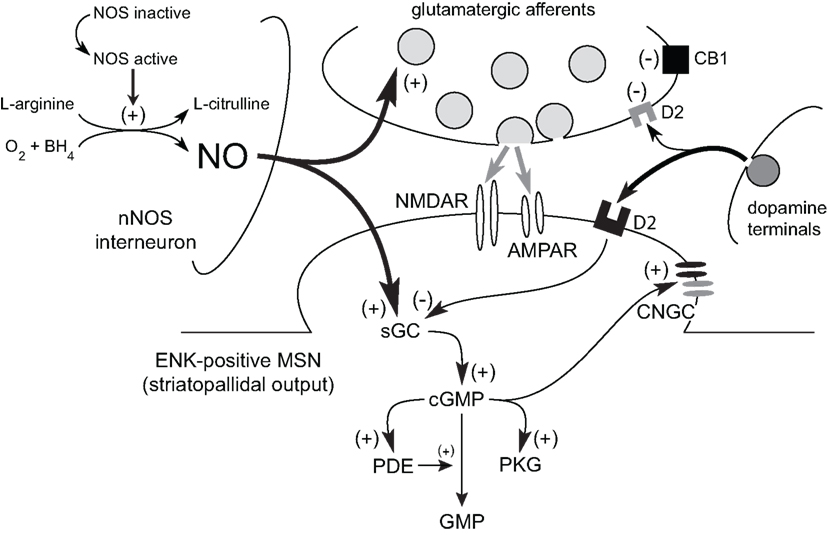
Figure 3. Model of the role of NO signaling in the short-term facilitation of corticostriatal transmission. Tonic NO signaling increases glutamatergic transmission across corticostriatal synapses via a sGC–cGMP-dependent mechanism. Similarly, phasic NO–sGC–cGMP signaling also increases corticostriatal synaptic transmission and the responsiveness of MSNs to this input. Transient increases in intracellular cGMP levels can affect MSN activity via activation of PKG and downstream targets (e.g., cyclic nucleotide gated channels; CNGC). Numerous phosphodiesterases (PDEs), which metabolize cyclic nucleotides, limit the effects of cGMP on MSN function. In striatopallidal output neurons (ENK-positive MSN), the facilitatory action of cGMP may be directly opposed via D2 receptor activation.
Our studies have shown that in both chloral hydrate and urethane anesthetized rats, tonic, and phasic NO–sGC–cGMP signaling acts to promote short-term excitatory influences on corticostriatal synaptic activity recorded in identified MSNs (Figure 3; West and Grace, 2004; Ondracek et al., 2008; Sammut et al., 2010). Our initial studies examined the impact of tonic NO signaling on short-term plasticity induced across corticostriatal synapses during paired-pulse stimulation of the frontal cortex (West and Grace, 2004). In these studies, MSNs were monitored using in vivo intracellular recordings during intrastriatal infusion of either vehicle or the NO scavenger CPT-IO. Interestingly, MSNs recorded in the presence of the NO scavenger were less responsive to the paired-pulse stimulation protocol and exhibited lower levels of synaptic facilitation during stimulation of corticostriatal pathways (West and Grace, 2004). More recently, we have examined the impact of high frequency train stimulation of the frontal cortex on evoked spike activity in striatal MSNs (Ondracek et al., 2008; Sammut et al., 2010). Importantly, the stimulation protocol used in our studies (train duration = 1 s, pulse frequency = 30 Hz, inter-train interval = 2 s) was designed to approximate the natural burst firing (spikes per burst, intra-burst frequency, and bursts per second) and up and down state activity of corticostriatal pyramidal neurons recorded in anesthetized rats (Cowan & Wilson, 1994). As we have discussed above, we have found that this protocol consistently produces an intensity-dependent and transient increase in striatal NO efflux (Sammut et al., 2007a; Ondracek et al., 2008; Park and West, 2009). Inhibition of this evoked NO efflux was shown to eliminate excitatory responses to stimulation and increase the short-term depression (STD) of cortically evoked spike activity (Ondracek et al., 2008).
Our laboratory has also examined the impact of phasic NO signaling on the spontaneous generation of local field potentials recorded in the intact rat striatum (Sammut et al., 2007a). These studies demonstrated that systemic administration of a non-specific NOS and sGC inhibitor (methylene blue) simultaneously decreased: (1) NO efflux evoked via cortical stimulation, and (2) the peak oscillation frequency (observed within the delta band) of local striatal field potential oscillations. These observations are consistent with studies using local application of NO–sGC inhibitors which were found to decrease the amplitude of spontaneous glutamate-driven up states (West and Grace, 2004). Additionally, stimulation of corticostriatal pathways facilitates electrotonic coupling between MSNs in rat striatal slices in a manner which is blocked by NOS inhibitors and mimicked by bath application of an NO generator (O’Donnell and Grace, 1997). NO signaling may therefore induce a functional coupling between MSNs and act to synchronize the oscillatory activity of related neuronal ensembles. When examined in vivo, disruption of nNOS activity increases the magnitude of D2 receptor-mediated STD of cortically evoked spike activity induced during phasic stimulation of frontal cortical afferents (Ondracek et al., 2008). Thus, in the intact striatum, corticostriatal transmission may be preferentially detected and amplified by nNOS interneurons in a feed-forward manner which may facilitate the synchronization of local network activity with glutamate-driven events. Interruption of NO neuromodulation, therefore, is likely to disrupt the integration of corticostriatal transmission, short-term plasticity, and functional coupling of MSNs in striatal networks (West and Grace, 2004; Sammut et al., 2007b; Ondracek et al., 2008). Consistent with this, it is clear that pharmacological or genetic downregulation of striatal NOS activity has profound effects on striatal output as measured in electrophysiological (West and Grace, 2000; West et al., 2002) and behavioral studies (Del Bel et al., 2005).
