Consumption of Substances of Abuse during Pregnancy Increases Consumption in Offspring: Possible Underlying Mechanisms
 Kinning Poon
Kinning Poon Sarah F. Leibowitz
Sarah F. Leibowitz- Laboratory of Behavioral Neurobiology, The Rockefeller University, New York, NY, USA
Correlative human observational studies on substances of abuse have been highly dependent on the use of rodent models to determine the neuronal and molecular mechanisms that control behavioral outcomes. This is particularly true for gestational exposure to non-illicit substances of abuse, such as excessive dietary fat, ethanol, and nicotine, which are commonly consumed in our society. Exposure to these substances during the prenatal period has been shown in offspring to increase their intake of these substances, induce other behavioral changes, and affect neurochemical systems in several brain areas that are known to control behavior. More importantly, emerging studies are linking the function of the immune system to these neurochemicals and ingestion of these abused substances. This review article will summarize the prenatal rodent models used to study developmental changes in offspring caused by prenatal exposure to dietary fat, ethanol, or nicotine. We will discuss the various techniques used for the administration of these substances into rodents and summarize the published outcomes induced by prenatal exposure to these substances. Finally, this review will cover some of the recent evidence for the role of immune factors in causing these behavioral and neuronal changes.
Introduction
Scientific research has relied heavily on the use of animal models to identify various characteristics of diseases and disorders found in humans. These animal models serve an important purpose when there is limited ability to ethically evaluate such disorders in humans. Most limiting in human research are studies of embryonic development and the effects produced by exposure to substances of abuse, such as alcohol, nicotine, and dietary fat, which occur as a result of voluntary maternal consumption. In humans, ingestion of alcohol during pregnancy triggers neurological disorders and increases the risk of fetal alcohol syndrome in the offspring (1, 2), effects subsequently confirmed and characterized using animal models (3–5). Also, smoking during pregnancy increases the risk of a decrease in birth weight (6, 7) and multiple behavioral problems (8), including attention deficit disorders (9) and increased propensity to abuse drugs (10, 11). In human observational studies, increased intake of dietary fats and obesity during pregnancy are found to increase the risk for dietary obesity in offspring (12–14).
Further testing of these physiological and behavioral changes using animal models exposed to substances of abuse have revealed disturbances in the development of neuronal circuits that modulate both homeostatic and reward pathways (15–17). The main players involved include a variety of neuropeptides that are found in various regions of the hypothalamus and forebrain and are shown to modulate neuronal function that may ultimately contribute to the behavioral changes in offspring. These behavioral changes include an increased propensity to ingest these substances of abuse (15–18), with a significant crossover effect from one substance to another (19). Although great strides have been taken to characterize these changes in neurochemical systems that control behavior, the molecular mechanisms involved in producing these disturbances in the brain have yet to be determined.
In addition to these neurochemicals, the field of ingestive behavior has recently focused attention on immunology. These new studies build on prior research of neurological disorders and neurodegenerative diseases, which show the immune system to play a large part in the health, function, and development of neurons and other cell types in the central nervous system (20–24), along with the development of embryos (25, 26). Recently, both human and animal observational studies have demonstrated that exposure to these substances of abuse, in addition to altering classical neuropeptide and neurotransmitter systems, also disturbs inflammatory systems in key regions of the brain that control ingestion and related behaviors (17, 27, 28). Prenatal inflammation itself has been shown to increase the risk of developing neurological disorders and diseases, such as autism (29, 30) and other psychiatric disorders (31), which the offspring are at a higher risk of developing when exposed during gestation to substances of abuse (32–35).
To understand these neurochemical and immune systems affected by prenatal substance exposure and their possible role in promoting consummatory and other behaviors in the offspring, the use of animal models involving prenatal manipulations is clearly essential. This review will cover the current techniques used to perform prenatal studies using rodent models and their general conclusions about the neuronal changes induced by embryonic exposure to environmental substances. It will also summarize the current research linking these neuronal and neurochemical changes to inflammatory systems, focusing on the three most commonly abused substances, dietary fat, alcohol, and nicotine.
Experimental Methods Used to Introduce Environmental Factors into Pregnant Rats
There are a few factors that must be taken into consideration when designing an experiment using a rodent model. The first is to choose the appropriate rodent strain to use in your model, with various strains having different preferences for different substances. Rat strains with a preference for dietary fats include Sprague-Dawley (36), Brattleboro (37, 38), and Zucker (39), with the latter having an obese phenotype. In alcohol studies, several different strains are used, including outbred rats such as Wistar (40) and Long-Evans (41), and also genetically modified rats that have increased alcohol intake, such as ALKO alcohol (42), high alcohol drinking or HAD (40), and Sardinian preferring (sP) (43) rats. In nicotine research, rats have been found to show differences in behavioral effects between different strains (44, 45) that are attributed to genetic variability. Some of the rat strains used in nicotine studies include Sprague-Dawley (46), Long-Evans (47), Lewis (48), Holtzman (49, 50), and Wistar (51).
The second factor is choosing the method for administering the substance of abuse. This can be broken down into two main paradigms, forced or choice. The forced paradigm does not give the animal a choice in intake, with the substance being the only option or its administration being forced. This is in contrast to a choice paradigm, whereby the animal has one or more competing substances to choose from, with one of the options generally being a control substance, such as chow or water. Studies on dietary fat have used both choice and forced paradigms, with some reports using a combination of the two. Generally, a high-fat and a low-fat diet are made available to the rat, with intake measured daily (52, 53). In combination paradigms, rats are exposed to the high-fat diet in conjunction with their usual diet for a period of several days until acclimation to the new diet is achieved, after which the high-fat diet is given as the only choice (52, 53). Under forced conditions, rats may be given an oil emulsion via oral gavage (54). Studies of ethanol and nicotine, in choice paradigms have used both methods of self-administration (55, 56) and two bottle conditions (57–59). Generally, the concentration of ethanol or nicotine is given in intervals, ranging from a low to high concentration, until the desired concentration is reached (60, 61), with some groups combining palatable sucrose with ethanol or nicotine until voluntary drinking of the drug is established (62). Forced exposure methods, in contrast, include oral gavage, direct injection into the peritoneal cavity (61, 63), intravenous infusion (64), a liquid diet (65), or having the substance as a sole liquid source (66–69).
In studies relating inflammation to ingestive behavior, a specific inflammatory mediator or an agent that induces inflammation, such as lipopolysaccharide, can be administered to any area of the rat through injection. This includes systemic infusion (70), intraperitoneal injection (71, 72), or use of an osmotic mini-pump (73–75).
These methods are only a brief summary, with a wide range of models and rodents used to study the effects of prenatal exposure. Once a particular model is well established, further measurements of behavior in tissues and cells of different type can be extensively performed. The sections below will focus on our current understanding of how prenatal exposure to substances of abuse affect neuronal systems that control behavior in offspring and how the inflammatory response may be a factor in promoting those changes (Figure 1).
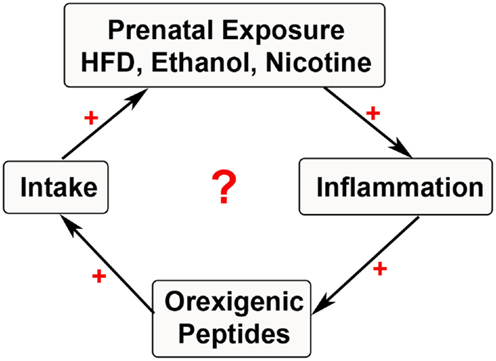
Figure 1. Cycle of substance exposure. The schematic depicts the current hypothesis of a simplified positive feedback loop involving prenatal exposure to substances of abuse that stimulate inflammatory systems. This inflammation may be involved in stimulating neuropeptides that further increase ingestive behavior, thus leading to a cyclical increase in exposure during the prenatal period with negative outcomes in the offspring.
Prenatal HFD Exposure
Animal models investigating the effects of excessive HFD intake during pregnancy have revealed several changes in both the physiology and behavior of offspring. Prenatal exposure to a HFD has been shown to induce several effects in offspring. These include increased body weight, faster weight gain, and larger fat pads (15, 76–78), as well as behavioral changes that include increased ingestion (15, 52, 76), autism spectrum disorders (32, 33), depression (79), and attention hyperactive disorders (80) along with a decrease in spatial memory acquisition (78, 81). Increased understanding of the neuronal systems involved in invoking these behavioral changes is made possible by the numerous animal models used to study these phenomena. These behavioral changes have been attributed to changes in the neurochemistry of various brain regions involved in homeostatic, reward, emotional, and memory processes (Figure 2) and, more recently, to changes in inflammatory processes.
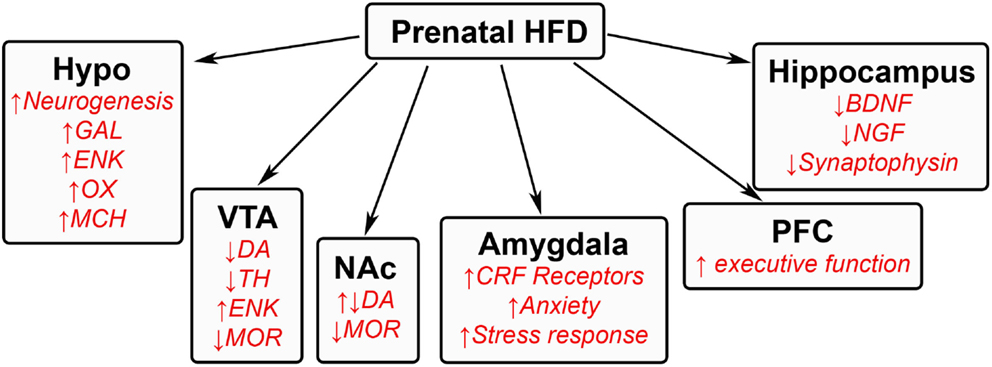
Figure 2. Effects of prenatal HFD exposure on offspring brain. A schematic summarizing some of the changes that occur in the brains of offspring after being exposed to a HFD during gestation. GAL, galanin; ENK, enkephalin; OX, orexin; MCH, melanin-concentrating hormone; DA, dopamine; TH, tyrosine hydroxylase; MOR, μ-opioid receptor; CRF, corticotrophin releasing factor; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor.
Prenatal HFD Exposure Alters Hypothalamic Neurocircuitry
Changes in specific brain areas caused by prenatal HFD exposure seem to control different aspects of HFD intake. The change in homeostatic processes occurs in the hypothalamus, a region important in controlling ingestive behavior. Several lines of evidence show prenatal exposure to a HFD to produce changes in both the developing embryo and in adolescent and adult offspring. These include an increase in the neurogenesis of hypothalamic orexigenic peptide neurons (15, 82), with increased synthesis of the peptides that further induce HFD intake (15). These neuropeptides include galanin and enkephalin in the medial paraventricular nucleus (15, 83), orexin and melanin-concentrating hormone in the perifornical lateral hypothalamus (15), and also ghrelin in the midbrain (84).
