 Fabrizio Tabbó1 Antonella Barreca1 Roberto Piva1,2 Giorgio Inghirami1,2* The European T-Cell Lymphoma Study Group#
Fabrizio Tabbó1 Antonella Barreca1 Roberto Piva1,2 Giorgio Inghirami1,2* The European T-Cell Lymphoma Study Group#
- 1 Department of Pathology, Center for Experimental Research and Medical Studies, University of Torino, Torino, Italy
- 2 Department of Pathology, NYU Cancer Center, New York University School of Medicine, New York, NY, USA
The discovery by Morris et al. (1994) of the genes contributing to the t(2;5)(p23;q35) translocation has laid the foundation for a molecular based recognition of anaplastic large cell lymphoma and highlighted the need for a further stratification of T-cell neoplasia. Likewise the detection of anaplastic lymphoma kinase (ALK) genetic lesions among many human cancers has defined unique subsets of cancer patients, providing new opportunities for innovative therapeutic interventions. The objective of this review is to appraise the molecular mechanisms driving ALK-mediated transformation, and to maintain the neoplastic phenotype. The understanding of these events will allow the design and implementation of novel tailored strategies for a well-defined subset of cancer patients.
Introduction
The concept of anaplastic large cell lymphoma (ALCLs) has changed over the years as a result of a fruitful stream of new information and novel understanding about the cell of origin, biology, genetics, and clinical features of these neoplasms. Most importantly, the discovery of anaplastic lymphoma kinase (ALK) fusion chimera, among ALCLs, has provided the basis for the definition of a unique subgroup among peripheral T-cell lymphoma (PTCL) and set the stage for the identification of new entities, some of which still remain provisional within the current WHO Classification of tumors of hematopoietic and lymphoid tissues.
Indeed, the discovery of the Nucleophosmin (NPM)–ALK fusion, in many but not entirely systemic ALCL, have led to the recognition that ALCLs are biologically and clinically heterogeneous. In fact, it has likewise become evident that ALK+ ALCLs, most commonly seen in children and young adults, display a more favorable prognosis. On the contrary, ALK− ALCLs have a more aggressive clinical course and could represent a distinct morphologic variant among PTCL, not otherwise specified, NOS (PTCL–NOS; Delsol et al., 2008).
Although the molecular stratification of ALCLs has provided objective criteria for precise diagnoses, many issues remain open. As a matter of fact, we do not know the mechanisms leading to the transformation of ALK− ALCLs, including those involving the skin primarily (cALCL), or whether various entities among ALCLs somehow share common pathogenetic features and if putative relationship(s) with other T-cell lymphoma may exist. These issues are critical for the design efficient therapies, tailored to unique subsets, targeting individual biological and genetics defects.
The main objective of this review is to undertake a systematic appraisal of ALK deregulation in human cancers and in particular in ALCLs, with the intent of discussing novel targeted therapies and/or personalized therapeutic modalities.
Peripheral T-Cell Lymphoma
T-non-Hodgkin lymphoma (NHLs) include a large number of neoplasia, most likely representing the neoplastic counterpart of functionally distinct T-cells, transformed by unique but still largely unknown genetic defects. Among those derived from mature T-lymphocytes, two subsets have been described, known otherwise as PTCL specified and NOS. These lymphoma are overall quite rare, ranging from 12 to 15% among all NHL in Western populations (Table 1; Vose et al., 2008). Many of them display a great variability in their clinical, morphological, immunophenotypic, cytogenetics, and molecular features. PTCL–NOS is the most common (30–40%), followed by the angioimmunoblastic (AITL) and ALCLs. ALCLs occur in ∼2–8% of NHL in adults and represent 15–30% of all high-grade lymphoma in children.
