
- 1 INSERM U624, Stress Cellulaire, Parc Scientifique et Technologique de Luminy, Marseille, France
- 2 Centre d’Investigation Clinique de Marseille, Marseille, France
- 3 Département d’Oncologie Médicale, Institut Paoli-Calmettes, Marseille, France
- 4 Département de Chirurgie Oncologique, Institut Paoli-Calmettes, Marseille, France
Pancreatic cancer is the fourth leading cause of cancer death with a median survival of 6 months and a dismal 5-year survival rate of 3–5%. The development and progression of pancreatic cancer are caused by the activation of oncogenes, the inactivation of tumor suppressor genes, and the deregulation of many signaling pathways. Therefore, the strategies targeting these molecules as well as their downstream signaling could be promising for the prevention and treatment of pancreatic cancer. However, although targeted therapies for pancreatic cancer have yielded encouraging results in vitro and in animal models, these findings have not been translated into improved outcomes in clinical trials. This failure is due to an incomplete understanding of the biology of pancreatic cancer and to the selection of poorly efficient or imperfectly targeted agents. In this review, we will critically present the current knowledge regarding the molecular, biochemical, clinical, and therapeutic aspects of pancreatic cancer.
Introduction
Pancreatic cancer is the fourth leading cause of cancer death with a median survival of 6 months and a dismal 5-year survival rate of 3–5% and this figure has remained relatively unchanged over the past 25 years (Jemal et al., 2006). Even for patients diagnosed with local disease, the 5-year survival rate is only 15%. Known risk factors for the disease include cigaret smoking, chronic and hereditary pancreatitis, late-onset diabetes mellitus, and familial cancer syndromes. The lethal nature of pancreatic cancer stems from its propensity to rapidly disseminate to the lymphatic system and distant organs. The presence of occult or clinical metastases at the time of diagnosis together with the lack of effective chemotherapies contributes to the high mortality in patients with pancreatic cancer. Pancreatic cancer is one of the most intrinsically drug-resistant tumors and resistance to chemotherapeutic agents is a major cause of treatment failure in pancreatic cancer. Gemcitabine is the standard chemotherapeutic drug for patients with advanced pancreatic cancer after a phase III trial in 1997 that demonstrated a modest survival advantage of this agent over 5-FU (median survival 5.65 versus 4.41 months, respectively), but surprisingly this treatment improved alleviation of disease-related symptoms (Burris et al., 1997). Very recently, a polychemotherapy regimen combining 5-FU, irinotecan, and oxaliplatin (FOLFIRINOX) was shown to nearly double overall survival compared to gemcitabine, at the expense of a manageable but increased toxicity, limiting its use to good performance status patients. In addition, overall survival was less than 12 months (Conroy et al., 2011). Therefore, there is a dire need for designing new and targeted therapeutic strategies that can overcome the drug-resistance and improve the clinical outcome for patients diagnosed with pancreatic cancer. For this purpose, the knowledge on the molecular aspects of pancreatic cancer is very important and is likely to be helpful in the design of new drugs and the molecular selection of existing drugs for targeted therapy. In the following sections we will summarize what is known regarding the precursor lesions of pancreatic cancer, the molecular aspects of pancreatic cancer, and how some of these molecular pathways could be exploited for the prevention or treatment of pancreatic cancer. In addition, we will describe the current knowledge on other major factors of pancreatic cancer development such as inflammation, telomerases, angiogenesis, miRNA, epigenetics and genetics factors, and pancreatic cancer stem cells, presently under study as possible therapeutic targets. We will also present the therapeutic strategies currently available. Finally, we will describe some promising chemopreventive agents and some new anticancer drugs which are in development.
Pancreatic Cancer Precursor Lesions
It is supposed, although not formally established, that like other epithelial cancers pancreatic cancers do not arise de novo but undergo a stepwise progression through histologically well-defined non-invasive precursor lesions, culminating in frank, invasive neoplasia. Although putative precursor lesions of pancreatic cancer were first documented over a century ago, it was only at the end of the last century that several lines of evidence began to associate invasive pancreatic cancer with these lesions. For example, autopsy studies confirmed that the prevalence of what is now recognized as precursor lesions increased with age, thus paralleling the frequency of invasive pancreatic cancer. Similarly, most surgically resected pancreas harboring invasive cancer also showed the presence of non-invasive intra-ductular lesions in the surrounding parenchyma, suggesting an etiologic association (Cubilla and Fitzgerald, 1976; Andea et al., 2003; Schwartz and Henson, 2007). Most importantly, careful molecular analyses over the last 10 years have unequivocally demonstrated that these precursor lesions share many of the underlying genetic alterations observed in the infiltrating cancer, underscoring their precursor status (Brat et al., 1998; Brockie et al., 1998). By the late 1990s, several terminologies were in use to describe these non-invasive ductal lesions, leading to considerable difficulties in comparing inter-institutional studies. Fortunately, in 1999 emerged a consensus nomenclature for precursor lesions of pancreatic cancer. The “pancreatic intraepithelial neoplasia” (PanIN) scheme for classifying these lesions, first proposed by Klimstra and Longnecker, has since become a gold standard at academic centers worldwide (Hruban et al., 2001, 2004). Briefly, PanINs are microscopic lesions in the smaller (less than 5 mm) pancreatic ducts. PanINs can be papillary or flat, and are composed of columnar to cuboidal cells with varying amounts of mucin. PanINs are classified into four groups, PanIN-1A, PanIN-1B (low-grade PanINs), PanIN-2 (intermediate grade PanINs), and PanIN-3 (high-grade PanINs), reflecting a progressive increase in histologic grade culminating in invasive neoplasia. PanIN lesions of the lowest grade can be flat (1A) or papillary (1B) but are characterized by absence of nuclear abnormalities and conserved nuclear polarity. PanIN-2 lesions are architecturally slightly more complex than PanIN-1 lesions and show more nuclear changes including loss of nuclear polarity, nuclear crowding, variation in nuclear size (pleomorphism), nuclear hyperchromasia, and nuclear pseudostratification. Mitoses are rarely seen. In contrast, PanIN-3 lesions, also referred to as “carcinoma-in situ,” demonstrate widespread loss of polarity, nuclear abnormalities, and frequent mitoses. However, as a pre-invasive lesion, PanIN-3 is still contained within the basement membrane.
If PanINs are the most common proposed precursor lesions for pancreatic cancer, they are not the only ones. Intraductal papillary mucinous neoplasms (IPMNs) are other precursor lesions, so called because they typically present as radiologically detectable cysts in the pancreas (Winter et al., 2006). IPMNs are mucin-producing epithelial neoplasms, which arise within the main pancreatic duct or one of its branches, and have often, although not always, a papillary architecture (Schmitz-Winnenthal et al., 2003; Hruban et al., 2004). By definition, IPMNs are found in the larger pancreatic ducts. Those that involve the main pancreatic ducts are designated “main duct type,” while those that involve the secondary branches of the main pancreatic duct are designated “branch duct type” (Hruban et al., 2004; Longnecker et al., 2005). Similar to PanINs, the cystic precursor lesions also demonstrate a multistep histological and genetic progression to invasive neoplasia.
Molecular Mechanisms Involved in the Pathogenesis of Pancreatic Cancer
Intensive investigation of molecular pathogenesis will help identifying useful molecules for diagnosis, treatment, and prognosis of pancreatic cancer. In the last years, considerable research has focused on identifying molecular events associated with pancreatic carcinogenesis and their correlation with the pathological status. Multiple subsets of genes were found to be activated or inactivated during the development and progression of pancreatic cancer. The activation of oncogenes and the inactivation of tumor suppressor genes are in part responsible for the initiation and progression of pancreatic cancers. Moreover, the deregulation of molecules in several signaling pathways, such as EGFR, Akt, NFκB, CCKR, Hedgehog, etc., and their molecular crosstalk also play important roles in the molecular pathogenesis of pancreatic cancer (see Figure 1).
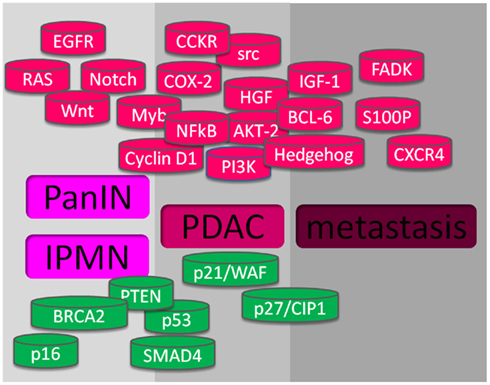
Figure 1. Altered genes in pancreatic cancer. At the upper part, in red, are shown the gene alterations with gain-of-function, and at the lower part, in green, are showed the genes with lost-of-function, that promote pre-cancerous lesions PanIN and IPMN, PDAC, and metastasis.
Activation of Oncogenes
The Ras Activated Pathway
Oncogenes can be activated through different mechanisms including point mutation and amplification. The activation of the ras oncogene has been observed in more than 90% of pancreatic cancers (Almoguera et al., 1988). The ras gene family encodes three 21-kDa membrane-bound proteins involved in signal transduction and mediating pleiotropic effects including cell proliferation and migration. Activated ras is involved in growth factor-mediated signal transduction pathways. About 80–90% of pancreatic cancers harbor point mutations at codons 12, 13, and 61 in K-ras (Almoguera et al., 1988). This is the highest level of K-ras alteration found in any human tumor type. The point mutation leads to the generation of a constitutively active form of ras. The constitutively activated ras binds to GTP and gives uncontrolled stimulation signals to downstream signaling cascades, promoting uncontrolled cell growth. K-ras mutations in pancreatic cancer typically develop during the early phase of carcinogenesis and patients with mutated K-ras have a shorter survival than patients with wild-type K-ras, suggesting that the mutation of K-ras participates in the initiation and progression of pancreatic cancer. In addition to point mutations, amplification of ras is also frequently observed in pancreatic cancers, suggesting that activation of the ras oncogene is an important molecular event in pancreatic cancers.
A peptide vaccine that aims to stimulate immunity against cancer cells with mutant Ras proteins has been tested as an adjuvant treatment in patients with pancreatic cancer (Toubaji et al., 2008). An extension to this research investigated the effects of combination therapy with mutant Ras peptide plus granulocyte–macrophage colony-stimulating factor (Gjertsen et al., 2001). For Ras to function, it must undergo post-translational modification so that it can attach to the cell membrane. One essential step involves the addition of a 15-carbon isoprenoid chain, mediated by farnesyltransferase. The therapeutic use of tipifarnib, a farnesyltransferase inhibitor (FTI), in combination with gemcitabine was disappointing in a phase III trial (Van Cutsem et al., 2004). This finding could be partly explained by the fact that K-ras can be alternatively prenylated by the addition of a 20-carbon isoprenoid moiety mediated by the enzyme geranylgeranyltransferase. Moreover, FTIs work largely by inhibition of the cell cycle, but gemcitabine needs cell cycle progression to be effective. To this end, a dual inhibitor of farnesyltransferase and geranylgeranyltransferase (L-778,123) was tested in a phase I trial in combination with radiotherapy for locally advanced pancreatic cancer (Martin et al., 2004). Inhibition of farnesylation and sensitivity to radiotherapy was demonstrated in a patient-derived cell line. Further development of this drug was, nevertheless, halted owing to adverse cardiac effects. Other compounds that are in early phases of clinical testing after yielding promising laboratory results include romidepsin, a histone deacetylase inhibitor that inhibits Ras-mediated signal transduction and thus causes cell cycle arrest, and farnesylthiosalicylic acid (salirasib), which disrupts Ras from its membrane-binding site. These compounds seem to have clinical activity in combination with gemcitabine and further studies are necessary. MAP2K, the principal downstream component of Ras signaling, has also been the subject of targeted inhibition. In a phase II trial, the inhibitor CI-1040 (PD184532) did not demonstrate enough anti-tumor activity to justify further development (Rinehart et al., 2004). Nevertheless, combined inhibition of MAP2K and other kinases (such as EGFR) has been effective in preclinical studies, which suggests that this approach might still have a role in therapy for pancreatic cancer (Jimeno et al., 2007; Takayama et al., 2008).
The Notch Pathway
Notch has also been considered as an oncogene involved in the pathogenesis of pancreatic cancer (Miyamoto et al., 2003). So far, four Notch genes have been identified (Notch-1, Notch-2, Notch-3, and Notch-4) and five Notch ligands (Dll-1, Dll-3, Dll-4, Jagged-1, and Jagged-2) have been found in mammals. Notch protein can be activated by interacting with its ligands. Upon activation, Notch protein is cleaved by the γ-secretase, releasing intracellular Notch which translocates into the nucleus. The intracellular Notch associates with transcriptional factors which regulate the expression of target genes and thus play an important role in both organ development and pancreatic carcinogenesis. In fact, Notch signaling is frequently deregulated in human pancreatic cancers (Buchler et al., 2005; Wang et al., 2006c,e). Notch signaling occurs downstream of Ras, EGFR, and TGF-α signaling in pancreatic tumorigenesis and promotes tumor vascularization. Down-regulation of Notch-1 with siRNA or curcumin (owing to the crosstalk between Notch and NFκB signaling pathways) can inhibit cell growth and induce apoptosis in pancreatic cancer cell lines in vitro (Wang et al., 2006a,d).
The Cyclooxygenase Pathway
The cyclooxygenase (COX) enzymes promote the formation of prostaglandins, leading to the induction of cell growth. There are two isoforms of the COX enzyme. COX-1 is produced at a constant rate and the prostaglandins formed are involved in several normal physiologic events. COX-2, in contrast, is an inducible enzyme, absent from most normal tissues. However, its synthesis is stimulated in inflammatory and carcinogenic processes by cytokines, growth factors, and other cancer promoters. COX-2 has been shown to be increased in a variety of cancers including pancreatic cancers. Several pancreatic cancer cell lines strongly express COX-2. Immunohistochemical studies have shown that about 50% of human pancreatic cancers over-express COX-2 and that COX-2 mRNA expression is much higher in tumors than in normal surrounding tissue (Okami et al., 1999). Moreover, there is a positive association between ras mutation and COX-2 level because activated ras increases the stability of COX-2 mRNA. Therefore, COX-2 appears to be of significance in pancreatic carcinogenesis and a crosstalk between ras, NFκB, Notch, and COX-2 in cellular signaling might contribute to the molecular pathogenesis of pancreatic cancer.
As indicated above, COX-2 and its metabolic product (PGE2) play important roles in pancreatic cancer, suggesting that targeting COX-2 could provide a therapeutic benefit. Several COX-2 inhibitors have shown activity in reducing tumor growth with different mechanisms. Indomethacin, one of the COX-2 inhibitors, inhibits both isoforms of the COX enzyme while newer agents, such as celecoxib, inhibit only COX-2 and are more desirable for clinical use. In an orthotopic pancreatic cancer animal model, celecoxib treatment showed inhibition of tumor growth, angiogenesis, and metastasis (Wei et al., 2004). Non-steroidal anti-inflammatory drugs (NSAIDs) also show their inhibitory effect on COX-2 and PGE2, leading to a reduced incidence of tumor formation and a reduced number of tumors per animal. Experiments using two NSAIDs, sulindac and NS398, in pancreatic cancer cell lines, have shown that both agents cause a dose-dependent inhibition of cancer cell growth. A combination of the agents, celecoxib and Zyflo (a 5-lipoxygenase inhibitor), has shown a reduction in the incidence and size of pancreatic tumors, and a reduced number of liver metastases in animal model. It has been reported that celecoxib potentiates gemcitabine-induced growth inhibition through the down-regulation of NFκB activation and induction of apoptosis in pancreatic cancer cells (El-Rayes et al., 2004). In a clinical trial, patients with advanced pancreatic adenocarcinoma were given celecoxib in combination with 5-fluorouracil. Such association was capable of inducing durable and objective responses, even in gemcitabine-resistant pancreatic cancer (Milella et al., 2004). These results collectively suggest that COX-2 inhibition with other novel agents could be useful in future clinical trials for pancreatic cancer.
The Hepatocyte Growth Factor Receptor Pathway
The MET oncogene, which encodes the receptor for hepatocyte growth factor (HGF), is overexpressed in 78% of pancreatic cancers (Furukawa et al., 1995). HGF is normally produced by mesenchymal cells and acts on epithelial cells to promote tissue regeneration. In hypoxic conditions, however, tumor-associated fibroblasts produce HGF, which stimulates angiogenesis and tumor growth, enhances cell motility and extracellular matrix breakdown and leads to invasion and metastasis. Targeting the HGF pathway with use of a synthetic competitive antagonist of HGF (Tomioka et al., 2001) and an antibody against the MET receptor (Jin et al., 2008) has yielded encouraging results in the laboratory setting. ARQ 197 is a MET receptor tyrosine kinase inhibitor that is currently being tested in a phase II trial. A phase I study showed that it was well-tolerated by patients (Munshi et al., 2010).
The Insulin-Like Growth Factor Pathway
The insulin-like growth factor I (IGF-I) receptor, a transmembrane tyrosine kinase receptor, is overexpressed in 64% of pancreatic cancers (Hakam et al., 2003). The IGF-I receptor has antiapoptotic and growth-promoting effects and acts via multiple signaling cascades, including the PI3-Akt, MAPK, and STAT pathways. Inhibition of the IGF-I receptor by the tyrosine kinase inhibitor NVP-AEW541, a dominant-negative mutant, and RNA interference have all been shown to reduce the growth of pancreatic cancer cells in vitro and in vivo, and increase chemotherapy-induced or radiation-induced apoptosis (Piao et al., 2008). Concomitant inhibition of K-ras increases the therapeutic effect of IGF-I receptor antisense oligonucleotide (Shen et al., 2008). Human anti-IGF-I receptor antibodies have been reported to enhance the anti-tumor effects of gemcitabine and EGFR inhibition in vivo (Beltran et al., 2009).
The Focal Adhesion Kinase Pathway
Focal adhesion kinase (FADK) is a cytoplasmic non-receptor tyrosine kinase that mediates functions involved in cell motility and survival and is closely related to the integrin signaling pathway. Close to 80% of pancreatic cancers (Furuyama et al., 2006) express FADK and, importantly, it shares a pathway with the IGF-I receptor (Liu et al., 2008). The dual IGF-I receptor–FADK inhibitor NVP-TAE226 has shown significant tumor-suppressive activity in vivo (Liu et al., 2007).
The Src Pathway
Src is one of the nine members of the Src family of non-receptor protein tyrosine kinases. In normal conditions, Src is maintained in a phosphorylated and inactive form but it is activated in a number of malignancies, including in 70% of pancreatic cancers (Hakam et al., 2003). Src has diverse roles in cell proliferation, survival, motility, invasiveness, resistance to chemotherapy, and angiogenesis. This protein acts via multiple signaling pathways and, therefore, is an ideal target for therapeutic intervention. Src kinase inhibitors have been effective in suppressing pancreatic tumor growth and metastasis in vivo (Baker et al., 2006; Trevino et al., 2006; Ischenko et al., 2007). Dasatinib is an orally active multitargeted kinase inhibitor of Src, BCR–ABL, platelet-derived growth factor receptor (PDGFR), ephrin type A receptor 2, and SCFR, and is licensed for the treatment of chronic myelogenous leukemias and acute lymphoblastic leukemias. Dasatinib is being examined in a phase II trial in patients with metastatic pancreatic cancer (Morton et al., 2010).