Prenatal HFD Exposure Alters VTA–NA System in Offspring
The centers controlling the rewarding aspects of intake consist of the ventral tegmental area (VTA) and the nucleus accumbens (NA) core and shell, which contain the dopaminergic signaling system, μ-opioid receptors, and glutamatergic inputs that are activated by rewarding substances (85, 86). Similar to drug addiction (85), prolonged intake of a HFD has been shown to block dopamine reuptake and enhance dopaminergic function (87). Similarly, exposure to this diet during the prenatal period has been found in adult offspring to increase the levels and expression of dopamine in the NA core and decrease the expression of tyrosine hydroxylase in the VTA, thus decreasing the formation of dopamine (88, 89). Reduced expression of the μ-opioid receptor (89) and increased levels of enkephalin are also found in the VTA and NA regions, with injection of an enkephalin analog into the NA shown to increase HFD intake (90, 91). Similar changes in dopamine, dopamine transporter, and μ-opioid receptor have been found in other studies using maternal junk food or obesity-prone offspring (92, 93), in addition to a reduction in dopamine release in the nucleus accumbens and other terminal sites of dopamine release (92). These studies suggest that prenatal HFD exposure markedly alters the reward pathway, inducing a compensatory mechanism that leads the offspring to ingest excessive amounts of dietary fat to obtain a rewarding feeling caused by the reduced dopamine function (88, 92). Epigenetic changes involving hypomethylation are also found for the dopamine transporter, μ-opioid receptor, and enkephalin, suggesting long-term changes and consequences in offspring (89). While studies in the VTA–NA system have mostly focused on dopamine and agonists of the μ-opioid receptor, other targets are also involved. These include ghrelin, a neuropeptide, known to stimulate the rewarding effects of food intake (94) and promote the rewarding feeling of food intake (95), which is also abundantly expressed in the VTA and found to increase HFD feeding after injection into the VTA (84).
Prenatal HFD Has Global Effects on Other Areas of the Brain in Offspring
Other brain regions also show permanent changes that affect behavior. In the hippocampus, prenatal HFD exposure in offspring decreases expression and levels of proteins that are involved in memory function, such as brain-derived neurotrophic factor, nerve growth factor, and synaptophysin, suggesting a delay in memory acquisition (78, 81). The transcription of genes controlling executive function in the prefrontal cortex is also markedly increased by dietary fat in offspring (96). The emotional aspect of feeding, controlled by the amygdala, has been found in adult rats to evoke several changes in neurochemical pathways (97, 98) that in turn may induce changes in anxiety as well as feeding. Although there are only a few studies of prenatal HFD exposure that have examined the amygdala, there is some evidence that altered functioning of this brain region is involved in emotional changes in offspring that may further promote consumption. Exposure to a fat-rich diet during the prenatal period causes in offspring an increase in corticosteroid receptors in the amygdala (99). This exposure also increases anxiety in an open field, an elevated plus maze, and during light–dark transition tasks, while increasing corticosteroid levels in response to stress (99), suggesting an overall increase in the stress response and thus anxiety. These responses have been reported to increase ingestive behavior in attempt to reduce stress (100–102). These global brain changes affecting decision-making may be involved in both the impulsive and rational choice to overeat.
Prenatal HFD Induces Epigenetic Changes in Offspring
The effect of a HFD during the prenatal period on gene expression in developing neurons is thought to be attributed to epigenetic changes. In human adults, several studies in peripheral tissue reveal alterations in histone modification at promoters of proteins that are affected by dietary fat (103) and in methylation in specific tissue such as skeletal muscle (104). Prenatal exposure to a HFD has also been shown to alter methylation or microRNA expression in placental tissue (105) and adipose tissue (106, 107). That epigenetic changes may be transmitted to offspring is indicated by studies showing a generational effect on specific genes during dietary protein restriction (108, 109). While there is little evidence on the epigenetic effects of prenatal HFD exposure in neurons of embryos and postnatal offspring, several reviews exist that describe global metabolic epigenetic changes in the periphery (110, 111), indicating the need for more such studies in the brain.
Relationship between Dietary Fat and Inflammation
While several studies examining the effects of acute and chronic inflammatory mediators in adult obese animals have revealed an increase in fat intake and weight gain (112), evidence from prenatal inflammatory studies is more limited. Early findings show chronic HFD intake to induce a systemic low-grade inflammation characterized by an increase in cytokines and chemokines (113, 114). This HFD intake also increases the activation of several inflammatory signaling pathways, such as jun amino-terminal kinases, nuclear factor kappa light-chain enhancer, inhibitor of nuclear factor kappa-B kinase subunit beta, peroxisome proliferator-activator receptor, and toll-like receptors (115–118). Chronic treatment with an agent, such as lipopolysaccharide, that induces inflammation can increase body fat mass and caloric intake, and these effects are exacerbated by a HFD (119), suggesting a strong link between HFD and inflammation. More recent studies have uncovered a major role for chemokines, specifically CCL2, which is affected by a HFD and may also mediate neuronal function. This chemokine has been found early on to be increased in obese animals and during HFD intake (120) and, along with its receptor CCR2, is found in all of the key brain areas involved in HFD ingestion (121, 122). Furthermore, blocking the CCR2 receptor with an antagonist is shown to improve symptoms of obesity and decrease food intake (123, 124). In limited studies, prenatal HFD exposure has been found to increase CCL2 in peripheral organs, such as the liver, in offspring (125).
Recent studies from our lab have found a positive relationship between CCL2 and both the migration and expression of orexigenic peptide neurons in primary hypothalamic neurons (126). Exposing cultured embryonic hypothalamic neurons to increasing levels of CCL2 revealed a dose-dependent increase in migration as well as expression of the orexigenic peptides, enkephalin, and galanin in neurons (126). These hypothalamic enkephalin-expressing neurons are found to co-express the receptor, CCR2 (Figure 3), with CCL2 treatment increasing the number of colocalized neurons (126), suggesting an important role for this chemokine in neuronal growth during the prenatal period. In addition, in rats exposed to a HFD during gestation, this chemokine system is found to be greatly altered (127). Prenatal HFD exposure decreases expression of CCL2 while increasing the expression of its receptors, CCR2 and CCR4, in the hypothalamus, and these HFD-exposed neurons are found to exhibit markedly reduced sensitivity to the actions of CCL2 on neuronal migration and peptide expression. With this limited evidence raising new but interesting questions, future studies using the prenatal HFD and prenatal inflammation models should shed light on the molecular mechanisms leading to the neuronal changes and, in turn, altering ingestive behavior in the offspring.
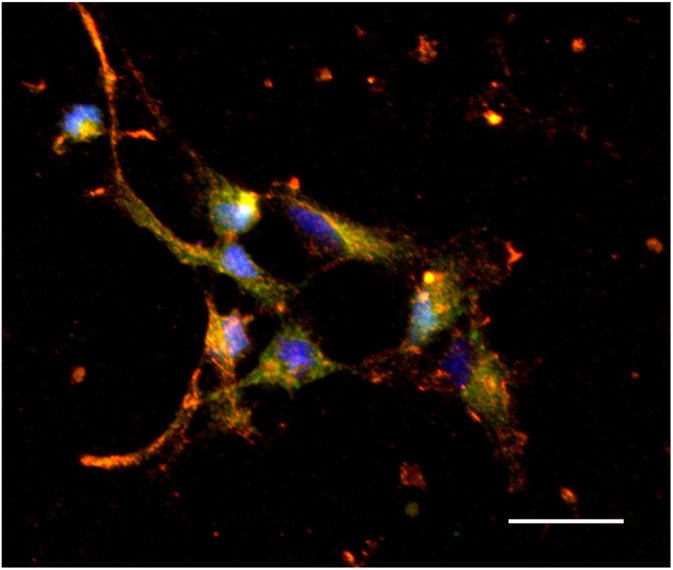
Figure 3. Colocalization of CCR2 and enkephalin in hypothalamic neurons. Hypothalamic neurons extracted from chow-exposed embryos showing CCR2 to colocalize with the orexigenic peptide, enkephalin (orange). Scale: 25 μm. Green: enkephalin, red: CCR2, and blue: dapi.
Prenatal Ethanol Exposure
Original ethanol studies have shown that exposure during the prenatal period to high levels of alcohol is associated with developing fetal alcohol spectrum disorder in offspring (1–5), with many negative developmental, behavioral, and physiological outcomes (128, 129). These high levels of alcohol, within 20–30% or 6 g/kg/day range, decrease the development of neurons in several brain areas (40, 130–132) and additionally induce epigenetic changes in fetal DNA (133–135). Currently unknown are the effects of low levels of alcohol consumption, within 5% or 1–2 g/kg/day range (16, 136–138), on fetal development and ultimately on offspring behavior. Recent studies have demonstrated that low levels of ethanol exposure during gestation induce several behavioral, neurochemical, and developmental effects, similar to prenatal HFD exposure that are caused by changes in brain regions involved in homeostasis, reward, emotional, memory, and inflammatory processes. These changes are thought to induce excessive drinking (16, 137–139), increased preference (17, 139, 140), and reinforcement (141, 142) for ethanol in offspring during the adolescent period to adulthood. These low levels have also been linked to other behavioral changes, such as hyperactivity (74). While ethanol has several targets in many brain regions, studies of low ethanol levels are lacking. This review will summarize some of the current findings in the field (Figure 4).
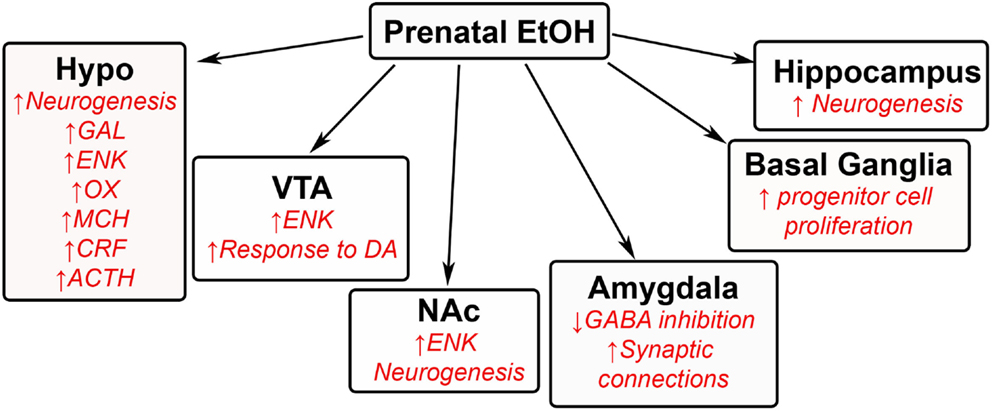
Figure 4. Effects of prenatal ethanol exposure on offspring brain. A schematic summarizing some of the changes that occur in the brains of offspring after being exposed to low levels of ethanol during gestation. GAL, galanin; ENK, enkephalin; OX, orexin; MCH, melanin-concentrating hormone; CRF, corticotrophin releasing factor; ACTH, adrenocorticotropic hormone; DA, dopamine; GABA, γ-aminobutyric acid.
