In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full-length (YAC128) transgenic mouse models of Huntington’s disease
- Center for Molecular and Behavioral Neuroscience, Rutgers, The State University of New Jersey, Newark, NJ, USA
Huntington’s disease (HD) is characterized by numerous alterations within the corticostriatal circuitry. The striatum is innervated by a dense array of dopaminergic (DA) terminals and these DA synapses are critical to the proper execution of motor functions. As motor disturbances are prevalent in HD we examined DA neurotransmission in the striatum in transgenic (tg) murine models of HD. We used in vivo microdialysis to compare extracellular concentrations of striatal DA in both a fragment (R6/2) model, which displays a rapid and severe phenotype, and a full-length (YAC128) model that expresses a more progressive phenotype. Extracellular striatal DA concentrations were significantly reduced in R6/2 mice and decreased concomitantly with age-dependent increasing motor impairments on the rotarod task (7, 9, and 11 weeks). In a sample of 11-week-old R6/2 mice, we also measured tissue concentrations of striatal DA and found that total levels of DA were significantly depleted. However, the loss of total DA content (<50%) was insufficient to account for the full extent of DA depletion in the extracellular fluid (ECF; ∼75%). We also observed a significant reduction in extracellular DA concentrations in the striatum of 7-month-old YAC128 mice. In a separate set of experiments, we applied D-amphetamine (AMPH; 10 μm) locally into the striatum to stimulate the release of intracellular DA into the ECF. The AMPH-induced increase in extracellular DA levels was significantly blunted in 9-week-old R6/2 mice. There also was a decrease in AMPH-stimulated DA efflux in 7-month-old YAC128 mice in comparison to WT controls, although the effect was milder. In the same cohort of 7-month-old YAC128 mice we observed a significant reduction in the total locomotor activity in response to systemic AMPH (2 mg/kg). Our data demonstrate that extracellular DA release is attenuated in both a fragment and full-length tg mouse model of HD and support the concept of DA involvement in aspects of the syndrome.
Introduction
Huntington’s disease (HD) is a genetically inherited neurodegenerative disorder that results in motor, cognitive, and psychiatric disturbances. One out of every 10,000 people is affected with the disorder and an even greater proportion remains at risk. HD is caused by a polyglutamine (CAG) trinucleotide repeat expansion in the IT-15 (HTT) gene, located on the short arm of chromosome 4 (The Huntington’s Disease Collaborative Research Group, 1993). The mutation is autosomal dominant and onset occurs between 30 and 50 years of age, although in a rare juvenile variant of the disease, symptoms can emerge as early as 5 years old. The severity of symptoms progressively worsens as a function of age and the illness is ultimately fatal (for review see Walker, 2007). No cure exists for HD, however, therapeutics that target the dopamine system have shown promise for managing the motor syndromes that are involved with the disorder (Bonelli and Wenning, 2006; Bonelli and Hofmann, 2007).
Since the discovery of the mutation in the HTT gene, several transgenic mouse models expressing the CAG repeat expansion have been developed which recapitulate characteristics of the disorder, including reductions in total brain volume, mutant huntingtin (mHTT) protein aggregation, transcriptional dysregulation, and neurotransmitter receptor alterations (Davies et al., 1997; Cha et al., 1998; Luthi-Carter et al., 2000; Stack et al., 2005). Critically, these pathological changes are accompanied by progressive motor and cognitive deficits (Menalled and Chesselet, 2002; Levine et al., 2004) that closely mimic the human condition. Transgenic mouse models of HD can be sorted into two broad categories depending on whether they possess a fragment (e.g., exon 1) of the HTT gene or the full-length composition. Mouse strains from fragment models express only a truncated mHTT protein fraction and in vitro studies have demonstrated that this abbreviated fragment may be more toxic than the full-length protein (Hackam et al., 1998; Lunkes and Mandel, 1998). As such, fragment models typically exhibit an accelerated phenotype in comparison to full-length models that includes motor deficits, altered gait, hypoactivity, and weight loss (Carter et al., 1999). While fragment models display an HD-like phenotype, the obvious weakness is that they lack the natural genomic and protein context of the polyglutamine expansion. This raises the potential for alterations in protein dynamics and therefore necessitates the use of full-length models to investigate any resulting changes in disease features.
R6 transgenic mice are the most widely used strain derived from the fragment models and were the first established transgenic line in HD research. The most extensively characterized variant of this transgenic strain is the R6/2 line, which express the first of 67 exons and carry a ∼160 CAG repeat expansion (Mangiarini et al., 1996). These mice develop an aggressive HD-like phenotype that advances rapidly until the occurrence of spontaneous morbidity by 14–16 weeks of age (Carter et al., 1999). In contrast to the R6 models, the YAC128 transgenic mouse model incorporates the entire HD gene, using a yeast artificial chromosome (YAC) vector system to express the full-length human HTT gene with a 128 CAG repeat expansion (Hodgson et al., 1999). The lifespan, as well as the progression of the HD-like phenotype in YAC128 mice is more prolonged than in R6/2 mice, with neuropathological and behavioral abnormalities manifesting at later ages (Slow et al., 2003; Van Raamsdonk et al., 2007).
Evidence implicates abnormal nigrostriatal dopaminergic neurotransmission in HD. In both human cases and animal models, early reductions in the striatal expression of dopamine receptors, attenuated receptor binding, and loss of the dopamine- and cAMP-regulated phosphoprotein (DARPP-32) have been reported (Joyce et al., 1988; Sedvall et al., 1994; Cha et al., 1999; Bibb et al., 2000; Miller and Bezprozvanny, 2010). Reduced dopamine concentrations in striatal tissue and the degeneration of nigrostriatal terminals emerge in late stages of the disorder in both human cases and transgenic models of HD (Kish et al., 1987; Reynolds et al., 1999; Suzuki et al., 2001; Petersén et al., 2002). Additionally, studies utilizing striatal slice preparations have demonstrated that electrically evoked dopamine efflux is reduced in both R6/1 and R6/2 mice (Johnson et al., 2007; Ortiz et al., 2011; respectively), suggesting that dopamine release may be compromised in these models. As of yet, the synaptic release of dopamine has not been studied during awake, behaving conditions in transgenic mouse models of HD.
The goal of the present study was to examine, and compare where possible, nigrostriatal dopamine release dynamics in the fragment R6/2 and the full-length YAC128 mouse models of HD, using in vivo microdialysis. Both R6/2 and YAC128 mice develop deficits on the rotarod treadmill task and we measured the levels of striatal dopamine during these periods of motor dysfunction. As analogous neurophysiological abnormalities have been shown to exist across fragment and full-length transgenic models (for review see Cummings et al., 2010), we speculated that neurochemical abnormalities may also be a common feature of such models. Specifically, we hypothesized that extracellular striatal dopamine release would be attenuated in R6/2 and YAC128 mice in awake, behaving conditions concomitant with motor decline and as a function of age. In vitro studies have indicated that the intracellular storage of dopamine in nerve terminals may be compromised in HD as well and such a deficiency could contribute to deficits in extracellular dopamine release (Ortiz et al., 2010). In a separate set of experiments, we therefore measured the effects of amphetamine, a dopamine-releasing agent, on local striatal neurochemistry and on locomotor activity in order to investigate the integrity of intracellular dopamine stores in R6/2 and YAC128 mice.
Experimental Procedures
Animals
Transgenic male mice expressing a truncated human HTT gene with a 160 ± 10 CAG repeat expansion (R6/2) or a full-length human HTT gene with a 128 CAG repeat expansion (YAC128) in exon 1 and their respective wild-type controls were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Animals were housed individually in plastic microisolator cages with food and water available ad libitum. Animals were kept under conditions of constant temperature (21°C) and humidity (40%) and maintained on a 12-h light/dark cycle (lights on from 7:00 am to 7:00 pm). All efforts were made to minimize animal suffering and to limit the number of animals utilized for these experiments. Animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Rutgers University Institutional Animal Care and Use Committee.