The Wnt Pathway
Wnt signaling is involved in normal embryonic development and homeostatic self-renewal of a number of adult tissues. Three Wnt signaling cascades have been described, the canonical Wnt–β-catenin, the planar-cell polarity, and the Wnt–Ca2+ pathways. The former is the best known and has been implicated in a variety of cancers including liver, colorectal, breast, prostate, renal, and hematological malignancies. Normally, β-catenin is phosphorylated and targeted for degradation. However, binding of Wnt proteins results in activation of intracellular pathways that cause β-catenin to enter the nucleus, where its interaction with the T-cell factor (TCF) and lymphoid enhancer factor (LEF) families of transcription factors leads to targeted gene expression. Any gain-of-function mutation of activators or loss-of-function mutation of inhibitors of Wnt signaling could lead to aberrant activation of these signaling pathways, which could result in carcinogenesis and tumor progression. Aberrant activation occurs in 65% of pancreatic cancers (Zeng et al., 2006). Inhibition of Wnt signaling to reduce proliferation and increase apoptosis of pancreatic cancer cells has been achieved in the laboratory setting by a variety of methods, including the use of β-catenin-interacting protein 1, a dominant-negative mutant of LEF-1, and siRNA against β-catenin or extracellular sulfatases (Nawroth et al., 2007; Pasca di Magliano et al., 2007). Wnt signaling is positively regulated by the hedgehog and SMAD4 signaling pathways (Romero et al., 2008), which could be targets for a combined inhibitory therapeutic strategy.
The Chemokine Receptor 4 and Its Ligand SDF-1
The chemokine receptor 4 (CXCR4) and its ligand, SDF-1 have a role in tumor growth, angiogenesis, and particularly in metastatic spread. In vitro blockade of CXCR4 inhibits pancreatic cancer growth through inhibition of the canonical Wnt pathway (Wang et al., 2008). Furthermore, plerixafor, an antagonist of CXCR4, reduces metastasis from pancreatic cancer cells positive for the CXCR4 and CD133 markers (the latter being a marker of pancreatic cancer stem cells) in vivo (Hermann et al., 2007).
Other Oncogenes
Amplification of other oncogenes also plays a very important role in the development and progression of pancreatic cancer. The Akt-2 gene is amplified in 15% of pancreatic cancers while Myb gene is amplified in 10% of pancreatic cancers. The amplification of the oncogenes contributes to the stimulation of cell growth and the progression of pancreatic cancers. There are experimental evidences of up-regulation of other oncogenes including Bcl-6, S100P, and Cyclin D1 (Mimeault et al., 2005). Up-regulation of cyclin D1 has been found in pancreatic cancers and overexpression of cyclin D1 is associated with poor prognosis. Inhibiting cyclin D1 in pancreatic cancer cell lines leads to growth inhibition and loss of tumorigenicity in nude mice (Kornmann et al., 1998). Recent studies have shown that overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase–myc transgene-expressing pancreatic tumor cell line, suggesting that cyclin D1 influences the progression of pancreatic cancer (Biliran et al., 2005).
Inactivation of Tumor Suppressor Genes
Inactivation of tumor suppressor genes is another important event for the initiation of pancreatic cancer. Tumor suppressor genes can be activated by mutation, deletion, or hypermethylation. The tumor suppressor genes targeted in pancreatic cancer include p16, p53, SMAD4, and PTEN (see Figure 1).
The P16/INK4a Pathway
It has been shown that p16/INK4a inhibits the activity of cyclin D and of the CDK4/6 complex. CDK4 and CDK6 normally interact with cyclin D to phosphorylate the retinoblastoma (Rb) protein. The phosphorylation of Rb allows its dissociation from a complex formed with the elongation factor 2 (E2F), allowing E2F to activate genes required for DNA synthesis along the cell cycle. p16/INK4a controls cell cycle progression through G1/S transition by inhibiting cyclin D and CDK4/6 mediated phosphorylation of Rb, and therefore inhibiting cell growth. Approximately 95% of pancreatic cancer patients have inactivated p16/INK4a in their tumors (about 40% by deletion, 40% by mutation, and 15% by hypermethylation of its promoter; Schutte et al., 1997). Experimental studies have demonstrated that transfection of wild-type p16/INK4a into human pancreatic cancer cells results in decreased tumor cell proliferation in vitro and in vivo. Moreover, in pancreatic cancer patients the tumor size is significantly larger and the survival time is significantly shorter when the p16/INK4a mutation is present, compared to patients with wild-type p16/INK4a. These evidences suggest that p16/INK4a alterations participate in the aggressiveness of pancreatic cancer through its interaction with various cellular signaling pathways.
The P53 Pathway
In addition to p16/INK4a, another tumor suppressor gene, named p53, is well known to be involved in the control of cell cycle. p53 Binds to the p21/WAF1 promoter and stimulates the production of p21/WAF1, which negatively regulates the complex consisting of cyclin D1 and CDK2, thereby arresting the cell at the G1 phase and inhibiting cell growth. The p53 also plays important roles in the induction of apoptotic cell death. Inactivation of p53 during carcinogenesis can lead to uncontrolled cell growth and increased cell survival. The p53 gene is inactivated in about 50% of pancreatic cancers through gene mutation and deletion. It had been shown that p53 mutation results in alteration of the 3D structure of the p53 protein (Li et al., 1998), associated with shorter survival in patients with pancreatic cancer. In addition, alterations in the p53 gene are associated with K-ras mutations, suggesting a crosstalk and cooperative activity between p53 and K-ras in the molecular pathogenesis of pancreatic cancers. Moreover, the status of p53 is important in mediating the specificity of chemotherapeutic agents on cancers. Loss of p53 function could result in decreased sensitivity to certain types of chemotherapeutic agents. Therefore, the status of p53 could be a useful guide for those patients who are likely to respond to adjuvant chemotherapy.
The TGF-β and SMAD4 Pathway
TGF-β is a cytokine secreted by epithelial, endothelial, hematopoietic, and mesenchymal cells. Binding of TGF-β to type I and type II TGF-β receptors forms a heteromeric complex that triggers the phosphorylation of cytoplasmic SMAD2 and SMAD3. In turn, SMAD proteins form a complex with SMAD4, which translocates into the nucleus to activate gene transcription. TGF-β can also signal via SMAD-independent pathways that involve Ras, PI3K, and MAPK. TGF-β mediates a wide range of physiological processes, such as embryonic development, tissue repair, angiogenesis, and immunosuppression. TGF-β also has a complex role in tumorigenesis, as it is tumor-suppressive in epithelial cells, but promotes invasion and metastasis during the late stages of cancer progression. Mutations of the TGFBR1 and TGFBR2 genes are found in about 1 and 4% of pancreatic cancers, respectively (Goggins et al., 1998b).
The inactivation of DPC4 (Deleted in Pancreatic Cancer locus 4, Smad4) tumor suppressor gene is another common genetic alteration identified in pancreatic cancer. The DPC4 gene encodes a 64-kDa protein, Smad 4, which plays roles in the inhibition of cell growth and angiogenesis. The inactivation of DPC4 is relatively specific to pancreatic cancer although it occurs with low incidence in other cancers. It has been found that the DPC4 tumor suppressor gene is deleted in approximately 50% of pancreatic cancers (Cowgill and Muscarella, 2003). Up to 90% of pancreatic adenocarcinomas could harbor loss of heterozygosity. DPC4 alterations occur relatively late in pancreatic carcinogenesis. The frequency of loss of DPC4 expression is significantly higher in poorly differentiated pancreatic adenocarcinoma and the pancreatic cancer patients with intact DPC4 gene have significantly longer survival after resection compared to patients with mutant DPC4 gene. Furthermore, DPC4 inactivation is always accompanied by inactivation of p16/INK4a, supporting its importance in the pathogenesis of pancreatic cancer. It is noteworthy that inactivation of SMAD4 abolishes TGF-β-mediated tumor-suppressive functions while it maintains some tumor-promoting TGF-β responses, such as epithelial–mesenchymal transition, which makes cells more invasive and more prone to migrate (Levy and Hill, 2005).
TGF-β-based therapeutic strategies are currently in development, including inhibitors of TGFBR1 and TGFBR2. AP 12009, an antisense oligonucleotide specific to TGF-β2, is currently being tested in a phase I–II study of malignant melanoma, pancreatic cancer, and colorectal carcinomas (Bonafoux and Lee, 2009).
Other Tumor Suppressor Genes
As indicated earlier, p21/WAF1 is an inhibitor of CDK. It forms complexes with cyclinA/CDK2 or cyclinD1/CDK4 and inhibits their activity, causing cell cycle arrest in G1 phase. Loss of p21WAF1 activity has been observed in approximately 50% of pancreatic cancers (Garcea et al., 2005). p27/CIP1 is another CDK inhibitor which regulates cell cycle progression from G1 to S phases. The loss of p27/CIP1 expression has also been observed in pancreatic cancers (Garcea et al., 2005). Another tumor suppressor gene, BRCA2, has been found to participate in DNA damage repair and mutations in BRCA2 have been linked to a significantly increased risk of pancreatic cancer. These evidences demonstrate that inactivation of p21/WAF1, p27/CIP1, and BRCA2 tumor suppressor genes is involved in the pathogenesis of pancreatic cancer.
Intracellular Signaling Pathways Involved in Pancreatic Cancer
The EGFR Signaling
EGFR consists of an extracellular ligand-binding domain, a hydrophobic transmembrane region, and an intracellular tyrosine kinase domain. EGFR is a member of the ErbB family of receptor tyrosine kinases, which include ErbB-1 (EGFR), ErbB-2 (HER-2), ErbB-3, and ErbB-4. The principal ligands of EGFR are EGF and TGF-α. Binding of a ligand to EGFR induces receptor dimerization, which results in intracellular transphosphorylation of tyrosine residues. Phosphorylation of EGFR activates molecules in different cell signaling pathways including PI3K, Src, MAPK, and STAT, inducing cell cycle progression, cell division, survival, motility, invasion, and metastasis. The genomic alterations of EGFR that occur in cancers include overexpression, mutation, deletion, and rearrangement. These alterations of EGFR induce the activity of tyrosine kinase receptors and may promote the development and progression of pancreatic cancer. Experimental studies have shown that EGFR activation plays important roles in proliferation, apoptosis inhibition, angiogenesis, metastasis, and resistance to chemotherapy or radiation therapy. Overexpression of EGF and EGFR is a common feature of human pancreatic cancer (Talar-Wojnarowska and Malecka-Panas, 2006). HER-2/neu amplification and p185 overexpression have been observed in more than 50% of pancreatic cancers. Moreover, EGFR overexpression has been found significantly more often in advanced clinical stages of pancreatic cancer and thus is associated with shorter survival in pancreatic cancer patients (Talar-Wojnarowska and Malecka-Panas, 2006), suggesting that deregulation of the EGFR pathway participates in the development and progression of pancreatic cancer. The EGFR signaling and its downstream signaling is therefore an important signaling pathway to be targeted for pancreatic cancer therapy.
Two classes of EGFR inhibitors, monoclonal antibodies, and small molecule tyrosine kinase inhibitors have been considered in the treatment of pancreatic cancer but results of comparative clinical trials have been relatively disappointing (see above). Because of the importance of EGFR in pancreatic cancer a novel negative regulator of EGFR, termed EGFR-related protein (ERRP), whose expression was found to attenuate EGFR activation was developed. ERRP significantly inhibits cell proliferation and induces apoptosis in BxPC3, HPAC, and Panc1 pancreatic cancer cells (Zhang et al., 2005, 2006). ERRP also inhibits ligand-induced activation of EGFR, HER-2, and HER-3. Most importantly, ERRP was found to inhibit pancreatic tumor growth in a SCID mouse xenograft model. The anti-tumor activity of ERRP correlated well with down-regulation of NFκB, MAPK, Akt, and Notch-1 (Zhang et al., 2005, 2006; Wang et al., 2006b). ERRP also down-regulates genes downstream from NFκB such as vascular endothelial growth factor (VEGF) and MMP-9, and inhibits cancer cell invasion, suggesting that ERRP could be a very potent agent for the treatment of pancreatic cancer by inhibiting cell survival signaling, tumor growth, invasion, and angiogenesis. These results suggest that further development of ERRP for clinical use is mandatory.
The Akt Signaling
EGF binding and subsequent EGFR, ras, or Src activation lead to the activation of the PI3K pathway. Activated PI3K phosphorylates phosphatidylinositides (PIP3), which eventually phosphorylate and activate Akt which in turn has multiple downstream targets, including the mammalian target of rapamycin (mTOR) and the transcription factor NFκB. Phosphorylated Akt (p-Akt) promotes cell survival by inhibiting apoptosis and activating NFκB. p-Akt is known to inhibit apoptosis through its ability to phosphorylate and inactivate several targets including Bad, Forkhead transcription factors, and caspase-9, all of which are involved in the apoptotic pathway. Akt also regulates the NFκB pathway via phosphorylation and activation of molecules in the NFκB pathway, suggesting that there is a cross talk between these two signaling pathways. AKT2 is amplified and the PI3K–Akt pathway is activated in 20 and 60% of pancreatic cancers, respectively (Ruggeri et al., 1998; Schlieman et al., 2003). Deregulation of this pathway through aberrant expression of PTEN (phosphatase and tensin homolog, a natural antagonist of PI3K) is frequently observed in pancreatic cancer (Asano et al., 2004). Furthermore, an architectural transcription factor, HMGA1, is overexpressed in pancreatic cancer (Abe et al., 2000). This transcription factor activates PI3K–Akt signaling and seems to mediate resistance to gemcitabine (Liau and Whang, 2008), which, therefore, provides another target for inhibition therapy (Trapasso et al., 2004; Liau et al., 2006). Furthermore, the inhibition of Akt decreases the function of NFκB, and has been shown to sensitize MiaPaCa2 pancreatic cancer cells to chemotherapy (Fahy et al., 2004), suggesting that both Akt and NFκB are potential targets for the treatment of pancreatic cancers.
Temsirolimus is an mTOR inhibitor approved for the treatment of renal-cell carcinoma, but use of this agent in pancreatic cancer has been limited. Other agents, including everolimus and sirolimus, are currently in phase II clinical trials (Wolpin et al., 2009). A combination of an mTOR inhibitor with other standard or targeted therapies might be needed, as mTOR expression does not correlate with survival of patients.
The NFκB Signaling
The NFκB signaling pathway plays important roles in the control of cell growth, differentiation, apoptosis, inflammation, stress response, and many other physiological processes in cellular signaling (Karin, 2006). In human cells without specific signal, NFκB is sequestered in the cytoplasm through tight association with its inhibitors: IκBα which acts as an NFκB inhibitor and p100 proteins which serves as both an inhibitor and precursor of NFκB DNA-binding subunits. NFκB can be activated through phosphorylation of IκBα by IKKβ and/or phosphorylation of p100 by IKKα, leading to degradation of IκBα and/or the processing of p100 into a smaller form (p52). This process allows two forms of NFκB (p50–p65 and p52–RelB) to become free, resulting in the translocation of active NFκB into the nucleus for binding to NFκB-specific DNA-binding sites and, in turn, regulating gene transcription. By binding to the promoters of target genes, NFκB controls the expression of many genes [i.e., survivin, MMP-9, urokinase-type plasminogen activator (Upa), and VEGF] that are involved in cell survival, apoptosis, invasion, metastasis, and angiogenesis.
NFκB is constitutively activated in most human pancreatic cancer tissues and cell lines but not in normal pancreatic tissues and cells, suggesting that the activation of NFκB is involved in the carcinogenesis of pancreas. Inhibition of NFκB by a super-inhibitor of NFκB results in impaired proliferation and induction of apoptosis (Liptay et al., 2003), suggesting an important role of NFκB in pancreatic tumorigenesis. Moreover, the inhibition of constitutive NFκB activity completely suppresses metastasis in liver of the pancreatic cancer cell line ASPC1 (Fujioka et al., 2003). An experimental study also demonstrates that uPA, one of the critical proteases involved in tumor invasion and metastasis, is overexpressed in pancreatic cancer cells, and its overexpression is induced by constitutive NFκB activity (Wang et al., 1999). These results suggest that constitutively activated NFκB is tightly related to the invasion and metastasis frequently observed in pancreatic cancers. Moreover, the deregulation of NFκB could also be due to Notch signaling as discussed earlier and as such the crosstalk between Notch and NFκB appears to be an important signaling event that regulates the processes of tumor invasion and angiogenesis in pancreatic cancer.
De novo resistance to chemotherapeutic agents is a major cause of treatment failure in pancreatic cancer, which could be due to the constitutive activation of NFκB among others. Moreover, chemotherapeutic agents can activate NFκB in pancreatic and other cancer cells, leading to cancer cell resistance to chemotherapy (acquired resistance). Therefore, the strategies by which NFκB could be inactivated represent a novel approach for the treatment of pancreatic cancer. Some studies have demonstrated that the anti-tumor effects of chemotherapeutic agents can be enhanced by combination treatment involving inhibition of NFκB. It was shown that NFκB activity was significantly increased by cisplatin, docetaxel, doxorubicin, and gemcitabine treatment. When the NFκB inducing activity of these agents was completely abrogated by pre-treatment of the pancreatic cancer cells with NFκB inhibitors, growth inhibition, and apoptosis were clearly enhanced (Muerkoster et al., 2003; Zhang et al., 2003; Li et al., 2004b, 2005; Banerjee et al., 2005; Mohammad et al., 2006). Recently, it was also shown that anti-tumor and anti-metastatic activities of docetaxel could be enhanced by NFκB inhibitors through regulation of osteoprotegerin/receptor activator of NFκB (RANK)/RANK ligand/MMP-9 signaling in vitro and in vivo (Li et al., 2006).
The Gastrin and Cholecystokinin Receptors
The peptide hormone gastrin is secreted by G cells in the gastric antrum and duodenum and it can act as a growth factor for gastric, colonic, and pancreatic cancers. CCK-BR (the gastrin and cholecystokinin receptor), gastrin precursors, and the fully amidated gastrin are expressed in 95, 55–91, and 23% of pancreatic cancers, respectively (Caplin et al., 2000). A selective CCK-BR antagonist, gastrazole, was tested in two small, randomized, controlled trials in patients with advanced pancreatic cancer (Chau et al., 2006). Gastrazole was superior to placebo, but not to 5-fluorouracil. Another inhibitor, the orally active Z-360, has demonstrated promising laboratory results and, in combination with gemcitabine, is well-tolerated by patients (Kawasaki et al., 2008). An alternative approach to a blockade of this pathway involves the use of gastrimmune, an immunogen that stimulates the formation of antibodies against gastrin 17 and its precursors. This agent was, however, not successful in a phase III trial.
The Hedgehog Signaling
Hedgehog (Hh) signaling is an essential pathway for embryonic pancreatic development. Hh signaling plays important roles in adequate tissue morphogenesis and organ formation during gastrointestinal tract development. Hedgehog ligands including sonic hedgehog (Shh) are expressed throughout the endodermal epithelium at early embryonic stages but excluded from the region that forms the pancreas. Deregulation of the Hh pathway has been implicated in a variety of cancers including pancreatic cancer. Overexpression of Shh was reported to contribute to pancreatic tumorigenesis. Thayer et al. (2003) found that no Shh was detected in the islets, acini, or ductal epithelium of normal pancreas while Shh was aberrantly expressed in 70% of specimens from patients with pancreatic adenocarcinoma, suggesting that Shh is a mediator of pancreatic cancer tumorigenesis. Importantly, the down-regulation of Shh by cyclopamine, a specific inhibitor of Shh, can reduce the growth and viability of pancreatic cancer cells, suggesting that targeting Shh signaling may be an effective novel approach for the treatment of pancreatic cancer (Berman et al., 2003). Cyclopamine can enhance sensitivity to radiotherapy and chemotherapy and suppress metastatic spread (Shafaee et al., 2006; Feldmann et al., 2007) as well as improving anti-tumor activity when combined with an EGFR inhibitor (Hu et al., 2007). A downstream target of the Shh pathway, the transcription factor GLI 1, can also be inhibited by miRNA (Tsuda et al., 2006).