Low Levels of Ethanol Exposure Alters Hypothalamic Neurocircuitry in Offspring
In the hypothalamus, the same orexigenic neuropeptides known to stimulate HFD intake, namely enkephalin, galanin, orexin, and melanin-concentrating hormone, are also found to stimulate ethanol intake [for review, see Ref. (143); (17)]. While different neurochemical systems in the brain are known to be altered by prenatal exposure to ethanol (144–146), the stimulatory effects of prenatal ethanol on these specific neuropeptides are particularly notable, given the potency of their effects on behavior and the sensitivity of the peptide neurons to low doses of ethanol (16, 147). A study from our group has also found low levels of ethanol to increase the genesis of hypothalamic neurons containing enkephalin, orexin, galanin, and melanin-concentrating hormone (16, 17). Additionally, prenatal ethanol exposure is shown to affect stress hormones in the hypothalamus, causing in adolescent and adult offspring an increase in the expression of corticotropin-releasing factor (CRF) in the hypothalamic paraventricular nucleus (144, 148–149) along with levels of corticosterone (144) and also adrenocorticotropic hormone in this same region (144, 150). Prenatal ethanol also increases the levels of these peptides and hormones in response to stress (151–154), with increased stress linked to further consummatory behavior (155–157). Not surprisingly, the CRF system has been linked to addiction of other substances of abuse (158), including dietary fat.
Low Levels of Ethanol Exposure Alters VTA–NA Center in Offspring
Several studies have linked low levels of ethanol during the prenatal period to changes in the mesolimbic area. Increased neurogenesis of enkephalin neurons is found in the NA shell (16), with overall increased levels of enkephalin in both the VTA (159) and NA core (147, 160). These changes may significantly increase ethanol intake in offspring, as high levels of enkephalin have been shown to activate dopamine terminals in the NA (161, 162). The effects of prenatal ethanol on the dopaminergic system in these brain regions are also significant, with the VTA having an increased response to dopaminergic agonists and the NA having increased sensitivity to the stimulatory effects of alcohol in offspring (137, 163, 164). Although ghrelin has been found to be involved in the rewarding feeling of alcohol (165), there are currently no studies on how low ethanol levels during the prenatal period affects this peptide and other neurochemical systems in these brain regions of offspring.
Prenatal Ethanol Has Global Effects on Other Areas of the Brain in Offspring
While there exists plenty of research describing the effects of high gestational ethanol exposure on the developing brain, there are only a few studies measuring the effects of low ethanol exposure on other brain areas not discussed above. Some of the findings include an ethanol-induced increase in progenitor cell proliferation in the basal ganglia (166) and a decrease in neural activity in the infralimbic cortex (164). They also include increased neurogenesis in regions of the hippocampus (167). The amygdala has been suggested to be affected by low levels of ethanol during the prenatal period. Offspring exposed to low ethanol display anxiety-like behavior when exposed to stressful conditions, and this behavior has been related to both an increase in synaptic connectivity in the basolateral amygdala (168) and a decrease in GABA inhibition (169), both of which stimulate the excitability of the amygdala (168, 169). Further studies on the effects of low ethanol concentrations in these other brain regions are needed to determine the extent of ethanol’s action on neuronal development throughout the brain.
Prenatal Ethanol Induces Epigenetic Changes in Offspring
There are several studies that reveal high ethanol exposure during the prenatal period to induce dramatic epigenetic changes in offspring. The adult liver provides a clear example, with high levels of ethanol exposure found to alter DNA methylation related to alcoholic liver disease (170–172). Also, chronic maternal ethanol exposure is shown to decrease methylation at a gene called agouti viable yellow, which affects the color of their coat, that is passed down to offspring (173), while acute prenatal exposure to high levels of ethanol globally causes hypomethylation of DNA in embryos (133). Long-term prenatal exposure to high ethanol levels also induces changes in methylation and microRNA in hippocampal neurons (174). In light of these studies of high ethanol exposure, further investigations of epigenetic effects are clearly needed involving low concentrations of ethanol, which as described above have strong, stimulatory effects on neuronal development in the brain.
Relationship between Ethanol and Inflammation
Although only a few studies exist, ethanol intake has also been linked to inflammatory systems. The most commonly studied peripheral organ is the liver, with excessive drinking linked to alcoholic liver disease that increases inflammatory mediators (175, 176). More recent studies in adult animals have also shown ethanol exposure to stimulate inflammatory systems in the central nervous system. Endotoxin treatment after ethanol exposure has been found to induce a long-term inflammatory state in the brain (177) and increase nitric oxide synthase and cyclooxygenase, which lead to inflammation (178). This increase in inflammation has also been detected in offspring after prenatal exposure. Similar to prenatal HFD exposure, our lab recently found prenatal ethanol to induce several changes in the CCL2 chemokine system. We found low levels of ethanol during gestation to increase in the offspring the genesis of neurons that co-express CCR2 and melanin-concentrating hormone in the lateral hypothalamus (17), a neuropeptide implicated in excessive ethanol drinking (179). With current research showing low levels of ethanol exposure to increase drinking in offspring and produce changes in the immune system that ultimately affects neuronal function, future research on inflammatory systems could be very informative and important.
Prenatal Nicotine Exposure
The effects of prenatal nicotine exposure are broad in nature, affecting both behavioral and neuronal development in several regions of offspring brain. Human studies show that children exposed to tobacco during gestation exhibit an increased risk for tobacco use, craving, and withdrawal (180), as well as dependence (181). Animal studies similarly reveal increased nicotine self-administration and consumption in adolescent and adult offspring (182–185), along with increased ingestion of other substances including fat and ethanol (18). Additional behavioral problems include an increased risk of hyperactivity (186), impulsivity (185), and anxiety (34, 35). High levels of nicotine exposure are also associated with detrimental effects, such as growth retardation (187). While these nicotine studies lead one to question whether these changes are attributed to certain chemicals from the tobacco (188, 189) rather than to nicotine itself and result from social smoking as well as chronic smoking, the overall evidence clearly demonstrates that prenatal nicotine exposure negatively affects offspring. Similar to prenatal HFD and ethanol exposure, these changes in physiology and behavior induced by nicotine or smoking may be attributed to neuronal changes in the offspring brain (Figure 5).
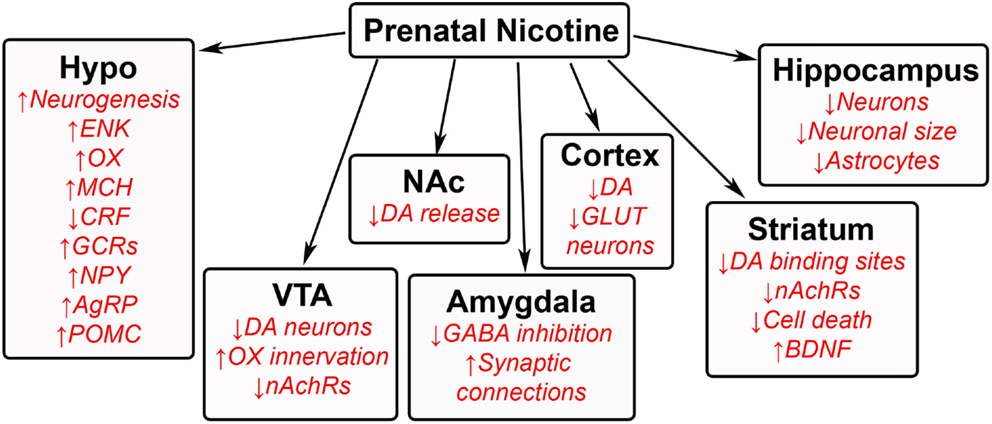
Figure 5. Effects of prenatal nicotine exposure on offspring brain. A schematic summarizing some of the changes that occur in the brains of offspring after being exposed to nicotine during gestation. GAL, galanin; ENK, enkephalin; OX, orexin; MCH, melanin-concentrating hormone; CRF, corticotrophin releasing factor; GCRs, glucocorticoid receptors; NPY, neuropeptide Y; AgRP, agouti-related protein; POMC, proopiomelanocortin; DA, dopamine; nAchRs, nicotinic acetylcholine receptors; GABA, γ-aminobutyric acid; GLUT, glutamate; BDNF, brain-derived neurotrophic factor.
Prenatal Nicotine Exposure Alters Hypothalamic Neurocircuitry
Similar to dietary fat and ethanol, prenatal nicotine exposure has been found to affect the neuronal architecture and function of the hypothalamus. Several neuropeptides have been found to be altered in offspring during exposure to both low and high levels of nicotine. Some of the findings include a decrease in CRF and an increase in glucocorticoid receptors in the hypothalamus (190). They also show an increase in several orexigenic peptides, including neuropeptide Y, agouti-related peptide, and proopiomelanocortin in the arcuate nucleus (191), enkephalin in the medial hypothalamic paraventricular nucleus, and orexin and melanin-concentrating hormone in the perifornical lateral hypothalamus (18, 192). One of the more important findings from our lab shows that exposure to nicotine actually stimulates the genesis of neurons that express enkephalin, orexin, and melanin-concentrating hormone in the offspring hypothalamus (18), with these peptides positively related to the intake of nicotine (193). A small number of epigenetic studies also show changes in DNA methylation of the gene encoding brain-derived neurotrophic factor in human studies (194, 195), revealing the need for further epigenetic studies of specific cell types.
Prenatal Nicotine Exposure Alters VTA–NA Center in Offspring
Prenatal nicotine exposure has been found to varying degrees to change neurons in the VTA and NA in offspring. With regards to the mesolimbic dopamine system, prenatal nicotine exposure decreases the number of dopaminergic neurons in the VTA (196), dopamine release from the NA (197, 198), and the number of dopamine binding sites in the striatum (199), altering the rewarding effects of nicotine in offspring. Neuronal connections to the VTA are also affected, with orexin innervation from the lateral hypothalamus to the VTA found to be increased (192). Additionally, prenatal nicotine reduces the number of nicotinic cholinergic receptor expression in both the VTA and NA core (196). Similar to the neurogenesis effect in the hypothalamus, prenatal nicotine increases cell survival in the NA and inhibits cell death related pathways (200), with this increase in cell survival consistent with the finding that prenatal nicotine exposure increases the nerve growth factor, BDNF (201). Further studies on this reward region in offspring will shed more light on how prenatal exposure reprograms offspring to become more prone to abusing nicotine.