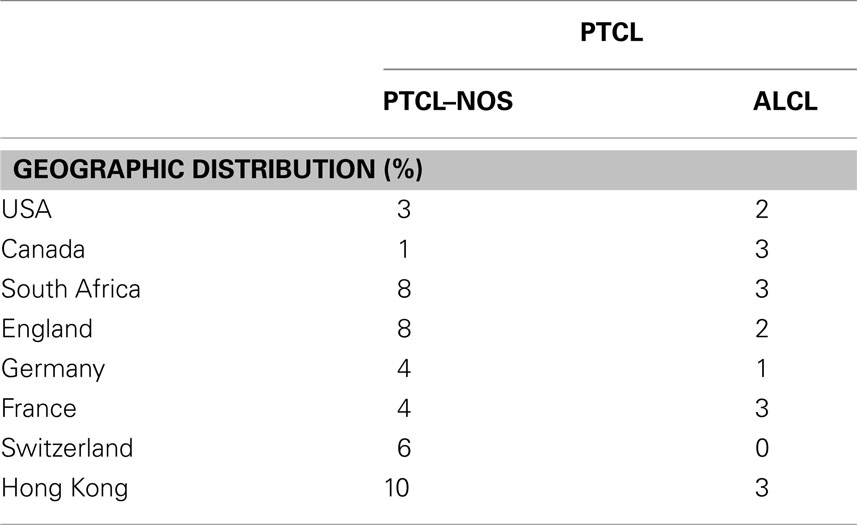
Table 1. Geographic distribution of anaplastic large cell lymphoma compared to peripheral T-cell lymphoma, not-otherwise specified (PTCL–NOS).
Systemic ALCLs are classified as de novo and secondary (anaplastic transformation of other lymphoma). De novo ALK+ ALCLs are clinically aggressive lymphoma that frequently occur within the first three decades of life, with a typical male predominance. They often present as a stage III–IV disease, with systemic symptoms, and extranodal involvement (60%; Falini et al., 1999; Stein et al., 2000). ALK− ALCLs arise in older patients (most commonly at sixth decade of life), with a slight predominance in males (Savage et al., 2008), and high International prognostic index (IPI) scores (Tilly et al., 1997; see Table 2). Extranodal involvement is relatively common with cutaneous, hepatic, or gastrointestinal sites most frequently engaged. ALK− ALCLs, when compared to ALK+ ALCLs, display a worse clinical presentation and outcome, meanwhile they have a relatively better overall survival rate than PTCL–NOS (36 versus 20%, respectively; Vose et al., 2008). The more favorable clinical outcome of the ALK+ ALCLs (Savage et al., 2008) has been associated to the age of occurrence and to the driving molecular defects, in particular to ALK fusions. When ALCL patients are however studied taking into account the age and/or other clinical parameters, ALK+ and ALK− ALCLs display analogous clinical features, suggesting that relevant clinical factors may have critical implications on their outcome. Nevertheless, it is unclear whether these clinical features are due to and/or associated to restricted defects occurring at different ages and/or accumulate overtime. Besides the well-defined primary chromosomal translocations, ALK+ ALCLs carry frequent secondary chromosomal imbalances, although their complex karyotype is substantially less complex than those observed in ALK− ALCL, suggesting that the more heterogeneous genetic setting of the latter group may be associated to a worse clinical outcome. If this hypothesis is correct, one could postulate that the ectopic activation of ALK provides dominant oncogenic inputs (strong oncogenic addiction), sufficient to induce cell transformation possibly in association with a limited number of additional synergizing defects. On the contrary, ALK− ALCLs might require the acquisition of a large number of tumorigenic defects, which could accumulate overtime resulting in a highly heterogeneous karyotype.