A recent report shows that NFκB contributes to Hh signaling pathway activation through Shh induction in pancreatic cancer (Nakashima et al., 2006), demonstrating a crosstalk between NFκB and Hh signaling pathways in pancreatic cancer.
Other Mechanisms Involved in the Pathogenesis of Pancreatic Cancer
Inflammation
There is a growing feeling that inflammation contributes to pancreatic cancer development. The initiation, promotion, and progression of tumors may be influenced by numerous components including cytokines such as TNF-α, IL-6, IL-8 and interferon γ, the COX-2, and the peroxisome proliferator-activated receptor-γ, that function in the inflammatory response. Other mechanisms involved in the inflammatory response may also contribute to neoplasia, such as free radicals which may particularly contribute to cancer initiation. Free radicals can induce genetic alterations and post-translational modifications of key cancer-related proteins. Reactive oxygen intermediates and reactive nitrogen intermediates are capable of causing oxidative damage and nitration of DNA bases, thus increasing the likelihood that DNA mutations occur (Hussain et al., 2003). Interestingly, recognized risk factors for pancreatic cancer such as cigaret smoking, chronic hereditary pancreatitis, obesity, and type II diabetes are linked by the fact that inflammation significantly drives these pathologies. By itself, chronic pancreatitis is associated with increased risk of pancreatic cancer while obesity and cigaret smoking require additional co-factors to induce pancreatic carcinogenesis. The role of inflammation in the promotion of pancreatic cancer from PanIN lesions has been experimentally confirmed by Guerra et al. (2007, 2011) using an elegant mutated K-ras-dependent mouse model in which the animals develop PanIN lesions spontaneously. In these works, repeated episodes of caerulein-induced acute pancreatitis are able to promote cell transformation, apparently by blocking the mutated K-ras-induced senescence. Activation of STAT3 in pancreas cancer is related to tumor growth directly through mechanisms affecting the tumor and indirectly by modulating tumor-associated stroma and the immune system. Activation depends on the phosphorylation of a conserved tyrosine residue (Y705) by upstream kinases, such as Janus kinase 2 (Jak2). Jak2 activation requires activation of the ubiquitously expressed gp130 receptor by specific ligands (IL-6, LIF, IL-11, oncostatin M, CNTF, and IL-27). Importantly, it was recently reported that STAT3 activity plays a critical role in K-ras-induced pancreatic tumorigenesis (Corcoran et al., 2011) supporting the link between STAT3 activation and cell transformation (Figure 2).
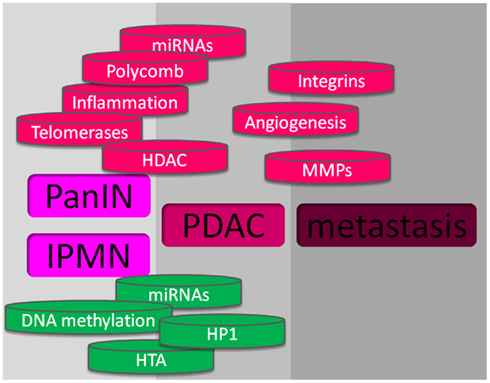
Figure 2. Mechanisms involved in pancreas cancer. At the upper part, in red, are shown the mechanisms that promote pancreatic cancer, and at the lower part, in green, are showed the mechanisms against pancreatic cancer development.
The Telomerases
The telomeres located at the end of chromosomes normally shrink with each cell division and thereby impose a finite lifespan to the cell. Most malignant cells have detectable activity of telomerase, a reverse transcriptase that contains an RNA template and elongates telomeres. Telomerase is overexpressed in 95% of pancreatic cancers (Hiyama et al., 1997) which provides a rationale for the development of antitelomerase agents. GV1001 is a telomerase peptide vaccine that has shown some promising results in phase I/II studies (Bernhardt et al., 2006). This vaccine is being tested in the large (>1000 patients), phase III, TeloVac trial with gemcitabine and capecitabine in locally advanced and metastatic pancreatic cancers (Figure 2).
Angiogenesis
Angiogenesis is essential for solid tumor growth, and is principally mediated by the VEGF family of proteins and receptors. Stimuli that upregulate VEGF expression include hypoxia, other growth factors, and oncogenic proteins such as TGF-β, EGF, and Ras. VEGF is overexpressed in >90% of pancreatic cancers (Seo et al., 2000) and is, therefore, an appealing target for therapy. Bevacizumab is a humanized antibody against VEGF approved for use in patients with colorectal cancer. However, a phase III trial in advanced pancreatic cancer failed to show any survival benefit for bevacizumab in combination with gemcitabine (Kindler et al., 2010). The AVITA (BO17706) phase III study of patients with metastatic pancreatic cancer reported that the addition of bevacizumab to gemcitabine and erlotinib did not significantly prolong overall survival, although a significant improvement in progression-free survival was seen (Van Cutsem et al., 2009). A number of other trials are being conducted to examine bevacizumab in combination with other agents or treatment modalities for pancreatic cancer; however, this agent seems unlikely to confer sufficient benefit to justify its licensing for this condition. The failure of bevacizumab in therapeutic trials for pancreatic cancer highlighted the need for angiogenic inhibitors that could target other non-VEGF pathways and have better access to the tumor environment than an antibody. Sorafenib is a multitargeted kinase inhibitor that inhibits the VEGF receptor (VEGFR), PDGFR, SCFR (formerly c-KIT), Raf1, and FLT3, which are all implicated in tumor growth and angiogenesis. Sorafenib was approved in 2005 for the treatment of advanced renal-cell carcinoma. However, a phase II study concluded that, although well-tolerated, it was inactive in patients with advanced pancreatic cancer. A randomized phase III study comparing gemcitabine plus sorafenib versus gemcitabine plus placebo was recently reported and revealed no advantage for the experimental arm (Gonçalves et al., 2011). Axitinib is an orally active inhibitor of both VEGFR and related tyrosine kinase receptors at high concentrations. A median survival of 6.9 months was reported for axitinib combined with gemcitabine compared with 5.6 months for gemcitabine alone in a phase II trial in patients with advanced pancreatic cancer, but this finding was not statistically significant (Spano et al., 2008). Phase III trials of axitinib combined with gemcitabine are currently in progress. Aflibercept, a recombinant fusion protein that functions as a soluble decoy receptor and thereby inhibits VEGF, is another novel agent being tested in a phase III trial of patients treated with gemcitabine for metastatic pancreatic cancer (Figure 2).
Signaling by Integrin Receptors
Integrin receptors on the cell surface interact with the extracellular matrix and mediate various signaling pathways. These receptors are involved in many neoplastic processes, including tumor survival, invasion, and metastasis. The αVβ3 and αVβ5 integrins induce angiogenesis, principally via basic fibroblast growth factor and VEGF, respectively. Cilengitide inhibits these integrins, but in a phase II trial in patients with advanced pancreatic cancer it did not show significant benefit compared to gemcitabine alone (Friess et al., 2006). Other anti-integrin agents, including an antibody against α5β1 (volociximab; Ricart et al., 2008) and an inhibitor of α2 (E7820) are in early phase clinical trials (Funahashi et al., 2002; Figure 2).
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteolytic enzymes that degrade the extracellular matrix and are essential for tumor spread and neovascularization. We should not, therefore, be surprised that imbalance between MMPs and their natural inhibitors is a frequent event in pancreatic cancer. Despite promising laboratory results, MMP inhibitors have failed to live up to their initial therapeutic expectation in three phase III clinical trials (Bramhall et al., 2001, 2002; Moore et al., 2003), although critics argued that the trials included a large number of patients with metastatic disease, which contradicts the rationale of exploiting the cytostatic effect of MMP inhibitors (Figure 2).
Cancer Stem Cells
Cancer stem cells possess important properties associated with their normal counterparts, namely the ability for self-renewal and differentiation. Pancreatic cancer stem cells are identified by their surface markers, such as CD133, CD44, CD24, and flotillin 2 epithelial specific-antigen. Evidence suggests that such cells form a small subset in the heterogenous tumor population, and contribute to neoplastic progression, metastasis, and resistance to chemotherapy and radiotherapy (Hermann et al., 2007; Lee et al., 2008). For this reason, cancer stem cells are thought to be responsible for relapse of disease after clinical remission. Dysregulation of various signaling cascades, including the PTEN, Shh, Notch, and Wnt pathways, are frequently observed in cancer stem cells, which provides further rationale for use of these pathways as a target for therapeutic purposes. Further studies are still needed to understand the genetic and biological properties of cancer stem cells for the development of effective treatment modalities.
miRNA Expression Profiling in Pancreatic Ductal Adenocarcinoma
Analysis of miRNA expression in PDAC revealed a specific miRNA signature that can be monitored by profiling miRNAs at different stages of cancer (Table 1). Over the past years, a number of different approaches, including DNA microchips and RT-PCR have been described to quantify miRNAs. By these techniques, several studies demonstrated the tissue-specificity of miRNA expression and the deregulation of miRNA expression in pancreatic cancer (Roldo et al., 2006; Szafranska et al., 2007). Monitoring differential miRNA expression might therefore be useful in the differential diagnosis of pancreatic cancer. Expression of the miR-376 precursor was highest in the human pancreatic cancer cell line Panc1, compared to other cell lines studied (Jiang et al., 2005; Volinia et al., 2006). Large-scale miRNA profiling in 540 samples of solid tumors (breast, colon, lung, pancreas, prostate, and stomach) showed that the spectrum of miRNA expression varied in different solid tumors and was different from that of normal cells (43 of 137 miRNAs, 31%). miR-21, miR-191, and miR-17-5p were significantly overexpressed in all six tumor types, whereas miR-218-2 was consistently down-regulated in colon, stomach, prostate, and pancreatic cancers, but not in lung and breast carcinomas. This observation indicates that colon, pancreas, prostate, and stomach have similar miRNA signatures, different from those of breast and lung cancer. Similarly, up-regulation of miR-142-3p, miR-142-5p, miR-155, and miR-146a expressions was observed in human pancreatic neuroendocrine tumors (PNETs) as compared with normal human islets (Olson et al., 2009). Lee et al. (2007) reported the aberrant expression of 100 miRNA precursors in pancreatic cancer or desmoplasia, including the miRNAs previously reported in other human cancers (miR-155, miR-21, miR-221, and miR-222) as well as the first reported miR-376a and miR-301 for the differential expression of cancer. A significant up-regulation of miR-196a, miR-190, miR-186, miR-221, miR-222, miR-200b, miR-15b, and miR-95 in most pancreatic cancer tissues and cell lines was reported. miR-155 and miR-21 were significantly up-regulated in 15 IPMNs versus matched controls (Habbe et al., 2009). miR-155 was up-regulated in 53 of 64 IPMNs (83%) compared to 4 of 54 normal ducts only (7%), and miR-21 was up-regulated in 52 of 64 IPMNs (81%) compared to 1 of 54 normal ducts (2%). The expression of miR-216 has also shown to be specific to the pancreas (Sood et al., 2006). Let-7 miRNA was found expressed in pancreatic cancer cells but did not inhibit the epithelial–mesenchymal transition (Watanabe et al., 2009). Therefore, miRNAs play vital roles not only in different kinds and stages of pancreatic tumors but also in many diseases, including cancers, cardiovascular diseases, and immune disorders. Epigenetic modifications, DNA copy number changes, and genetic mutations may regulate the expression of miRNAs. The conclusion of these data is that PDAC miRNAomes associated with normal and tumor tissues are different. Differences are tumor-specific and, in some cases, indicators of prognosis. These findings suggest that miRNA expression patterns constitute a signature of the disease which could offer new clues about pancreatic cancer occurrence and also provide new molecular markers that would improve diagnosis and orient the treatment. A very promising diagnostic strategy could arise from miRNAs if they are found in serum and can be detected by RT-PCR. In a remarkable study Lu et al. (2005) showed that expression data for 217 miRNAs only performed better at identifying cancer types than analysis of 16000 mRNAs. They concluded that miRNAs might help detecting cancer better than other strategies presently available because miRNAs are only several hundred, compared to tens of thousands for mRNAs and proteins. One single miRNA can modulate the expression of many genes rendering miRNAs powerful molecules regulating several related pathways, each miRNA playing multiple but coherent roles in the cell.
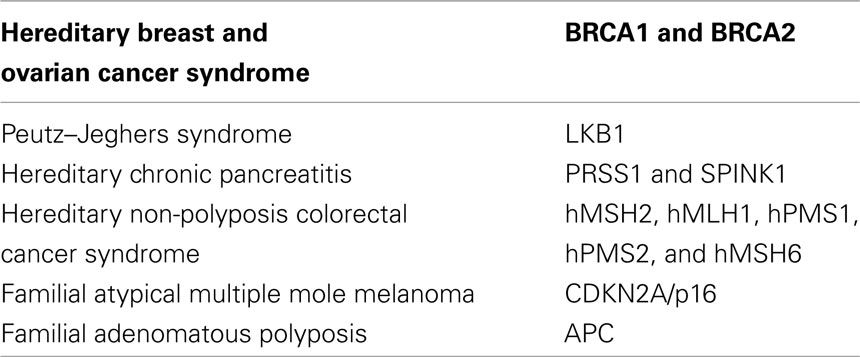
Table 1. Genes implicated in the familiar forms of pancreas cancer.
Epigenetics of Pancreatic Cancer
The revolution of somatic genetics in the field of cancer brought about by the model developed by Fearon and Vogelstein (1990) in colon, which later led to an adaptation to the pancreas by Hruban et al. (2000), opened a fruitful era for pancreatic cancer research. The basic premise of somatic genetics in cancer is that if a gene, which is suspected to play a role related to cancer, is over-amplified or mutated with gain-of-function, it behaves as an oncogene, but if it is deleted or mutated with loss-of-function, it behaves as a tumor suppressor. However, in light of the modern biology we can assume that regulation of genes activity is more complex and it is therefore necessary to explain how changes in chromatin dynamics can silence tumor suppressor genes via mechanisms that are totally independent from either deletions or mutations of these genes. Some epigenetic mechanisms can silence gene function such as:
DNA Methylation
DNA methylation was the first type of epigenetic change to be studied as a mechanism for the inactivation of tumor suppressors (Esteller, 2008). DNA methylation occurs on dinucleotide CpGs. The process of DNA methylation entails the addition of a methyl group to carbon number 5 of the cytosine pyrimidine ring, which ultimately silences gene expression. In pancreatic cancer, DNA methylation has been known for a long time as a mechanism for inactivating tumor suppressor genes, a well known example being the inactivation of the p16 promoter via methylation (Singh and Maitra, 2007). In addition, loss of methylation of a normally silenced promoter in pancreatic cells, such as in the gene encoding the hematopoietic-specific guanine nucleotide exchange factor, VAV1, can alter its expression (Fernandez-Zapico et al., 2005). Although several genes were discovered to be methylated in advanced pancreatic cancer, current evidence supports the idea that aberrant methylation occurs very early during the histopathological progression of this neoplasia. Interestingly, in a study involving large-scale methylation analysis, Sato et al. (2003) analyzed DNA samples from 65 PanIN lesions for methylation status of eight genes, identified beforehand by a large-scale microarray approach as aberrantly hypermethylated in invasive pancreatic cancer. Methylation of these genes was identified in 68% of PanINs. More importantly, aberrant methylation was present in approximately 70% of PanINs-1A, the earliest lesions. The prevalence of methylation increased from PanIN-1 to PanIN-2 for the NPTX2 gene, and from PanIN-2 to PanIN-3 for SARP2, Reprimo, and LHX1.
Histone Acetylation and Deacetylation
Another important mechanism underlying the epigenetic regulation of gene expression is the acetylation and deacetylation of lysine residues within histone tails. Regarding acetylation, the process occurs via HATs, such as CBP, P300, and PCAF, to result in activation of gene expression, whereas deacetylation is mediated by two different families of HDACs, resulting in gene silencing. Together, these enzymes provide a fine-tuned mechanism which, upon alteration, can promote the activation of oncogenic pathways and the silencing of tumor suppressors. Studies have demonstrated that HDAC activity is increased in various tumors compared with normal tissue, and this increase in HDAC activity has been associated with transcriptional repression of tumor suppressor genes that cause growth inhibition and apoptosis (Cress and Seto, 2000). In a study performed by Ouaïssi et al. (2008) ∼80% of pancreatic adenocarcinoma samples examined showed a significant increase in HDAC7 RNA and protein levels (Ouaïssi et al., 2008). Thus, it is clear that HDACs play an important role in the maintenance of the proper balance of chromatin marks on a given promoter. If this balance is altered, such as with HDAC overexpression in pancreatic cancer, the expected global effect on promoters is daunting.
Histone H3–Methyl-K27 and Polycomb
Polycomb proteins silence gene expression by specifically methylating histone H3, on K27 (Cao et al., 2002; Ringrose, 2007). At the core of this pathway, the polycomb group of proteins (PcG) act via the stepwise recruitment of PRC2, containing the histone H3 K27 methylase activity, to chromatin. Subsequently, the trimethyl-K27–H3 mark deposited by PRC2 recruits the PRC1 complex, thereby completing the formation of the gene silencing complex. The enzymatic activity of the PCR2 complex involves the K27–H3 histone methylase, EZH2, but requires formation of a complex with Suz12 and EED to function. The PCR1 complex contains the oncogene BMI1, as well as HPC1-3, HPH1-3, SCMH1, and the methyl-K27–H3-binding proteins, Cbx 2, 4, 6, 7, and 8. The role of polycomb proteins in pancreatic cancer is an emerging area of research. Interesting studies have demonstrated that loss of trimethylation at lysine 27 of histone H3, which is achieved by EZH2, is a predictor of poor outcome in pancreatic cancers (Wei et al., 2008). In fact, together with tumor size and lymph node status, the level of trimethyl-K27–H3 was found to have a strong and independent prognostic influence in pancreatic cancer. In another recent study, nuclear accumulation of EZH2 was identified as a hallmark of poorly differentiated pancreatic adenocarcinoma and nuclear overexpression of EZH2 contributes to pancreatic cancer cell proliferation, suggesting EZH2 as a potential therapeutic target for the treatment of pancreatic cancer (Ougolkov et al., 2008). Mechanistically, one of the outcomes of aberrant polycomb regulation is the silencing of the p16 gene, which could occur prior to DNA methylation, directly via altered recruitment of members of this family to the p16 promoter sequence (Kotake et al., 2007). Upon recent studies in human cells, EZH2, and DNA methyltransferases (DNMTs) were found to physically and functionally interact, evidenced by the PRC2 subunits, EZH2, and EED, co-immunoprecipitating with all three human DNMTs and the co-dependency of certain target gene silencing requiring both EZH2 and DNMTs (Vire et al., 2006). Therefore, the presence of polycomb proteins on the p16 promoter can recruit DNA methylases which then further inactivate the expression of p16 via DNA methylation.
Histone H3–Methyl-K9 and Heterochromatin Protein 1
HP1 binds to methylated K9 of histone H3, causing transcriptional repression (Lomberk et al., 2006). This occurs through the N-terminal chromodomain of HP1, while the highly related C-terminal chromo shadow domain allows for dimerization of these HP1 molecules and serves as a docking site for various factors involved in a wide array of functions, from transcription to maintaining nuclear architecture. To mediate gene silencing via the formation of heterochromatin, HP1 isoforms must interact with two different H3–K9 histone methylases namely G9a and Suv39h1 (Lomberk et al., 2006). These methylases work in concert with HP1 in a circular manner to form silenced chromatin. When either of the methylases adds methyl groups to K9, this, in turn, forms an HP1 docking site on chromatin. Since HP1 also recruits the methylases, this cycle repeats, and the HP1-methylase pair can spread the formation of silenced chromatin to adjacent nucleosomes, causing long-term silencing of entire genes.