Prenatal Nicotine Has Global Effects on Other Areas of the Brain in Offspring
Prenatal nicotine exposure has been found to affect several other brain regions in offspring. In the hippocampus, this exposure decreases the number of neurons (202) while increasing the number of astrocytes (202), and it also decreases the neuronal area and cell size (203, 204), suggesting decreased hippocampal function. Similar effects are also found in the cortex of early postnatal rats (205), pre-weaned rats (204), and embryos (206), with studies revealing fewer glutamatergic neurons (207). These changes in the cortex induced by prenatal exposure have been linked to cognitive deficits and impaired executive control, causing rats to be more impulsive (208). Similar to the VTA and NA, dopamine levels are also decreased in the cortex of postnatal offspring (209). In the amygdala, one study found nicotine exposure to reduce the size of the amygdala in adolescent offspring (210), while a recent study from our lab has described an increase in neurogenesis and expression of enkephalin neurons in the central amygdala (18). With nicotine intake shown to generally reduce anxiety, future studies with prenatal exposure that relate behavior to amygdaloid function in offspring, as well as to other brain regions involved in decision making, would be interesting.
Prenatal Nicotine Induces Epigenetic Changes in Offspring
Several studies show prenatal nicotine exposure to have epigenetic effects on peripheral organs in offspring. Prenatal nicotine has been found to decrease methylation on the promoter-expressing angiotensin receptor type 1a (211) and increase histone acetylation of the protein and fatty acid synthase in liver (212). Human studies have also reveal global changes in DNA methylation in offspring (213, 214). Evidence of a generational effect has also been shown in rat models, with maternal nicotine use and exposure during the prenatal period found to induce asthma and epigenetic changes in lungs of offspring that are two generations past the original exposure (215). This evidence suggests that the changes induced by prenatal nicotine exposure on brain neurochemical systems may be related to epigenetic changes occurring during development.
Relationship between Nicotine and Inflammation
While reports of pure nicotine on adult systems generally reveal a reduction in inflammation (216, 217), several studies in humans show cigarette smoking to cause an increase in inflammation (218). Also, in a rat model, exposure to pure nicotine during the gestational period is found to increase the inflammatory mediators, IL-6 and TNF-alpha, in newborn blood serum (219). While this evidence is limited, it suggests the possibility that prenatal nicotine may have similar effects to HFD and ethanol exposure on inflammatory mediators, including CCL2.
General Conclusion
The current knowledge of the neural control of ingestive behavior in offspring that are prenatally exposed to substances of abuse has come a long way from observational human studies. We are now only beginning to piece together how these changes in specific brain regions affect the overall neuronal communication within the brain. In addition, other systems of the central nervous system, such as glial cells, astrocytes, and oligodendrocytes, may also play a major role in this disturbed communication. More importantly is the emerging function of the immune system in the development of these neuronal systems in offspring and how substances of abuse disturb its actions. Future studies using these prenatal animal models will provide much insight in both the molecular and neuronal network changes as well as the mechanisms leading to these changes.
Author Contributions
KP and SL prepared and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the National Institutes of Health, F32DK100058 (KP) and R01AA12882 (SL).
References
1. Jones KL, Smith DW, Ulleland CN, Streissguth P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet (1973) 1:1267–71. doi: 10.1016/S0140-6736(73)91291-9
2. Lemoine P, Harousseau H, Borteyru JP, Menuet JC. Children of alcoholic parents – observed anomalies: discussion of 127 cases. Ther Drug Monit (2003) 25:132–6. doi:10.1097/00007691-200304000-00002
3. Chernoff GF. The fetal alcohol syndrome in mice: an animal model. Teratology (1977) 15:223–9. doi:10.1002/tera.1420150303
4. Randall CL, Taylor J, Walker DW. Ethanol-induced malformations in mice. Alcohol Clin Exp Res (1977) 1:219–24. doi:10.1111/j.1530-0277.1977.tb05876.x
5. Abel EL, Dintcheff BA. Effects of prenatal alcohol exposure on growth and development in rats. J Pharmacol Exp Ther (1978) 207:916–21.
6. Simpson WJ. A preliminary report on cigarette smoking and the incidence of prematurity. Am J Obstet Gynecol (1957) 73:807–15.
7. Butler NR, Goldstein H, Ross EM. Cigarette smoking in pregnancy: its influence on birth weight and perinatal mortality. Br Med J (1972) 2:127–30. doi:10.1136/bmj.2.5806.127
8. Weitzman M, Gortmaker S, Sobol A. Maternal smoking and behavior problems of children. Pediatrics (1992) 90:342–9.
9. Thapar A, Fowler T, Rice F, Scourfield J, van den Bree M, Thomas H, et al. Maternal smoking during pregnancy and attention deficit hyperactivity disorder symptoms in offspring. Am J Psychiatry (2003) 160:1985–9. doi:10.1176/appi.ajp.160.11.1985
10. Kandel DB, Wu P, Davies M. Maternal smoking during pregnancy and smoking by adolescent daughters. Am J Public Health (1994) 84:1407–13. doi:10.2105/AJPH.84.9.1407
11. Porath AJ, Fried PA. Effects of prenatal cigarette and marijuana exposure on drug use among offspring. Neurotoxicol Teratol (2005) 27:267–77. doi:10.1016/j.ntt.2004.12.003
12. Edwards LE, Dickes WF, Alton IR, Hakanson EY. Pregnancy in the massively obese: course, outcome, and obesity prognosis of the infant. Am J Obstet Gynecol (1978) 131:479–83.
13. Gross T, Sokol RJ, King KC. Obesity in pregnancy: risks and outcome. Obstet Gynecol (1980) 56:446–50.
14. Curhan GC, Chertow GM, Willett WC, Spiegelman D, Colditz GA, Manson JE, et al. Birth weight and adult hypertension and obesity in women. Circulation (1996) 94:1310–5. doi:10.1161/01.CIR.94.6.1310
15. Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci (2008) 28:12107–19. doi:10.1523/JNEUROSCI.2642-08.2008
16. Chang GQ, Karatayev O, Liang SC, Barson JR, Leibowitz SF. Prenatal ethanol exposure stimulates neurogenesis in hypothalamic and limbic peptide systems: possible mechanism for offspring ethanol overconsumption. Neuroscience (2012) 222:417–28. doi:10.1016/j.neuroscience.2012.05.066
17. Chang GQ, Karatayev O, Leibowitz SF. Prenatal exposure to ethanol stimulates hypothalamic CCR2 chemokine receptor system: possible relation to increased density of orexigenic peptide neurons and ethanol drinking in adolescent offspring. Neuroscience (2015) 310:163–75. doi:10.1016/j.neuroscience.2015.09.020
18. Chang GQ, Karatayev O, Leibowitz SF. Prenatal exposure to nicotine stimulates neurogenesis of orexigenic peptide-expressing neurons in hypothalamus and amygdala. J Neurosci (2013) 33:13600–11. doi:10.1523/JNEUROSCI.5835-12.2013
19. Chang GQ, Karatayev O, Barson JR, Liang SC, Leibowitz SF. Common effects of fat, ethanol, and nicotine on enkephalin in discrete areas of the brain. Neuroscience (2014) 277:665–78. doi:10.1016/j.neuroscience.2014.07.050
20. Skrzydelski D, Guyon A, Dauge V, Rovere C, Apartis E, Kitabgi P, et al. The chemokine stromal cell-derived factor-1/CXCL12 activates the nigrostriatal dopamine system. J Neurochem (2007) 102:1175–83. doi:10.1111/j.1471-4159.2007.04639.x
21. Guyon A, Massa F, Rovere C, Nahon JL. How cytokines can influence the brain: a role for chemokines? J Neuroimmunol (2008) 198:46–55. doi:10.1016/j.jneuroim.2008.04.009
22. Guyon A, Skrzydelski D, Rovere C, Apartis E, Rostene W, Kitabgi P, et al. Stromal-cell-derived factor 1alpha/CXCL12 modulates high-threshold calcium currents in rat substantia nigra. Eur J Neurosci (2008) 28:862–70. doi:10.1111/j.1460-9568.2008.06367.x
23. Park MH, Lee YK, Lee YH, Kim YB, Yun YW, Nam SY, et al. Chemokines released from astrocytes promote chemokine receptor 5-mediated neuronal cell differentiation. Exp Cell Res (2009) 315:2715–26. doi:10.1016/j.yexcr.2009.06.017
24. Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JL. CCL2/MCP-1 modulation of microglial activation and proliferation. J Neuroinflammation (2011) 8:77. doi:10.1186/1742-2094-8-77
25. McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol (1999) 213:442–56. doi:10.1006/dbio.1999.9405
26. Saito S. Cytokine cross-talk between mother and the embryo/placenta. J Reprod Immunol (2001) 52:15–33. doi:10.1016/S0165-0378(01)00112-7
27. Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology (2010) 151:1622–32. doi:10.1210/en.2009-1019
28. Lussier AA, Stepien KA, Neumann SM, Pavlidis P, Kobor MS, Weinberg J. Prenatal alcohol exposure alters steady-state and activated gene expression in the adult rat brain. Alcohol Clin Exp Res (2015) 39:251–61. doi:10.1111/acer.12622
29. Meldrum SJ, Strunk T, Currie A, Prescott SL, Simmer K, Whitehouse AJ. Autism spectrum disorder in children born preterm-role of exposure to perinatal inflammation. Front Neurosci (2013) 7:123. doi:10.3389/fnins.2013.00123
30. Le Belle JE, Sperry J, Ngo A, Ghochani Y, Laks DR, Lopez-Aranda M, et al. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Reports (2014) 3:725–34. doi:10.1016/j.stemcr.2014.09.004
31. Cannon M, Clarke MC, Cotter DR. Priming the brain for psychosis: maternal inflammation during fetal development and the risk of later psychiatric disorder. Am J Psychiatry (2014) 171:901–5. doi:10.1176/appi.ajp.2014.14060749
32. Leonard H, Nassar N, Bourke J, Blair E, Mulroy S, de Klerk N, et al. Relation between intrauterine growth and subsequent intellectual disability in a ten-year population cohort of children in Western Australia. Am J Epidemiol (2008) 167:103–11. doi:10.1093/aje/kwm245
33. Schendel D, Bhasin TK. Birth weight and gestational age characteristics of children with autism, including a comparison with other developmental disabilities. Pediatrics (2008) 121:1155–64. doi:10.1542/peds.2007-1049
34. Eppolito AK, Bachus SE, McDonald CG, Meador-Woodruff JH, Smith RF. Late emerging effects of prenatal and early postnatal nicotine exposure on the cholinergic system and anxiety-like behavior. Neurotoxicol Teratol (2010) 32:336–45. doi:10.1016/j.ntt.2009.12.009
35. Parameshwaran K, Buabeid MA, Karuppagounder SS, Uthayathas S, Thiruchelvam K, Shonesy B, et al. Developmental nicotine exposure induced alterations in behavior and glutamate receptor function in hippocampus. Cell Mol Life Sci (2012) 69:829–41. doi:10.1007/s00018-011-0805-4
36. Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol (1997) 273:R725–30.