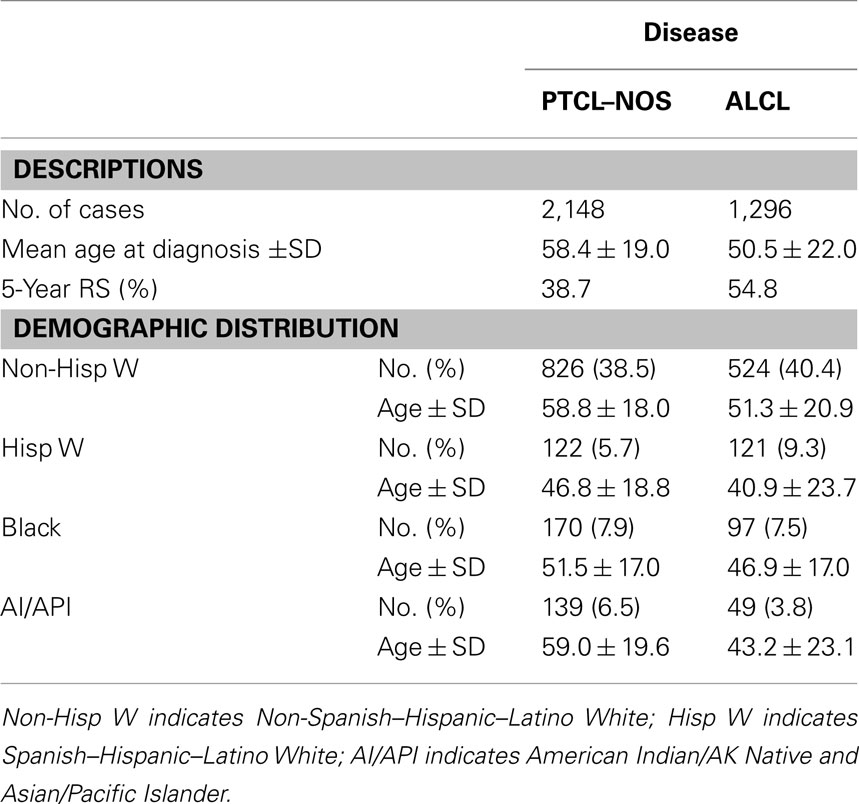
Table 2. Clinical features of anaplastic large cell lymphoma compared to peripheral T-cell lymphoma, not-otherwise specified (PTCL–NOS).
Morphologically ALCLs can be divided in five morphologic variants (a common, giant cell-rich, lympho-histiocytic, small-cell type, and Hodgkin-like, with the common variants corresponding to the lesions originally described by Stein et al., 2000). Cytologically, the neoplastic cells show a wide spectrum, although all cases have a variable proportion of “hallmark cells” (Benharroch et al., 1998; Jaffe, 2001; Figure 1). These are large lymphocytes with “horse shoe” or “kidney shaped” nuclei and abundant cytoplasm. ALCLs have a unique immunophenotype characterized by the strong expression of the CD30 antigen, a cytokine receptor of the tumor necrosis factor receptor superfamily, with a membranous pattern often associated with reinforcement staining within the Golgi area. The neoplastic cells, which are of T-cell origin, commonly lack many of the T-cell markers, although they frequently express cytotoxic molecules such as granzyme B, perforin, and T-cell-restricted intracellular antigen-1 (TIA1). Moreover, ALK+ ALCLs frequently display phosphorylated peptides including pSHP2 and pSTAT3 (Table 3).
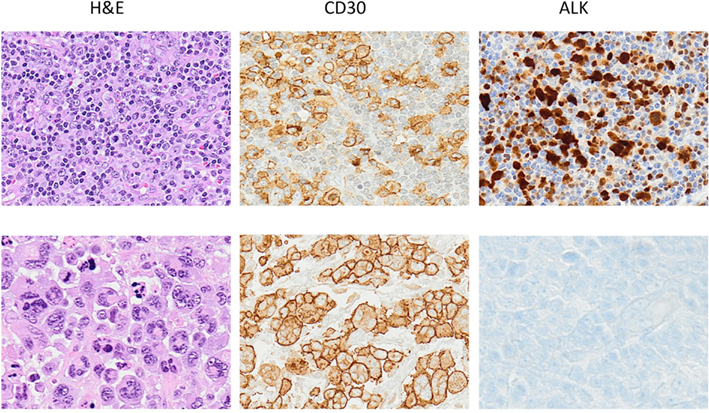
Figure 1. Anaplastic large cell lymphoma. The neoplastic cells of ALCL typically show a broad spectrum of morphologic features, and virtually all cases display a variable proportion of “hallmark cells.” The CD30 antigen, a cytokine receptor of the tumor necrosis factor receptor superfamily, is strongly expressed by all ALCL often in association with perforin, granzymes and T1A1 antigens. Alk expression is detected only in a subset of ALCL as result of translocations involving the ALK gene.