Motor Phenotype
Motor coordination was assessed using an Economex rotarod apparatus (Columbus Instruments, Columbus, OH, USA). R6/2 mice and wild-type controls were trained on the rotarod treadmill task at 10 rpm for three consecutive trials (60 s maximum duration) 1 day prior to testing. The following day, mice were tested at five fixed speeds (5, 10, 15, 20, 25 rpm) for two separate trials. Fall latency (s) at each speed was averaged across trials and used for statistical comparison. Testing was carried out at 7, 9, and 11 weeks in separate groups of R6/2 mice and 7 months in YAC128 mice and their respective wild-type controls.
Microdialysis Procedure
Microdialysis probes were of a vertical, concentric design, similar to that previously described in our laboratory (Cobb and Abercrombie, 2002). Probes were constructed such that the probe inlet consisted of a piece of PE-10 tubing (Clay Adams, Parsippany, NJ, USA) and a piece of fused silica capillary tubing (I.D. 75 μm and O.D. 150 μm; Polymicro Technologies, Phoenix, AZ, USA) served as the outlet. A semi-permeable microdialysis membrane (molecular weight cut-off = 13 kD; O.D. = 216 μm; Spectrum Laboratories, Rancho Dominguez, CA, USA) was placed over the end of the exposed silica tubing, glued to the PE-10 tubing and coated with a thin epoxy layer, leaving a 2-mm long active exchange area at the end of the probe. Probes were continuously perfused with artificial cerebrospinal fluid (aCSF; NaCl 147 mM, KCl 2.5 mM, CaCl2 1.3 mM, MgCl2 0.9 mM, pH 7.4) using a microliter infusion pump (Harvard Apparatus, Holliston, MA, USA) at a flow rate of 1.5 μl/min. Prior to implantation, probes were calibrated in vitro to determine their relative recovery rates. Only probes with recovery rates between 10 and 15% were used.
R6/2 mice and their wild-type controls were anesthetized with 40–50 mg/kg pentobarbital and 250 mg/kg chloral hydrate (i.p.). YAC128 mice and their wild-type controls were anesthetized with 80–100 mg/kg ketamine and 10 mg/kg xylazine (i.p.). The mice were placed into a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) and onto a heating pad to prevent hypothermia. With the skull flat, the microdialysis probe was implanted into the striatum at the following coordinates: AP: +0.5 mm, ML: ±1.95 mm relative to bregma and DV: −4.0 mm from dura (Franklin and Paxinos, 2008). Probes were anchored to the skull by two small screws (Small Parts, Miami Lakes, FL, USA) and dental cement. The probe inlet and outlet lines were then fed through a metal tether that attached to the head-post at one end, and to a single-channel fluid swivel (Instech Laboratories, Plymouth Meeting, PA, USA) at the other end. The mice were allowed to recover for at least 18 h before experiments began. Post-operative care consisted of administering lactated Ringer’s solution (1.0–2.0 ml/30 g body weight, s.c.) and placing the animal under a heat lamp for 1–3 h after surgery. All microdialysis experiments were conducted during the light portion of the diurnal cycle.
Dopamine Quantification
Microdialysis experiments were conducted in round plastic test chambers (14 cm × 20 cm) equipped with a counter-balanced arm and a swivel assembly (Instech, Plymouth Meeting, PA, USA). The HPLC–EC system consisted of an injector (Rheodyne, Cotati, CA, USA), a VeloSep RP-18 column (100 × 3.2 mm; PerkinElmer, Waltham, MA, USA) and a Shimadzu LC-10AD VP solvent delivery pump (Shimadzu Scientific Instruments, Inc., Columbia, MD, USA), that delivered the mobile phase at a flow rate of 0.7 ml/min. The mobile phase was composed of 0.1 M sodium acetate buffer (pH 4.2), 0.1 mM EDTA, 1.2 mM sodium octyl sulfate, and 8% (v/v) methanol. An electrochemical detector (Coulochem II; ESA Inc., Chelmsford, MA, USA) with a flow cell electrode set at an applied potential of +260 mV was used. The detector output was connected to a computerized data acquisition system (PowerChrom, Denistone East, NSW, Australia). Dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) were identified by retention time and quantified based on peak height relative to the peak height of a 10-nM standard prepared in 0.1 M perchloric acid that was made up fresh daily.
A separate cohort of 11-week-old R6/2 mice and their wild-type controls were sacrificed by decapitation and the striata were rapidly dissected on ice, wrapped in aluminum foil, labeled, and frozen at −80°C until analysis. The weight of each striatum was measured and then tissue was homogenized in 0.1 M perchloric acid containing 100 μM EDTA (20 μl/mg wet tissue weight). Homogenates were centrifuged at 29,200 g for 25 min at 2–8°C. The amount of dopamine in 20 μl samples of the resulting supernatant was quantified by HPLC–EC using the protocol described above for the analysis of dopamine in dialysate samples.
Experimental Procedures
Prior to pharmacological manipulations, baseline dialysate samples were assayed for dopamine until three consecutive samples differed by less than 15%. In experiments involving the local application of d-amphetamine into the striatum via reverse microdialysis, a glass syringe containing a 10-μM solution of amphetamine dissolved in aCSF was connected to the inlet of the probe following the initial baseline determination. Collection of dialysate samples resumed 15 min later to allow for the fluid to flow through the probe and equilibrate to the drug solution. Samples were collected every 10 min for a 60-min duration. The effect of local amphetamine on extracellular dopamine was investigated in 9-week-old R6/2 mice, 7-month-old YAC128 mice and their respective wild-type controls. All experiments involving the local application of amphetamine were conducted 48 h after probe implantation.
In experiments involving systemic administration of d-amphetamine (2 mg/kg, i.p.), the drug was dissolved in sterile 0.9% saline vehicle. The effect of systemic amphetamine on locomotor activity was investigated in 7-month-old YAC128 mice and their wild-type controls. Prior to testing, mice were untethered and the polyethylene tubing emanating from the probes was clipped so that only the acrylic head cap remained. Mice were allowed to habituate in their home cages with this new arrangement for at least 1 week preceding amphetamine treatment. Based on qualitative observations, animals showed no signs of impairment due to either the surgery or the presence of head caps. One day prior to amphetamine administration, mice were habituated to plastic open field chambers (51 cm × 32 cm × 20 cm) overnight. Following habituation, mice were administered drug and spontaneous locomotor activity was monitored for 60 min via automated activity chambers equipped with infrared photobeams (Flexfield, San Diego Instruments, Inc., San Diego, CA, USA).
Data Analysis
Data are presented as mean ± SEM. Dialysate values represent picograms per 20 μl microdialysis sample. Factorial ANOVA was used to assess main effect in each data set examined. Data then were subdivided according to the interactions found in the global test and separate lower order analyses carried out. Where appropriate, the Bonferroni post hoc test was used for multiple comparisons. Differences were considered statistically significant when p < 0.05.
Histology
Upon completion of each experiment, animals were administered a lethal dose of sodium pentobarbital and were perfused intracardially with 0.9% saline followed by 10% formaldehyde. Brains were extracted and post-fixed in fresh fixative overnight at 4°C and transferred to 30% sucrose in phosphate buffer solution overnight at 4°C for cryoprotection. Coronal sections of 50 μm thickness were obtained from striata and stained with Cresyl Violet to verify probe placement in the striatum. A representative histological section showing a microdialysis probe track in the mouse striatum is shown in Figure 1.
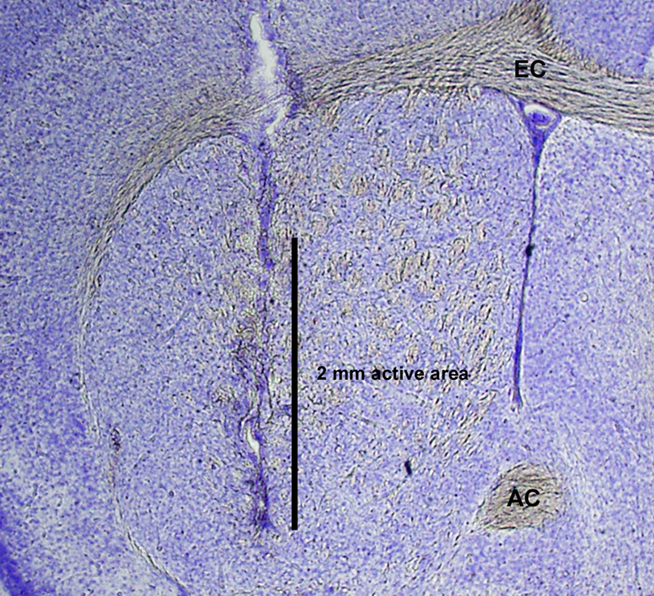
Figure 1. Photomicrograph of Nissl-stained histological section demonstrating representative microdialysis probe placement with a 2-mm active area in mouse striatum. AC, anterior commissure; EC, external capsule.