Interestingly, studies performed in brain slice preparations from both rats and mice have frequently reported inhibitory effects (i.e., LTD) of NO–sGC–cGMP signaling on excitatory corticostriatal transmission (reviewed in Calabresi et al., 2007). A parsimonious explanation for this apparent flip flop of the impact of NO on corticostriatal plasticity observed between in vivo and in vitro preparations is that corticostriatal pathway stimulation can be processed differently in the intact versus reduced striatum, and that in both preparations, NO may promote this differential processing. Indeed, similar mechanisms are implicated in studies using in vivo and in vitro preparations (e.g., sGC and PDEs play a key role in NO-mediated effects in all of these studies). Furthermore, the former tenet is supported by studies showing that stimulation protocols known to produce LTD of corticostriatal neurotransmission in vitro, produce LTP in vivo (Charpier and Deniau, 1997). A similar switch from LTD to LTP is observed in vitro following removal of magnesium from the bath perfusate (Calabresi et al., 1992). Thus, it is likely that with most common protocols cortical stimulation delivered in vivo results in greater activation of glutamatergic drive onto postsynaptic AMPA receptors and more effective removal of the voltage-dependent magnesium block of NMDA receptors, leading to a state that generally favors LTP. In contrast, similar stimulation of corticostriatal signaling in vitro favors LTD in the absence of the removal of magnesium block of NMDA receptors. In support of this, most studies show that LTD-induction in the mature striatum is not NMDA receptor dependent, whereas LTP requires activation of these channels (Reviewed in Surmeier et al., 2009). Like LTP, stimulation of striatal NOS activity also requires NMDA receptor activation in both in vivo and in vitro preparations (Nishi et al., 2005; Sammut et al., 2007a; Park and West, 2009). Given this, it is more readily understandable how corticostriatal pathway activation could lead to NO-dependent facilitation of synaptic efficacy (i.e., an LTP-like phenomenon). However, studies by Calabresi et al. (2007) have produced compelling evidence that facilitation of signaling at any number of key sites in the NO–sGC–cGMP–PKG cascade mediates LTD and occludes further LTD induced via corticostriatal stimulation. Because these studies stimulated cortical areas close to the recording electrode or white matter between cortex and striatum (Calabresi et al., 1999), it is possible that NMDA receptor stimulation was not required for NOS activation in this preparation as these interneurons may have been activated by direct current spread within striatum. In any event, the information available at this time suggests that, in addition to promoting short-term increases in excitatory synaptic transmission, NO signaling may act to facilitate and stabilize the dominant state of long-term synaptic plasticity occurring across corticostriatal synapses (i.e., primarily potentiation when postsynaptic NMDA receptors are activated, or depression in the absence of this activation). Because NO is a potent vasodilator, it may also function to couple changes in synaptic plasticity and blood flow in striatal microcircuits. Future studies using new genetic and optical approaches will have to determine the role of NO in LTP and LTD of synaptic transmission (i.e., bidirectional plasticity) at both excitatory and inhibitory synapses onto identified striatonigral and striatopallidal MSNs.
Striatal Pathophysiology in Parkinson’s Disease: Involvement of No–sGC–cGMP Signaling Pathways
Parkinson’s disease is associated with a preferential degeneration of the nigrostriatal DA pathway (Hornykiewicz, 1975). The loss of DA modulation triggers a complex series of neurochemical, anatomical, and electrophysiological alterations that lead to persistent changes in striatal neurons and their signaling pathways. For instance, striatonigral MSNs develop D1 receptor supersensitivity and reduced expression of D1 receptors (Gerfen et al., 1990). Elevations in D2 receptor protein and mRNA are also observed together with increases in enkephalin expression in striatopallidal MSNs (Gerfen et al., 1990). A substantial population of corticostriatal terminals also expresses D2 receptors (Wang and Pickel, 2002) which become hypersensitive in the absence of DA innervation (Calabresi et al., 1993; Bamford et al., 2004; Picconi et al., 2004). The loss of DA tone on these D2 heteroreceptors is likely to result in enhanced glutamatergic transmission and altered NMDA receptor function (Meshul et al., 1999; Nash et al., 1999; Betarbet et al., 2004). Alterations in dendritic spine morphology and complexity have also been described in both DA-depleted rats and patients with PD (Ingham et al., 1989; Stephens et al., 2005) which may occur preferentially in striatopallidal MSNs (Day et al., 2006).