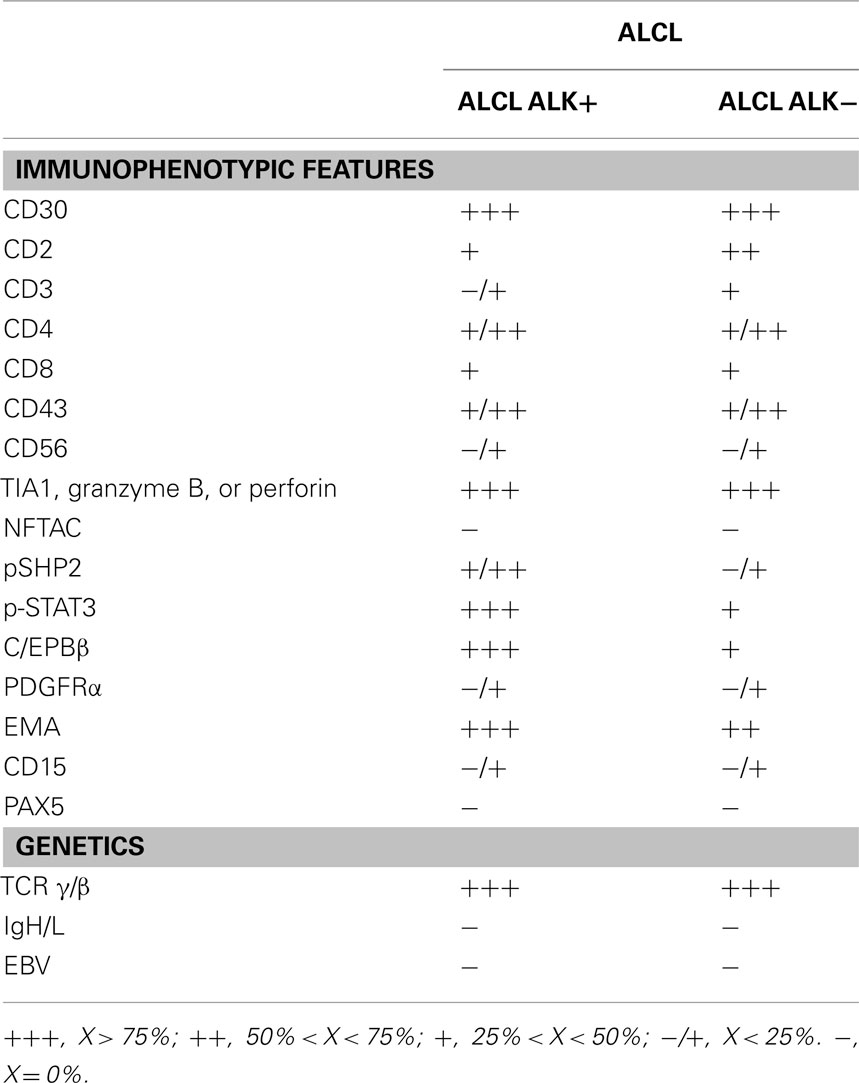
Table 3. Immunophenotypic and genomic features of ALCL.
Anaplastic Lymphoma Kinase
Anaplastic lymphoma kinase is a tyrosine kinase receptor (TKR), with an extracellular ligand-binding domain, a transmembrane-spanning region and an intracellular tyrosine kinase domain. It belongs to the Insulin Receptor superfamily and is closely related to the leukocyte tyrosine kinase (LTK). The overall structure of ALK has been conserved through evolution; this is particularly the case for the kinase domain, which is highly homologous among a variety of species, even though some differences, including the Tyr1604, have been found, suggesting peculiar functions in different species and/or in unique pathological settings (Bai et al., 1998).
The physiological role of ALK has been only partially defined. In C. elegans and Drosophila, the loss of ALK results in defects in midgut development, neuronal wiring, and plasticity. In adult mice, the genomic deletion of ALK has subtle or no phenotype, although its expression is upregulated after neuronal injuries enhancing the regeneration of myelinated axons (Mi et al., 2007). Recent data have been confirmed that loss of ALK signaling results in a dramatic decrease in newborn neurons in young as well as adult mice, as in Drosophila where ALK protects larval neurogenesis from nutrient stress (Cheng et al., 2011). Interestingly, ALK in mammals appears to enhance cognitive performance as well (Weiss et al., 2012).