Epigenetics as Target for Chemoprevention and Chemotherapies
The study of epigenetics also impacts the field of therapeutics. In stark contrast with genetic alterations, epigenetic changes have been proven to be reversible. For instance, methylation can be reversed through the use of 5-Aza-2′-Deoxycytidine or DNA methyltransferase inhibitors. Two types of DNMT inhibitors exist, namely, nucleoside and non-nucleoside (small molecule) inhibitors (Mund et al., 2006; Griffiths and Gore, 2008). These types of pharmacologic DNMT inhibitors are currently being tested in phase I–III clinical trials. Currently, among the most promising epigenetic treatments for cancer are the potential therapeutic agents that target histone deacetylation HDAC inhibitors (HDACIs; Berger, 2002; Glaser et al., 2003; Bruserud et al., 2007; Hildmann et al., 2007; Pan et al., 2007). Intuitively, the proposed mechanisms of action for HDACs attempt to harness their ability to reactivate transcription of multiple genes found to be silenced in human tumors. However, their pleiotropic anti-tumor effects, specific for cancer cells, along with their potential activity in other diseases, elicits the possibility that they also act in other biological processes, currently poorly understood. In addition to being well-tolerated, several HDACIs show promising anti-tumor activity with more than 50 naturally occurring or synthetic HDACIs currently developed. Hydroxamic acid compounds, trichostatin A (TSA), and suberoylanilide hydroxamic acid (SAHA), are the best known among these agents. By the same token, however, other equally promising, although less known, drugs can be classified into wider groups such as short-chain fatty acids (e.g., valproic acid), epoxides (e.g., trapoxin), cyclic peptides (e.g., apicidin), benzamides (e.g., CI-994, Nacetyldinaline), and hybrid compounds (e.g., SK-7068). Recent studies have shown that certain HDACIs can induce death of cultured pancreatic cells (Ouaïssi et al., 2008). Additionally, conventional chemotherapeutic drugs, such as gemcitabine, in combination of some of these agents, can achieve a synergistic inhibition of pancreatic adenocarcinoma cell growth (Donadelli et al., 2007). As a result of these promising studies, new agents belonging to this family of drugs, such as FR235222, could potentially be used as therapeutic tools in this cancer (Singh et al., 2008). HDACIs are in advanced phases of clinical trials and may soon become part of the therapeutic regimen for oncologists treating this disease. It is interesting to note that HDACIs are already potential therapeutic agents against pancreatic cancer, in spite of the fact that the characterization of histone deacetylases in pancreatic cancer is far from being completed and their role is not fully understood.
Genetics of the Familial Pancreatic Adenocarcinoma
In addition to sporadic adenocarcinoma, an increased risk of pancreatic cancer has been demonstrated among persons with a family history of pancreatic cancer. Coughlin et al. (2000) reported an increased risk of developing pancreatic cancer for individuals who reported a positive family history of pancreatic cancer at baseline, with a relative risk of 1.5 after adjustment for age. Furthermore, a population-based cohort study demonstrated that the risk of pancreatic cancer increased 1.72-fold for individuals with a parent who developed a pancreatic cancer. The risk was not elevated when a more distant relative had been diagnosed with pancreatic cancer. Thus, several studies strongly support the hypothesis that familial aggregation and genetic susceptibility play an important role in the development of pancreatic cancer. Discovering the genetic basis of inherited pancreatic cancer is an active area of research. In 2001, a multicenter linkage consortium, PACGENE, was established to conduct linkage studies aiming at localizing and identifying pancreatic cancer susceptibility genes (Petersen et al., 2006). Other groups have used linkage studies to suggest that mutations in the paladin gene (PALD) on chromosome 4q32 predisposes to pancreatic cancer (Pogue-Geile et al., 2006); however, this finding has not been validated in subsequent studies (Earl et al., 2006). The complex nature of pedigree data makes it difficult to accurately assess risk based upon the simple counting of the number of affected family members, as it does not account for family size, current age, or age of onset of pancreatic cancer, and the exact relationship between affected family members. Computer-based, risk assessment tools have been developed to integrate this complex risk factor and pedigree data into risk assessment. These models can provide more precise risk assessment than guidelines or models that rely on counts of affected family members, such as the Bethesda Guidelines for hereditary non-polyposis colorectal cancer (HNPCC) or myriad tables for hereditary breast and ovarian cancer. In April 2007, the first risk prediction tool for pancreatic cancer, PancPRO, was released (Wang et al., 2007). Although the genetic basis for most instances of aggregation of pancreatic cancer in families is unknown, the genes responsible for a small portion of familial pancreatic cancer are known (Table 2). Germline mutations in the BRCA2, CDKN2A/p16, STK11/LKB1, and PRSS1 genes have all been shown to increase the risk of pancreatic cancer (Lowenfels et al., 1993; Goldstein et al., 1995; Whitcomb et al., 1996; Giardiello et al., 2000). Additionally, some studies have described pancreatic cancers developing among individuals with HNPCC; however, the association between HNPCC syndromes and pancreatic cancer is not as well-defined as it is for some of the other syndromes (Abraham et al., 2001; Yamamoto et al., 2001). While most patients with sporadic and familial pancreatic cancer have classic infiltrating ductal (tubular) adenocarcinoma, some inherited syndromes are associated with a specific histologic type. Although rare, these cases provide a unique opportunity to correlate genetics with histology. For example, many pancreatic cancers that develop in patients with HNPCC syndrome have a medullary phenotype (Goggins et al., 1998a; Wilentz et al., 2000; Banville et al., 2006) and individuals with the Peutz–Jeghers syndrome appear to be predisposed to IPMNs (Su et al., 1999; Furukawa, 2007). These associations between phenotype and genotype are important because tumor phenotype can be used to identify at-risk families.

Table 2. microRNAs frequently misregulated in pancreatic cancers.
Germline DNA Mutations that Increase Susceptibility to Develop Pancreatic Adenocarcinoma
Most pancreatic cancers are sporadic. In sporadic forms, association with polymorphic somatic mutations, spontaneous, or generated by environmental factors is of paramount importance. It is well known that sporadic forms of cancers result from the accumulation of genetic modifications or mutations. Besides particular syndromes whose association with pancreatic cancer is well known, a lot of work remains to identify genetic variations associated with cancer predisposition. Recent advances in large-scale genotyping (SNP chips) increased considerably the number of markers that can be simultaneously genotyped. As a consequence, haplotype blocks can be determined much more precisely than with microsatellite markers. These methodologies allowed recent studies of associations covering all intragenic regions of the whole human genome, these studies being called “genome-wide association studies” (GWAS). Several GWAS led to the identification of susceptibility markers of several types of cancers (review in Manolio, 2010). More than 700 GWAS are presently registered but only four concern pancreatic cancer (Amundadottir et al., 2009; Diergaarde et al., 2010; Low et al., 2010; Petersen et al., 2010). Two GWAS for pancreatic cancer (Amundadottir et al., 2009; Petersen et al., 2010), have been conducted within the framework of the “pancreatic cancer cohort consortium,” or PCCC, an international consortium comprising Centers from Europe, China, and North America. In these studies, 12 cohorts representing more than 3800 cases of cancer and 3900 controls allowed detection of four new genes associated with pancreatic cancer. The first locus, harboring several SNPs strongly associated with cancer occurrence, was localized on chromosome 9q34 and contains the first intron of the ABO gene. That gene encodes the glycosyl-transferase which catalyzes the transfer of sugars to antigen H (Histo-blood group ABO system transferase). By this mechanism, antigen H becomes antigen A or B, depending on the nature of the sugar transferred by the ABO enzyme. In individuals from group O, the gene is mutated and generates a non-functional protein. Association of the ABO gene with pancreatic cancer has already been suggested 50 years ago in studies showing a higher frequency of pancreatic or gastric cancer in individuals from groups A, B, or AB than from group O (Aird et al., 1953; Marcus, 1969). That gene is also found altered in primary tumors and metastases of pancreatic cancer (Itzkowitz et al., 1987). More recently, the PCCC added three other regions strongly associated with cancer (Petersen et al., 2010). The first one is an intergenic region of 600 kb in chromosome 13q22.1, located between genes KLF5 and KLF12. These two members of the Kruppel transcription factor family are involved in regulating cell growth and transformation (Dong and Chen, 2009; Nakamura et al., 2009). The second region is located on 1q32.1. That region harbors gene NR5A2 (Nuclear Receptor subfamily 5 group A member 2), 91 kb upstream from the gene. The receptor encoded by gene NR5A2 is present, in adults, in the exocrine pancreas, liver, gut, and ovaries. Its function is not completely understood. In pancreas, NR5A2 would contribute to the regulation of the expression of several genes (Fayard et al., 2003). The last locus identified by this consortium is localized at 5p15.33. SNP markers that border this locus are within intron 13 of the CLPTM1L gene (Cleft lip and palate transmembrane 1-like). CLPTM1L is poorly described. One of the critical factors in the interpretation of GWAS is the ethnicity of the studied group. PCCC studies mentioned above have been conducted on Caucasian populations and do not necessarily reflect risk factors for other ethnic groups. In fact, a GWAS conducted by Biobank Japan (BJJ) has evidenced a different set of markers of pancreatic cancer (Low et al., 2010). Among loci strongly associated with the disease, three are specific to the Japanese population. The first one, in 6p25.3, is a 75-bp linkage disequilibrium block containing the FOXQ1 gene [Forkhead-box (Fox) Q1]. The second SNP showing significant association is located within gene BICD1 (Bicaudal-D homolog 1). The last locus with a high odd-ratio corresponds to several SNPs that fall in the first intron of the DPP6 gene (Dipeptidyl-peptidase 6, a member of the DPP family devoid of the catalytic residues required for enzymatic activity but which acts through protein–protein interactions).
Two studies have extended the results obtained by analyzing tumor exome, including a study comparing the exomes of the primary tumor and corresponding metastases (Campbell et al., 2010; Yachida et al., 2010). In the first one, Yachida et al. (2010) gathered data on the exomes of seven tumors previously characterized by Jones et al. (2008), and compared them to data on the exomes of the corresponding metastases. The second study was conducted on 13 tumors and corresponding metastases (Campbell et al., 2010). Information produced by these studies allowed to fill in a high resolution framework of the various modifications and rearrangements that occur in the DNA of metastases, of the various underlying mechanisms, on the genes potentially involved in the metastatic process and finally on the time between the appearance of the cancerous cell and the occurrence of a clone with a metastatic profile. Genomic instability is apparently the main characteristic of pancreatic cancer (Campbell et al., 2010). DNA sequencing in tumors and in cells at different stages of progression toward metastases revealed that the major chromosomal rearrangement is a duplication of fragments and their insertion in opposite directions, a phenomenon called “fold-back inversions” by the authors. The origin of fold-back inversions is not known. A possible hypothesis is that early in tumor progression telomerase activity is altered or inhibited (Bardeesy and DePinho, 2002) generating a progressive shortening of telomeres and successive cycles of breaking and abnormal fusion of chromatids by a mechanism called “breakage–fusion–bridge” (McClintock, 1941). These rearrangements will determine the gain or loss of DNA and, as consequence, genomic instability. Interestingly, these results suggest that fold-back inversions arise early in tumor evolution and would trigger progression toward metastasis (Campbell et al., 2010). These results have shown unequivocally, for the first time, that the pancreatic tumor evolves from a first (parental) clone which generates progressively other sub-clones. Each sub-clone will evolve independently by accumulating specific genetic modifications and, possibly, a tissue-specific metastatic affinity profile. Establishment of a phylogenetic correlation between genomic alterations of tumors and their metastases allowed drawing a kinetic of tumor progression (Yachida et al., 2010). Unexpectedly, the mean time between arising of the founding cell and the occurrence of the first parent clone (non-metastasis) is 11.8 years. Another 6.8 years will be required for one of the sub-clone to acquire the metastatic profile, which occurs about 3 years before the patient dies. These findings, if confirmed, have major clinical consequences. They show that, if early detection of pancreatic cancer is possible, there is a large window of about 10 years for therapeutic intervention.
Pancreatic Cancer Protection by Dietary Chemopreventive Agents
More than two-thirds of human cancers could be prevented by modification of lifestyle including dietary modification. The dietary factors associated with increased risk of pancreatic cancer are meat, red meat in particular, and energy. Protection is mainly provided by fruit, vegetables, and vitamins. In recent years, more dietary compounds have been recognized as cancer chemopreventive agents because of their anti-carcinogenic activity. Therefore, early invasion and metastasis of pancreatic cancer could be preventable by these dietary compounds. In addition to dietary factors, there are some chemopreventive agents such as:
Curcumin
Curcumin is a compound from Curcuma longa (tumeric). Curcumin has recently received considerable attention due to its pronounced anti-inflammatory, anti-oxidative, immunomodulating, anti-atherogenic, and anti-carcinogenic activities. Curcumin suppresses the activation of NFκB through inhibition of IKK activity in pancreatic cancer cells (Li et al., 2004a). Phase II trials of curcumin with and without gemcitabine showed that curcumin was well-tolerated and might have some biological activity in patients with pancreatic cancer (Dhillon et al., 2008).
Soy Isoflavones
Genistein is the main isoflavone found in a relatively high concentration in soybeans and most soy-protein products. Genistein inhibits cell growth and induces apoptosis in various cancers including pancreatic cancer. Genistein treatment significantly inhibited NFκB DNA-binding activity, inhibited Akt activity (Banerjee et al., 2005; Li et al., 2005) and significantly down-regulated Notch signaling, leading to the inhibition of NFκB and induction of apoptosis in pancreatic cancer cells (Wang et al., 2006c,e).
Indole-3-Carbinol and 3,3′-Diindolylmethane
Indole-3-carbinol (I3C) is produced from all kinds of cabbages, broccoli, cauliflower, and Brussels sprouts. I3C is biologically active and it is easily converted in vivo to its dimeric product 3,3′-diindolylmethane (DIM), which is also biologically active. Both I3C and DIM inhibit cell proliferation and induce apoptotic cell death in a variety of cancers including pancreatic cancer (Abdelrahim et al., 2006). I3C induces apoptosis in pancreatic cancer cells through the inhibition of STAT3 whose activation has been observed in human pancreatic carcinoma specimens and pancreatic cell line but not in normal pancreatic tissues (Lian et al., 2004).
Green Tea
Consumption of green tea has been implicated in better human health including the prevention of cancers. Green tea contains several catechins including epicatechin (EC), epigallocatechin (EGC), epicatechin-3-gallate (ECG), and epigallocatechin-3-gallate (EGCG). However, EGCG is among catechins the most potent for oncogenesis inhibition and reduction of oxidative stress (Mukhtar and Ahmad, 1999). By targeting multiple signaling pathways including MAPK, EGFR, and NFκB, EGCG inhibits the malignant transformation of epidermal cell lines, inhibits cell growth, and induces apoptosis in a number of cancer cells including pancreatic cancer cells (Mukhtar and Ahmad, 1999; Qanungo et al., 2005; Khan et al., 2006).
Resveratrol
Relatively high quantities of resveratrol are found in grape juice and red wine. Resveratrol has been shown to have beneficial effects on the reduction of oxidative stress and the prevention of cancers. Resveratrol was first noted to be a cancer chemopreventive agent having antioxidant and anti-tumorigenic properties (Jang et al., 1997). Like EGCG, it is a polyphenol which can cause cell cycle arrest in G1 in various tumor cell lines including pancreatic cancer (Ding and Adrian, 2002).
Lycopene and Vitamins
Tomato products including ketchup, tomato juice, and pizza sauce, are the richest sources of lycopene in the occidental diet. Dietary intake of lycopene is associated with reduced pancreatic cancer risk, suggesting a role in the prevention of pancreatic cancer (Nkondjock et al., 2005).
Current Treatments
Due to delayed diagnosis and aggressiveness of pancreatic cancer, only 10% of patients are eligible for curative treatment and 90% of patients undergo palliative therapies. Surgery, chemotherapy, and radiotherapy are the weapons used to fight pancreatic cancer, with disappointing impact on survival though.
Surgery
To date, surgery remains the most efficient treatment. However, only 10% of patients are eligible for resection: no distant disease (hepatic or lung metastasis) and no local vascular invasion (mainly mesenteric vessels and celiac trunk) must be present to consider curative resection. Palliative surgery can be performed in selected patients but is not recommended: endoscopic derivation (duodenal prosthesis and biliary duct stenting) has reduced the field of the historical twin derivation. The last two decades have shown a dramatic reduction in postoperative morbidity and mortality of pancreatic surgery. Now, pancreatic surgery performed by experienced surgeons in high volume centers results in less than 5% mortality. Due to reduced morbidity, pancreatic surgery can be performed safely in elderly patients. Moreover, vascular resection/reconstruction (only recommended for venous involvement; arterial resection remains a contra indication to surgery) is becoming safe and increases the proportion of patients elected for pancreatic surgery. Despite appropriate surgery and perioperative treatments, the 5-year overall survival after resection remains poor and long-term survivors are rare. Yet, some progress has been made. In the 80s, a new type of pancreatic tumor was identified and named IPMNs (Sohn et al., 2001). IPMN is defined by a benign cystic tumor connected with the pancreatic duct network. However, transformation in pancreatic cancer occurs in 5–20% of IPMN according to sub-IPMN types (side branch or main duct IPMNs). Thus, patients diagnosed with IPMN (fortuitous diagnosis, pancreatitis, etc.) had to be strictly followed up or their pancreas resected. Resection of a pre-cancerous tumor should be considered the most efficient advance in pancreatic cancer management.
Chemotherapy
Chemotherapy may be used either in the palliative setting for metastatic or locally advanced unresectable tumors, or in resectable disease before (neoadjuvant) or after surgery (adjuvant treatment).
Palliative Treatment
Gemcitabine, a nucleoside analog, became the reference treatment for advanced pancreatic cancer after a randomized trial in which 126 chemotherapy-naïve patients with advanced disease were randomized to receive either weekly gemcitabine or 5-FU (Burris et al., 1997). No statistically significant difference was found in the overall response rate between the two groups (5.4% for gemcitabine and 0% for 5-FU), but the number of patient who experienced improved clinical response (improvement in pain or performance status) was significantly greater in the gemcitabine group than in the 5-FU group (23.8 versus 4.8%, P = 0.0022). Gemcitabine treatment was associated with improvement in the median overall survival (5.65 versus 4.41 months, P = 0.0025) and 1-year survival (18 versus 2%). Following this trial, several studies have evaluated in phase III randomized trials the impact of adding a second cytotoxic compound to gemcitabine, notably 5-FU and analogs (such as capecitabine, an oral 5-FU pro-drug; Berlin et al., 2002; Herrmann et al., 2007; Cunningham et al., 2009) or platinum (cisplatin or oxaliplatin) derivatives (Colucci et al., 2002, 2010; Louvet et al., 2005; Heinemann et al., 2006; Poplin et al., 2009). No clear survival advantage was demonstrated in these trials, even though some of them have suggested a benefit in patients with good performance status (Heinemann et al., 2008). Several classes of targeted agents have also been tested in combination with gemcitabine and were compared to gemcitabine alone. In a phase III trial, the combination of erlotinib, an orally administered EGFR tyrosine kinase inhibitor, plus gemcitabine versus gemcitabine alone showed statistically significant improvements in overall survival [6.2 versus 5.9 months; hazard ratio (HR): 0.82; P = 0.038] and 1-year survival (23 versus 17%, P = 0.023), leading to its approval by health authorities for first-line treatment of patients with advanced pancreatic cancer. However, the small improvement in survival, only 2 weeks, was associated with increased toxicity and therefore not considered as clinically meaningful (Moore et al., 2007), explaining why it is barely used in the routine setting. Cetuximab, a chimeric monoclonal antibody targeting EGFR, was also evaluated in combination with gemcitabine versus gemcitabine alone, but no difference in term of response, progression-free survival, or overall survival was observed (Philip et al., 2010). Two phase III trials investigated the potential impact of bevacizumab, a humanized monoclonal antibody targeting VEGF, in advanced pancreatic cancer patients (gemcitabine plus bevacizumab versus gemcitabine alone; gemcitabine plus erlotinib plus bevacizumab versus gemcitabine plus erlotinib plus placebo) but failed to demonstrate any overall survival advantage (Van Cutsem et al., 2009; Kindler et al., 2010). Other tested targeted agents to date, such as tipifarnib (farnesyl transferase inhibitor), axitinib (multitargeted tyrosine kinase inhibitor), matrix metalloprotease inhibitors have generated disappointing results.