Most pathophysiological models and metabolic studies of PD predict that the net effect of these alterations is an imbalance in striatal output in which the indirect pathway becomes functionally hyperactive and the direct pathway is hypoactive (Marsden, 1982; Albin et al., 1989; Alexander et al., 1990; Hirsch et al., 2000; see Figure 1C). Recent studies using optogenetic control of striatonigral and striatopallidal MSNs have provided substantial evidence supporting the validity of this pioneering basal ganglia model (Kravitz et al., 2010). These studies by Kravitz and colleagues showed that bilateral activation of striatopallidal MSNs elicits a parkinsonian state characterized by freezing, bradykinesia, and decreased locomotion. Stimulation of striatonigral MSNs reduced freezing and facilitated locomotion. Additionally, stimulation of striatonigral MSNs reversed motor deficits observed in parkinsonian mice (Kravitz et al., 2010).
The above model has also served as the framework for recent studies aimed at understanding how changes in striatal DA transmission impact the temporal dynamics and plasticity of cortico-basal ganglia transmission. For instance, recent studies have suggested that striatopallidal neurons in DA-depleted animals become more responsive to corticostriatal inputs and as a result, exhibit bursts of spike activity which correlates with cortical oscillations (Tseng et al., 2001; Mallet et al., 2006; Walters et al., 2007). Thus, loss of D2 receptor-mediated inhibition of striatopallidal neurons and their corticostriatal inputs results in the unfiltered spreading of cortical rhythms to the GP and other components of the basal ganglia and some of the modifications in neuron activity that may underlie the pathophysiology of PD (Murer et al., 2002). In addition to increased spike activity (Mallet et al., 2006), striatopallidal neurons recorded in DA-depleted mice also exhibit a selective loss of endocannabinoid-dependent LTD (Kreitzer and Malenka, 2007). Further studies by Surmeier et al. (2009) have shown that following DA depletion, the pairing of presynaptic and postsynaptic activity, in any order, induced LTD in D1 receptor-expressing MSNs and LTP in D2 receptor-expressing MSNs in an exclusive manner which prohibited bidirectional plasticity (Shen et al., 2008). These observations indicate that, following striatal DA depletion, activity-dependent changes in the strength of corticostriatal synaptic transmission occur in parallel with alterations in intrinsic excitability (Surmeier et al., 2009).
While the role of NO–sGC–cGMP signaling in the above pathological changes induced in the parkinsonian striatum remains to be fully characterized, it is clear that disruption of striatal NO–sGC–cGMP signaling cascades results in profound changes in behavioral, electrophysiological, and molecular responses to pharmacological manipulations of DA and glutamate transmission (Morris, 1995; Greengard, 2001; West et al., 2002; Del Bel et al., 2005; Ondracek et al., 2008; Threlfell et al., 2009; Sammut et al., 2010; West et al., 2009). For example, studies by Del Bel et al. (2005) have shown that striatal nNOS interneurons play a critical role in the generation of motor behavior. Systemic and intrastriatal exposure to NOS and sGC inhibitors has been shown to depress basal locomotion and induce catalepsy (Stewart et al., 1994; Del Bel et al., 2004; Echeverry et al., 2007). Pharmacological disruption of NO function also potentiates catalepsy induced via D2-class receptor antagonists (Del Bel and Guimaraes, 2000; Cavas and Navarro, 2002). Furthermore, motor activation stimulated by substance P (Mancuso et al., 1994), NMDA receptor antagonists (Deutsch et al., 1996), and DA receptor agonists (Starr and Starr, 1995) is suppressed following systemic administration of NOS inhibitors.
The above behavioral studies demonstrate that in animals with an intact DA system, striatal NO–sGC–cGMP transmission is likely to play an important role in facilitating locomotor activity. It is less clear how this role for NO–sGC–cGMP signaling may change in the parkinsonian striatum. Various measures of striatal NOS activity have indicated that NO signaling may be disrupted in patients with PD (Bockelmann et al., 1994; Eve et al., 1998) and DA-depleted rats (De Vente et al., 2000; Sahach et al., 2000; Barthwal et al., 2001; Sancesario et al., 2004). With regard to NOS activity measured in DA-depleted rodents, however, conflicting outcomes have been reported between the above studies performed in rats and studies of mouse models (see Chalimoniuk and Langfort, 2007; Chalimoniuk et al., 2004). Moreover, conflicting results have been reported in studies measuring cGMP levels in striatal tissue from DA-depleted rodents (De Vente et al., 2000; Chalimoniuk et al., 2004; Sancesario et al., 2004; Chalimoniuk and Langfort, 2007; Giorgi et al., 2008). Interestingly, PDE mRNA, protein, and activity are elevated in DA-depleted rats (Sancesario et al., 2004; Giorgi et al., 2008), indicating that cyclic nucleotide metabolism is elevated in PD. While speculative, this may be a compensatory homeostatic change induced in MSNs in response to overactivity resulting from striatal DA depletion (i.e., an attempt by the MSNs to reverse cyclic nucleotide over-production). In support of this, a subpopulation of MSNs (40%) has been reported to be hyper-responsive to NO generators (Galati et al., 2008). Therefore, upregulation of PDE activity would be expected to compensate for this abnormal responsivity of these MSNs to NO. A parallel down-regulation of nNOS activity and NO signaling would also facilitate the normalization of cyclic nucleotide levels following DA depletion. As indicated above, there is strong evidence for this in both DA-depleted rats (De Vente et al., 2000; Sahach et al., 2000; Barthwal et al., 2001; Sancesario et al., 2004) and post-mortem tissue from patients with PD (Bockelmann et al., 1994; Eve et al., 1998). However, studies examining how parallel measures of nNOS, sGC, cGMP, PKG, and various PDEs change across time following DA depletion are needed to clarify the complex pathophysiological and homeostatic changes in this signaling pathway in PD.