In ALCLs, the ALK gene is inappropriately expressed in lymphoid cells as a result of several translocations, in which the intracytoplasmic region of the gene, coded by the exons 20–29 (NM_004304.3), is fused with different partners that provide dimerization domains (Table 4; Pulford et al., 2001; Chiarle et al., 2008b). The most common of these translocations is the t(2;5; p23;q35) that leads to the nucleophosmin (NPM)–ALK fusion protein (Chiarle et al., 2008b). Alternative translocations of ALCL have been discovered involving various partners, including the TPM (Lamant et al., 1999), TFG (Hernandez et al., 1999, 2002), ATIC (Colleoni et al., 2000), TSPYL2 (Touriol et al., 2000), MSN (Tort et al., 2001), KIAA1618 (Cools et al., 2002), VCL (Debelenko et al., 2003), MYH9 (Lamant et al., 2003), KIFB5 (Wong et al., 2011; Takeuchi et al., 2009). Notably, ALK+ ALCL often display additional alterations involving many chromosomes (deletions:4, 9p, 10p, 11q, and 13q, and gains: 1q, 7p, 6p, 17p, and 17q; Ott et al., 1998; Zettl et al., 2004; Salaverria et al., 2008), although common discrete secondary lesions have not been discovered yet. Moreover, using conventional genomic approaches, and more recent next generation sequencing from fresh and/or paraffin embedded tissue samples have shown new translocation of ALK in many types of human cancers (Table 4; Jung et al., 2012; Lipson et al., 2012; Takeuchi et al., 2012; Togashi et al., 2012).
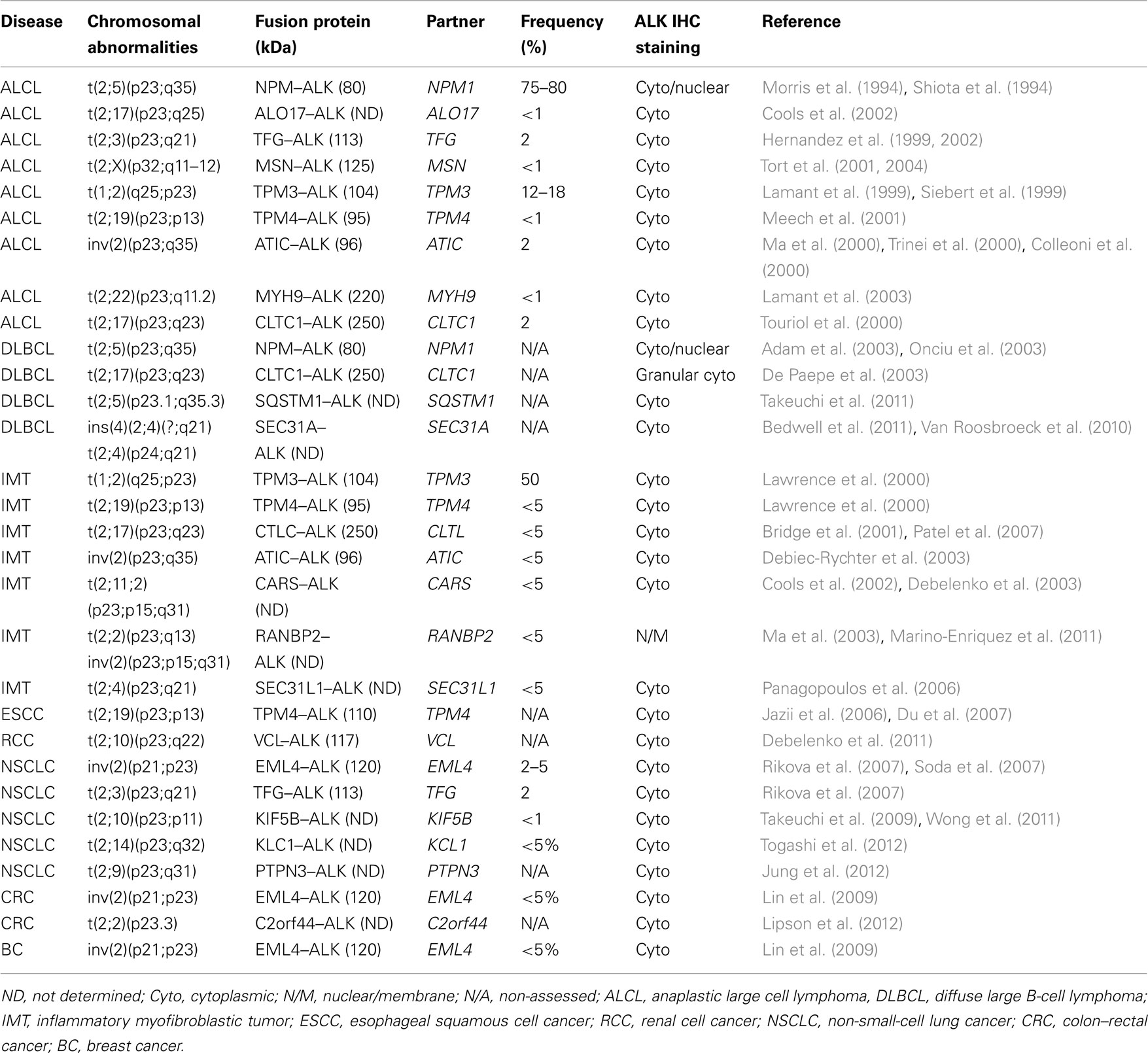
Table 4. Recurrent chromosomal translocations involving the ALK gene in human cancers.