37. Laycock JF. Review: the Brattleboro rat with hereditary hypothalamic diabetes insipidus as an ideal experimental model. Lab Anim (1976) 10:261–70. doi:10.1258/002367776781035332
38. Odorizzi M, Max JP, Tankosic P, Burlet C, Burlet A. Dietary preferences of Brattleboro rats correlated with an overexpression of galanin in the hypothalamus. Eur J Neurosci (1999) 11:3005–14. doi:10.1046/j.1460-9568.1999.00722.x
39. Cleary MP, Vasselli JR, Greenwood MR. Development of obesity in Zucker obese (fafa) rat in absence of hyperphagia. Am J Physiol (1980) 238:E284–92.
40. Li TK, Lumeng L, Doolittle DP. Selective breeding for alcohol preference and associated responses. Behav Genet (1993) 23:163–70. doi:10.1007/BF01067421
41. Barson JR, Fagan SE, Chang GQ, Leibowitz SF. Neurochemical heterogeneity of rats predicted by different measures to be high ethanol consumers. Alcohol Clin Exp Res (2013) 37(Suppl 1):E141–51. doi:10.1111/j.1530-0277.2012.01858.x
42. Sommer W, Hyytia P, Kiianmaa K. The alcohol-preferring AA and alcohol-avoiding ANA rats: neurobiology of the regulation of alcohol drinking. Addict Biol (2006) 11:289–309. doi:10.1111/j.1369-1600.2006.00037.x
43. Colombo G, Agabio R, Diaz G, Fa M, Lobina C, Reali R, et al. Sardinian alcohol-preferring rats prefer chocolate and sucrose over ethanol. Alcohol (1997) 14:611–5. doi:10.1016/S0741-8329(97)00075-X
44. Rosecrans JA. The psychopharmacological basis of nicotine’s differential effects on behavior: individual subject variability in the rat. Behav Genet (1995) 25:187–96. doi:10.1007/BF02196927
45. Chen H, Hiler KA, Tolley EA, Matta SG, Sharp BM. Genetic factors control nicotine self-administration in isogenic adolescent rat strains. PLoS One (2012) 7:e44234. doi:10.1371/journal.pone.0044234
46. Fudala PJ, Teoh KW, Iwamoto ET. Pharmacologic characterization of nicotine-induced conditioned place preference. Pharmacol Biochem Behav (1985) 22:237–41. doi:10.1016/0091-3057(85)90384-3
47. Corrigall WA, Coen KM. Fixed-interval schedules for drug self-administration in the rat. Psychopharmacology (Berl) (1989) 99:136–9. doi:10.1007/BF00634468
48. Horan B, Smith M, Gardner EL, Lepore M, Ashby CR Jr. (-)-Nicotine produces conditioned place preference in Lewis, but not Fischer 344 rats. Synapse (1997) 26:93–4. doi:10.1002/(SICI)1098-2396(199705)26:1<93::AID-SYN10>3.0.CO;2-W
49. Jorenby DE, Steinpreis RE, Sherman JE, Baker TB. Aversion instead of preference learning indicated by nicotine place conditioning in rats. Psychopharmacology (Berl) (1990) 101:533–8. doi:10.1007/BF02244233
50. Valentine JD, Hokanson JS, Matta SG, Sharp BM. Self-administration in rats allowed unlimited access to nicotine. Psychopharmacology (Berl) (1997) 133:300–4. doi:10.1007/s002130050405
51. Laviolette SR, van der Kooy D. Blockade of mesolimbic dopamine transmission dramatically increases sensitivity to the rewarding effects of nicotine in the ventral tegmental area. Mol Psychiatry (2003) 8(50–59):59. doi:10.1038/sj.mp.4001197
52. Dourmashkin JT, Chang GQ, Hill JO, Gayles EC, Fried SK, Leibowitz SF. Model for predicting and phenotyping at normal weight the long-term propensity for obesity in Sprague-Dawley rats. Physiol Behav (2006) 87:666–78. doi:10.1016/j.physbeh.2006.01.008
53. Chang GQ, Karatayev O, Ahsan R, Gaysinskaya V, Marwil Z, Leibowitz SF. Dietary fat stimulates endogenous enkephalin and dynorphin in the paraventricular nucleus: role of circulating triglycerides. Am J Physiol Endocrinol Metab (2007) 292:E561–70. doi:10.1152/ajpendo.00087.2006
54. Carrillo CA, Leibowitz SF, Karatayev O, Hoebel BG. A high-fat meal or injection of lipids stimulates ethanol intake. Alcohol (2004) 34:197–202. doi:10.1016/j.alcohol.2004.08.009
55. Hanson HM, Ivester CA, Morton BR. Nicotine self-administration in rats. NIDA Res Monogr (1979) 23:70–90.
56. Corcoran ME, Lewis J, Fibiger HC. Forebrain noradrenaline and oral self-administration of ethanol by rats. Behav Brain Res (1983) 8:1–21. doi:10.1016/0166-4328(83)90168-7
57. Sinclair JD. Motivation for alcohol in rats: position and bottle preferences do not cause drinking. Drug Alcohol Depend (1976) 1:357–66. doi:10.1016/0376-8716(76)90038-7
58. Flynn FW, Webster M, Ksir C. Chronic voluntary nicotine drinking enhances nicotine palatability in rats. Behav Neurosci (1989) 103:356–64. doi:10.1037/0735-7044.103.2.356
59. Dadmarz M, Vogel WH. Individual self-administration of nicotine by rats. Pharmacol Biochem Behav (2003) 76:425–32. doi:10.1016/j.pbb.2003.08.014
60. Sandbak T, Murison R. Voluntary alcohol consumption in rats: relationships to defensive burying and stress gastric erosions. Physiol Behav (1996) 59:983–9. doi:10.1016/0031-9384(95)02173-6
61. Morganstern I, Chang GQ, Barson JR, Ye Z, Karatayev O, Leibowitz SF. Differential effects of acute and chronic ethanol exposure on orexin expression in the perifornical lateral hypothalamus. Alcohol Clin Exp Res (2010) 34:886–96. doi:10.1111/j.1530-0277.2010.01161.x
62. Samson HH. Initiation of ethanol reinforcement using a sucrose-substitution procedure in food- and water-sated rats. Alcohol Clin Exp Res (1986) 10:436–42. doi:10.1111/j.1530-0277.1986.tb05120.x
63. Morganstern I, Chang GQ, Chen YW, Barson JR, Zhiyu Y, Hoebel BG, et al. Role of melanin-concentrating hormone in the control of ethanol consumption: region-specific effects revealed by expression and injection studies. Physiol Behav (2010) 101:428–37. doi:10.1016/j.physbeh.2010.07.009
64. Morganstern I, Lukatskaya O, Moon SH, Guo WR, Shaji J, Karatayev O, et al. Stimulation of nicotine reward and central cholinergic activity in Sprague-Dawley rats exposed perinatally to a fat-rich diet. Psychopharmacology (Berl) (2013) 230:509–24. doi:10.1007/s00213-013-3178-6
65. Lumeng L, Li TK. The development of metabolic tolerance in the alcohol-preferring P rats: comparison of forced and free-choice drinking of ethanol. Pharmacol Biochem Behav (1986) 25:1013–20. doi:10.1016/0091-3057(86)90079-1
66. Lucchi L, Moresco RM, Govoni S, Trabucchi M. Effect of chronic ethanol treatment on dopamine receptor subtypes in rat striatum. Brain Res (1988) 449:347–51. doi:10.1016/0006-8993(88)91051-7
67. Pietzak ER, Wilce PA, Shanley BC. The effect of chronic ethanol consumption on muscarinic receptors in rat brain. Neurochem Int (1988) 12:447–52. doi:10.1016/0197-0186(88)90027-7
68. Lograno DE, Matteo F, Trabucchi M, Govoni S, Cagiano R, Lacomba C, et al. Effects of chronic ethanol intake at a low dose on the rat brain dopaminergic system. Alcohol (1993) 10:45–9. doi:10.1016/0741-8329(93)90052-P
69. Schramm-Sapyta NL, Kingsley MA, Rezvani AH, Propst K, Swartzwelder HS, Kuhn CM. Early ethanol consumption predicts relapse-like behavior in adolescent male rats. Alcohol Clin Exp Res (2008) 32:754–62. doi:10.1111/j.1530-0277.2008.00631.x
70. Lin HC, Wan FJ, Kang BH, Wu CC, Tseng CJ. Systemic administration of lipopolysaccharide induces release of nitric oxide and glutamate and c-fos expression in the nucleus tractus solitarii of rats. Hypertension (1999) 33:1218–24. doi:10.1161/01.HYP.33.5.1218
71. Yoshioka T, Goto M, Gottschalk ME, Anderson CL, Zeller WP. Plasma endotoxin concentration after an intraperitoneal injection of endotoxin in fed and fasted suckling rats. Shock (1994) 1:362–5. doi:10.1097/00024382-199405000-00008
72. Porter MH, Hrupka BJ, Langhans W, Schwartz GJ. Vagal and splanchnic afferents are not necessary for the anorexia produced by peripheral IL-1beta, LPS, and MDP. Am J Physiol (1998) 275:R384–9.