Most recently, a polychemotherapy regimen including 5-FU, leucovorin, oxaliplatin, and irinotecan was compared to gemcitabine as first-line therapy in 342 metastatic pancreatic cancer patients, with good performance status, normal liver function and age under 76. The median overall survival, which was the primary endpoint, was 11.1 months in the FOLFIRINOX group as compared with 6.8 months in the gemcitabine group [HR for death, 0.57; 95% confidence interval (CI), 0.45–0.73; P < 0.001]. Median progression-free survival was 6.4 months in the FOLFIRINOX group and 3.3 months in the gemcitabine group (HR for disease progression, 0.47; 95% CI, 0.37–0.59; P < 0.001). The objective response rate was 31.6% in the FOLFIRINOX group versus 9.4% in the gemcitabine group (P < 0.001). More adverse events were noted in the FOLFIRINOX group with a higher incidence of grade 3 or 4 neutropenia, febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy, as well as grade 2 alopecia. At 6 months, 31% of the patients in the FOLFIRINOX group had a definitive degradation of the quality of life versus 66% in the gemcitabine group (HR, 0.47; 95% CI, 0.30–0.70; P < 0.001; Conroy et al., 2011). These results suggest that FOLFIRINOX should be considered as the recommended first-line option for patients with metastatic pancreatic cancer and judged fit to receive it (Performance status 0–1, normal, or nearly normal bilirubin level, age < 76).
In patients with non-metastatic locally advanced pancreatic tumor (i.e., non-resectable), primary chemoradiation was the preferred treatment in the 90s. However, some patients received a local treatment and were diagnosed with distant disease after completion of chemoradiation. In addition, a French randomized trial comparing gemcitabine alone to chemoradiation (5-FU + cisplatin) followed by gemcitabine, showed a lower overall survival in the radiotherapy arm (Chauffert et al., 2008). Thus, unresectable tumors are usually treated with short period chemotherapy followed by chemoradiation in patients with non-progressive disease. In addition, some of these patients may be secondarily brought to surgery.
Neoadjuvant Chemotherapy
As judged from its impact on survival, neoadjuvant treatment is not recommended for resectable pancreatic cancer. However, two uncontrolled phase II trials have evaluated neoadjuvant gemcitabine-based chemotherapy alone (without radiation), with promising results. Palmer et al. (2007) randomized 50 patients with potentially resectable pancreatic lesions to be treated with a short phase of weekly gemcitabine plus or minus cisplatin. Overall, 54% of patients underwent resection, 9 (38%) in the gemcitabine arm, and 18 (70%) in the combination arm, with no increase in surgical complications. Twelve-month survival for the gemcitabine and combination groups was 42 and 62% (Palmer et al., 2007). In a prospective phase II study, Heinrich et al. (2008) tested neoadjuvant gemcitabine–cisplatin biweekly, for four cycles in 28 patients with resectable adenocarcinoma of the pancreatic head. Resection rate R0 was 80% and survival was increased (Heinrich et al., 2008). However, these studies were small-sized and further data are required before integrating preoperative chemotherapy in the routine setting. Of course, because of the results reported with FOLFIRINOX in advanced disease, this regimen should be imperatively evaluated in this strategy. Neoadjuvant chemoradiation was also used in phase II studies but showed poor impact on disease progression (Gillen et al., 2010).
Adjuvant Treatment
Till the mid-2000s, resection of pancreatic cancer was not routinely associated with postoperative chemotherapy. In 2004, the final results of a randomized trial (ESPAC-1) were reported, comparing surgery alone (n = 69) to chemotherapy alone (5-FU bolus plus folinic acid, 6 months; n = 75), radiochemotherapy (5-FU-based; n = 73), or both (n = 72). The estimated 5-year survival rate was 10% among patients assigned to receive chemoradiotherapy and 20% among patients who did not receive chemoradiotherapy (P = 0.05). The 5-year survival rate was 21% among patients who received chemotherapy and 8% among patients who did not receive chemotherapy (P = 0.009). These results indicate that adjuvant chemotherapy provided significant survival benefit in patients with resected pancreatic cancer, whereas adjuvant chemoradiotherapy had a deleterious effect on survival (Ghaneh and Neoptolemos, 2004). In 2007, Oettle et al. (2007) compared weekly gemcitabine (6 months) to observation after curative-intent resection in 368 pancreatic cancer patients (CONKO-001 trial). Median disease-free survival, the primary endpoint, was 13.4 months in the gemcitabine group and 6.9 months in the control group (P < 0.001, log-rank; Oettle et al., 2007). Final results of this trial showed that this disease-free survival difference translated into an overall survival advantage (Oettle et al., 2007). Thus, both 5-FU- and gemcitabine-adjuvant chemotherapy improve outcome in resected pancreatic cancer. A face to face comparative study between 5-FU and gemcitabine in this setting revealed no difference in efficacy, but 5-FU appeared more toxic (Neoptolemos et al., 2009). Therefore, gemcitabine is the recommended choice for adjuvant treatment, but 5-FU may be used as an alternative in case of intolerance. Again, after recent exciting results reported in the palliative setting, FOLFIRINOX will have to be evaluated as adjuvant treatment after surgery.
Radiotherapy
Radiotherapy is an efficient weapon against pancreatic cancer. However, appropriate use/timing of radiotherapy was not precisely identified. Radiotherapy alone was not used in pancreatic cancer. Neoadjuvant chemoradiation was performed with uneven results in patients with resectable tumor. Several drugs were associated to radiotherapy (5-FU, cisplatine, gemcitabine…) with similar results. Patients who had had tumor resection after chemoradiation showed histologic modifications in the removed tumor. Moreover, sterilized specimens were sometimes observed. Thus, chemoradiation is certainly efficient in a small proportion of patients but no factor could be identified at diagnosis to determine which group of patient had to receive chemoradiation. To date, chemoradiation should not be recommended in resectable tumor. Finally, in patients with locally advanced tumor, chemoradiation is used after inductive chemotherapy in patients with still non-metastatic disease. However, a small proportion of patients with locally advanced tumors are eligible for resection after completion of chemotherapy followed by chemoradiation.
New Strategies for the Treatment of Pancreatic Cancers
Using Gene Therapy to Enhance Cancer Chemotherapeutic Efficacy
In recent years, gene therapy based on the knowledge of molecular pathogenesis has been actively developed as a novel therapeutic strategy to be used alone or in combination with conventional chemotherapy in pancreatic cancer. Strategies for gene therapy in pancreatic cancer include antisense and RNA interference strategies whereby the function of activated oncogenes (K-ras, H-ras, Notch, LSM1, etc.) is inhibited, and strategies to restore the function of tumor suppressor genes (p53, p16/INK4a, p21/WAF1, DPC4/Smad4, etc.). These therapeutic strategies have shown their promising effects on the inhibition of pancreatic cancer in vitro and in vivo (Bhattacharyya and Lemoine, 2006). In addition, gene-directed pro-drug activation therapy is another gene therapy system in which a gene that encodes an enzyme is delivered to the tumor cell. Then, a pro-drug administered as chemotherapy is metabolized by the enzyme to release cytotoxic drug at the site of the tumor. A recent report has shown that introduction of a fusion gene combining deoxycytidine kinase and uridine monophosphate kinase, which converts gemcitabine into its toxic phosphorylated metabolite, sensitizes pancreatic cancer cells to gemcitabine by reducing dramatically both in vitro cell viability and in vivo tumor volume (Vernejoul et al., 2006). Moreover, the combinations of gemcitabine treatment with introduction of IFN-α, NK4, or p53 significantly suppress the growth and the metastasis of human pancreatic cancer cells, suggesting that the efficacy of chemotherapy in pancreatic cancer can actually be enhanced by gene therapy (Bhattacharyya and Lemoine, 2006; Ogura et al., 2006).
Promising Triazole Nucleoside Compounds to Fight Drug-Resistant Pancreatic Cancer
We initiated, several years ago, a research program to develop novel triazole nucleoside analogs with appended aromatic systems on the triazole base, in the view to searching for biologically active nucleoside analogs as drug candidates against pancreatic cancer, particularly for drug-resistant pancreatic cancer. The rationale for molecular design was based on the following considerations. First, nucleoside analogs show important anticancer activity; several successful nucleoside drugs such as gemcitabine, cladribine, capecitabine, among others, are currently used as anticancer therapy (Galmarini et al., 2002). Second, triazole is an unnatural heterocycle, and nucleosides containing triazole as nucleobase may be more resistant to nucleos(t)ide metabolizing enzymes and thus offer better in vivo stability. Third, triazole is considered as a universal base capable to form base-pairs with all five natural nucleobases, it can therefore impart triazole nucleosides much better interaction with their biological receptors (Loakes, 2001). Finally, appending aromatic systems to a triazole nucleobase may yield an expanded and enlarged conjugated with advantageous binding properties to the corresponding nucleosides for interaction with biological targets through stronger and more efficient binding via larger aromatic systems. Based on all these rationales, we have designed various triazole nucleoside analogs with appended aromatic groups on the triazole nucleobase. Their sugar moiety is either ribose or acyclic glycogen and the triazole nucleobase is connected to the sugar moiety in such a way that either 3- or 5-aryltriazole nucleoside analogs can be obtained. The aromatic moieties are introduced on the triazole ring via direct connection (type A), triple bond bridge (type B), triazolyl ring linker (type C), or N-arylation (type D). Appending aromatic moieties directly on the tiazole ring provides biaryl motifs as nucleobase in type A compounds. It is well known that biaryl motifs are very important in medicinal chemistry. Biaryl motifs as nucleobase in nucleoside analogs are of great interest in the view to exploring new nucleoside mimics. Connection of aromatic moieties via triple bond bridge or triazolyl ring on the triazole ring can offer enlarged conjugated aromatic systems as nucleobases, and may lead to better and stronger binding with biological targets. Amine linkage between aromatic moieties and triazole ring in type D is expected to provide some flexibility to the corresponding nucleobases. Altogether, these compounds provide not only considerable structural deviation from gemcitabine on one hand, but also important structural diversity on the other hand. They should be a good starting point to search for novel anticancer candidates against pancreatic cancer.
All the synthesized triazole nucleosides types A–D were first assessed in vitro using MTT test to evaluate their antiproliferative activity on drug-sensitive (Capan2) and drug-resistant (MiaPaCa2) human pancreatic cancer cells. Among more than 600 compounds tested, dozens of compounds showed effective antiproliferative activity against drug-sensitive pancreatic cancer Capan2 cells, whereas a smaller amount of compounds emerged with effective anticancer activity against drug-resistant pancreatic cancer MiaPaCa2 cells. Since we are particularly interested in identifying leads against drug-resistant pancreatic cancer, we focused our attention on the most promising active compounds named 1 and 2, which exhibited much better in vitro activity than gemcitabine (Wan et al., 2009; Xia et al., 2009). Moreover, both compounds 1 and 2 could efficiently inhibit tumor growth in MiaPaCa2-xenograft nude mice after a 2-week treatment only, with no apparent adverse effect. Further studies revealed that the induction of caspase-dependent apoptosis represented the main eason for the observed anticancer activity of compounds 1 and 2. Although both compounds are originally designed as nucleoside analogs to interfere with DNA synthesis, neither compound 1 nor compound 2 could significantly inhibit DNA synthesis, contrary to gemcitabine. Gemcitabine has been also reported to down-regulate p8, a stress-associated protein with antiapoptotic properties. However, neither compound 1 nor compound 2 could significantly down-regulate p8 in a way similar similar to gemcitabine. On the contrary, both of them rather upregulate p8 expression. These findings suggest that the mechanisms of action of the active Triazole nucleosides compounds 1 and 2 have mechanisms of action very different from those of gemcitabine. In fact, preliminary investigations demonstrated that compound 2 could effectively down-regulate heat shock protein 27 (Hsp27) at both mRNA and protein levels. Hsp27 is an ATP-independent small molecular chaperone recently recognized as a novel target for anticancer therapy (Concannon et al., 2003; Arrigo et al., 2007). It plays an important role in the resistance shown by patients with pancreatic cancer to gemcitabine and its function is to protect against drug toxicity (Mori-Iwamoto et al., 2007). Recent studies with antisense oligonucleotides and siRNA molecules down-regulating Hsp27 demonstrated potent caspase-dependent apoptotic anticancer activity on prostate cancer (Rocchi et al., 2004, 2005). These antisense oligonucleotides are currently in clinical trial for use in the treatment of several human cancers, including lung, breast, prostate, bladder, and ovarian cancers. An additional study with IFN-γ also showed that down-regulation of Hsp27 via IFN-γ could generate effective anticancer activity in drug-resistant pancreatic cancer cells, further validating the general utility and importance of down-regulating Hsp27 in treating drug-resistant cancers (Mori-Iwamoto et al., 2009). This represents the first small molecular anticancer lead with such a novel mode of action and effective anticancer consequence, opening a new avenue in the search for novel anticancer drug candidates to fight drug-resistant pancreatic cancer.
Conclusion
The development and progression of pancreatic cancer are closely associated with the activation of oncogenes, the inactivation of tumor suppressor genes, the deregulation of EGFR, Akt, NFκB, and their downstream signaling pathways. Although targeted therapies for pancreatic cancer have yielded encouraging results in vitro and in animal models, these findings have not been translated to improved outcomes in clinical trials. Reasons for this failure might include an incomplete understanding of the biology of pancreatic cancer, the selection of poor active agents, problems with trial design (such as inappropriate therapeutic end points or patient selection) and the rapidity with which agents move into randomized, controlled trials without the extensive early testing necessary to optimize treatment regimens. Furthermore, preclinical studies performed on mouse models do not always recapitulate the human condition, a particularly important limitation with human pancreatic cancer xenografts in immunodeficient mice. Despite these setbacks, lessons have been learnt, and our collective effort has generated a substantial platform of knowledge from which further work could spring. Genetically engineered immunocompetent mice, such as those with K-RAS or TP53 mutations, have been developed and they hold promise for future studies of the disease. The bioavailability of compounds such as antisense oligonucleotides and siRNAs in humans remains a big hurdle, which will require further improvement of gene-delivery strategies.
The individualization of therapy for patients is possible if factors that predict treatment response, such as biological markers, can be determined accurately. These approaches are likely to comprise a mixture of targeted agents in combination with conventional chemotherapy and radiotherapy. For a clinically significant effect to be achieved, treatment strategies should either be in the form of a “horizontal” approach, in which several oncogenic pathways are inhibited, or a “vertical” approach, whereby multiple levels of a major pathway are targeted. Combined therapies, together with improved diagnostic tools and predictive markers, are ultimately hoped to improve the bleak outlook of patients diagnosed with pancreatic cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from INSERM, Ligue Contre le Cancer, and INCA.
References
Abdelrahim, M., Newman, K., Vanderlaag, K., Samudio, I., and Safe, S. (2006). 3,3′-Diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis 27, 717–728.
Abe, N., Watanabe, T., Masaki, T., Mori, T., Sugiyama, M., Uchimura, H., Fujioka, Y., Chiappetta, G., Fusco, A., and Atomi, Y. (2000). Pancreatic duct cell carcinomas express high levels of high mobility group I(Y) proteins. Cancer Res. 60, 3117–3122.
Abraham, S. C., Wu, T. T., Klimstra, D. S., Finn, L. S., Lee, J. H., Yeo, C. J., Cameron, J. L., and Hruban, R. H. (2001). Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/β-catenin pathway and chromosome 11p. Am. J. Pathol. 159, 1619–1627.
Aird, I., Bentall, H. H., and Roberts, J. A. (1953). A relationship between cancer of stomach and the ABO blood groups. Br. Med. J. 1, 799–801.
Almoguera, C., Shibata, D., Forrester, K., Martin, J., Arnheim, N., and Perucho, M. (1988). Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549–554.
Amundadottir, L., Kraft, P., Stolzenberg-Solomon, R. Z., Fuchs, C. S., Petersen, G. M., Arslan, A. A., Bueno-de-Mesquita, H. B., Gross, M., Helzlsouer, K., Jacobs, E. J., LaCroix, A., Zheng, W., Albanes, D., Bamlet, W., Berg, C. D., Berrino, F., Bingham, S., Buring, J. E., Bracci, P. M., Canzian, F., Clavel-Chapelon, F., Clipp, S., Cotterchio, M., de Andrade, M., Duell, E. J., Fox, J. W. Jr., Gallinger, S., Gaziano, J. M., Giovannucci, E. L., Goggins, M., González, C. A., Hallmans, G., Hankinson, S. E., Hassan, M., Holly, E. A., Hunter, D. J., Hutchinson, A., Jackson, R., Jacobs, K. B., Jenab, M., Kaaks, R., Klein, A. P., Kooperberg, C., Kurtz, R. C., Li, D., Lynch, S. M., Mandelson, M., McWilliams, R. R., Mendelsohn, J. B., Michaud, D. S., Olson, S. H., Overvad, K., Patel, A. V., Peeters, P. H., Rajkovic, A., Riboli, E., Risch, H. A., Shu, X. O., Thomas, G., Tobias, G. S., Trichopoulos, D., Van Den Eeden, S. K., Virtamo, J., Wactawski-Wende, J., Wolpin, B. M., Yu, H., Yu, K., Zeleniuch-Jacquotte, A., Chanock, S. J., Hartge, P., and Hoover, R. N. (2009). Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat. Genet. 41, 986–990.
Andea, A., Sarkar, F., and Adsay, V. N. (2003). Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod. Pathol. 16, 996–1006.
Arrigo, A. P., Simon, S., Gibert, B., Kretz-Remy, C., Nivon, M., Czekalla, A., Guillet, D., Moulin, M., Diaz-Latoud, C., and Vicart, P. (2007). Hsp27 (HspB1) and αB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 581, 3665–3674.
Asano, T., Yao, Y., Zhu, J., Li, D., Abbruzzese, J. L., and Reddy, S. A. (2004). The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-κB and c-Myc in pancreatic cancer cells. Oncogene 23, 8571–8580.
Baker, C. H., Trevino, J. G., Summy, J. M., Zhang, F., Caron, A., Nesbit, M., Gallick, G. E., and Fidler, I. J. (2006). Inhibition of PDGFR phosphorylation and Src and Akt activity by GN963 leads to therapy of human pancreatic cancer growing orthotopically in nude mice. Int. J. Oncol. 29, 125–138.