ALK Signaling
Understanding how ALK signals and defining the mechanisms responsible for its deregulation are critical to dissecting how ALK mediates cellular transformation and provides the basis for “intelligent” therapeutic approaches.
An area of debate remains on how the ALK receptor physiologically signals in mammals. Contrary to Drosophila in which the dALK ligands have clearly been identified (Palmer et al., 2009), in mammals several hypothetical ligands have been considered, among them are: PTN (pleiotrophin or HB-GAM), OSF-1(osteoblast-specific factor-1), HARP (heparin affinity regulatory peptide), HBNF (heparin-binding neurotrophic factor), and MK (midkine). Nevertheless the role of these molecules remains controversial and it is plausible that other, so far unknown ALK ligands, may exist. It can also be contemplated that some of the current ligands, such as PTN, may function indirectly inhibiting alternative molecules like the phosphatase RPTPβ/ζ, providing a higher steady state phosphorylation status of the receptor (Perez-Pinera et al., 2007).
The ectopic expression of ALK chimera leads to their hetero- and homo-dimerization, via the dimerization domains of ALK partners resulting in a constitutive activation of the intracytoplasmic catalytic domain. This results in the constitutive activation of multiple signaling pathways, which lead to cell transformation and sustains the neoplastic phenotype (Chiarle et al., 2008b). In NPM–ALK ALCL, the N-terminus region of NPM1 provides the dimerization domain essential for the chimera’s auto-phosphorylation. NPM1 is a multifunctional protein, which acts as a molecular chaperone in the transport of pre-ribosomal particles from the nucleus to the cytoplasm, but it functions in addition to DNA repair, transcription, and genomic stability regulation (Okuwaki, 2008).
It is well established that the forced expression of ALK chimeras has oncogenic potential in in vivo as well in vitro models (Kuefer et al., 1997; Bai et al., 1998; Chiarle et al., 2005; Turner and Alexander, 2005). In vivo the constitutive expression of ALK transgenic cassettes under hematopoietic promoters can drive B- or T-cell transformation, providing a valuable tool for the study of ALCL (Chiarle et al., 2003; Turner and Alexander, 2005) and for the set up of innovative chemotherapeutic modalities (Cheng et al., 2012). Similar findings have been recently obtained expressing EML4–ALK (Soda et al., 2008) or TFG-1–ALK fusion proteins under lung-restricted promoters.
The transforming properties and the signaling elicited by ALK fusion proteins have been extensively studied using a variety of genetic, proteomics, and pharmacological approaches. Overall, as in many other oncogenic kinases, the deregulated expression of ALK leads to the simultaneous activation of many signaling pathways, which have been proven to synergistically provide the required signals for the maintenance of the neoplastic phenotype. Indeed, the ablation of the ALK signaling, at least in lymphoid cells, demonstrates the absolute ALK oncogenic “addiction” of these tumors (Piva et al., 2006; Wan et al., 2006; Christensen et al., 2007; Galkin et al., 2007). It is unclear which further defects may contribute in ALK+ ALCL, although it is proven that the forced expression of ALK chimera in normal cells leads to senescence (Martinelli et al., 2011) and that ALK+ lymphoma in Tg mice require a few months to occur, suggesting that a combination of lesions is acquired. Remarkably, the activation of IGF-IR can contribute to the neoplastic phenotype of ALK+ ALCL cell lines (Shi et al., 2009). More importantly, in the case of ALK+ NSCLC cell lines, ALK addiction has been proven to be variable and the pathogenetic role of other kinase receptors have been suggested in these settings (Koivunen et al., 2008). The determination of the ALK dependency and the contribution of other co-signaling kinase(s) is critical for defining the most effective chemotherapeutic regimen in each patient. Thus in selected settings, it may be necessary not only to dissect the presence of any given oncogenetic defect (i.e., EGRF, k-Ras mutation etc.) but to also quantify the levels of the activation of critical targetable molecules as well.