Materials
Ketamine was purchased from Phoenix Pharmaceutical Inc. (St. Joseph, MO, USA). Xylazine, chloral hydrate, sodium pentobarbital, and d-amphetamine were purchased from Sigma (St. Louis, MO, USA). All other reagents and chemicals were of the highest purity commercially available (Fisher Scientific, Suwanee, GA, USA).
Results
Rotarod Assessment in R6/2 and YAC128 Mice
Performance on a fixed speed rotarod paradigm was assessed in R6/2, YAC128 mice and their respective wild-type controls (Figure 2A). Mice were placed on an elevated, rotating platform for a maximum of 60 s and the latency that it took for each to fall off was measured at 5, 10, 15, 20, and 25 rpm. The latency to stay on the rotating platform was significantly reduced as a function of genotype in 7-week-old [F (1, 172) = 18.78; p < 0.0001), 9-week-old [F (1, 104) = 50.84; p < 0.0001], and 11-week-old [F (1, 72) = 98.22; p < 0.0001] R6/2 mice compared to wild-type controls. Post hoc tests revealed that R6/2 mice fell off the rotarod at significantly shorter latencies than wild-type controls at every speed across all ages tested. One-way ANOVA tests showed that the latency to remain on the rotating platform significantly decreased as a function of age in R6/2 mice [F (2, 39) = 8.074; p < 0.01] but not in wild-type controls [F (2, 48) = 2.013; n.s.]. The latency to stay on the rotating platform also was significantly diminished as a function of genotype in 7-month-old YAC128 mice compared to age-matched wild-type controls [F (1, 56) = 11.88; p < 0.01; Figure 2B]. Post hoc tests indicated that 7-month-old YAC128 mice fell at significantly shorter latencies than wild-type controls at every speed faster than 5 rpm. Data are only presented in graphical form for 10 and 20 rpm as these speeds are most representative of overall performance in both genotypes.
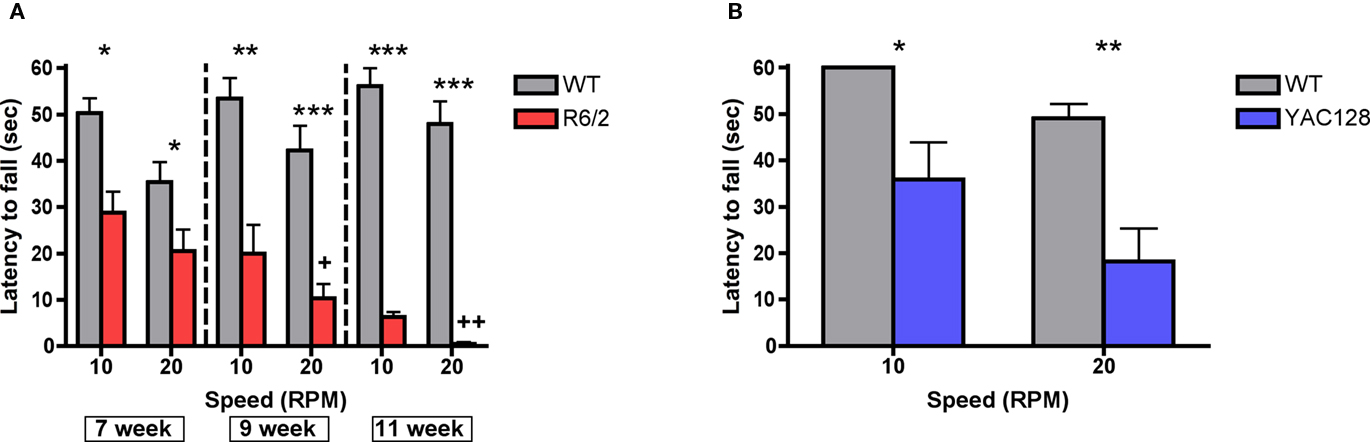
Figure 2. Rotarod performance in (A) R6/2 transgenic mice (7 weeks, n = 22 WT, 23 R6/2; 9 weeks, n = 15 WT, 13 R6/2; 11 weeks, n = 14 WT, 6 R6/2) and (B) 7-month-old YAC128 transgenic mice (n = 7 WT, 9 YAC128). All data are mean ± SEM. * Indicates significantly different from WT, p < 0.05; ** indicates p < 0.001; *** indicates p < 0.005; +, ++ R6/2 week 7 vs. weeks 9 and 11 (p < 0.05, 0.01, respectively).
Extracellular Striatal Dopamine and DOPAC Concentrations
R6/2 mice
Extracellular striatal dopamine was significantly attenuated as a function of genotype [F (1, 34) = 46.96; p < 0.0001; Figure 3A]. Post hoc tests revealed that there was a significant reduction in extracellular dopamine in R6/2 mice and levels were diminished at 7 weeks (WT: 8.4 ± 0.9 pg/20 μl; R6/2: 5.6 ± 0.6 pg/20 μl), 9 weeks (WT: 8.1 ± 1.4 pg/20 μl; R6/2: 3.3 ± 0.7 pg/20 μl), and 11 weeks of age (WT: 11.4 ± 1.2 pg/20 μl; R6/2: 2.7 ± 0.5 pg/20 μl). One-way ANOVA indicated that extracellular levels of striatal dopamine significantly decreased in R6/2 mice as a function of age [F (2, 17) = 6.812; p < 0.01]. Specifically, post hoc tests demonstrated that extracellular dopamine was significantly reduced in 9- and 11-week old R6/2 mice in comparison to 7-week-old R6/2 mice. There was no change in striatal extracellular dopamine concentrations as a function of age in wild-type controls [F (2, 17) = 2.303; n.s.]. Extracellular striatal DOPAC was significantly attenuated as a function of genotype [F (1, 34) = 50.96; p < 0.0001; Figure 3B]. Post hoc tests indicated that although extracellular concentrations of striatal DOPAC were not significantly different at 7 weeks (WT: 1569 ± 137 pg/20 μl; R6/2: 1191 ± 114 pg/20 μl) they were significantly reduced by 9 weeks (WT: 1589 ± 127 pg/20 μl; R6/2: 647 ± 122 pg/20 μl) and 11 weeks of age (WT: 1544 ± 191 pg/20 μl; R6/2: 503 ± 453 pg/20 μl) in R6/2 mice in comparison to wild-type controls. A one-way ANOVA revealed that extracellular DOPAC significantly decreased in R6/2 mice as a function of age [F (2, 17) = 15.32; p < 0.001]. There was no change in extracellular DOPAC amounts as a function of age in wild-type controls [F (2, 17) = 0.20; n.s.].