Banerjee, S., Zhang, Y., Ali, S., Bhuiyan, M., Wang, Z., Chiao, P. J., Philip, P. A., Abbruzzese, J., and Sarkar, F. H. (2005). Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 65, 9064–9072.
Banville, N., Geraghty, R., Fox, E., Leahy, D. T., Green, A., Keegan, D., Geoghegan, J., O’Donoghue, D., Hyland, J., and Sheahan, K. (2006). Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum. Pathol. 37, 1498–1502.
Bardeesy, N., and DePinho, R. A. (2002). Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2, 897–909.
Beltran, P. J., Mitchell, P., Chung, Y. A., Cajulis, E., Lu, J., Belmontes, B., Ho, J., Tsai, M. M., Zhu, M., Vonderfecht, S., Baserga, R., Kendall, R., Radinsky, R., and Calzone, F. J. (2009). AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol. Cancer Ther. 8, 1095–1105.
Berger, S. L. (2002). Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12, 142–148.
Berlin, J. D., Catalano, P., Thomas, J. P., Kugler, J. W., Haller, D. G., and Benson, A. B. III. (2002). Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J. Clin. Oncol. 20, 3270–3275.
Berman, D. M., Karhadkar, S. S., Maitra, A., Montes De Oca, R., Gerstenblith, M. R., Briggs, K., Parker, A. R., Shimada, Y., Eshleman, J. R., Watkins, D. N., and Beachy, P. A. (2003). Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846–851.
Bernhardt, S. L., Gjertsen, M. K., Trachsel, S., Moller, M., Eriksen, J. A., Meo, M., Buanes, T., and Gaudernack, G. (2006). Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br. J. Cancer 95, 1474–1482.
Bhattacharyya, M., and Lemoine, N. R. (2006). Gene therapy developments for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 20, 285–298.
Biliran, H. Jr., Wang, Y., Banerjee, S., Xu, H., Heng, H., Thakur, A., Bollig, A., Sarkar, F. H., and Liao, J. D. (2005). Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin. Cancer Res. 11, 6075–6086.
Bonafoux, D., and Lee, W. C. (2009). Strategies for TGF-β modulation: a review of recent patents. Expert Opin. Ther. Pat. 19, 1759–1769.
Bramhall, S. R., Rosemurgy, A., Brown, P. D., Bowry, C., and Buckels, J. A. (2001). Marimastat as first-line therapy for patients with unresectable pancreatic cancer: a randomized trial. J. Clin. Oncol. 19, 3447–3455.
Bramhall, S. R., Schulz, J., Nemunaitis, J., Brown, P. D., Baillet, M., and Buckels, J. A. (2002). A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 87, 161–167.
Brat, D. J., Lillemoe, K. D., Yeo, C. J., Warfield, P. B., and Hruban, R. H. (1998). Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 22, 163–169.
Brockie, E., Anand, A., and Albores-Saavedra, J. (1998). Progression of atypical ductal hyperplasia/carcinoma in situ of the pancreas to invasive adenocarcinoma. Ann. Diagn. Pathol. 2, 286–292.
Bruserud, O., Stapnes, C., Ersvaer, E., Gjertsen, B. T., and Ryningen, A. (2007). Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr. Pharm. Biotechnol. 8, 388–400.
Buchler, P., Gazdhar, A., Schubert, M., Giese, N., Reber, H. A., Hines, O. J., Giese, T., Ceyhan, G. O., Muller, M., Buchler, M. W., and Friess, H. (2005). The Notch signaling pathway is related to neurovascular progression of pancreatic cancer. Ann. Surg. 242, 791–800; discussion 800–801.
Burris, H. A. III, Moore, M. J., Andersen, J., Green, M. R., Rothenberg, M. L., Modiano, M. R., Cripps, M. C., Portenoy, R. K., Storniolo, A. M., Tarassoff, P., Nelson, R., Dorr, F. A., Stephens, C. D., and Von Hoff, D. D. (1997). Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 15, 2403–2413.
Campbell, P. J., Yachida, S., Mudie, L. J., Stephens, P. J., Pleasance, E. D., Stebbings, L. A., Morsberger, L. A., Latimer, C., McLaren, S., Lin, M. L., McBride, D. J., Varela, I., Nik-Zainal, S. A., Leroy, C., Jia, M., Menzies, A., Butler, A. P., Teague, J. W., Griffin, C. A., Burton, J., Swerdlow, H., Quail, M. A., Stratton, M. R., Iacobuzio-Donahue, C., and Futreal, P. A. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113.
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., Jones, R. S., and Zhang, Y. (2002). Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043.
Caplin, M., Savage, K., Khan, K., Brett, B., Rode, J., Varro, A., and Dhillon, A. (2000). Expression and processing of gastrin in pancreatic adenocarcinoma. Br. J. Surg. 87, 1035–1040.
Chau, I., Cunningham, D., Russell, C., Norman, A. R., Kurzawinski, T., Harper, P., Harrison, P., Middleton, G., Daniels, F., Hickish, T., Prendeville, J., Ross, P. J., Theis, B., Hull, R., Walker, M., Shankley, N., Kalindjian, B., Murray, G., Gillbanks, A., and Black, J. (2006). Gastrazole (JB95008), a novel CCK2/gastrin receptor antagonist, in the treatment of advanced pancreatic cancer: results from two randomised controlled trials. Br. J. Cancer 94, 1107–1115.
Chauffert, B., Mornex, F., Bonnetain, F., Rougier, P., Mariette, C., Bouche, O., Bosset, J. F., Aparicio, T., Mineur, L., Azzedine, A., Hammel, P., Butel, J., Stremsdoerfer, N., Maingon, P., and Bedenne, L. (2008). Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann. Oncol. 19, 1592–1599.
Colucci, G., Giuliani, F., Gebbia, V., Biglietto, M., Rabitti, P., Uomo, G., Cigolari, S., Testa, A., Maiello, E., and Lopez, M. (2002). Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer 94, 902–910.
Colucci, G., Labianca, R., Di Costanzo, F., Gebbia, V., Carteni, G., Massidda, B., Dapretto, E., Manzione, L., Piazza, E., Sannicolo, M., Ciaparrone, M., Cavanna, L., Giuliani, F., Maiello, E., Testa, A., Pederzoli, P., Falconi, M., Gallo, C., Di Maio, M., Perrone, F., Gruppo Oncologico Italia Meridionale (GOIM), Gruppo Italiano per lo Studio dei Carcinomi dell’Apparato Digerente (GISCAD), and Gruppo Oncologico Italiano di Ricerca Clinica (GOIRC). (2010). Randomized phase III trial of gemcitabine plus cisplatin compared with single-agent gemcitabine as first-line treatment of patients with advanced pancreatic cancer: the GIP-1 study. J. Clin. Oncol. 28, 1645–1651.
Concannon, C. G., Gorman, A. M., and Samali, A. (2003). On the role of Hsp27 in regulating apoptosis. Apoptosis 8, 61–70.
Conroy, T., Desseigne, F., Ychou, M., Bouche, O., Guimbaud, R., Becouarn, Y., Adenis, A., Raoul, J. L., Gourgou-Bourgade, S., de la Fouchardiere, C., Bennouna, J., Bachet, J. B., Khemissa-Akouz, F., Péré-Vergé, D., Delbaldo, C., Assenat, E., Chauffert, B., Michel, P., Montoto-Grillot, C., Ducreux, M., Groupe Tumeurs Digestives of Unicancer, and PRODIGE Intergroup. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825.
Corcoran, R. B., Contino, G., Deshpande, V., Tzatsos, A., Conrad, C., Benes, C. H., Levy, D. E., Settleman, J., Engelman, J. A., and Bardeesy, N. (2011). STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 71, 5020–5029.
Coughlin, S. S., Calle, E. E., Patel, A. V., and Thun, M. J. (2000). Predictors of pancreatic cancer mortality among a large cohort of United States adults. Cancer Causes Control 11, 915–923.
Cowgill, S. M., and Muscarella, P. (2003). The genetics of pancreatic cancer. Am. J. Surg. 186, 279–286.
Cress, W. D., and Seto, E. (2000). Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 184, 1–16.
Cubilla, A. L., and Fitzgerald, P. J. (1976). Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res. 36, 2690–2698.
Cunningham, D., Chau, I., Stocken, D. D., Valle, J. W., Smith, D., Steward, W., Harper, P. G., Dunn, J., Tudur-Smith, C., West, J., Falk, S., Crellin, A., Adab, F., Thompson, J., Leonard, P., Ostrowski, J., Eatock, M., Scheithauer, W., Herrmann, R., and Neoptolemos, J. P. (2009). Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 27, 5513–5518.
Dhillon, N., Aggarwal, B. B., Newman, R. A., Wolff, R. A., Kunnumakkara, A. B., Abbruzzese, J. L., Ng, C. S., Badmaev, V., and Kurzrock, R. (2008). Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 14, 4491–4499.
Diergaarde, B., Brand, R., Lamb, J., Cheong, S. Y., Stello, K., Barmada, M. M., Feingold, E., and Whitcomb, D. C. (2010). Pooling-based genome-wide association study implicates γ-glutamyltransferase 1 (GGT1) gene in pancreatic carcinogenesis. Pancreatology 10, 194–200.
Ding, X. Z., and Adrian, T. E. (2002). Resveratrol inhibits proliferation and induces apoptosis in human pancreatic cancer cells. Pancreas 25, e71–e76.
Donadelli, M., Costanzo, C., Beghelli, S., Scupoli, M. T., Dandrea, M., Bonora, A., Piacentini, P., Budillon, A., Caraglia, M., Scarpa, A., and Palmieri, M. (2007). Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim. Biophys. Acta 1773, 1095–1106.
Dong, J. T., and Chen, C. (2009). Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell. Mol. Life Sci. 66, 2691–2706.
Earl, J., Yan, L., Vitone, L. J., Risk, J., Kemp, S. J., McFaul, C., Neoptolemos, J. P., Greenhalf, W., European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer Kress, R., Sina-Frey, M., Hahn, S. A., Rieder, H., Bartsch, D. K., and German National Case Collection for Familial Pancreatic Cancer. (2006). Evaluation of the 4q32-34 locus in European familial pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 15, 1948–1955.
El-Rayes, B. F., Ali, S., Sarkar, F. H., and Philip, P. A. (2004). Cyclooxygenase-2-dependent and -independent effects of celecoxib in pancreatic cancer cell lines. Mol. Cancer Ther. 3, 1421–1426.
Fahy, B. N., Schlieman, M. G., Virudachalam, S., and Bold, R. J. (2004). Inhibition of AKT abrogates chemotherapy-induced NF-κB survival mechanisms: implications for therapy in pancreatic cancer. J. Am. Coll. Surg. 198, 591–599.
Fayard, E., Schoonjans, K., Annicotte, J. S., and Auwerx, J. (2003). Liver receptor homolog 1 controls the expression of carboxyl ester lipase. J. Biol. Chem. 278, 35725–35731.
Fearon, E. R., and Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767.
Feldmann, G., Dhara, S., Fendrich, V., Bedja, D., Beaty, R., Mullendore, M., Karikari, C., Alvarez, H., Iacobuzio-Donahue, C., Jimeno, A., Gabrielson, K. L., Matsui, W., and Maitra, A. (2007). Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 67, 2187–2196.
Fernandez-Zapico, M. E., Gonzalez-Paz, N. C., Weiss, E., Savoy, D. N., Molina, J. R., Fonseca, R., Smyrk, T. C., Chari, S. T., Urrutia, R., and Billadeau, D. D. (2005). Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 7, 39–49.
Friess, H., Langrehr, J. M., Oettle, H., Raedle, J., Niedergethmann, M., Dittrich, C., Hossfeld, D. K., Stoger, H., Neyns, B., Herzog, P., Piedbois, P., Dobrowolski, F., Scheithauer, W., Hawkins, R., Katz, F., Balcke, P., Vermorken, J., van Belle, S., Davidson, N., Esteve, A. A., Castellano, D., Kleeff, J., Tempia-Caliera, A. A., Kovar, A., and Nippgen, J. (2006). A randomized multi-center phase II trial of the angiogenesis inhibitor cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer 6, 285. doi:10.1186/1471-2407-6-285
Fujioka, S., Sclabas, G. M., Schmidt, C., Frederick, W. A., Dong, Q. G., Abbruzzese, J. L., Evans, D. B., Baker, C., and Chiao, P. J. (2003). Function of nuclear factor κB in pancreatic cancer metastasis. Clin. Cancer Res. 9, 346–354.
Funahashi, Y., Sugi, N. H., Semba, T., Yamamoto, Y., Hamaoka, S., Tsukahara-Tamai, N., Ozawa, Y., Tsuruoka, A., Nara, K., Takahashi, K., Okabe, T., Kamata, J., Owa, T., Ueda, N., Haneda, T., Yonaga, M., Yoshimatsu, K., and Wakabayashi, T. (2002). Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res. 62, 6116–6123.
Furukawa, T. (2007). Molecular genetics of intraductal papillary-mucinous neoplasms of the pancreas. J. Hepatobiliary Pancreat. Surg. 14, 233–237.
Furukawa, T., Duguid, W. P., Kobari, M., Matsuno, S., and Tsao, M. S. (1995). Hepatocyte growth factor and Met receptor expression in human pancreatic carcinogenesis. Am. J. Pathol. 147, 889–895.
Furuyama, K., Doi, R., Mori, T., Toyoda, E., Ito, D., Kami, K., Koizumi, M., Kida, A., Kawaguchi, Y., and Fujimoto, K. (2006). Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J. Surg. 30, 219–226.
Galmarini, C. M., Mackey, J. R., and Dumontet, C. (2002). Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3, 415–424.
Garcea, G., Neal, C. P., Pattenden, C. J., Steward, W. P., and Berry, D. P. (2005). Molecular prognostic markers in pancreatic cancer: a systematic review. Eur. J. Cancer 41, 2213–2236.
Ghaneh, P., and Neoptolemos, J. P. (2004). Conclusions from the European Study Group for Pancreatic Cancer adjuvant trial of chemoradiotherapy and chemotherapy for pancreatic cancer. Surg. Oncol. Clin. N. Am. 13, 567–587.
Giardiello, F. M., Brensinger, J. D., Tersmette, A. C., Goodman, S. N., Petersen, G. M., Booker, S. V., Cruz-Correa, M., and Offerhaus, J. A. (2000). Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 119, 1447–1453.
Gillen, S., Schuster, T., Meyer Zum Buschenfelde, C., Friess, H., and Kleeff, J. (2010). Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 7, e1000267. doi: 10.1371/journal.pmed.1000267
Gjertsen, M. K., Buanes, T., Rosseland, A. R., Bakka, A., Gladhaug, I., Soreide, O., Eriksen, J. A., Moller, M., Baksaas, I., Lothe, R. A., Saeterdal, I., and Gaudernack, G. (2001). Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 92, 441–450.
Glaser, K. B., Staver, M. J., Waring, J. F., Stender, J., Ulrich, R. G., and Davidsen, S. K. (2003). Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2, 151–163.
Goggins, M., Offerhaus, G. J., Hilgers, W., Griffin, C. A., Shekher, M., Tang, D., Sohn, T. A., Yeo, C. J., Kern, S. E., and Hruban, R. H. (1998a). Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 152, 1501–1507.
Goggins, M., Shekher, M., Turnacioglu, K., Yeo, C. J., Hruban, R. H., and Kern, S. E. (1998b). Genetic alterations of the transforming growth factor β receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 58, 5329–5332.
Goldstein, A. M., Fraser, M. C., Struewing, J. P., Hussussian, C. J., Ranade, K., Zametkin, D. P., Fontaine, L. S., Organic, S. M., Dracopoli, N. C., Clark, W. H. Jr., and Tucker, M. A. (1995). Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N. Engl. J. Med. 333, 970–974.
Gonçalves, A., Viret, F., François, E., Dahan, L., Perrier, H., Lamy, R., Re, D., Largillier, R., Gasmi, M., Tchiknavorian, X., Turrini, O., Moureau-Zabotto, L., Delpero, J., and Viens, P. (2011). BAYPAN study: a double-blind, phase III randomized trial of gemcitabine plus sorafenib versus gemcitabine plus placebo in patients with advanced pancreatic cancer. J. Clin. Oncol. 29(suppl.), abstr 4028.
Griffiths, E. A., and Gore, S. D. (2008). DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 45, 23–30.
Guerra, C., Collado, M., Navas, C., Schuhmacher, A. J., Hernandez-Porras, I., Canamero, M., Rodriguez-Justo, M., Serrano, M., and Barbacid, M. (2011). Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 19, 728–739.
Guerra, C., Schuhmacher, A. J., Canamero, M., Grippo, P. J., Verdaguer, L., Perez-Gallego, L., Dubus, P., Sandgren, E. P., and Barbacid, M. (2007). Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302.
Habbe, N., Koorstra, J. B., Mendell, J. T., Offerhaus, G. J., Ryu, J. K., Feldmann, G., Mullendore, M. E., Goggins, M. G., Hong, S. M., and Maitra, A. (2009). MicroRNA miR-155 is a biomarker of early pancreatic neoplasia. Cancer Biol. Ther. 8, 340–346.
Hakam, A., Fang, Q., Karl, R., and Coppola, D. (2003). Coexpression of IGF-1R and c-Src proteins in human pancreatic ductal adenocarcinoma. Dig. Dis. Sci. 48, 1972–1978.
Heinemann, V., Boeck, S., Hinke, A., Labianca, R., and Louvet, C. (2008). Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer 8, 82. doi:10.1186/1471-2407-8-82
Heinemann, V., Quietzsch, D., Gieseler, F., Gonnermann, M., Schonekas, H., Rost, A., Neuhaus, H., Haag, C., Clemens, M., Heinrich, B., Vehling-Kaiser, U., Fuchs, M., Fleckenstein, D., Gesierich, W., Uthgenannt, D., Einsele, H., Holstege, A., Hinke, A., Schalhorn, A., and Wilkowski, R. (2006). Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 24, 3946–3952.
Heinrich, S., Pestalozzi, B. C., Schafer, M., Weber, A., Bauerfeind, P., Knuth, A., and Clavien, P. A. (2008). Prospective phase II trial of neoadjuvant chemotherapy with gemcitabine and cisplatin for resectable adenocarcinoma of the pancreatic head. J. Clin. Oncol. 26, 2526–2531.
Hermann, P. C., Huber, S. L., Herrler, T., Aicher, A., Ellwart, J. W., Guba, M., Bruns, C. J., and Heeschen, C. (2007). Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323.
Herrmann, R., Bodoky, G., Ruhstaller, T., Glimelius, B., Bajetta, E., Schuller, J., Saletti, P., Bauer, J., Figer, A., Pestalozzi, B., Köhne, C. H., Mingrone, W., Stemmer, S. M., Tàmas, K., Kornek, G. V., Koeberle, D., Cina, S., Bernhard, J., Dietrich, D., Scheithauer, W., Swiss Group for Clinical Cancer Research, and Central European Cooperative Oncology Group. (2007). Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J. Clin. Oncol. 25, 2212–2217.
Hildmann, C., Riester, D., and Schwienhorst, A. (2007). Histone deacetylases – an important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 75, 487–497.
Hiyama, E., Kodama, T., Shinbara, K., Iwao, T., Itoh, M., Hiyama, K., Shay, J. W., Matsuura, Y., and Yokoyama, T. (1997). Telomerase activity is detected in pancreatic cancer but not in benign tumors. Cancer Res. 57, 326–331.
Hruban, R. H., Adsay, N. V., Albores-Saavedra, J., Compton, C., Garrett, E. S., Goodman, S. N., Kern, S. E., Klimstra, D. S., Kloppel, G., Longnecker, D. S., Lüttges, J., and Offerhaus, G. J. (2001). Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 25, 579–586.