Mutagenesis and functional studies have identified a plethora of NPM–ALK interacting molecules which ultimately lead to the activation of key pathways including RAS/Erk, PLC-γ, PI3K, and Jak/signal transducers and activators of transcription (STAT) pathways, which in turn control cell proliferation and survival and cytoskeletal rearrangements (Figure 2).
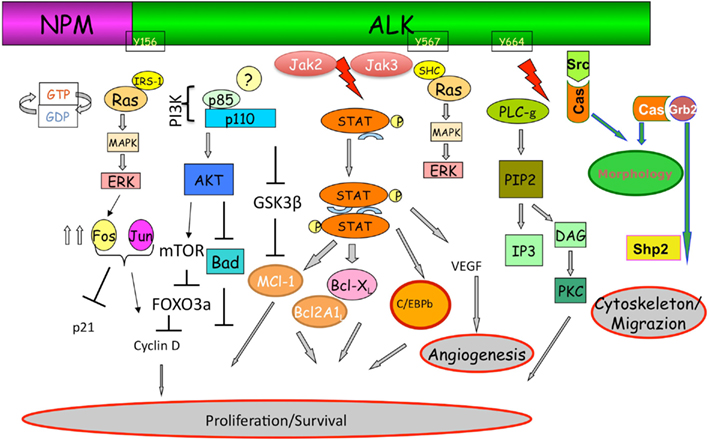
Figure 2. Anaplastic lymphoma kinase signaling. The ectopic expression of ALK fusion proteins leads to the constitutive activation of the tyrosine kinase, and concomitant activation of multiple signaling pathways. ALK, via a direct binding of IRS1, Shc, and Src on specific tyrosine residues, is a strong activator of the Ras/Mek/Erk-1/2 extracellular signal-regulated kinase (Erk) pathway that provides key mitogenic signaling. Ras/Erk pathways, via multiple AP-1 transcription factors, up regulates the CD30 expression. Mitogen-induced extracellular kinase (MEK)/ERK signaling pathway results in phosphorylation of mTOR which in turn leads to the phosphorylation of several critical targets (p70S6K and S6rp, eukaryotic initiation factor 4E (eiF4E)–binding protein-1 (4E-BP1). Activation of mTOR is also achieved through the synergist activation of the phosphatidylinositol 3-kinase (PI3K) pathway as result of the activation of Akt1/2 molecules. In the case of PLC-γ, this protein is directly activated by ALK (Y664) and capable of triggering pro-mitogenic stimuli generating diacylglycerol and IP3, which leads to the activation of PKC, and mobilization of calcium stores from the endoplasmic reticulum as well. As for many other cells, activated Akt1/2 enhance ALCL cell survival by blocking the function of pro-apoptotic proteins, such as BAD and indirectly regulate key cell cycle regulators (BIM-1, p27, and cyclinD2) via FOXO3A. Negative regulation of p-GSK3β leads to the up-regulation of Mcl-1 and CDC25A which confers the advantage of growth and protection from apoptosis. Stat3 regulated molecules are essential and required for the maintenance of the ALK-mediated neoplastic phenotype of ALCL cells. ALK fusion proteins can directly phosphorylate STAT3, alternatively JAK2/3 can also synergize. Negative regulators of this pathway may impose their action by dephosphorylating ALK itself and/or receptor-associated kinases (Src and JAK). Notably, methylation of the promoter region of SHP1 has been reported in ALK+ ALCL cells, resulting in the down-regulation of its expression and thus an overall enhancement of STAT3 signaling. Activation of STAT3 is associated with a specific signature (∼1500 genes), which includes several transcription factors (i.e., CEBP/β), cell cycle (i.e., Cyclin D, c-myc etc.), survival/apoptosis molecules (Bcl-A2, Bcl-XL, Survivin, MCL-1) and cell adhesion, and mobility proteins.