Given the above findings, it is likely that striatal DA denervation results in transient and dynamic alterations in the synthesis (NOS and sGC dependent) and degradation (PDE dependent) of striatal cyclic nucleotides. These complex changes are likely to be complicated further by L-DOPA treatment. Indeed, in patients with PD, serum levels of cGMP are reportedly increased following L-DOPA therapy (Chalimoniuk and Stepien, 2004). However, studies in DA-depleted rats have shown that L-DOPA-induced dyskinesias are associated with decreased striatal cyclic nucleotide levels (Giorgi et al., 2008; Picconi et al., 2011). Moreover, the non-selective PDE inhibitor zaprinast was shown to reverse decreases in striatal cyclic nucleotide levels and abnormal involuntary movements induced by L-DOPA administration (Giorgi et al., 2008; Picconi et al., 2011). Calabresi and colleagues also showed that PDE inhibition can rescue abnormal synaptic plasticity observed in dyskinetic rats (Picconi et al., 2011). On the other hand, studies aimed at decreasing NO signaling (and presumably cGMP levels) have shown that co-administration of NOS inhibitors with L-DOPA attenuates L-DOPA-induced dyskinesias (Padovan-Neto et al., 2009). NOS inhibition also improved the motor performance of the same animals on a rotorod test (Padovan-Neto et al., 2009). Taken together, the above studies suggest that under some circumstances, drugs with opposite pharmacological profiles (PDE inhibitors increase cGMP, nNOS inhibitors decrease cGMP) may both be beneficial for reversing L-DOPA-induced dyskinesias. Further studies determining the time course of L-DOPA-induced changes in cyclic nucleotide levels in the absence and presence of these inhibitors should open new avenues which will be essential for understanding and treating PD and side effects associated with L-DOPA therapy.
Conclusion
Studies reported to date indicate that it will be important to clarify how NO–sGC–cGMP signaling is dysregulated in hypo- and hyper-dopaminergic states and how this can be normalized to restore function within striatal output pathways. Taken together, the above studies indicate that cGMP synthesis and catabolism, as well as the temporal and spatial patterning of NO–sGC–cGMP signaling may be perturbed in MSNs in the parkinsonian striatum. Moreover, these signaling abnormalities are likely to be a reflection of functional disturbances in DA and glutamate interactions and iatrogenic effects of chronic L-DOPA treatment. Definitive characterization of the impact of NO–sGC–cGMP signaling and downstream targets in animal models of parkinsonism and L-DOPA-induced dyskinesia should provide key information on how cyclic nucleotide signaling cascades can be modulated as an approach for treating motor symptoms in PD and other neurological disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by United States Public Health grants NS047452 and NS047452-S1 to Anthony R. West, and DA004093 and Rosalind Franklin University Start-up funds to Kuei Y. Tseng.
References
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375.
Alderton, W. K., Cooper, C. E., and Knowles, R. G. (2001). Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357, 593–615.
Alexander, G. E., and Crutcher, M. D. (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13, 266–271.
Alexander, G. E., Crutcher, M. D., and DeLong, M. R. (1990). Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog. Brain Res. 85, 119–146.
Altar, C. A., Boyar, W. C., and Kim, H. S. (1990). Discriminatory roles for D1 and D2 dopamine receptor subtypes in the in vivo control of neostriatal cyclic GMP. Eur. J. Pharmacol. 181, 17–21.
Ariano, M. A. (1983). Distribution of components of the guanosine 3′,5′-phosphate system in rat caudate-putamen. Neuroscience 10, 707–723.
Bamford, N. S., Robinson, S., Palmiter, R. D., Joyce, J. A., Moore, C., and Meshul, C. K. (2004). Dopamine modulates release from corticostriatal terminals. J. Neurosci. 24, 9541–9552.
Barthwal, M. K., Srivastava, N., and Dikshit, M. (2001). Role of nitric oxide in a progressive neurodegeneration model of Parkinson’s disease in the rat. Redox Rep. 6, 297–302.
Betarbet, R., Poisik, O., Sherer, T. B., and Greenamyre, J. T. (2004). Differential expression and ser897 phosphorylation of striatal N-methyl-D-aspartate receptor subunit NR1 in animal models of Parkinson’s disease. Exp. Neurol. 187, 76–85.
Bockelmann, R., Wolf, G., Ransmayr, G., and Riederer, P. (1994). NADPH-diaphorase/nitric oxide synthase containing neurons in normal and Parkinson’s disease putamen. J. Neural Transm. 7, 115–121.
Bredt, D. S. (2003). Nitric oxide signaling in brain: potentiating the gain with YC-1. Mol. Pharmacol. 63, 1206–1208.
Bredt, D. S., Hwang, P. M., and Snyder, S. H. (1990). Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347, 768–770.