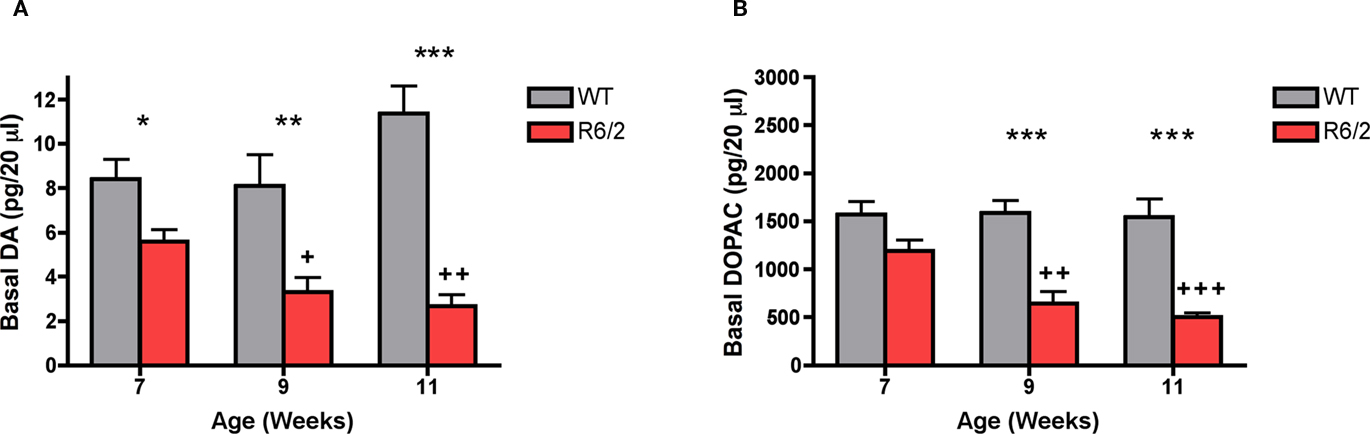
Figure 3. In vivo microdialysis measurements of basal levels of extracellular (A) dopamine and (B) DOPAC in striatum of R6/2 transgenic mice (7 weeks, n = 6 WT, 6 R6/2; 9 weeks, n = 6 WT, 6 R6/2; 11 weeks, n = 8 WT, 8 R6/2). Data are picograms per 20 μl sample. All data are mean ± SEM. * Indicates significantly different from WT, p < 0.05; ** indicates p < 0.001; *** indicates p < 0.005; +, ++ R6/2 week 7 vs. weeks 9 and 11 (p < 0.05, 0.01, respectively).
In the 11-week-old cohort, striatal tissue concentrations of dopamine were measured in R6/2 mice and wild-type controls (Figure 4A). R6/2 mice exhibited a significant abatement in striatal tissue dopamine content (5478 ± 487 pg/20 μl) in comparison to wild-type controls (10680 ± 833 pg/20 μl) [t (10) = 5.39; p < 0.001]. The reduction in striatal tissue content of dopamine (47%) was not as dramatic as the reduction in striatal extracellular concentrations of dopamine (76%) in 11-week-old R6/2 mice (Figure 4B).
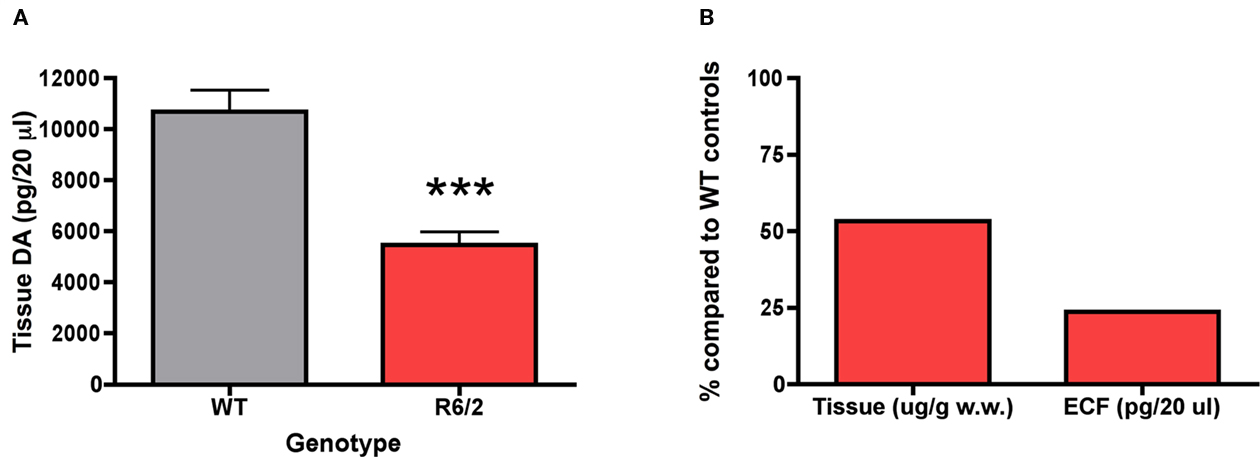
Figure 4. Dopamine content in (A) striatal tissue of 11-week-old R6/2 transgenic mice (n = 6 WT, 6 R6/2) and (B) comparison of the percentage of dopamine levels in relation to WT mice in tissue and the extracellular fluid (ECF). Striatal tissue dopamine content data are μg/g wet weight of tissue and striatal ECF dopamine data are picograms per 20 μl sample of dialysate. All data are mean ± SEM. *** Indicates significantly different from WT, P < 0.005.
YAC128 mice
Extracellular dopamine concentrations in 7-month-old YAC128 mice were significantly reduced (5.0 ± 1.4 pg/20 μl) in comparison to wild-type controls (10.5 ± 1.9 pg/20 μl) [t (14) = 2.39; p < 0.05; Figure 5A]. Although extracellular DOPAC levels were slightly lower in 7-month-old YAC128 mice (1834 ± 245 pg/20 μl), there was no significant difference between wild-type controls (2358 ± 299 pg/20 μl) [t (14) = 1.37; n.s.; Figure 5B].
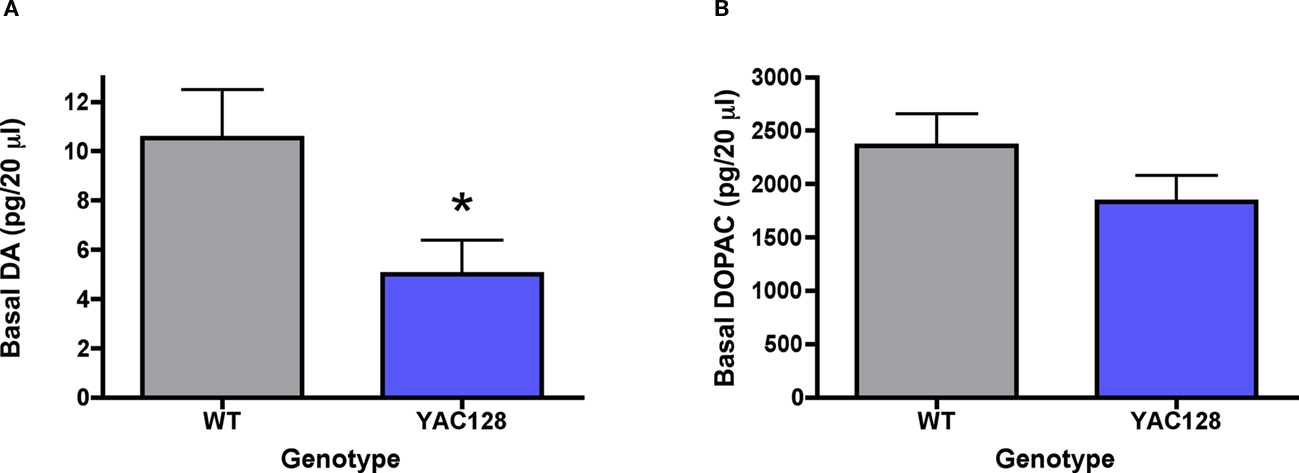
Figure 5. In vivo microdialysis measurements of basal levels of extracellular (A) dopamine and (B) DOPAC in striatum of 7-month-old YAC128 mice (n = 7 WT, 9 YAC128). Data are picograms per 20 μl sample. All data are mean ± SEM. * Indicates significantly different from WT, p < 0.05.
Effect of Local Striatal Amphetamine (10 μM) Application on Extracellular Dopamine Concentrations
R6/2 mice
Local amphetamine induced a significant increase in extracellular dopamine levels across time [F (6, 24) = 75.42; p < 0.0001]. The amphetamine induced increase in dopamine efflux was significantly attenuated as a function of genotype [F (1,24) = 42.25; p < 0.01; Figure 6A]. Post hoc tests indicated that amphetamine induced dopamine release was significantly diminished in 9-week-old R6/2 mice in comparison to wild-type controls at all time intervals measured. The maximum peak response in amphetamine induced dopamine efflux was significantly diminished in 9-week-old R6/2 mice (38.9 ± 7.4 pg/20 μl) vs. wild-type controls (114.4 ± 10.7 pg/20 μl) [t (4) = 5.82; p < 0.01; Figure 6B].