73. Lindros KO, Jarvelainen HA. Chronic systemic endotoxin exposure: an animal model in experimental hepatic encephalopathy. Metab Brain Dis (2005) 20:393–8. doi:10.1007/s11011-005-7924-2
74. Kim P, Park JH, Choi CS, Choi I, Joo SH, Kim MK, et al. Effects of ethanol exposure during early pregnancy in hyperactive, inattentive and impulsive behaviors and MeCP2 expression in rodent offspring. Neurochem Res (2013) 38:620–31. doi:10.1007/s11064-012-0960-5
75. Fischer CW, Elfving B, Lund S, Wegener G. Behavioral and systemic consequences of long-term inflammatory challenge. J Neuroimmunol (2015) 288:40–6. doi:10.1016/j.jneuroim.2015.08.011
76. Rothwell NJ, Stock MJ. Body weight and brown fat activity in hyperphagic cafeteria-fed female rats and their offspring. Biol Neonate (1986) 49:284–91. doi:10.1159/000242543
77. Salvado J, Segues T, Alemany M, Arola L. Effects of lactation on circulating plasma metabolites in ‘cafeteria-fed’ rats. Br J Nutr (1986) 55:139–47. doi:10.1079/BJN19860017
78. Page KC, Jones EK, Anday EK. Maternal and postweaning high-fat diets disturb hippocampal gene expression, learning, and memory function. Am J Physiol Regul Integr Comp Physiol (2014) 306:R527–37. doi:10.1152/ajpregu.00319.2013
79. Alati R, Najman JM, O’Callaghan M, Bor W, Williams GM, Clavarino A. Fetal growth and behaviour problems in early adolescence: findings from the Mater University Study of Pregnancy. Int J Epidemiol (2009) 38:1390–400. doi:10.1093/ije/dyp252
80. Rodriguez A, Miettunen J, Henriksen TB, Olsen J, Obel C, Taanila A, et al. Maternal adiposity prior to pregnancy is associated with ADHD symptoms in offspring: evidence from three prospective pregnancy cohorts. Int J Obes (Lond) (2008) 32:550–7. doi:10.1038/sj.ijo.0803741
81. Tozuka Y, Kumon M, Wada E, Onodera M, Mochizuki H, Wada K. Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem Int (2010) 57:235–47. doi:10.1016/j.neuint.2010.05.015
82. Poon K, Barson JR, Fagan SE, Leibowitz SF. Developmental changes in embryonic hypothalamic neurons during prenatal fat exposure. Am J Physiol Endocrinol Metab (2012) 303:E432–41. doi:10.1152/ajpendo.00238.2012
83. Naleid AM, Grace MK, Chimukangara M, Billington CJ, Levine AS. Paraventricular opioids alter intake of high-fat but not high-sucrose diet depending on diet preference in a binge model of feeding. Am J Physiol Regul Integr Comp Physiol (2007) 293:R99–105. doi:10.1152/ajpregu.00675.2006
84. Wei XJ, Sun B, Chen K, Lv B, Luo X, Yan JQ. Ghrelin signaling in the ventral tegmental area mediates both reward-based feeding and fasting-induced hyperphagia on high-fat diet. Neuroscience (2015) 300:53–62. doi:10.1016/j.neuroscience.2015.05.001
85. Koob GF, Nestler EJ. The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci (1997) 9:482–97. doi:10.1176/jnp.9.3.482
86. Bassareo V, Di Chiara G. Modulation of feeding-induced activation of mesolimbic dopamine transmission by appetitive stimuli and its relation to motivational state. Eur J Neurosci (1999) 11:4389–97. doi:10.1046/j.1460-9568.1999.00843.x
87. Cone JJ, Chartoff EH, Potter DN, Ebner SR, Roitman MF. Prolonged high fat diet reduces dopamine reuptake without altering DAT gene expression. PLoS One (2013) 8:e58251. doi:10.1371/journal.pone.0058251
88. Naef L, Srivastava L, Gratton A, Hendrickson H, Owens SM, Walker CD. Maternal high fat diet during the perinatal period alters mesocorticolimbic dopamine in the adult rat offspring: reduction in the behavioral responses to repeated amphetamine administration. Psychopharmacology (Berl) (2008) 197:83–94. doi:10.1007/s00213-007-1008-4
89. Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology (2010) 151:4756–64. doi:10.1210/en.2010-0505
90. Bakshi VP, Kelley AE. Feeding induced by opioid stimulation of the ventral striatum: role of opiate receptor subtypes. J Pharmacol Exp Ther (1993) 265:1253–60.
91. Zhang M, Kelley AE. Opiate agonists microinjected into the nucleus accumbens enhance sucrose drinking in rats. Psychopharmacology (Berl) (1997) 132:350–60. doi:10.1007/s002130050355
92. Geiger BM, Behr GG, Frank LE, Caldera-Siu AD, Beinfeld MC, Kokkotou EG, et al. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J (2008) 22:2740–6. doi:10.1096/fj.08-110759
93. Ong ZY, Muhlhausler BS. Maternal “junk-food” feeding of rat dams alters food choices and development of the mesolimbic reward pathway in the offspring. FASEB J (2011) 25:2167–79. doi:10.1096/fj.10-178392
94. Hosoda H, Kojima M, Kangawa K. Biological, physiological, and pharmacological aspects of ghrelin. J Pharmacol Sci (2006) 100:398–410. doi:10.1254/jphs.CRJ06002X
95. Egecioglu E, Jerlhag E, Salome N, Skibicka KP, Haage D, Bohlooly YM, et al. Ghrelin increases intake of rewarding food in rodents. Addict Biol (2010) 15:304–11. doi:10.1111/j.1369-1600.2010.00216.x
96. Grissom NM, Herdt CT, Desilets J, Lidsky-Everson J, Reyes TM. Dissociable deficits of executive function caused by gestational adversity are linked to specific transcriptional changes in the prefrontal cortex. Neuropsychopharmacology (2015) 40:1353–63. doi:10.1038/npp.2014.313
97. Primeaux SD, York DA, Bray GA. Neuropeptide Y administration into the amygdala alters high fat food intake. Peptides (2006) 27:1644–51. doi:10.1016/j.peptides.2005.12.009
98. Chang GQ, Karatayev O, Barson JR, Chang SY, Leibowitz SF. Increased enkephalin in brain of rats prone to overconsuming a fat-rich diet. Physiol Behav (2010) 101:360–9. doi:10.1016/j.physbeh.2010.06.005
99. Sasaki A, de Vega WC, St-Cyr S, Pan P, McGowan PO. Perinatal high fat diet alters glucocorticoid signaling and anxiety behavior in adulthood. Neuroscience (2013) 240:1–12. doi:10.1016/j.neuroscience.2013.02.044
100. Prasad A, Prasad C. Short-term consumption of a diet rich in fat decreases anxiety response in adult male rats. Physiol Behav (1996) 60:1039–42. doi:10.1016/0031-9384(96)00135-7
101. Krolow R, Noschang CG, Arcego D, Andreazza AC, Peres W, Goncalves CA, et al. Consumption of a palatable diet by chronically stressed rats prevents effects on anxiety-like behavior but increases oxidative stress in a sex-specific manner. Appetite (2010) 55:108–16. doi:10.1016/j.appet.2010.03.013
102. Ortolani D, Oyama LM, Ferrari EM, Melo LL, Spadari-Bratfisch RC. Effects of comfort food on food intake, anxiety-like behavior and the stress response in rats. Physiol Behav (2011) 103:487–92. doi:10.1016/j.physbeh.2011.03.028
103. Suter MA, Ma J, Vuguin PM, Hartil K, Fiallo A, Harris RA, et al. In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am J Obstet Gynecol (2014) 210:.e1–463. doi:10.1016/j.ajog.2014.01.045
104. Jacobsen SC, Brons C, Bork-Jensen J, Ribel-Madsen R, Yang B, Lara E, et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia (2012) 55:3341–9. doi:10.1007/s00125-012-2717-8
105. Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS One (2010) 5:e14398. doi:10.1371/journal.pone.0014398
106. Takanabe R, Ono K, Abe Y, Takaya T, Horie T, Wada H, et al. Up-regulated expression of microRNA-143 in association with obesity in adipose tissue of mice fed high-fat diet. Biochem Biophys Res Commun (2008) 376:728–32. doi:10.1016/j.bbrc.2008.09.050
107. Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology (2012) 153:2823–30. doi:10.1210/en.2011-2161
108. Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr (2007) 97:435–9. doi:10.1017/S0007114507352392
109. Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr (2007) 97:1064–73. doi:10.1017/S000711450769196X
110. Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr (2014) 5:71–81. doi:10.3945/an.113.004705
111. Chango A, Pogribny IP. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients (2015) 7:2748–70. doi:10.3390/nu7042748
112. Iwasa T, Matsuzaki T, Kinouchi R, Fujisawa S, Murakami M, Kiyokawa M, et al. Neonatal LPS injection alters the body weight regulation systems of rats under non-stress and immune stress conditions. Int J Dev Neurosci (2010) 28:119–24. doi:10.1016/j.ijdevneu.2009.08.015
113. Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract (2005) 69:29–35. doi:10.1016/j.diabres.2004.11.007
114. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
115. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature (2002) 420:333–6. doi:10.1038/nature01137
116. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med (2005) 11:191–8. doi:10.1038/nm1185
117. Yan X, Zhu MJ, Xu W, Tong JF, Ford SP, Nathanielsz PW, et al. Up-regulation of toll-like receptor 4/nuclear factor-kappaB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology (2010) 151:380–7. doi:10.1210/en.2009-0849
118. Poon K, Alam M, Karatayev O, Barson JR, Leibowitz SF. Regulation of the orexigenic neuropeptide, enkephalin, by PPARdelta and fatty acids in neurons of the hypothalamus and forebrain. J Neurochem (2015) 135:918–31. doi:10.1111/jnc.13298
119. Dudele A, Fischer CW, Elfving B, Wegener G, Wang T, Lund S. Chronic exposure to low doses of lipopolysaccharide and high-fat feeding increases body mass without affecting glucose tolerance in female rats. Physiol Rep (2015) 3:e12584. doi:10.14814/phy2.12584
120. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116:1494–505. doi:10.1172/JCI26498
121. Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, et al. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem (2002) 81:257–69. doi:10.1046/j.1471-4159.2002.00809.x
122. Banisadr G, Gosselin RD, Mechighel P, Rostene W, Kitabgi P, Melik Parsadaniantz S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J Comp Neurol (2005) 492:178–92. doi:10.1002/cne.20729
123. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi:10.1172/JCI24335C1
124. Kang YS, Cha JJ, Hyun YY, Cha DR. Novel C-C chemokine receptor 2 antagonists in metabolic disease: a review of recent developments. Expert Opin Investig Drugs (2011) 20:745–56. doi:10.1517/13543784.2011.575359
125. Pruis MG, Lendvai A, Bloks VW, Zwier MV, Baller JF, de Bruin A, et al. Maternal western diet primes non-alcoholic fatty liver disease in adult mouse offspring. Acta Physiol (Oxf) (2014) 210:215–27. doi:10.1111/apha.12197