Calabresi, P., Centonze, D., Gubellini, P., Marfia, G. A., Pisani, A., Sancesario, G., and Bernardi, G. (2000). Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog. Neurobiol. 61, 231–265.
Calabresi, P., Gubellini, P., Centonze, D., Sancesario, G., Morello, M., Giorgi, M., Pisani, A., and Bernardi, G. (1999). A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J. Neurosci. 19, 2489–2499.
Calabresi, P., Mercuri, N. B., Sancesario, G., and Bernardi, G. (1993). Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson’s disease. Brain 116, 433–452.
Calabresi, P., Picconi, B., Tozzi, A., and Di Filippo, M. (2007). Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219.
Calabresi, P., Pisani, A., Mercuri, N. B., and Bernardi, G. (1992). Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur. J. Neurosci. 4, 929–935.
Cavas, M., and Navarro, J. F. (2002). Coadministration of L-NOARG and tiapride: effects on catalepsy in male mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 26, 69–73.
Cepeda, C., and Levine, M. S. (1998). Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev. Neurosci. 20, 1–18.
Cepeda, C., and Levine, M. S. (2006). Where do you think you are going? The NMDA-D1 receptor trap. Sci. STKE 2006, pe20.
Chalimoniuk, M., and Langfort, J. (2007). The effect of subchronic, intermittent L-DOPA treatment on neuronal nitric oxide synthase and soluble guanylyl cyclase expression and activity in the striatum and midbrain of normal and MPTP-treated mice. Neurochem. Int. 50, 821–833.
Chalimoniuk, M., and Stepien, A. (2004). Influence of the therapy with pergolide mesylate plus L-DOPA and with L-DOPA alone on serum cGMP level in PD patients. Pol. J. Pharmacol. 56, 647–650.
Chalimoniuk, M., Langfort, J., Lukacova, N., and Marsala, J. (2004). Upregulation of guanylyl cyclase expression and activity in striatum of MPTP-induced parkinsonism in mice. Biochem. Biophys. Res. Commun. 324, 118–126.
Charpier, S., and Deniau, J. M. (1997). In vivo activity-dependent plasticity at cortico-striatal connections: evidence for physiological long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 94, 7036–7040.
Chepkova, A. N., Fleischer, W., Kazmierczak, T., Doreulee, N., Haas, H. L., and Sergeeva, O. A. (2009). Developmental alterations of DHPG-induced long-term depression of corticostriatal synaptic transmission: switch from NMDA receptor-dependent towards CB1 receptor- dependent plasticity. Pflugers Arch. 459, 131–141.
Cowan, R. L., and Wilson, C. J. (1994). Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J. Neurophysiol. 71, 17–32.
Dawson, T. M., Bredt, D. S., Fotuhi, M., Hwang, P. M., and Snyder, S. H. (1991). Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 88, 7797–7801.
Day, M., Wang, Z., Ding, J., An, Z., Ingham, C. A., Shering, A. F., Wokosin, D., Ilijic, E., Sun, Z., Sampson, A. R., Mugnaini, E., Deutch, A. Y., Sesack, S. R., Arbuthnott, G. W., and Surmeier, D. J. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259.
De Vente, J., Markerink-van Ittersum, M., Van Abeelen, J., Emson, P. C., Axer, H., and Steinbusch, H. W. M. (2000). NO-mediated cGMP synthesis in cholinergic neurons in the rat forebrain: effects of lesioning dopaminergic or serotonergic pathways on nNOS and cGMP synthesis. Eur. J. Neurosci. 12, 507–519.
Del Bel, E. A., da Silva, C. A., Guimaraes, F. S., and Bermudez-Echeverry, M. (2004). Catalepsy induced by intra-striatal administration of nitric oxide synthase inhibitors in rats. Eur. J. Pharmacol. 485, 175–181.
Del Bel, E. A., and Guimaraes, F. S. (2000). Sub-chronic inhibition of nitric-oxide synthesis modifies haloperidol-induced catalepsy and the number of NADPH-diaphorase neurons in mice. Psychopharmacology (Berl.) 147, 356–361.
Del Bel, E. A., Guimaraes, F. S., Bermudez-Echeverry, M., Gomes, M. Z., Schiaveto-de-souza, A., Padovan-Neto, F. E., Tumas, V., Barion-Cavalcanti, A. P., Lazzarini, M., Nucci-da-Silva, L. P., and de Paula-Souza, D. (2005). Role of nitric oxide on motor behavior. Cell. Mol. Neurobiol. 25, 371–392.
DeLong, M. R., and Wichmann, T. (2007). Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 64, 20–24.
Deutsch, S. I., Rosse, R. B., Paul, S. M., Tomasino, V., Koetzner, L., Morn, C. B., and Mastropaolo, J. (1996). 7-Nitroindazole and methylene blue, inhibitors of neuronal nitric oxide synthase and NO-stimulated guanylate cyclase, block MK-801-elicited behaviors in mice. Neuropsychopharmacology 15, 37–43.