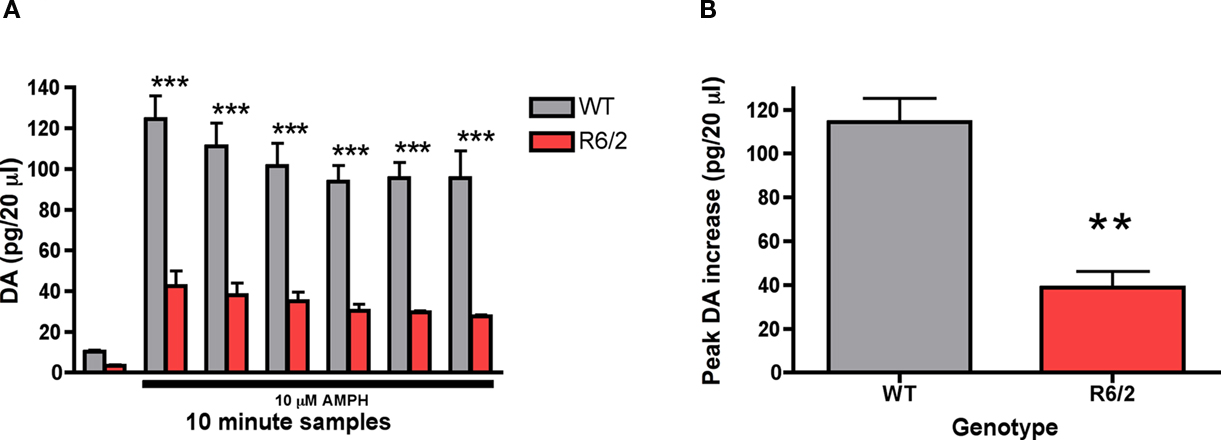
Figure 6. Effect of intrastriatal application of amphetamine (10 μM) on striatal (A) dopamine release and the (B) maximum peak increase in dopamine efflux in 9-week-old R6/2 transgenic mice (n = 3 WT, 3 R6/2). Presence of amphetamine in perfusate is indicated by black bar. Data are picograms per 20 μl sample. All data are mean ± SEM. * Indicates significantly different from WT, P < 0.05; ** indicates P < 0.001; *** indicates P < 0.005.
YAC128 mice
Local amphetamine induced a significant increase in extracellular dopamine across time [F (6, 72) = 23.04; p < 0.0001; Figure 7A]. Although amphetamine induced dopamine efflux was diminished in 7-month-old YAC128 mice in comparison to wild-type controls, there was no significant effect of genotype [F (1, 72) = 1.93, n.s.]. Similarly, while the maximum peak response in amphetamine induced dopamine release was diminished in 7-month-old YAC128 mice (31.8 ± 9.6 pg/20 μl) vs. wild-type controls (50.2 ± 12.0 pg/20 μl) the effect was not significant [t (12) = 1.18; n.s.; Figure 7B].
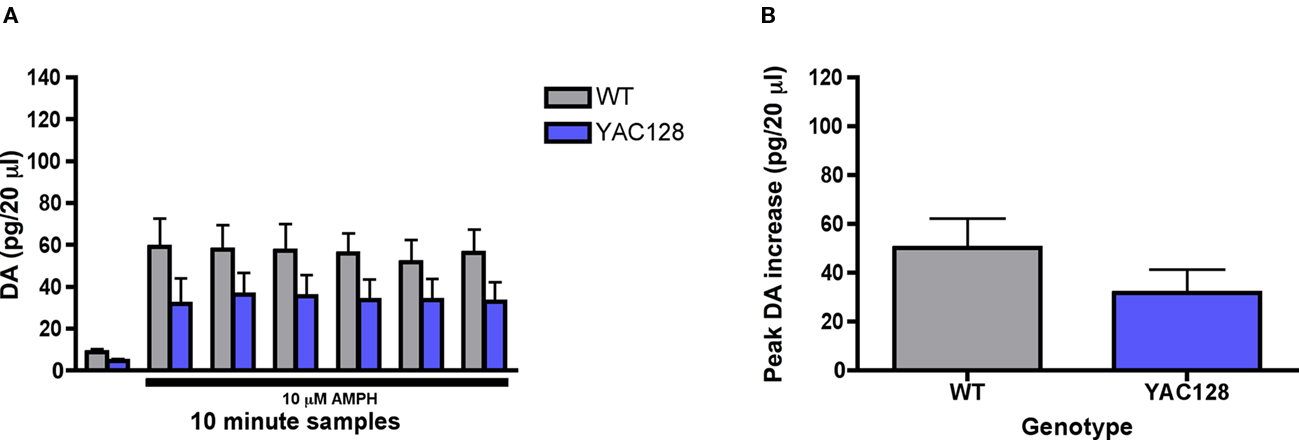
Figure 7. Effect of intrastriatal application of amphetamine (10 μM) on striatal (A) dopamine release and the (B) maximum peak increase in dopamine efflux in 7-month-old YAC128 transgenic mice (n = 5 WT, 9 YAC128). Presence of amphetamine in perfusate is indicated by black bar. Data are picograms per 20 μl sample. All data are mean ± SEM. * Indicates significantly different from WT, P < 0.05; ** indicates P < 0.001; *** indicates P < 0.005.
In 7-month-old YAC128 mice, the effect of systemic amphetamine (2 mg/kg, i.p.) on spontaneous locomotor activity was investigated. There was no significant difference in the basal activity counts between genotypes in 7-month-old YAC128 mice and wild-type controls [t (9) = 0.39; n.s.; Figure 8A]. Amphetamine induced a significant increase in locomotor activity across time [F (6, 54) = 0.96; p < 0.05]. Although amphetamine induced locomotor activity was diminished in 7-month-old YAC128 mice in comparison to wild-type controls, there was no significant effect of genotype [F (1, 54) = 3.80, n.s.]. Collapsing the locomotor counts across all time bins revealed that the total activity in response to amphetamine over 60 min post-drug was significantly attenuated in 7-month-old YAC128 mice vs. wild-type controls [t (9) = 1.88; p < 0.05; Figure 8B].
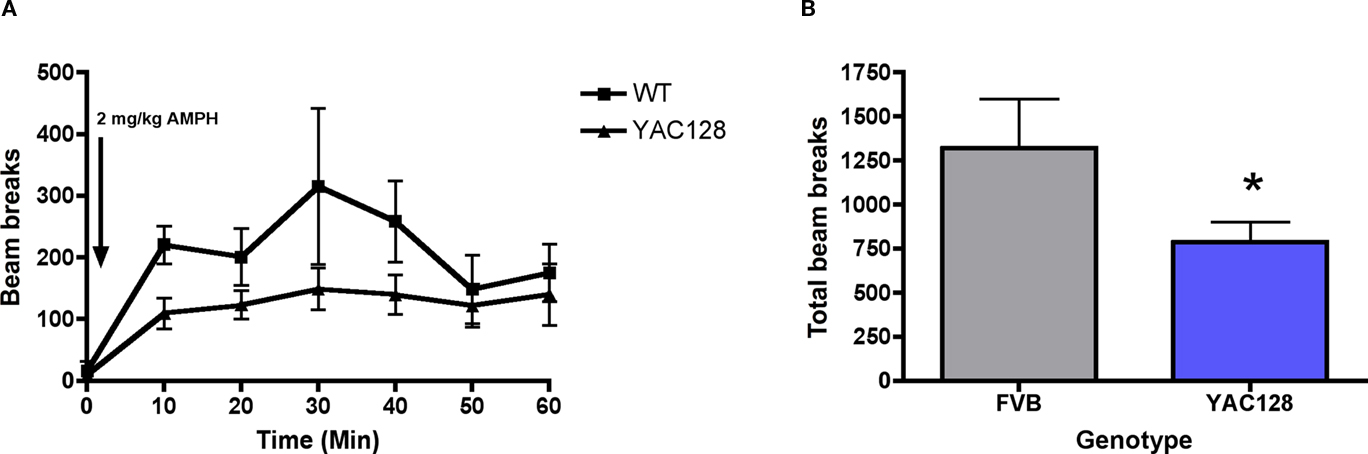
Figure 8. Effect of systemic injection of amphetamine (2 mg/kg; injection indicated by arrow) on (A) 10 min bins of locomotor activity and (B) the total beam break counts during a 60-min period in 7-month-old YAC128 transgenic mice (n = 5 WT, 6 YAC128). * Indicates significantly different from WT, P < 0.05.