126. Poon K, Ho HT, Barson JR, Leibowitz SF. Stimulatory role of the chemokine CCL2 in the migration and peptide expression of embryonic hypothalamic neurons. J Neurochem (2014) 131:509–20. doi:10.1111/jnc.12827
127. Poon K, Abramova D, Ho HT, Leibowitz SF. Prenatal fat-rich diet exposure alters responses of embryonic neurons to the chemokine, CCL2, in the hypothalamus. Neuroscience (2016) 324:407–19. doi:10.1016/j.neuroscience.2016.03.017
128. Mattson SN, Roebuck TM. Acquisition and retention of verbal and nonverbal information in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res (2002) 26:875–82. doi:10.1111/j.1530-0277.2002.tb02617.x
129. Roszel EL. Central nervous system deficits in fetal alcohol spectrum disorder. Nurse Pract (2015) 40:24–33. doi:10.1097/01.NPR.0000444650.10142.4f
130. Maier RV. Ethanol abuse and the trauma patient. Surg Infect (Larchmt) (2001) 2:133–41. doi:10.1089/109629601750469456
131. Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol (2003) 25:447–58. doi:10.1016/S0892-0362(03)00030-8
132. Margret CP, Li CX, Chappell TD, Elberger AJ, Matta SG, Waters RS. Prenatal alcohol exposure delays the development of the cortical barrel field in neonatal rats. Exp Brain Res (2006) 172:1–13. doi:10.1007/s00221-005-0319-0
133. Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol Clin Exp Res (1991) 15:395–8. doi:10.1111/j.1530-0277.1991.tb00536.x
134. Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics (2009) 4:500–11. doi:10.4161/epi.4.7.9925
135. Chen Y, Ozturk NC, Zhou FC. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS One (2013) 8:e60503. doi:10.1371/journal.pone.0060503
136. Miller MW. Migration of cortical neurons is altered by gestational exposure to ethanol. Alcohol Clin Exp Res (1993) 17:304–14. doi:10.1111/j.1530-0277.1993.tb00768.x
137. Chotro MG, Arias C. Prenatal exposure to ethanol increases ethanol consumption: a conditioned response? Alcohol (2003) 30:19–28. doi:10.1016/S0741-8329(03)00037-5
138. Youngentob SL, Glendinning JI. Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc Natl Acad Sci U S A (2009) 106:5359–64. doi:10.1073/pnas.0809804106
139. Arias C, Chotro MG. Increased palatability of ethanol after prenatal ethanol exposure is mediated by the opioid system. Pharmacol Biochem Behav (2005) 82:434–42. doi:10.1016/j.pbb.2005.09.015
140. Shea KM, Hewitt AJ, Olmstead MC, Brien JF, Reynolds JN. Maternal ethanol consumption by pregnant guinea pigs causes neurobehavioral deficits and increases ethanol preference in offspring. Behav Pharmacol (2012) 23:105–12. doi:10.1097/FBP.0b013e32834ed866
141. Nizhnikov ME, Molina JC, Varlinskaya EI, Spear NE. Prenatal ethanol exposure increases ethanol reinforcement in neonatal rats. Alcohol Clin Exp Res (2006) 30:34–45. doi:10.1111/j.1530-0277.2006.00009.x
142. Pautassi RM, Nizhnikov ME, Spear NE, Molina JC. Prenatal ethanol exposure leads to greater ethanol-induced appetitive reinforcement. Alcohol (2012) 46:585–93. doi:10.1016/j.alcohol.2012.05.004
143. Barson JR, Leibowitz SW. Hypothalamic neuropeptide signaling in alcohol addiction. Prog Neuropsychopharmacol Biol Psychiatry (2016) 65:321–9. doi:10.1016/j.pnpbp.2015.02.006
144. Angelogianni P, Gianoulakis C. Prenatal exposure to ethanol alters the ontogeny of the beta-endorphin response to stress. Alcohol Clin Exp Res (1989) 13:564–71. doi:10.1111/j.1530-0277.1989.tb00379.x
145. Lee S, Choi I, Kang S, Rivier C. Role of various neurotransmitters in mediating the long-term endocrine consequences of prenatal alcohol exposure. Ann N Y Acad Sci (2008) 1144:176–88. doi:10.1196/annals.1418.015
146. Wilson ME, Handa RJ. Gonadotropin secretion in infantile rats exposed to ethanol in utero. Alcohol (1997) 14:497–501. doi:10.1016/S0741-8329(97)00037-2
147. Lugo JN Jr, Marino MD, Gass JT, Wilson MA, Kelly SJ. Ethanol exposure during development reduces resident aggression and testosterone in rats. Physiol Behav (2006) 87:330–7. doi:10.1016/j.physbeh.2005.10.005
148. Gabriel KI, Cunningham CL. Effects of topiramate on ethanol and saccharin consumption and preferences in C57BL/6J mice. Alcohol Clin Exp Res (2005) 29:75–80. doi:10.1097/01.ALC.0000150014.79657.64
149. Glavas MM, Ellis L, Yu WK, Weinberg J. Effects of prenatal ethanol exposure on basal limbic-hypothalamic-pituitary-adrenal regulation: role of corticosterone. Alcohol Clin Exp Res (2007) 31:1598–610. doi:10.1111/j.1530-0277.2007.00460.x
150. Lee S, Rivier C. Long-term influence of an initial exposure to alcohol on the rat hypothalamic-pituitary axis. Alcohol Clin Exp Res (2003) 27:1463–70. doi:10.1097/01.ALC.0000086065.06203.DD
151. Gabriel KI, Yu CL, Osborn JA, Weinberg J. Prenatal ethanol exposure alters sensitivity to the effects of corticotropin-releasing factor (CRF) on behavior in the elevated plus-maze. Psychoneuroendocrinology (2006) 31:1046–56. doi:10.1016/j.psyneuen.2006.06.003
152. Lee S, Schmidt D, Tilders F, Rivier C. Increased activity of the hypothalamic-pituitary-adrenal axis of rats exposed to alcohol in utero: role of altered pituitary and hypothalamic function. Mol Cell Neurosci (2000) 16:515–28. doi:10.1006/mcne.2000.0890
153. Choi IY, Lee S, Rivier C. Novel role of adrenergic neurons in the brain stem in mediating the hypothalamic-pituitary axis hyperactivity caused by prenatal alcohol exposure. Neuroscience (2008) 155:888–901. doi:10.1016/j.neuroscience.2008.04.081
154. Wieczorek L, Fish EW, O’Leary-Moore SK, Parnell SE, Sulik KK. Hypothalamic-pituitary-adrenal axis and behavioral dysfunction following early binge-like prenatal alcohol exposure in mice. Alcohol (2015) 49:207–17. doi:10.1016/j.alcohol.2015.01.005
155. Varlinskaya EI, Truxell EM, Spear LP. Ethanol intake under social circumstances or alone in sprague-dawley rats: impact of age, sex, social activity, and social anxiety-like behavior. Alcohol Clin Exp Res (2015) 39:117–25. doi:10.1111/acer.12604
156. Lopez MF, Laber K. Impact of social isolation and enriched environment during adolescence on voluntary ethanol intake and anxiety in C57BL/6J mice. Physiol Behav (2015) 148:151–6. doi:10.1016/j.physbeh.2014.11.012
157. Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci (2007) 30:399–406. doi:10.1016/j.tins.2007.06.006
158. Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol (2014) 35:234–44. doi:10.1016/j.yfrne.2014.01.001
159. Bordner K, Deak T. Endogenous opioids as substrates for ethanol intake in the neonatal rat: the impact of prenatal ethanol exposure on the opioid family in the early postnatal period. Physiol Behav (2015) 148:100–10. doi:10.1016/j.physbeh.2015.02.013
160. Abate P, Hernandez-Fonseca K, Reyes-Guzman AC, Barbosa-Luna IG, Mendez M. Prenatal ethanol exposure alters met-enkephalin expression in brain regions related with reinforcement: possible mechanism for ethanol consumption in offspring. Behav Brain Res (2014) 274:194–204. doi:10.1016/j.bbr.2014.08.022
161. Joyce EM, Iversen SD. The effect of morphine applied locally to mesencephalic dopamine cell bodies on spontaneous motor activity in the rat. Neurosci Lett (1979) 14:207–12. doi:10.1016/0304-3940(79)96149-4
162. Kalivas PW, Widerlov E, Stanley D, Breese G, Prange AJ Jr. Enkephalin action on the mesolimbic system: a dopamine-dependent and a dopamine-independent increase in locomotor activity. J Pharmacol Exp Ther (1983) 227:229–37.
163. Morikawa H, Morrisett RA. Ethanol action on dopaminergic neurons in the ventral tegmental area: interaction with intrinsic ion channels and neurotransmitter inputs. Int Rev Neurobiol (2010) 91:235–88. doi:10.1016/S0074-7742(10)91008-8
164. Fabio MC, Vivas LM, Pautassi RM. Prenatal ethanol exposure alters ethanol-induced Fos immunoreactivity and dopaminergic activity in the mesocorticolimbic pathway of the adolescent brain. Neuroscience (2015) 301:221–34. doi:10.1016/j.neuroscience.2015.06.003
165. Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A (2009) 106:11318–23. doi:10.1073/pnas.0812809106
166. Zharkovsky T, Kaasik A, Jaako K, Zharkovsky A. Neurodegeneration and production of the new cells in the dentate gyrus of juvenile rat hippocampus after a single administration of ethanol. Brain Res (2003) 978:115–23. doi:10.1016/S0006-8993(03)02796-3
167. Aberg E, Hofstetter CP, Olson L, Brene S. Moderate ethanol consumption increases hippocampal cell proliferation and neurogenesis in the adult mouse. Int J Neuropsychopharmacol (2005) 8:557–67. doi:10.1017/S1461145705005286
168. Cullen CL, Burne TH, Lavidis NA, Moritz KM. Low dose prenatal ethanol exposure induces anxiety-like behaviour and alters dendritic morphology in the basolateral amygdala of rat offspring. PLoS One (2013) 8:e54924. doi:10.1371/journal.pone.0054924
169. Zhou R, Wang S, Zhu X. Prenatal ethanol exposure attenuates GABAergic inhibition in basolateral amygdala leading to neuronal hyperexcitability and anxiety-like behavior of adult rat offspring. Neuroscience (2010) 170:749–57. doi:10.1016/j.neuroscience.2010.07.055
170. Park PH, Miller R, Shukla SD. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem Biophys Res Commun (2003) 306:501–4. doi:10.1016/S0006-291X(03)01040-4
171. Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology (2007) 45:1306–12. doi:10.1002/hep.21650
172. Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res (2008) 32:1525–34. doi:10.1111/j.1530-0277.2008.00729.x
173. Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, et al. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet (2010) 6:e1000811. doi:10.1371/journal.pgen.1000811
174. Zhang CR, Ho MF, Vega MC, Burne TH, Chong S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin (2015) 8:40. doi:10.1186/s13072-015-0032-6
175. Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med (2000) 343:1467–76. doi:10.1056/NEJM200011163432007
176. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143:1158–72. doi:10.1053/j.gastro.2012.09.008
177. Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation (2008) 5:10. doi:10.1186/1742-2094-5-10
178. Blanco AM, Perez-Arago A, Fernandez-Lizarbe S, Guerri C. Ethanol mimics ligand-mediated activation and endocytosis of IL-1RI/TLR4 receptors via lipid rafts caveolae in astroglial cells. J Neurochem (2008) 106:625–39. doi:10.1111/j.1471-4159.2008.05425.x