Di Stefano, A., Sozio, P., Cacciatore, I., Cocco, A., Giorgioni, G., Costa, B., Montali, M., Lucacchini, A., Martini, C., Spoto, G., Di Pietrantonio, F., Di Matteo, E., and Pinnen, F. (2005). Preparation and pharmacological characterization of trans-2-amino-5(6)-fluoro-6(5)-hydroxy-1-phenyl-2,3- dihydro-1H-indenes as D2-like dopamine receptor agonists. J. Med. Chem. 48, 2646–2654.
Ding, J. D., Burette, A., Nedvetsky, P. I., Schmidt, H. H., and Weinberg, R. J. (2004). Distribution of soluble guanylyl cyclase in the rat brain. J. Comp. Neurol. 472, 437–448.
Doreulee, N., Sergeeva, O. A., Yanovsky, Y., Chepkova, A. N., Selbach, O., Godecke, A., Schrader, J., and Haas, H. L. (2003). Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res. 964, 159–163.
Echeverry, M. B., Salgado, M. L., Ferreira, F. R., da-Silva, C. A., and Del Bel, E. A. (2007). Intracerebroventricular administration of nitric oxide-sensitive guanylyl cyclase inhibitors induces catalepsy in mice. Psychopharmacology (Berl.) 194, 271–278.
Eve, D. J., Nisbet, A. P., Kingsbury, A. E., Hewson, E. L., Daniel, S. E., Lees, A. J., Marsden, C. D., and Foster, O. J. (1998). Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Mol. Brain Res. 63, 62–71.
Fujiyama, F., and Masuko, S. (1996). Association of dopaminergic terminals and neurons releasing nitric oxide in the rat striatum: an electron microscopic study using NADPH-diaphorase histochemistry and tyrosine hydroxylase immunohistochemistry. Brain Res. Bull. 40, 121–127.
Galati, S., D’Angelo, V., Scarnati, E., Stanzione, P., Martorana, A., Procopio, T., Sancesario, G., and Stefani, A. (2008). In vivo electrophysiology of dopamine-denervated striatum: focus on the nitric oxide/cGMP signaling pathway. Synapse 62, 409–420.
Garthwaite, J. (2008). Concepts of neural nitric oxide-mediated transmission. Eur. J. Neurosci. 27, 2783–2802.
Gerfen, C. R., Engber, T. M., Mathan, L. C., Susel, Z., Chase, T. N., Monsma, F. J., and Sibley, D. R. (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432.
Giorgi, M., D’Angelo, V., Esposito, Z., Nuccetelli, V., Sorge, R., Martorana, A., Stefani, A., Bernardi, G., and Sancesario, G. (2008). Lowered cAMP and cGMP signalling in the brain during levodopa- induced dyskinesias in hemiparkinsonian rats: new aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 28, 941–950.
Gracy, K. N., and Pickel, V. M. (1997). Ultrastructural localization and comparative distribution of nitric oxide synthase and N-methyl-D-aspartate receptors in the shell of the rat nucleus accumbens. Brain Res. 747, 259–272.
Hidaka, S., and Totterdell, S. (2001). Ultrastructural features of the nitric oxide synthase-containing interneurons in the nucleus accumbens and their relationship with tyrosine hydroxylase-containing terminals. J. Comp. Neurol. 431, 139–154.
Hirsch, E. C., Perier, C., Orieux, G., Francois, C., Feger, J., Yelnik, J., Vila, M., Levy, R., Tolosa, E. S., Marin, C., Trinidad Herrero, M., Obeso, J. A., and Agid, Y. (2000). Metabolic effects of nigrostriatal denervation in basal ganglia. Trends Neurosci. 23, S78–S85.
Hofmann, M., Spano, P. F., Trabucchi, M., and Kumakura, K. (1977). Guanylate cyclase activity in various rat brain areas. J. Neurochem. 29, 395–396.
Hope, B. T., Michael, G. J., Knigge, K. M., and Vincent, S. R. (1991). Neuronal NADPH diaphorase is a nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 88, 2811–2814.
Hoque, K. E., Indorkar, R. P., Sammut, S., and West, A. R. (2010). Impact of dopamine-glutamate interactions on striatal neuronal nitric oxide synthase activity. Psychopharmacology (Berl.) 207, 571–581.
Hornykiewicz, O. (1975). Brain monoamines and parkinsonism. Natl. Inst. Drug Abuse Res. Monogr. Ser. 00, 13–21.
Ingham, C. A., Hood, S. H., and Arbuthnott, G. W. (1989). Spine density on neostriatal neurons changes with 6-hydroxydopamine lesions and with age. Brain Res. 503, 334–338.
Kawaguchi, Y. (1993). Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J. Neurosci. 13, 4908–4923.
Kravitz, A. V., Freeze, B. S., Parker, P. R. L., Kay, K., Thwin, M. T., Deisseroth, K., and Kreitzer, A. C. (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kuo, H., Grant, S., Muth, N., Hengemihle, J., and Ingram, D. K. (1994). The correlation between neuron counts and optical density of NADPH-diaphorase histochemistry in the rat striatum: a quantitative study. Brain Res. 660, 57–65.
Mallet, N., Ballion, B., Le Moine, C., and Gonon, F. (2006). Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J. Neurosci. 26, 3875–3884.
Mallet, N., Le Moine, C., Charpier, S., and Gonon, F. (2005). Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J. Neurosci. 25, 3857–3869.