Discussion
The present studies utilized in vivo microdialysis to assess striatal dopamine release dynamics in two transgenic mouse models of HD. We report that the concentrations of extracellular striatal dopamine are reduced in R6/2 and YAC128 transgenic mice relative to their respective wild-type controls. These data are consistent with previous studies demonstrating alterations in nigrostriatal function in both animal models and human cases of HD (e.g., Kish et al., 1987; Petersén et al., 2002; Johnson et al., 2006; Ortiz et al., 2011). Importantly, the present studies extend these findings and represent the first demonstration of attenuated striatal dopamine release across transgenic mouse models of HD constructed using different genetic approaches.
Performance deficits on the rotarod treadmill task are reliably documented across multiple HD transgenic strains, including R6/2 and YAC128 mice (Carter et al., 1999; Slow et al., 2003; Menalled et al., 2009). In the present studies, significant impairments in the rotarod assay were apparent in R6/2 and YAC128 mice, with rotarod performance being roughly comparable at 7 weeks and 7 months of age, respectively. In both R6/2 and YAC128 mice, decline in motor function were associated with a significant reduction in the levels of extracellular striatal dopamine. Interestingly, while the 7-month age point in YAC128 mice is characterized by motor disturbances, blunt neurodegeneration does not emerge until older ages (Slow et al., 2003) suggesting the presence of alternative pathological alterations. Motor performance and striatal dopamine release were further assessed in R6/2 mice at 9 and 11 weeks of age, with both measures progressively diminishing as a function of group time point. Masuzawa et al. (2003) demonstrated that pentobarbital can attenuate dopamine release and while we used this compound as a general anesthetic during surgical procedures, all experiments took place at least 18 h subsequent to surgery making it unlikely that there was an interaction between anesthesia and extracellular dopamine at the time experiments were conducted. Taken together, these findings lend support to the notion that alterations in synaptic function, including reduced levels of extracellular dopamine, may contribute to the disease phenotype across different transgenic mouse models of HD. Indeed, a wide range of dopamine related abnormalities have been reported, including dopamine receptor sensitization, attenuated striatal dopamine-dependent plasticity and altered dopamine-dependent modulation of corticostriatal currents, that may reflect the attenuated extracellular dopamine concentrations presently reported (Pineda et al., 2005; Cummings et al., 2006; Kung et al., 2007; André et al., 2011).
In addition to extracellular dopamine concentrations, dopamine content in striatal tissue was assessed in 11-week old R6/2 mice. In agreement with previous reports (Reynolds et al., 1999; Mochel et al., 2011), it was found that striatal tissue levels of dopamine were reduced by roughly half, relative to wild-type control animals. Importantly, reductions in tissue concentrations of dopamine were not as severe as the reductions observed in the extracellular fluid (ECF; 45 vs. 75% reduction, respectively), suggesting the presence of an available intracellular pool of dopamine that is not being adequately utilized under spontaneous in vivo conditions. This finding is particularly intriguing when viewed in the context of animal models of Parkinson’s disease (PD) in which the opposite pattern emerges. In PD models, tissue levels of dopamine must be depleted by greater than 80% in order for deficits in extracellular dopamine release to emerge (Abercrombie et al., 1990). The loss of tissue dopamine content and reductions in the density of postsynaptic dopamine receptors has been reported extensively in HD, however, this finding accentuates the need to account for presynaptic release deficits in HD pathogenesis as well.
There are multiple factors that could contribute to exocytotic release dysfunctions in HD. Several synaptic proteins that are involved in exocytotic processes, including complexin II, snare complex, synaptobrevin 2 and rab3a, have been shown to be abnormal in both transgenic mouse models and human cases of HD (Morton et al., 2001; Freeman and Morton, 2004; Glynn et al., 2007). In line with our results, Morton and Edwardson (2001) demonstrated that there was a progressive decline in complexin II from 7 weeks onward in R6/2 mice. Another potential mechanism may involve alterations in calcium homeostasis and signaling, which could lead to changes in vesicular release probabilities (Bibb et al., 2000; Hansson et al., 2001). Moreover, the mHTT protein also disrupts vesicular transport and such an abnormality could alter vesicular pool sizes (Velier et al., 1998). Indeed, Ortiz et al. (2010) demonstrated that storage vesicles in acute nigrostriatal slices are significantly diminished in R6/2 mice.
Intriguingly, the concentrations of extracellular striatal DOPAC (the main catabolite of dopamine) were significantly decreased in R6/2 mice and showed a tendency to be decreased in YAC128 mice, although this effect was not significant. Extracellular DOPAC derives from, and reflects the metabolism of, newly synthesized dopamine in the cytosol (Zetterström et al., 1988; Yadid et al., 2000). In the present studies, the observation of diminished amounts of striatal DOPAC coupled to the finding that tissue levels of striatal dopamine are reduced lends support to the possibility that cytosolic dopamine pools are depleted as well in transgenic mouse models of HD.
In order to more directly quantify cytosolic dopamine concentrations, we examined the effect of intrastiatal application of amphetamine on striatal dopamine efflux via reverse microdialysis. Amphetamine is a psychostimulant that promotes the release of newly synthesized dopamine from cytosolic pools into the ECF via an impulse-independent, exchange diffusion method and selective inhibition of the dopamine transporter (Fischer and Cho, 1979). Previous work has confirmed the mechanism of action of amphetamine by demonstrating that its stimulant properties are abolished (except at very high doses) with the pretreatment of alpha-methyl-para-tyrosine (AMPT), a compound that inhibits the synthesis of dopamine in the cytosol, but not by reserpine, which depletes dopamine stored in vesicles (Chiueh and Moore, 1974; Niddam et al., 1985; Parker and Cubeddu, 1986; Callaway et al., 1989; Heeringa and Abercrombie, 1995). Alterations in the levels and/or kinetics of the dopamine transporter could obscure the comparisons between genotypes, however, Petersén et al. (2002) demonstrated that the expression patterns of striatal dopamine transporters were comparable and correspondingly Johnson et al. (2006) exhibited that uptake through the dopamine transporter was similar between R6/2 and wild-type controls.
The local application of amphetamine into the striatum produced an increase in extracellular dopamine release in both 9-week-old R6/2 mice and 7-month-old YAC128 mice, as well as in their respective wild-type controls. However, overall dopamine efflux was significantly attenuated across all time points in R6/2 mice and considerably reduced in 7-month-old YAC128 mice, although this effect fell short of significance. Similarly, the maximal effect of amphetamine on dopamine release was attenuated in both R6/2 and YAC128 mice (66 and 37% reductions, respectively). It should be noted that amphetamine-stimulated dopamine levels were considerably lower in the wild-type controls for the YAC128 mice, compared to those for the R6/2 mice. It is plausible that the different background strains and/or the divergent ages of the respective wild-type mice (7 months vs. 9 weeks) may account for the differences in amphetamine-stimulated dopamine release. Alternatively, it is possible that cytosolic pools of dopamine in 7-month-old YAC128 mice are not as depleted as in 9-week-old R6/2 mice. In any case, further experimentation at different age points, both younger and older, is necessary to fully delineate the progression of changes in dopamine release dynamics in the extended phenotype YAC128 mice.
In addition to the effect of local amphetamine on striatal dopamine release, the effect of systemic amphetamine on locomotor activity was assessed in the same cohort of 7-month-old YAC128 mice after a 1-week drug washout period. Phenotype progression is too rapid to challenge the same cohort of R6/2 mice to systemic amphetamine after an extended washout period and as such these experiments were only performed in YAC128 mice. Overall, there was a significant effect of genotype on locomotion, with YAC128 mice showing significantly reduced amphetamine-induced locomotion. This finding is consistent with decreased amphetamine-stimulated dopamine release and parallels a study in which amphetamine-induced locomotor activity was reduced in R6/2 mice (Hickey et al., 2002). These data lend support to the notion that cytosolic dopamine is attenuated in R6/2 and YAC128 mice and may contribute to the deficits in extracellular dopamine concentrations that we report.