Hruban, R. H., Goggins, M., Parsons, J., and Kern, S. E. (2000). Progression model for pancreatic cancer. Clin. Cancer Res. 6, 2969–2972.
Hruban, R. H., Takaori, K., Klimstra, D. S., Adsay, N. V., Albores-Saavedra, J., Biankin, A. V., Biankin, S. A., Compton, C., Fukushima, N., Furukawa, T., Goggins, M., Kato, Y., Klöppel, G., Longnecker, D. S., Lüttges, J., Maitra, A., Offerhaus, G. J., Shimizu, M., and Yonezawa, S. (2004). An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am. J. Surg. Pathol. 28, 977–987.
Hu, W. G., Liu, T., Xiong, J. X., and Wang, C. Y. (2007). Blockade of sonic hedgehog signal pathway enhances antiproliferative effect of EGFR inhibitor in pancreatic cancer cells. Acta Pharmacol. Sin. 28, 1224–1230.
Hussain, S. P., Hofseth, L. J., and Harris, C. C. (2003). Radical causes of cancer. Nat. Rev. Cancer 3, 276–285.
Ischenko, I., Guba, M., Yezhelyev, M., Papyan, A., Schmid, G., Green, T., Fennell, M., Jauch, K. W., and Bruns, C. J. (2007). Effect of Src kinase inhibition on metastasis and tumor angiogenesis in human pancreatic cancer. Angiogenesis 10, 167–182.
Itzkowitz, S. H., Yuan, M., Ferrell, L. D., Ratcliffe, R. M., Chung, Y. S., Satake, K., Umeyama, K., Jones, R. T., and Kim, Y. S. (1987). Cancer-associated alterations of blood group antigen expression in the human pancreas. J. Natl. Cancer Inst. 79, 425–434.
Jang, M., Cai, L., Udeani, G. O., Slowing, K. V., Thomas, C. F., Beecher, C. W., Fong, H. H., Farnsworth, N. R., Kinghorn, A. D., Mehta, R. G., Moon, R. C., and Pezzuto, J. M. (1997). Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275, 218–220.
Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J., Smigal, C., and Thun, M. J. (2006). Cancer statistics, 2006. CA Cancer J. Clin. 56, 106–130.
Jiang, J., Lee, E. J., Gusev, Y., and Schmittgen, T. D. (2005). Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 33, 5394–5403.
Jimeno, A., Rubio-Viqueira, B., Amador, M. L., Grunwald, V., Maitra, A., Iacobuzio-Donahue, C., and Hidalgo, M. (2007). Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol. Cancer Ther. 6, 1079–1088.
Jin, H., Yang, R., Zheng, Z., Romero, M., Ross, J., Bou-Reslan, H., Carano, R. A., Kasman, I., Mai, E., Young, J., Zha, J., Zhang, Z., Ross, S., Schwall, R., Colbern, G., and Merchant, M. (2008). MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 68, 4360–4368.
Jones, S., Zhang, X., Parsons, D. W., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Kamiyama, H., Jimeno, A., Hong, S. M., Fu, B., Lin, M. T., Calhoun, E. S., Kamiyama, M., Walter, K., Nikolskaya, T., Nikolsky, Y., Hartigan, J., Smith, D. R., Hidalgo, M., Leach, S. D., Klein, A. P., Jaffee, E. M., Goggins, M., Maitra, A., Iacobuzio-Donahue, C., Eshleman, J. R., Kern, S. E., Hruban, R. H., Karchin, R., Papadopoulos, N., Parmigiani, G., Vogelstein, B., Velculescu, V. E., and Kinzler, K. W. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806.
Kawasaki, D., Emori, Y., Eta, R., Iino, Y., Hamano, H., Yoshinaga, K., Tanaka, T., Takei, M., and Watson, S. A. (2008). Effect of Z-360, a novel orally active CCK-2/gastrin receptor antagonist on tumor growth in human pancreatic adenocarcinoma cell lines in vivo and mode of action determinations in vitro. Cancer Chemother. Pharmacol. 61, 883–892.
Khan, N., Afaq, F., Saleem, M., Ahmad, N., and Mukhtar, H. (2006). Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 66, 2500–2505.
Kindler, H. L., Niedzwiecki, D., Hollis, D., Sutherland, S., Schrag, D., Hurwitz, H., Innocenti, F., Mulcahy, M. F., O’Reilly, E., Wozniak, T. F., Picus, J., Bhargava, P., Mayer, R. J., Schilsky, R. L., and Goldberg, R. M. (2010). Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 28, 3617–3622.
Kornmann, M., Arber, N., and Korc, M. (1998). Inhibition of basal and mitogen-stimulated pancreatic cancer cell growth by cyclin D1 antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J. Clin. Invest. 101, 344–352.
Kotake, Y., Cao, R., Viatour, P., Sage, J., Zhang, Y., and Xiong, Y. (2007). pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4α tumor suppressor gene. Genes Dev. 21, 49–54.
Lee, C. J., Dosch, J., and Simeone, D. M. (2008). Pancreatic cancer stem cells. J. Clin. Oncol. 26, 2806–2812.
Lee, E. J., Gusev, Y., Jiang, J., Nuovo, G. J., Lerner, M. R., Frankel, W. L., Morgan, D. L., Postier, R. G., Brackett, D. J., and Schmittgen, T. D. (2007). Expression profiling identifies microRNA signature in pancreatic cancer. Int. J. Cancer 120, 1046–1054.
Levy, L., and Hill, C. S. (2005). Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 25, 8108–8125.
Li, L., Aggarwal, B. B., Shishodia, S., Abbruzzese, J., and Kurzrock, R. (2004a). Nuclear factor-κB and IκB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer 101, 2351–2362.
Li, Y., Ellis, K. L., Ali, S., El-Rayes, B. F., Nedeljkovic-Kurepa, A., Kucuk, O., Philip, P. A., and Sarkar, F. H. (2004b). Apoptosis-inducing effect of chemotherapeutic agents is potentiated by soy isoflavone genistein, a natural inhibitor of NF-κB in BxPC-3 pancreatic cancer cell line. Pancreas 28, e90–e95.
Li, Y., Ahmed, F., Ali, S., Philip, P. A., Kucuk, O., and Sarkar, F. H. (2005). Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 65, 6934–6942.
Li, Y., Bhuiyan, M., Vaitkevicius, V. K., and Sarkar, F. H. (1998). Molecular analysis of the p53 gene in pancreatic adenocarcinoma. Diagn. Mol. Pathol. 7, 4–9.
Li, Y., Kucuk, O., Hussain, M., Abrams, J., Cher, M. L., and Sarkar, F. H. (2006). Antitumor and antimetastatic activities of docetaxel are enhanced by genistein through regulation of osteoprotegerin/receptor activator of nuclear factor-κB (RANK)/RANK ligand/MMP-9 signaling in prostate cancer. Cancer Res. 66, 4816–4825.
Lian, J. P., Word, B., Taylor, S., Hammons, G. J., and Lyn-Cook, B. D. (2004). Modulation of the constitutive activated STAT3 transcription factor in pancreatic cancer prevention: effects of indole-3-carbinol (I3C) and genistein. Anticancer Res. 24, 133–137.
Liau, S. S., Ashley, S. W., and Whang, E. E. (2006). Lentivirus-mediated RNA interference of HMGA1 promotes chemosensitivity to gemcitabine in pancreatic adenocarcinoma. J. Gastrointest. Surg. 10, 1254–1262; discussion 1263.
Liau, S. S., and Whang, E. (2008). HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin. Cancer Res. 14, 1470–1477.
Liptay, S., Weber, C. K., Ludwig, L., Wagner, M., Adler, G., and Schmid, R. M. (2003). Mitogenic and antiapoptotic role of constitutive NF-κB/Rel activity in pancreatic cancer. Int. J. Cancer 105, 735–746.
Liu, W., Bloom, D. A., Cance, W. G., Kurenova, E. V., Golubovskaya, V. M., and Hochwald, S. N. (2008). FAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cells. Carcinogenesis 29, 1096–1107.
Liu, T. J., LaFortune, T., Honda, T., Ohmori, O., Hatakeyama, S., Meyer, T., Jackson, D., de Groot, J., and Yung, W. K. (2007). Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 6, 1357–1367.
Loakes, D. (2001). Survey and summary: the applications of universal DNA base analogues. Nucleic Acids Res. 29, 2437–2447.
Lomberk, G., Wallrath, L., and Urrutia, R. (2006). The heterochromatin protein 1 family. Genome Biol. 7, 228.
Longnecker, D. S., Adsay, N. V., Fernandez-del Castillo, C., Hruban, R. H., Kasugai, T., Klimstra, D. S., Kloppel, G., Luttges, J., Memoli, V. A., Tosteson, T. D., Yanagisawa, A., Wilentz, R., and Zamboni, G. (2005). Histopathological diagnosis of pancreatic intraepithelial neoplasia and intraductal papillary-mucinous neoplasms: interobserver agreement. Pancreas 31, 344–349.
Louvet, C., Labianca, R., Hammel, P., Lledo, G., Zampino, M. G., Andre, T., Zaniboni, A., Ducreux, M., Aitini, E., Taieb, J., Faroux, R., Lepere, C., de Gramont, A., GERCOR, and GISCAD. (2005). Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 23, 3509–3516.
Low, S. K., Kuchiba, A., Zembutsu, H., Saito, A., Takahashi, A., Kubo, M., Daigo, Y., Kamatani, N., Chiku, S., Totsuka, H., Ohnami, S., Hirose, H., Shimada, K., Okusaka, T., Yoshida, T., Nakamura, Y., and Sakamoto, H. (2010). Genome-wide association study of pancreatic cancer in Japanese population. PLoS ONE 5, e11824. doi:10.1371/journal.pone.0011824
Lowenfels, A. B., Maisonneuve, P., Cavallini, G., Ammann, R. W., Lankisch, P. G., Andersen, J. R., Dimagno, E. P., Andren-Sandberg, A., and Domellof, L. (1993). Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 328, 1433–1437.
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B. L., Mak, R. H., Ferrando, A. A., Downing, J. R., Jacks, T., Horvitz, H. R., and Golub, T. R. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838.
Manolio, T. A. (2010). Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 363, 166–176.
Marcus, D. M. (1969). The ABO and Lewis blood-group system. Immunochemistry, genetics and relation to human disease. N. Engl. J. Med. 280, 994–1006.
Martin, N. E., Brunner, T. B., Kiel, K. D., DeLaney, T. F., Regine, W. F., Mohiuddin, M., Rosato, E. F., Haller, D. G., Stevenson, J. P., Smith, D., Pramanik, B., Tepper, J., Tanaka, W. K., Morrison, B., Deutsch, P., Gupta, A. K., Muschel, R. J., McKenna, W. G., Bernhard, E. J., and Hahn, S. M. (2004). A phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin. Cancer Res. 10, 5447–5454.
McClintock, B. (1941). The stability of broken ends of chromosomes in Zea mays. Genetics 26, 234–282.
Milella, M., Gelibter, A., Di Cosimo, S., Bria, E., Ruggeri, E. M., Carlini, P., Malaguti, P., Pellicciotta, M., Terzoli, E., and Cognetti, F. (2004). Pilot study of celecoxib and infusional 5-fluorouracil as second-line treatment for advanced pancreatic carcinoma. Cancer 101, 133–138.
Mimeault, M., Brand, R. E., Sasson, A. A., and Batra, S. K. (2005). Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas 31, 301–316.
Miyamoto, Y., Maitra, A., Ghosh, B., Zechner, U., Argani, P., Iacobuzio-Donahue, C. A., Sriuranpong, V., Iso, T., Meszoely, I. M., Wolfe, M. S., Hruban, R. H., Ball, D. W., Schmid, R. M., and Leach, S. D. (2003). Notch mediates TGF α-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3, 565–576.
Mohammad, R. M., Banerjee, S., Li, Y., Aboukameel, A., Kucuk, O., and Sarkar, F. H. (2006). Cisplatin-induced antitumor activity is potentiated by the soy isoflavone genistein in BxPC-3 pancreatic tumor xenografts. Cancer 106, 1260–1268.
Moore, M. J., Goldstein, D., Hamm, J., Figer, A., Hecht, J. R., Gallinger, S., Au, H. J., Murawa, P., Walde, D., Wolff, R. A., Campos, D., Lim, R., Ding, K., Clark, G., Voskoglou-Nomikos, T., Ptasynski, M., Parulekar, W., and National Cancer Institute of Canada Clinical Trials Group. (2007). Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966.
Moore, M. J., Hamm, J., Dancey, J., Eisenberg, P. D., Dagenais, M., Fields, A., Hagan, K., Greenberg, B., Colwell, B., Zee, B., Tu, D., Ottaway, J., Humphrey, R., Seymour, L., National Cancer Institute of Canada Clinical Trials Group. (2003). Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 21, 3296–3302.
Mori-Iwamoto, S., Kuramitsu, Y., Ryozawa, S., Mikuria, K., Fujimoto, M., Maehara, S., Maehara, Y., Okita, K., Nakamura, K., and Sakaida, I. (2007). Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 31, 1345–1350.
Mori-Iwamoto, S., Taba, K., Kuramitsu, Y., Ryozawa, S., Tanaka, T., Maehara, S., Maehara, Y., Okita, K., Nakamura, K., and Sakaida, I. (2009). Interferon-γ down-regulates heat shock protein 27 of pancreatic cancer cells and helps in the cytotoxic effect of gemcitabine. Pancreas 38, 224–226.
Morton, J. P., Karim, S. A., Graham, K., Timpson, P., Jamieson, N., Athineos, D., Doyle, B., McKay, C., Heung, M. Y., Oien, K. A., Frame, M. C., Evans, T. R., Sansom, O. J., and Brunton, V. G. (2010). Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 139, 292–303.
Muerkoster, S., Arlt, A., Witt, M., Gehrz, A., Haye, S., March, C., Grohmann, F., Wegehenkel, K., Kalthoff, H., Folsch, U. R., and Schafer, H. (2003). Usage of the NF-κB inhibitor sulfasalazine as sensitizing agent in combined chemotherapy of pancreatic cancer. Int. J. Cancer 104, 469–476.
Mukhtar, H., and Ahmad, N. (1999). Green tea in chemoprevention of cancer. Toxicol. Sci. 52, 111–117.
Mund, C., Brueckner, B., and Lyko, F. (2006). Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: basic concepts and clinical applications. Epigenetics 1, 7–13.
Munshi, N., Jeay, S., Li, Y., Chen, C. R., France, D. S., Ashwell, M. A., Hill, J., Moussa, M. M., Leggett, D. S., and Li, C. J. (2010). ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol. Cancer Ther. 9, 1544–1553.
Nakamura, Y., Migita, T., Hosoda, F., Okada, N., Gotoh, M., Arai, Y., Fukushima, M., Ohki, M., Miyata, S., Takeuchi, K., Imoto, I., Katai, H., Yamaguchi, T., Inazawa, J., Hirohashi, S., Ishikawa, Y., and Shibata, T. (2009). Kruppel-like factor 12 plays a significant role in poorly differentiated gastric cancer progression. Int. J. Cancer 125, 1859–1867.
Nakashima, H., Nakamura, M., Yamaguchi, H., Yamanaka, N., Akiyoshi, T., Koga, K., Yamaguchi, K., Tsuneyoshi, M., Tanaka, M., and Katano, M. (2006). Nuclear factor-κB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 66, 7041–7049.
Nawroth, R., van Zante, A., Cervantes, S., McManus, M., Hebrok, M., and Rosen, S. D. (2007). Extracellular sulfatases, elements of the Wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE 2, e392. doi:10.1371/journal.pone.0000392
Neoptolemos, J. P., Stocken, D. D., Tudur Smith, C., Bassi, C., Ghaneh, P., Owen, E., Moore, M., Padbury, R., Doi, R., Smith, D., and Buchler, M. W. (2009). Adjuvant 5-fluorouracil and folinic acid vs observation for pancreatic cancer: composite data from the ESPAC-1 and -3(v1) trials. Br. J. Cancer 100, 246–250.
Nkondjock, A., Ghadirian, P., Johnson, K. C., and Krewski, D. (2005). Dietary intake of lycopene is associated with reduced pancreatic cancer risk. J. Nutr. 135, 592–597.
Oettle, H., Post, S., Neuhaus, P., Gellert, K., Langrehr, J., Ridwelski, K., Schramm, H., Fahlke, J., Zuelke, C., Burkart, C., Gutberlet, K., Kettner, E., Schmalenberg, H., Weigang-Koehler, K., Bechstein, W. O., Niedergethmann, M., Schmidt-Wolf, I., Roll, L., Doerken, B., and Riess, H. (2007). Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 297, 267–277.
Ogura, Y., Mizumoto, K., Nagai, E., Murakami, M., Inadome, N., Saimura, M., Matsumoto, K., Nakamura, T., Maemondo, M., Nukiwa, T., and Tanaka, M. (2006). Peritumoral injection of adenovirus vector expressing NK4 combined with gemcitabine treatment suppresses growth and metastasis of human pancreatic cancer cells implanted orthotopically in nude mice and prolongs survival. Cancer Gene Ther. 13, 520–529.
Okami, J., Yamamoto, H., Fujiwara, Y., Tsujie, M., Kondo, M., Noura, S., Oshima, S., Nagano, H., Dono, K., Umeshita, K., Ishikawa, O., Sakon, M., Matsuura, N., Nakamori, S., and Monden, M. (1999). Overexpression of cyclooxygenase-2 in carcinoma of the pancreas. Clin. Cancer Res. 5, 2018–2024.
Olson, P., Lu, J., Zhang, H., Shai, A., Chun, M. G., Wang, Y., Libutti, S. K., Nakakura, E. K., Golub, T. R., and Hanahan, D. (2009). MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 23, 2152–2165.
Ouaïssi, M., Sielezneff, I., Silvestre, R., Sastre, B., Bernard, J. P., Lafontaine, J. S., Payan, M. J., Dahan, L., Pirrò, N., Seitz, J. F., Mas, E., Lombardo, D., and Ouaissi, A. (2008). High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann. Surg. Oncol. 15, 2318–2328.
Ougolkov, A. V., Bilim, V. N., and Billadeau, D. D. (2008). Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 14, 6790–6796.
Palmer, D. H., Stocken, D. D., Hewitt, H., Markham, C. E., Hassan, A. B., Johnson, P. J., Buckels, J. A., and Bramhall, S. R. (2007). A randomized phase 2 trial of neoadjuvant chemotherapy in resectable pancreatic cancer: gemcitabine alone versus gemcitabine combined with cisplatin. Ann. Surg. Oncol. 14, 2088–2096.
Pan, L. N., Lu, J., and Huang, B. (2007). HDAC inhibitors: a potential new category of anti-tumor agents. Cell. Mol. Immunol. 4, 337–343.