Mancuso, F., Calignano, A., and Sorrentino, L. (1994). Endogenous nitric oxide modulates behavioural effects elicited by substance P in rat. Eur. J. Pharmacol. 271, 329–333.
Marsden, C. D. (1982). The mysterious motor function of the basal ganglia: the Robert Wartenberg Lecture. Neurology 32, 514–539.
Matsuoka, I., Giuili, G., Poyard, M., Stengel, D., Parma, J., Guellaen, G., and Hanoune, J. (1992). Localization of adenylyl and guanylyl cyclase in rat brain by in situ hybridization: comparison with calmodulin mRNA distribution. J. Neurosci. 12, 3350–3360.
Meshul, C. K., Emre, N., Nakamura, C. M., Allen, C., Donohue, M. K., and Buckman, J. F. (1999). Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience 88, 1–16.
Morello, M., Reiner, A., Sancesario, G., Karle, E. J., and Bernardi, G. (1997). Ultrastructural study of nitric oxide synthase-containing striatal neurons and their relationship with parvalbumin-containing neurons in rats. Brain Res. 776, 30–39.
Morris, B. J. (1995). Stimulation of immediate-early gene expression in striatal neurones by nitric oxide. J. Biol. Chem. 270, 24740–24744.
Morris, B. J., Simpson, C. S., Mundell, S., Maceachern, K., Johnston, H. M., and Nolan, A. M. (1997). Dynamic changes in NADPH-diaphorase staining reflect activity of nitric oxide synthase: evidence for a dopaminergic regulation of striatal nitric oxide release. Neuropharmacology 36, 1589–1599.
Murer, M. G., Tseng, K. Y., Kasanetz, F., Belluscio, M., and Riquelme, L. A. (2002). Brain oscillations, medium spiny neurons, and dopamine. Cell. Mol. Neurobiol. 22, 611–632.
Nash, J. E., Hill, M. P., and Brotchie, J. M. (1999). Antiparkinsonian actions of blockade of NR2B-containing NMDA receptors in the reserpine-treated rat. Exp. Neurol. 155, 42–48.
Nishi, A., Watanabe, Y., Higashi, H., Tanaka, M., Nairn, A. C., and Greengard, P. (2005). Glutamate regulation of DARPP-32 phosphorylation in neostriatal neurons involves activation of multiple signaling cascades. Proc. Natl. Acad. Sci. U.S.A. 102, 1199–1204.
O’Donnell, P., and Grace, A. A. (1997). Cortical afferents modulate striatal gap junction permeability via nitric oxide. Neuroscience 76, 1–5.
Ondracek, J. M., Dec, A., Hoque, K. E., Lim, S. A., Rasouli, G., Indorkar, R. P., Linardakis, J., Klika, B., Mukherji, S. J., Burnazi, M., Threlfell, S., Sammut, S., and West, A. R. (2008). Feed-forward excitation of striatal neuron activity by frontal cortical activation of nitric oxide signaling in vivo. Eur. J. Neurosci. 27, 1739–1754.
Padovan-Neto, F. E., Echeverry, M. B., Tumas, V., and Del-Bel, E. A. (2009). Nitric oxide synthase inhibition attenuates L-DOPA-induced dyskinesias in a rodent model of Parkinson’s disease. Neuroscience 159, 927–935.
Palmer, R. M., Ferrige, A. G., and Moncada, S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327, 524–526.
Park, D. J., and West, A. R. (2009). Regulation of striatal nitric oxide synthesis by local dopamine and glutamate interactions. J. Neurochem. 111, 1457–1465.
Picconi, B., Bagetta, V., Ghiglieri, V., Paille’, V., Di Filippo, M., Pendolino, M., Tozzi, A., Giampa’, C., Fusco, F. R., Sgobio, C., and Calabresi, P. (2011). Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain 134; 375–387.
Picconi, B., Centonze, D., Rossi, S., Bernardi, G., and Calabresi, P. (2004). Therapeutic doses of L-dopa reverse hypersensitivity of corticostriatal D2-dopamine receptors and glutamatergic overactivity in experimental parkinsonism. Brain 127, 1661–1669.
Sahach, V. F., Baziliuk, O. V., Oleshko, M. M., Kotsiuruba, O. V., Bukhanevych, O. M., and Appenzeller, O. (2000). The nitric oxide system in a chronic deficiency of mesostriatal dopamine: the action of nitroglycerin. Fiziol. Zh. 46, 55–63.
Sammut, S., Dec, A., Mitchell, D., Linardakis, J., Ortiguela, M., and West, A. R. (2006). Phasic dopaminergic transmission increases NO efflux in the rat dorsal striatum via a neuronal NOS and a dopamine D(1/5) receptor-dependent mechanism. Neuropsychopharmacology 31, 493–505.
Sammut, S., Park, D. J., and West, A. R. (2007a). Frontal cortical afferents facilitate striatal nitric oxide transmission in vivo via a NMDA receptor and neuronal NOS-dependent mechanism. J. Neurochem. 103, 1145–1156.
Sammut, S., Bray, K. E., and West, A. R. (2007b). Dopamine D2 receptor-dependent modulation of striatal NO synthase activity. Psychopharmacology (Berl.) 191, 793–803.