Reductions in cytosolic dopamine levels could be caused by either deficits in the production of newly synthesized dopamine or the loss of nigrostriatal terminals. In light of the fact that tyrosine hydroxylase (TH) is the rate-limiting enzyme in the biosynthetic pathway for the production of dopamine, Yohrling et al. (2003) demonstrated a significant reduction in TH activity in the striatum of 12 week-old-R6/2 mice, raising the possibility that deficits in dopamine synthesis may underlie depleted cytosolic concentrations. Alternatively, accumulating evidence indicates that the depletion of dopamine may be the result of nigrostriatal degeneration that manifests in an age-dependent manner. In 16-week-old R6/1 mice there is no difference in the number of nigrostriatal projections or the number of dopamine neurons within the substantia nigra pars compacta (Petersén et al., 2002). By 30 weeks, however, nigrostriatal projections are decreased and the total number of cells within the SNc is reduced (Pineda et al., 2005; Rubio et al., 2009). Interestingly, neurons within the SNc of 16-week-old R6/1 mice were diminished in size and exhibited mHTT aggregate bodies, suggesting that these pathological abnormalities may precede blunt degeneration within this area (Petersén et al., 2002). Similarly, Ariano et al. (2002) demonstrated that there was a late stage reduction of nigrostriatal terminals in 11–12 week old R6/2 mice. Studies in human cases also indicate that there is a reduction in nigrostriatal terminals and decreased striatal dopamine content in late stages of the disease (Kish et al., 1987; Ginovart et al., 1997; Bohnen et al., 2000).
This is the first report comparing extracellular striatal dopamine release dynamics in a fragment and full-length HD transgenic mouse model under awake behaving conditions and distinctively reveals that extracellular dopamine concentrations are attenuated across models. We speculate that the dysfunctions in dopamine release stems from a multifactorial set of presynaptic abnormalities that include both impairments in the synaptic machinery required to release dopamine as well as deficits in dopamine production and perhaps dopamine storage. Future research will need to determine with more specificity the nature of such alterations, when they emerge and the extent each contributes to HD symptomology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Andrew Farrar, Ph.D., for his contributions to the manuscript and Anna Chavez, B.S., for her technical assistance. This research was supported by United States Public Health Service grant NS059921 and the Hereditary Disease Foundation.
References
Abercrombie, E. D., Bonatz, A. E., and Zigmond, M. J. (1990). Effects of L-dopa on extracellular dopamine in striatum of normal and 6 hydroxydopamine-treated rats. Brain Res. 13, 36–44.
André, V. M., Cepeda, C., Fisher, Y. E., Huynh, M., Bardakjian, N., Singh, S., Yang, X. W., and Levine, M. S. (2011). Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J. Neurosci. 31, 1170–1182.
Ariano, M. A., Aronin, N., Difiglia, M., Tagle, D. A., Sibley, D. R., Leavitt, B. R., Hayden, M. R., and Levine, M. S. (2002). Striatal neurochemical changes in transgenic models of Huntington’s disease. J. Neurosci. Res. 68, 716–729.
Bibb, J. A., Yan, Z., Svenningsson, P., Snyder, G. L., Pieribone, V. A., Horiuchi, A., Nairn, A. C., Messer, A., and Greengard, P. (2000). Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 97, 6809–6814.
Bohnen, N. I., Koeppe, R. A., Meyer, P., Ficaro, E., Wernette, K., Kilbourn, M. R., Kuhl, D. E., Frey, K. A., and Albin, R. L. (2000). Decreased striatal monoaminergic terminals in Huntington disease. Neurology 54, 1753–1759.
Bonelli, R. M., and Hofmann, P. (2007). A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin. Pharmacother. 8, 141–153.
Bonelli, R. M., and Wenning, G. K. (2006). Pharmacological management of Huntington’s disease: an evidence-based review. Curr. Pharm. Des. 12, 2701–2720.
Callaway, C. W., Kuczenski, R., and Segal, D. S. (1989). Reserpine enhances amphetamine stereotypies without increasing amphetamine-induced changes in striatal dialysate dopamine. Brain Res. 505, 83–90.
Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., Dunnett, S. B., and Morton, A. J. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 8, 3248–3257.
Cha, J. H., Frey, A. S., Alsdorf, S. A., Kerner, J. A., Kosinski, C. M., Mangiarini, L., Penney, J. B. Jr., Davies, S. W., Bates, G. P., and Young, A. B. (1999). Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 981–989.
Cha, J. H., Kosinski, C. M., Kerner, J. A., Alsdorf, S. A., Mangiarini, L., Davies, S. W., Penney, J. B., Bates, G. P., and Young, A. B. (1998). Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. U.S.A. 95, 6480–6485.
Chiueh, C. C., and Moore, K. E. (1974). Effects of alpha-methyltyrosine on d-amphetamine-induced release of endogenously synthesized and exogenously administered catecholamines from the cat brain in vivo. J. Pharmacol. Exp. Ther. 190, 100–108.
Cobb, W. S., and Abercrombie, E. D. (2002). Distinct roles for nigral GABA and glutamate receptors in the regulation of dendritic dopamine release under normal conditions and in response to systemic haloperidol. J. Neurosci. 15, 1407–1413.
Cummings, D. M., Cepeda, C., and Levine, M. S. (2010). Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington’s disease. ASN Neuro 18, e00036.
Cummings, D. M., Milnerwood, A. J., Dallérac, G. M., Waights, V., Brown, J. Y., Vatsavayai, S. C., Hirst, M. C., and Murphy, K. P. (2006). Aberrant cortical synaptic plasticity and dopaminergic dysfunction in a mouse model of Huntington’s disease. Hum. Mol. Genet. 15, 2856–2868.
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L., and Bates, G. P. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548.
Fischer, J. F., and Cho, A. K. (1979). Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J. Pharmacol. Exp. Ther. 208, 203–209.
Franklin, K., and Paxinos, G. (2008). The Mouse Brain in Stereotaxic Coordinates, 3rd Edn. New York, NY: Academic Press.
Freeman, W., and Morton, A. J. (2004). Regional and progressive changes in brain expression of complexin II in a mouse transgenic for the Huntington’s disease mutation. Brain Res. Bull. 63, 45–55.
Ginovart, N., Lundin, A., Farde, L., Halldin, C., Backman, L., Swahn, C. G., Pauli, S., and Sedvall, G. (1997). PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain 120, 503–514.
Glynn, D., Reim, K., Brose, N., and Morton, A. J. (2007). Depletion of complexin II does not affect disease progression in a mouse model of Huntington’s disease (HD); support for role for complexin II in behavioural pathology in a mouse model of HD. Brain Res. Bull. 72, 108–120.
Hackam, A. S., Singaraja, R., Wellington, C. L., Metzler, M., McCutcheon, K., Zhang, T., Kalchman, M., and Hayden, M. R. (1998). The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol. 141, 1097–1105.
Hansson, O., Guatteo, E., Mercuri, N. B., Bernardi, G., Li, Y.-J., Castilho, R. F., and Brundin, P. (2001). Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioral deficits and with changes in calcium homeostasis in mice transgenic for exon 1 of the Huntington gene. Eur. J. Neurosci. 14, 1494–1504.
Heeringa, M. J., and Abercrombie, E. D. (1995). Biochemistry of somatodendritic dopamine release in substantia nigra: an in vivo comparison with striatal dopamine release. J. Neurochem. 65, 192–200.
Hickey, M. A., Reynolds, G. P., and Morton, A. J. (2002). The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington’s disease. J. Neurochem. 81, 46–59.
Hodgson, J. G., Agopyan, N., Gutekunst, C. A., Leavitt, B. R., LePiane, F., Singaraja, R., Smith, D. J., Bissada, N., McCutcheon, K., Nasir, J., Jamot, L., Li, X. J., Stevens, M. E., Rosemond, E., Roder, J. C., Phillips, A. G., Rubin, E. M., Hersch, S. M., and Hayden, M. R. (1999). A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181–192.