Pasca di Magliano, M., Biankin, A. V., Heiser, P. W., Cano, D. A., Gutierrez, P. J., Deramaudt, T., Segara, D., Dawson, A. C., Kench, J. G., Henshall, S. M., Sutherland, R. L., Dlugosz, A., Rustgi, A. K., and Hebrok, M. (2007). Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS ONE 2, e1155. doi:10.1371/journal.pone.0001155
Petersen, G. M., Amundadottir, L., Fuchs, C. S., Kraft, P., Stolzenberg-Solomon, R. Z., Jacobs, K. B., Arslan, A. A., Bueno-de-Mesquita, H. B., Gallinger, S., Gross, M., Helzlsouer, K., Holly, E. A., Jacobs, E. J., Klein, A. P., LaCroix, A., Li, D., Mandelson, M. T., Olson, S. H., Risch, H. A., Zheng, W., Albanes, D., Bamlet, W. R., Berg, C. D., Boutron-Ruault, M. C., Buring, J. E., Bracci, P. M., Canzian, F., Clipp, S., Cotterchio, M., de Andrade, M., Duell, E. J., Gaziano, J. M., Giovannucci, E. L., Goggins, M., Hallmans, G., Hankinson, S. E., Hassan, M., Howard, B., Hunter, D. J., Hutchinson, A., Jenab, M., Kaaks, R., Kooperberg, C., Krogh, V., Kurtz, R. C., Lynch, S. M., McWilliams, R. R., Mendelsohn, J. B., Michaud, D. S., Parikh, H., Patel, A. V., Peeters, P. H., Rajkovic, A., Riboli, E., Rodriguez, L., Seminara, D., Shu, X. O., Thomas, G., Tjønneland, A., Tobias, G. S., Trichopoulos, D., Van Den Eeden, S. K., Virtamo, J., Wactawski-Wende, J., Wang, Z., Wolpin, B. M., Yu, H., Yu, K., Zeleniuch-Jacquotte, A., Fraumeni, J. F. Jr., Hoover, R. N., Hartge, P., and Chanock, S. J. (2010). A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat. Genet. 42, 224–228.
Petersen, G. M., de Andrade, M., Goggins, M., Hruban, R. H., Bondy, M., Korczak, J. F., Gallinger, S., Lynch, H. T., Syngal, S., Rabe, K. G., Seminara, D., and Klein, A. P. (2006). Pancreatic cancer genetic epidemiology consortium. Cancer Epidemiol. Biomarkers Prev. 15, 704–710.
Philip, P. A., Benedetti, J., Corless, C. L., Wong, R., O’Reilly, E. M., Flynn, P. J., Rowland, K. M., Atkins, J. N., Mirtsching, B. C., Rivkin, S. E., Khorana, A. A., Goldman, B., Fenoglio-Preiser, C. M., Abbruzzese, J. L., and Blanke, C. D. (2010). Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 28, 3605–3610.
Piao, W., Wang, Y., Adachi, Y., Yamamoto, H., Li, R., Imsumran, A., Li, H., Maehata, T., Ii, M., Arimura, Y., Lee, C. T., Shinomura, Y., Carbone, D. P., and Imai, K. (2008). Insulin-like growth factor-I receptor blockade by a specific tyrosine kinase inhibitor for human gastrointestinal carcinomas. Mol. Cancer Ther. 7, 1483–1493.
Pogue-Geile, K. L., Chen, R., Bronner, M. P., Crnogorac-Jurcevic, T., Moyes, K. W., Dowen, S., Otey, C. A., Crispin, D. A., George, R. D., Whitcomb, D. C., and Brentnall, T. A. (2006). Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Med. 3, e516. doi:10.1371/journal.pmed.0030516
Poplin, E., Feng, Y., Berlin, J., Rothenberg, M. L., Hochster, H., Mitchell, E., Alberts, S., O’Dwyer, P., Haller, D., Catalano, P., Cella, D., and Benson, A. B. III. (2009). Phase III, randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed-dose rate infusion) compared with gemcitabine (30-minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 27, 3778–3785.
Qanungo, S., Das, M., Haldar, S., and Basu, A. (2005). Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 26, 958–967.
Ricart, A. D., Tolcher, A. W., Liu, G., Holen, K., Schwartz, G., Albertini, M., Weiss, G., Yazji, S., Ng, C., and Wilding, G. (2008). Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 14, 7924–7929.
Rinehart, J., Adjei, A. A., Lorusso, P. M., Waterhouse, D., Hecht, J. R., Natale, R. B., Hamid, O., Varterasian, M., Asbury, P., Kaldjian, E. P., Gulyas, S., Mitchell, D. Y., Herrera, R., Sebolt-Leopold, J. S., and Meyer, M. B. (2004). Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456–4462.
Ringrose, L. (2007). Polycomb comes of age: genome-wide profiling of target sites. Curr. Opin. Cell Biol. 19, 290–297.
Rocchi, P., Beraldi, E., Ettinger, S., Fazli, L., Vessella, R. L., Nelson, C., and Gleave, M. (2005). Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer Res. 65, 11083–11093.
Rocchi, P., So, A., Kojima, S., Signaevsky, M., Beraldi, E., Fazli, L., Hurtado-Coll, A., Yamanaka, K., and Gleave, M. (2004). Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 64, 6595–6602.
Roldo, C., Missiaglia, E., Hagan, J. P., Falconi, M., Capelli, P., Bersani, S., Calin, G. A., Volinia, S., Liu, C. G., Scarpa, A., and Croce, C. M. (2006). MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 24, 4677–4684.
Romero, D., Iglesias, M., Vary, C. P., and Quintanilla, M. (2008). Functional blockade of Smad4 leads to a decrease in β-catenin levels and signaling activity in human pancreatic carcinoma cells. Carcinogenesis 29, 1070–1076.
Ruggeri, B. A., Huang, L., Wood, M., Cheng, J. Q., and Testa, J. R. (1998). Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol. Carcinog. 21, 81–86.
Sato, N., Fukushima, N., Maitra, A., Matsubayashi, H., Yeo, C. J., Cameron, J. L., Hruban, R. H., and Goggins, M. (2003). Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 63, 3735–3742.
Schlieman, M. G., Fahy, B. N., Ramsamooj, R., Beckett, L., and Bold, R. J. (2003). Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer 89, 2110–2115.
Schmitz-Winnenthal, F. H., Z’Graggen, K., Volk, C., Schmied, B. M., and Buchler, M. W. (2003). Intraductal papillary mucinous tumors of the pancreas. Curr. Gastroenterol. Rep. 5, 133–140.
Schutte, M., Hruban, R. H., Geradts, J., Maynard, R., Hilgers, W., Rabindran, S. K., Moskaluk, C. A., Hahn, S. A., Schwarte-Waldhoff, I., Schmiegel, W., Baylin, S. B., Kern, S. E., and Herman, J. G. (1997). Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 57, 3126–3130.
Schwartz, A. M., and Henson, D. E. (2007). Familial and sporadic pancreatic carcinoma, epidemiologic concordance. Am. J. Surg. Pathol. 31, 645–646.
Seo, Y., Baba, H., Fukuda, T., Takashima, M., and Sugimachi, K. (2000). High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 88, 2239–2245.
Shafaee, Z., Schmidt, H., Du, W., Posner, M., and Weichselbaum, R. (2006). Cyclopamine increases the cytotoxic effects of paclitaxel and radiation but not cisplatin and gemcitabine in Hedgehog expressing pancreatic cancer cells. Cancer Chemother. Pharmacol. 58, 765–770.
Shen, Y. M., Yang, X. C., Yang, C., and Shen, J. K. (2008). Enhanced therapeutic effects for human pancreatic cancer by application K-ras and IGF-IR antisense oligodeoxynucleotides. World J. Gastroenterol. 14, 5176–5185.
Singh, E. K., Ravula, S., Pan, C. M., Pan, P. S., Vasko, R. C., Lapera, S. A., Weerasinghe, S. V., Pflum, M. K., and McAlpine, S. R. (2008). Synthesis and biological evaluation of histone deacetylase inhibitors that are based on FR235222: a cyclic tetrapeptide scaffold. Bioorg. Med. Chem. Lett. 18, 2549–2554.
Singh, M., and Maitra, A. (2007). Precursor lesions of pancreatic cancer: molecular pathology and clinical implications. Pancreatology 7, 9–19.
Sohn, T. A., Yeo, C. J., Cameron, J. L., Iacobuzio-Donahue, C. A., Hruban, R. H., and Lillemoe, K. D. (2001). Intraductal papillary mucinous neoplasms of the pancreas: an increasingly recognized clinicopathologic entity. Ann. Surg. 234, 313–321; discussion 321–312.
Sood, P., Krek, A., Zavolan, M., Macino, G., and Rajewsky, N. (2006). Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. U.S.A. 103, 2746–2751.
Spano, J. P., Chodkiewicz, C., Maurel, J., Wong, R., Wasan, H., Barone, C., Letourneau, R., Bajetta, E., Pithavala, Y., Bycott, P., Trask, P., Liau, K., Ricart, A. D., Kim, S., and Rixe, O. (2008). Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study. Lancet 371, 2101–2108.
Su, G. H., Hruban, R. H., Bansal, R. K., Bova, G. S., Tang, D. J., Shekher, M. C., Westerman, A. M., Entius, M. M., Goggins, M., Yeo, C. J., and Kern, S. E. (1999). Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol. 154, 1835–1840.
Szafranska, A. E., Davison, T. S., John, J., Cannon, T., Sipos, B., Maghnouj, A., Labourier, E., and Hahn, S. A. (2007). MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene 26, 4442–4452.
Takayama, Y., Kokuryo, T., Yokoyama, Y., Nagino, M., Nimura, Y., Senga, T., and Hamaguchi, M. (2008). MEK inhibitor enhances the inhibitory effect of imatinib on pancreatic cancer cell growth. Cancer Lett. 264, 241–249.
Talar-Wojnarowska, R., and Malecka-Panas, E. (2006). Molecular pathogenesis of pancreatic adenocarcinoma: potential clinical implications. Med. Sci. Monit. 12, RA186–RA193.
Thayer, S. P., di Magliano, M. P., Heiser, P. W., Nielsen, C. M., Roberts, D. J., Lauwers, G. Y., Qi, Y. P., Gysin, S., Fernandez-del Castillo, C., Yajnik, V., Antoniu, B., McMahon, M., Warshaw, A. L., and Hebrok, M. (2003). Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856.
Tomioka, D., Maehara, N., Kuba, K., Mizumoto, K., Tanaka, M., Matsumoto, K., and Nakamura, T. (2001). Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res. 61, 7518–7524.
Toubaji, A., Achtar, M., Provenzano, M., Herrin, V. E., Behrens, R., Hamilton, M., Bernstein, S., Venzon, D., Gause, B., Marincola, F., and Khleif, S. N. (2008). Pilot study of mutant ras peptide-based vaccine as an adjuvant treatment in pancreatic and colorectal cancers. Cancer Immunol. Immunother. 57, 1413–1420.
Trapasso, F., Sarti, M., Cesari, R., Yendamuri, S., Dumon, K. R., Aqeilan, R. I., Pentimalli, F., Infante, L., Alder, H., Abe, N., Watanabe, T., Viglietto, G., Croce, C. M., and Fusco, A. (2004). Therapy of human pancreatic carcinoma based on suppression of HMGA1 protein synthesis in preclinical models. Cancer Gene Ther. 11, 633–641.
Trevino, J. G., Summy, J. M., Lesslie, D. P., Parikh, N. U., Hong, D. S., Lee, F. Y., Donato, N. J., Abbruzzese, J. L., Baker, C. H., and Gallick, G. E. (2006). Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol. 168, 962–972.
Tsuda, N., Ishiyama, S., Li, Y., Ioannides, C. G., Abbruzzese, J. L., and Chang, D. Z. (2006). Synthetic microRNA designed to target glioma-associated antigen 1 transcription factor inhibits division and induces late apoptosis in pancreatic tumor cells. Clin. Cancer Res. 12, 6557–6564.
Van Cutsem, E., van de Velde, H., Karasek, P., Oettle, H., Vervenne, W. L., Szawlowski, A., Schoffski, P., Post, S., Verslype, C., Neumann, H., Safran, H., Humblet, Y., Perez Ruixo, J., Ma, Y., and Von Hoff, D. (2004). Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 22, 1430–1438.
Van Cutsem, E., Vervenne, W. L., Bennouna, J., Humblet, Y., Gill, S., Van Laethem, J. L., Verslype, C., Scheithauer, W., Shang, A., Cosaert, J., and Moore, M. J. (2009). Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J. Clin. Oncol. 27, 2231–2237.
Vernejoul, F., Ghenassia, L., Souque, A., Lulka, H., Drocourt, D., Cordelier, P., Pradayrol, L., Pyronnet, S., Buscail, L., and Tiraby, G. (2006). Gene therapy based on gemcitabine chemosensitization suppresses pancreatic tumor growth. Mol. Ther. 14, 758–767.
Vire, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., Van Eynde, A., Bernard, D., Vanderwinden, J. M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y., and Fuks, F. (2006). The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874.
Volinia, S., Calin, G. A., Liu, C. G., Ambs, S., Cimmino, A., Petrocca, F., Visone, R., Iorio, M., Roldo, C., Ferracin, M., Prueitt, R. L., Yanaihara, N., Lanza, G., Scarpa, A., Vecchione, A., Negrini, M., Harris, C. C., and Croce, C. M. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261.
Wan, J., Xia, Y., Liu, Y., Wang, M., Rocchi, P., Yao, J., Qu, F., Neyts, J., Iovanna, J. L., and Peng, L. (2009). Discovery of novel arylethynyltriazole ribonucleosides with selective and effective antiviral and antiproliferative activity. J. Med. Chem. 52, 1144–1155.
Wang, W., Abbruzzese, J. L., Evans, D. B., and Chiao, P. J. (1999). Overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma is regulated by constitutively activated RelA. Oncogene 18, 4554–4563.
Wang, W., Chen, S., Brune, K. A., Hruban, R. H., Parmigiani, G., and Klein, A. P. (2007). PancPRO: risk assessment for individuals with a family history of pancreatic cancer. J. Clin. Oncol. 25, 1417–1422.
Wang, Z., Banerjee, S., Li, Y., Rahman, K. M., Zhang, Y., and Sarkar, F. H. (2006a). Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-κB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 66, 2778–2784.
Wang, Z., Sengupta, R., Banerjee, S., Li, Y., Zhang, Y., Rahman, K. M., Aboukameel, A., Mohammad, R., Majumdar, A. P., Abbruzzese, J. L., and Sarkar, F. H. (2006b). Epidermal growth factor receptor-related protein inhibits cell growth and invasion in pancreatic cancer. Cancer Res. 66, 7653–7660.
Wang, Z., Zhang, Y., Banerjee, S., Li, Y., and Sarkar, F. H. (2006c). Inhibition of nuclear factor κB activity by genistein is mediated via Notch-1 signaling pathway in pancreatic cancer cells. Int. J. Cancer 118, 1930–1936.
Wang, Z., Zhang, Y., Banerjee, S., Li, Y., and Sarkar, F. H. (2006d). Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer 106, 2503–2513.
Wang, Z., Zhang, Y., Li, Y., Banerjee, S., Liao, J., and Sarkar, F. H. (2006e). Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol. Cancer Ther. 5, 483–493.
Wang, Z., Ma, Q., Liu, Q., Yu, H., Zhao, L., Shen, S., and Yao, J. (2008). Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br. J. Cancer 99, 1695–1703.
Watanabe, S., Ueda, Y., Akaboshi, S., Hino, Y., Sekita, Y., and Nakao, M. (2009). HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 174, 854–868.
Wei, D., Wang, L., He, Y., Xiong, H. Q., Abbruzzese, J. L., and Xie, K. (2004). Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 64, 2030–2038.
Wei, Y., Xia, W., Zhang, Z., Liu, J., Wang, H., Adsay, N. V., Albarracin, C., Yu, D., Abbruzzese, J. L., Mills, G. B., Bast, R. C. Jr., Hortobagyi, G. N., and Hung, M. C. (2008). Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol. Carcinog. 47, 701–706.
Whitcomb, D. C., Gorry, M. C., Preston, R. A., Furey, W., Sossenheimer, M. J., Ulrich, C. D., Martin, S. P., Gates, L. K. Jr., Amann, S. T., Toskes, P. P., Liddle, R., McGrath, K., Uomo, G., Post, J. C., and Ehrlich, G. D. (1996). Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145.
Wilentz, R. E., Goggins, M., Redston, M., Marcus, V. A., Adsay, N. V., Sohn, T. A., Kadkol, S. S., Yeo, C. J., Choti, M., Zahurak, M., Johnson, K., Tascilar, M., Offerhaus, G. J., Hruban, R. H., and Kern, S. E. (2000). Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am. J. Pathol. 156, 1641–1651.
Winter, J. M., Cameron, J. L., Lillemoe, K. D., Campbell, K. A., Chang, D., Riall, T. S., Coleman, J., Sauter, P. K., Canto, M., Hruban, R. H., Schulick, R. D., Choti, M. A., and Yeo, C. J. (2006). Periampullary and pancreatic incidentaloma: a single institution’s experience with an increasingly common diagnosis. Ann. Surg. 243, 673–680; discussion 680–673.
Wolpin, B. M., Hezel, A. F., Abrams, T., Blaszkowsky, L. S., Meyerhardt, J. A., Chan, J. A., Enzinger, P. C., Allen, B., Clark, J. W., Ryan, D. P., and Fuchs, C. S. (2009). Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J. Clin. Oncol. 27, 193–198.
Xia, Y., Liu, Y., Wan, J., Wang, M., Rocchi, P., Qu, F., Iovanna, J. L., and Peng, L. (2009). Novel triazole ribonucleoside down-regulates heat shock protein 27 and induces potent anticancer activity on drug-resistant pancreatic cancer. J. Med. Chem. 52, 6083–6096.
Yachida, S., Jones, S., Bozic, I., Antal, T., Leary, R., Fu, B., Kamiyama, M., Hruban, R. H., Eshleman, J. R., Nowak, M. A., Velculescu, V. E., Kinzler, K. W., Vogelstein, B., and Iacobuzio-Donahue, C. A. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117.
Yamamoto, H., Itoh, F., Nakamura, H., Fukushima, H., Sasaki, S., Perucho, M., and Imai, K. (2001). Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 61, 3139–3144.
Zeng, G., Germinaro, M., Micsenyi, A., Monga, N. K., Bell, A., Sood, A., Malhotra, V., Sood, N., Midda, V., Monga, D. K., Kokkinakis, D. M., and Monga, S. P. (2006). Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia 8, 279–289.
Zhang, H., Morisaki, T., Nakahara, C., Matsunaga, H., Sato, N., Nagumo, F., Tadano, J., and Katano, M. (2003). PSK-mediated NF-κB inhibition augments docetaxel-induced apoptosis in human pancreatic cancer cells NOR-P1. Oncogene 22, 2088–2096.
Zhang, Y., Banerjee, S., Wang, Z., Xu, H., Zhang, L., Mohammad, R., Aboukameel, A., Adsay, N. V., Che, M., Abbruzzese, J. L., Majumdar, A. P., and Sarkar, F. H. (2006). Antitumor activity of epidermal growth factor receptor-related protein is mediated by inactivation of ErbB receptors and nuclear factor-κB in pancreatic cancer. Cancer Res. 66, 1025–1032.
Keywords: pancreas, oncogenes, suppressor genes, signaling, cancer, surgery, neoadjuvants, chemoprevention
Citation: Iovanna J, Mallmann MC, Gonçalves A, Turrini O and Dagorn J-C (2012) Current knowledge on pancreatic cancer. Front. Oncol. 2:6. doi: 10.3389/fonc.2012.00006
Received: 19 September 2011;
Accepted: 11 January 2012;
Published online: 31 January 2012.
Edited by:
Eric Solary, University Paris-Sud, FranceReviewed by:
Agnès Noël, University of Liège, BelgiumFrancisco X. Real, Centro Nacional de Investigaciones Oncológicas, Spain
Jean-Yves Scoazec, Université Lyon 1, France
Copyright: © 2012 Iovanna, Mallmann, Gonçalves, Turrini and Dagorn. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Juan Iovanna, INSERM U624, Stress Cellulaire, 163 Avenue de Luminy, CP 915, 13288 Marseille Cedex 9, France. e-mail: juan.iovanna@inserm.fr