Sammut, S., Threlfell, S., and West, A. R. (2010). Nitric oxide-soluble guanylyl cyclase signaling regulates corticostriatal transmission and short-term synaptic plasticity of striatal projection neurons recorded in vivo. Neuropharmacology 58, 624–631.
Sancesario, G., Giorgi, M., D’Angelo, V., Modica, A., Martorana, A., Morello, M., Bengtson, C. P., and Bernardi, G. (2004). Down-regulation of nitrergic transmission in the rat striatum after chronic nigrostriatal deafferentation. Eur. J. Neurosci. 20, 989–1000.
Sancesario, G., Morello, M., Reiner, A., Giacomini, P., Massa, R., Schoen, S., and Bernardi, G. (2000). Nitrergic neurons make synapses on dual-input dendritic spines of neurons in the cerebral cortex and the striatum of the rat: implication for a postsynaptic action of nitric oxide. Neuroscience 99, 627–642.
Shen, W., Flajolet, M., Greengard, P., and Surmeier, D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851.
Siuciak, J. A., McCarthy, S. A., Chapin, D. S., Fujiwara, R. A., James, L. C., Williams, R. D., Stock, J. L., McNeish, J. D., Strick, C. A., Menniti, F. S., and Schmidt, C. J. (2006). Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology 51, 374–385.
Starr, M. S., and Starr, B. S. (1995). Do NMDA receptor-mediated changes in motor behaviour involve nitric oxide? Eur. J. Pharmacol. 272, 211–217.
Stephens, B., Mueller, A. J., Shering, A. F., Hood, S. H., Taggart, P., Arbuthnott, G. W., Bell, J. E., Kilford, L., Kingsbury, A. E., Daniel, S. E., and Ingham, C. A. (2005). Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience 132, 741–754.
Stewart, J., Deschamps, S. E., and Amir, S. (1994). Inhibition of nitric oxide synthase does not block the development of sensitization to the behavioral activating effects of amphetamine. Brain Res. 641, 141–144.
Surmeier, D. J., Plotkin, J., and Shen, W. (2009). Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr. Opin. Neurobiol. 19, 621–628.
Tepper, J. M., Koos, T., and Wilson, C. J. (2004). GABAergic microcircuits in the neostriatum. Trends Neurosci. 27, 662–669.
Threlfell, S., Sammut, S., Menniti, F. S., Schmidt, C. J., and West, A. R. (2009). Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to stimulation of frontal cortical afferents. J. Pharm. Exp. Ther. 328, 785–795.
Tseng, K. Y., Kasanetz, F., Kargieman, L., Riquelme, L. A., and Murer, M. G. (2001). Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J. Neurosci. 21, 6430–6439.
Vincent, S. R. (1994). Nitric oxide: a radical neurotransmitter in the central nervous system. Prog. Neurobiol. 42, 129–160.
Walters, J. R., Hu, D., Itoga, C. A., Parr-Brownlie, L. C., and Bergstrom, D. A. (2007). Phase relationships support a role for coordinated activity the indirect pathway in organizing slow oscillations in basal ganglia output after loss of dopamine. Neuroscience 144, 762–776.
Wang, H., and Pickel, V. M. (2002). Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J. Comp. Neurol. 442, 392–404.
West, A. R. (2010). “Nitric oxide signaling in the striatum,” in Handbook of Basal Ganglia Structure and Function, eds H. Steiner and K. Y. Tseng (New York, NY: Academic Press/Elsevier), 187–200.
West, A. R., Galloway, M. P., and Grace, A. A. (2002). Regulation of striatal dopamine neurotransmission by nitric oxide: effector pathways and signaling mechanisms. Synapse 44, 227–245.
West, A. R., and Grace, A. A. (2000). Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J. Neurophysiol. 83, 1796–1808.
West, A. R., and Grace, A. A. (2004). The nitric oxide-guanylyl cyclase signaling pathway modulates membrane activity States and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J. Neurosci. 24, 1924–1935.
Keywords: basal ganglia, striatum, dopamine, nitric oxide, Parkinson’s disease
Citation: West AR and Tseng KY (2011) Nitric oxide–soluble guanylyl cyclase–cyclic GMP signaling in the striatum: new targets for the treatment of Parkinson’s disease?. Front. Syst. Neurosci. 5:55. doi: 10.3389/fnsys.2011.00055
Received: 24 December 2010;
Paper pending published: 27 March 2011;
Accepted: 16 June 2011;
Published online: 30 June 2011.
Edited by:
Elizabeth Abercrombie, Rutgers-Newark: The State University of New Jersey, USAReviewed by:
Dalton J. Surmeier, Northwestern University, USAAlessandro Stefani, University of Rome, Italy
M. Gustavo Murer, Universidad de Buenos Aires, Argentina
Copyright: © 2011 West and Tseng. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Anthony R. West, Department of Neuroscience, Rosalind Franklin University of Medicine and Science, 3333 Green Bay Road, North Chicago, IL 60064, USA. e-mail: Anthony.West@rosalindfranklin.edu; Kuei Y. Tseng, Department of Cellular and Molecular Pharmacology, The Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA. e-mail: kuei-yuan.tseng@rosalindfranklin.edu