Johnson, M. A., Rajan, V., Miller, C. E., and Wightman, R. M. (2006). Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 97, 737–746.
Johnson, M. A., Villanueva, M., Haynes, C. L., Seipel, A. T., Buhler, L. A., and Wightman, R. M. (2007). Catecholamine exocytosis is diminished in R6/2 Huntington’s disease model mice. J. Neurochem. 103, 2102–2110.
Joyce, J. N., Lexow, N., Bird, E., and Winokur, A. (1988). Organization of dopamine D1 and D2 receptors in human striatum: receptor autoradiographic studies in Huntington’s disease and schizophrenia. Synapse 2, 546–557.
Kish, S. J., Shannak, K., and Hornykiewicz, O. (1987). Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 22, 386–389.
Kung, V. W., Hassam, R., Morton, A. J., and Jones, S. (2007). Dopamine-dependent long term potentiation in the dorsal striatum is reduced in the R6/2 mouse model of Huntington’s disease. Neuroscience 146, 1571–1580.
Levine, M. S., Cepeda, C., Hickey, M. A., Fleming, S. M., and Chesselet, M. F. (2004). Genetic mouse models of Huntington’s and Parkinson’s disease: illuminating but imperfect. Trends Neurosci. 27, 691–697.
Lunkes, A., and Mandel, J. L. (1998). A cellular model that recapitulates major pathogenic steps of Huntington’s disease. Hum. Mol. Genet. 7, 1355–1361.
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., Frey, A. S., Spektor, B. S., Penney, E. B., Schilling, G., Ross, C. A., Borchelt, D. R., Tapscott, S. J., Young, A. B., Cha, J. H., and Olson, J. M. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S. W., and Bates, G. P. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506.
Masuzawa, M., Nakao, S., Miyamoto, E., Yamada, M., Murao, K., Nishi, K., and Shingu, K. (2003). Pentobarbital inhibits ketamine-induced dopamine release in the rat nucleus accumbens: a microdialysis study. Anesth. Analg. 96, 148–152.
Menalled, L. B., and Chesselet, M. F. (2002). Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 23, 32–39.
Menalled, L., El-Khodor, B. F., Patry, M., Suárez-Fariñas, M., Orenstein, S. J., Zahasky, B., Leahy, C., Wheeler, V., Yang, X. W., MacDonald, M., Morton, A. J., Bates, G., Leeds, J., Park, L., Howland, D., Signer, E., Tobin, A., and Brunner, D. (2009). Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 35, 319–336.
Miller, B., and Bezprozvanny, I. (2010). Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine, and calcium. Future Neurol. 5, 735–756.
Mochel, F., Durant, B., Durr, A., and Schiffmann, R. (2011). Altered dopamine and serotonin metabolism in motorically asymptomatic r6/2 mice. PLoS ONE 31, e18336. doi: 10.1371/journal.pone.0018336
Morton, A. J., and Edwardson, J. M. (2001). Progressive depletion of complexin II in a transgenic mouse model of Huntington’s disease. J. Neurochem. 76, 166–172.
Morton, A. J., Faull, R. L., and Edwardson, J. M. (2001). Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res. Bull. 56, 111–117.
Niddam, R., Arbilla, S., Scatton, B., Dennis, T., and Langer, S. Z. (1985). Amphetamine induced release of endogenous dopamine in vitro is not reduced following pretreatment with reserpine. Naunyn Schmiedebergs Arch. Pharmacol. 329, 123–127.
Ortiz, A. N., Kurth, B. J., Osterhaus, G. L., and Johnson, M. A. (2010). Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J. Neurochem. 112, 755–761.
Ortiz, A. N., Kurth, B. J., Osterhaus, G. L., and Johnson, M. A. (2011). Impaired dopamine release and uptake in R6/1 Huntington’s disease model mice. Neurosci. Lett. 492, 11–14.
Parker, E. M., and Cubeddu, L. X. (1986). Effects of d-amphetamine and dopamine synthesis inhibitors on dopamine and acetylcholine neurotransmission in the striatum. I. Release in the absence of vesicular transmitter stores. J. Pharmacol. Exp. Ther. 237, 179–192.
Petersén, A., Puschban, Z., Lotharius, J., NicNiocaill, B., Wiekop, P., O’Connor, W. T., and Brundin, P. (2002). Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington’s disease mice. Neurobiol. Dis. 11, 134–146.
Pineda, J. R., Canals, J. M., Bosch, M., Adell, A., Mengod, G., Artigas, F., Ernfors, P., and Alberch, J. (2005). Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington’s disease. J. Neurochem. 93, 1057–1068.
Reynolds, G. P., Dalton, C. F., Tillery, C. L., Mangiarini, L., Davies, S. W., and Bates, G. P. (1999). Brain neurotransmitter deficits in mice transgenic for the Huntington’s disease mutation. J. Neurochem. 72, 1773–1776.
Rubio, I., Rodríguez-Navarro, J. A., Tomás-Zapico, C., Ruíz, C., Casarejos, M. J., Perucho, J., Gómez, A., Rodal, I., Lucas, J. J., Mena, M. A., and de Yébenes, J. G. (2009). Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 1281, 91–100.
Sedvall, G., Karlsson, P., Lundin, A., Anvret, M., Suhara, T., Halldin, C., and Farde, L. (1994). Dopamine D1 receptor number – a sensitive PET marker for early brain degeneration in Huntington’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 243, 249–255.
Slow, E. J., Van Raamsdonk, J., Rogers, D., Coleman, S. H., Graham, R. K., Deng, Y., Oh, R., Bissada, N., Hossain, S. M., Yang, Y. Z., Li, X. J., Simpson, E. M., Gutekunst, C. A., Leavitt, B. R., and Hayden, M. R. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567.
Stack, E. C., Kubilus, J. K., Smith, K., Cormier, K., Del Signore, S. J., Guelin, E., Ryu, H., Hersch, S. M., and Ferrante, R. J. (2005). Chronology of behavioral symptoms and neuropathological sequel in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 490, 354–370.
Suzuki, M., Desmond, T. J., Albin, R. L., and Frey, K. A. (2001). Vesicular neurotransmitter transporters in Huntington’s disease: initial observations and comparison with traditional synaptic markers. Synapse 15, 329–336.
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Van Raamsdonk, J. M., Warby, S. C., and Hayden, M. R. (2007). Selective degeneration in YAC mouse models of Huntington disease. Brain Res. Bull. 72, 124–131.
Velier, J., Kim, M., Schwarz, C., Kim, T. W., Sapp, E., Chase, K., Aronin, N., and DiFiglia, M. (1998). Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 152, 34–40.
Yadid, G., Harvey-White, J. D., Kopin, I. J., and Goldstein, D. S. (2000). Estimation of striatal dopamine spillover and metabolism in vivo. Neuroreport 11, 3367–3373.
Yohrling, G. J. IV, Jiang, G. C., DeJohn, M. M., Miller, D. W., Young, A. B., Vrana, K. E., and Cha, J. H. (2003). Analysis of cellular, transgenic and human models of Huntington’s disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res. Mol. Brain Res. 119, 28–36.
Keywords: microdialysis, neurodegeneration, dopaminergic, basal ganglia, neostriatum, transgenic, DOPAC, substantia nigra
Citation: Callahan JW and Abercrombie ED (2011) In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full-length (YAC128) transgenic mouse models of Huntington’s disease. Front. Syst. Neurosci. 5:61. doi: 10.3389/fnsys.2011.00061
Received: 02 May 2011;
Accepted: 01 July 2011;
Published online: 15 July 2011.
Edited by:
Jose Bargas, Universidad Nacional Autónoma de México, MexicoReviewed by:
Michael S. Levine, Brain Research Institute, USAJohn D. Salamone, University of Connecticut, USA
George V. Rebec, Indiana University, USA
Copyright: © 2011 Callahan and Abercrombie. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Joshua W. Callahan, Center for Molecular and Behavioral Neuroscience, Rutgers, The State University of New Jersey, 197 University Avenue, Newark, NJ 07102, USA. e-mail: joshcall@pegasus.rutgers.edu