179. Cippitelli A, Karlsson C, Shaw JL, Thorsell A, Gehlert DR, Heilig M. Suppression of alcohol self-administration and reinstatement of alcohol seeking by melanin-concentrating hormone receptor 1 (MCH1-R) antagonism in Wistar rats. Psychopharmacology (Berl) (2010) 211:367–75. doi:10.1007/s00213-010-1891-y
180. Rydell M, Cnattingius S, Granath F, Magnusson C, Galanti MR. Prenatal exposure to tobacco and future nicotine dependence: population-based cohort study. Br J Psychiatry (2012) 200:202–9. doi:10.1192/bjp.bp.111.100123
181. O’Callaghan FV, Al Mamun A, O’Callaghan M, Alati R, Najman JM, Williams GM, et al. Maternal smoking during pregnancy predicts nicotine disorder (dependence or withdrawal) in young adults – a birth cohort study. Aust N Z J Public Health (2009) 33:371–7. doi:10.1111/j.1753-6405.2009.00410.x
182. Klein LC, Stine MM, Pfaff DW, Vandenbergh DJ. Laternal nicotine exposure increases nicotine preference in periadolescent male but not female C57B1/6J mice. Nicotine Tob Res (2003) 5:117–24. doi:10.1080/1462220021000060446
183. Matta SG, Elberger AJ. Combined exposure to nicotine and ethanol throughout full gestation results in enhanced acquisition of nicotine self-administration in young adult rat offspring. Psychopharmacology (Berl) (2007) 193:199–213. doi:10.1007/s00213-007-0767-2
184. Chistyakov V, Patkina N, Tammimaki A, Talka R, Salminen O, Belozertseva I, et al. Nicotine exposure throughout early development promotes nicotine self-administration in adolescent mice and induces long-lasting behavioural changes. Eur J Pharmacol (2010) 640:87–93. doi:10.1016/j.ejphar.2010.04.044
185. Schneider T, Bizarro L, Asherson PJ, Stolerman IP. Hyperactivity, increased nicotine consumption and impaired performance in the five-choice serial reaction time task in adolescent rats prenatally exposed to nicotine. Psychopharmacology (Berl) (2012) 223:401–15. doi:10.1007/s00213-012-2728-7
186. Newman MB, Shytle RD, Sanberg PR. Locomotor behavioral effects of prenatal and postnatal nicotine exposure in rat offspring. Behav Pharmacol (1999) 10:699–706. doi:10.1097/00008877-199911000-00017
187. Slotkin TA, Cho H, Whitmore WL. Effects of prenatal nicotine exposure on neuronal development: selective actions on central and peripheral catecholaminergic pathways. Brain Res Bull (1987) 18:601–11. doi:10.1016/0361-9230(87)90130-4
188. Bluhm AL, Weinstein J, Sousa JA. Free radicals in tobacco smoke. Nature (1971) 229:500. doi:10.1038/229500a0
189. Ding YS, Zhang L, Jain RB, Jain N, Wang RY, Ashley DL, et al. Levels of tobacco-specific nitrosamines and polycyclic aromatic hydrocarbons in mainstream smoke from different tobacco varieties. Cancer Epidemiol Biomarkers Prev (2008) 17:3366–71. doi:10.1158/1055-9965.EPI-08-0320
190. Xu D, Liang G, Yan YE, He WW, Liu YS, Chen LB, et al. Nicotine-induced over-exposure to maternal glucocorticoid and activated glucocorticoid metabolism causes hypothalamic-pituitary-adrenal axis-associated neuroendocrine metabolic alterations in fetal rats. Toxicol Lett (2012) 209:282–90. doi:10.1016/j.toxlet.2012.01.006
191. Grove KL, Sekhon HS, Brogan RS, Keller JA, Smith MS, Spindel ER. Chronic maternal nicotine exposure alters neuronal systems in the arcuate nucleus that regulate feeding behavior in the newborn rhesus macaque. J Clin Endocrinol Metab (2001) 86:5420–6. doi:10.1210/jcem.86.11.8033
192. Morgan AJ, Harrod SB, Lacy RT, Stanley EM, Fadel JR. Intravenous prenatal nicotine exposure increases orexin expression in the lateral hypothalamus and orexin innervation of the ventral tegmental area in adult male rats. Drug Alcohol Depend (2013) 132:562–70. doi:10.1016/j.drugalcdep.2013.04.003
193. Morganstern I, Barson JR, Leibowitz SF. Regulation of drug and palatable food overconsumption by similar peptide systems. Curr Drug Abuse Rev (2011) 4:163–73. doi:10.2174/1874473711104030163
194. Toledo-Rodriguez M, Lotfipour S, Leonard G, Perron M, Richer L, Veillette S, et al. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am J Med Genet B Neuropsychiatr Genet (2010) 153B:1350–4. doi:10.1002/ajmg.b.31109
195. Fuchikami M, Morinobu S, Segawa M, Okamoto Y, Yamawaki S, Ozaki N, et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One (2011) 6:e23881. doi:10.1371/journal.pone.0023881
196. Chen H, Parker SL, Matta SG, Sharp BM. Gestational nicotine exposure reduces nicotinic cholinergic receptor (nAChR) expression in dopaminergic brain regions of adolescent rats. Eur J Neurosci (2005) 22:380–8. doi:10.1111/j.1460-9568.2005.04229.x
197. Kane VB, Fu Y, Matta SG, Sharp BM. Gestational nicotine exposure attenuates nicotine-stimulated dopamine release in the nucleus accumbens shell of adolescent Lewis rats. J Pharmacol Exp Ther (2004) 308:521–8. doi:10.1124/jpet.103.059899
198. Gold AB, Keller AB, Perry DC. Prenatal exposure of rats to nicotine causes persistent alterations of nicotinic cholinergic receptors. Brain Res (2009) 1250:88–100. doi:10.1016/j.brainres.2008.10.076
199. Fung YK, Lau YS. Effects of prenatal nicotine exposure on rat striatal dopaminergic and nicotinic systems. Pharmacol Biochem Behav (1989) 33:1–6. doi:10.1016/0091-3057(89)90419-X
200. Wei J, Wang J, Dwyer JB, Mangold J, Cao J, Leslie FM, et al. Gestational nicotine treatment modulates cell death/survival-related pathways in the brains of adolescent female rats. Int J Neuropsychopharmacol (2011) 14:91–106. doi:10.1017/S1461145710000416
201. Harrod SB, Lacy RT, Zhu J, Hughes BA, Perna MK, Brown RW. Gestational IV nicotine produces elevated brain-derived neurotrophic factor in the mesocorticolimbic dopamine system of adolescent rat offspring. Synapse (2011) 65:1382–92. doi:10.1002/syn.20975
202. Abdel-Rahman A, Dechkovskaia AM, Sutton JM, Chen WC, Guan X, Khan WA, et al. Maternal exposure of rats to nicotine via infusion during gestation produces neurobehavioral deficits and elevated expression of glial fibrillary acidic protein in the cerebellum and CA1 subfield in the offspring at puberty. Toxicology (2005) 209:245–61. doi:10.1016/j.tox.2004.12.037
203. Roy TS, Sabherwal U. Effects of gestational nicotine exposure on hippocampal morphology. Neurotoxicol Teratol (1998) 20:465–73. doi:10.1016/S0892-0362(97)00137-2
204. Roy TS, Seidler FJ, Slotkin TA. Prenatal nicotine exposure evokes alterations of cell structure in hippocampus and somatosensory cortex. J Pharmacol Exp Ther (2002) 300:124–33. doi:10.1124/jpet.300.1.124
205. Santiago SE, Huffman KJ. Postnatal effects of prenatal nicotine exposure on body weight, brain size and cortical connectivity in mice. Neurosci Res (2012) 73:282–91. doi:10.1016/j.neures.2012.05.005
206. Slotkin TA, Orband-Miller L, Queen KL, Whitmore WL, Seidler FJ. Effects of prenatal nicotine exposure on biochemical development of rat brain regions: maternal drug infusions via osmotic minipumps. J Pharmacol Exp Ther (1987) 240:602–11.
207. Aoyama Y, Toriumi K, Mouri A, Hattori T, Ueda E, Shimato A, et al. Prenatal nicotine exposure impairs the proliferation of neuronal progenitors, leading to fewer glutamatergic neurons in the medial prefrontal cortex. Neuropsychopharmacology (2016) 41:578–89. doi:10.1038/npp.2015.186
208. Bryden DW, Burton AC, Barnett BR, Cohen VJ, Hearn TN, Jones EA, et al. Prenatal nicotine exposure impairs executive control signals in medial prefrontal cortex. Neuropsychopharmacology (2016) 41:716–25. doi:10.1038/npp.2015.197
209. Navarro HA, Seidler FJ, Whitmore WL, Slotkin TA. Prenatal exposure to nicotine via maternal infusions: effects on development of catecholamine systems. J Pharmacol Exp Ther (1988) 244:940–4.
210. Haghighi A, Schwartz DH, Abrahamowicz M, Leonard GT, Perron M, Richer L, et al. Prenatal exposure to maternal cigarette smoking, amygdala volume, and fat intake in adolescence. JAMA Psychiatry (2013) 70:98–105. doi:10.1001/archgenpsychiatry.2012.1101
211. Xiao D, Dasgupta C, Li Y, Huang X, Zhang L. Perinatal nicotine exposure increases angiotensin II receptor-mediated vascular contractility in adult offspring. PLoS One (2014) 9:e108161. doi:10.1371/journal.pone.0108161
212. Ma N, Nicholson CJ, Wong M, Holloway AC, Hardy DB. Fetal and neonatal exposure to nicotine leads to augmented hepatic and circulating triglycerides in adult male offspring due to increased expression of fatty acid synthase. Toxicol Appl Pharmacol (2014) 275:1–11. doi:10.1016/j.taap.2013.12.010
213. Taki FA, Pan X, Lee MH, Zhang B. Nicotine exposure and transgenerational impact: a prospective study on small regulatory microRNAs. Sci Rep (2014) 4:7513. doi:10.1038/srep07513
214. Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet (2015) 24:2201–17. doi:10.1093/hmg/ddu739
215. Leslie FM. Multigenerational epigenetic effects of nicotine on lung function. BMC Med (2013) 11:27. doi:10.1186/1741-7015-11-27
216. Sopori ML, Kozak W, Savage SM, Geng Y, Soszynski D, Kluger MJ, et al. Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology (1998) 23:189–204. doi:10.1016/S0306-4530(97)00076-0
217. Kalra R, Singh SP, Pena-Philippides JC, Langley RJ, Razani-Boroujerdi S, Sopori ML. Immunosuppressive and anti-inflammatory effects of nicotine administered by patch in an animal model. Clin Diagn Lab Immunol (2004) 11:563–8. doi:10.1128/CDLI.11.3.563-568.2004
218. Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med (2002) 166:849–54. doi:10.1164/rccm.200202-097OC
Keywords: prenatal fat, prenatal ethanol, prenatal nicotine, inflammation, ingestive behavior
Citation: Poon K and Leibowitz SF (2016) Consumption of Substances of Abuse during Pregnancy Increases Consumption in Offspring: Possible Underlying Mechanisms. Front. Nutr. 3:11. doi: 10.3389/fnut.2016.00011
Received: 30 January 2016; Accepted: 04 April 2016;
Published: 20 April 2016
Edited by:
Patrick Christian Even, AgroParisTech, FranceReviewed by:
Emmanuel N. Pothos, Tufts University School of Medicine, USAAnna Aronis, The Hebrew University of Jerusalem, Israel
Copyright: © 2016 Poon and Leibowitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah F. Leibowitz, leibow@rockefeller.edu