Ras/Erk Pathway
The increased growth of ALK+ ALCL cells is believed to be largely dependent on the activation of the Ras/Erk pathway (Fujimoto et al., 1996; Pulford et al., 2001). In NPM–ALK several adaptors or scaffolding molecules with Src-homology 2 (SH2) or phosphotyrosine binding (PTB) domains can bind and activate the Ras/Erk pathway. Although, SHC, IRS1, and Grb2 bind directly to NPM–ALK, neither SHC nor IRS1 are essential for transformation, since loss of their ALK docking sites does not preclude cell transformation (Fujimoto et al., 1996). Grb2 binds to different regions of NPM–ALK, mainly Tyr(152–156), Tyr(567), and a proline-rich region, Pro(415–417), and primarily regulates ALK-mediated phosphorylation of SHP2, which plays a key role in ALCL cell growth (Riera et al., 2010). The concomitant down-regulation of ERK-1 and ERK-2 in ALK+ ALCL using RNAi or specific Mek inhibitors leads to a cell cycle arrest in absence of an increased rate of apoptotic cells (Marzec et al., 2007). Ras activation via MAPK (mitogen-activated protein kinases), and ERK-1 and 2, regulates several AP-1 transcription factors, which are believed to contribute to the ALCL neoplastic phenotype (CD30; Watanabe et al., 2005; Hsu et al., 2006; Staber et al., 2007), regulate cytotoxic molecule expression (Pearson et al., 2011) and they are required for ALK-mediated transformation in mouse models.
PCL-γ Pathway
PLC-γ, docking in position Y664 of NPM–ALK, induces the hydrolysis of phosphatidylinositol (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG), molecules that can modulate the release of Ca2+ from intracellular compartments and activates the serine/threonine protein kinase C (PKC). The pathogenetic role of this pathway has been documented in Ba/F3 cells (a pro-B line that requires IL-3 for survival and growth), which can overcome IL-3 requirements in presence of the ectopic NPM–ALKwt but not NPM–ALKY664F (Bai et al., 1998). Interestingly, the PLC-γ docking site (Y664) is present only in the human ALK but not in the mouse gene, suggesting a unique function in human ALK-mediated transformation.
PI3K Pathway
NPM–ALK interact directly and most likely indirectly with PI3K (Bai et al., 2000). The ALK–PI3K interaction (catalytic p110 subunit) leads to the activation of the PKB/AKT and the subsequent downstream signaling events. These play a critical role in ALK-mediated transformation, as originally demonstrated by the dominant negative PKB/Akt approach (Slupianek et al., 2001) and by up-regulation of cyclin D2 and down-regulation of Bim-1 and p27 via hyperphosphorylation of the transcription factor FOXO3a after ALK forced expression (Gu et al., 2004). Finally the PI3K/AKT signaling pathway has been shown recently to control the phosphorylation levels of GSK3β, upregulating the expression of MCL-1 and Cdc25A, which may contribute to the neoplastic phenotype (Mcdonnell et al., 2011).
C-SCR Pathway
c-Src, a TKR, plays a relevant role in ALCL cell migration, as well as in cell proliferation and growth. c-Src that is normally maintained in a catalytically inactive conformation can be activated by NPM–ALK and it is required to sustain ALCL cell growth (Cussac et al., 2004). Src-family kinases may also contribute to VAV-1 signaling, leading to a sustained activation state of Cdc42. Cdc42 and p130C as in this context regulates ALCL cell shape and migration (Ambrogio et al., 2008).
Jak/STAT Pathway
Signal transducers and activators of transcription proteins are a family of transcription factors first characterized for their role in cytokine signaling. These proteins contain a site for specific tyrosine phosphorylation, which after modification results in a conformational rearrangement and dimerization through phosphotyrosine–SH2 domain interactions. Once STATs are phosphorylated, they dimerize and accumulate in the nucleus (Levy and Inghirami, 2006). Alternative non-canonical properties have recently been described for STAT3 (Reich, 2009). The pathogenetic role of both STAT3 and STAT5 have been demonstrated in ALK+ ALCL, although the oncogenic contribution of STAT5 is less clear and most likely linked to the unique experimental models (Nieborowska-Skorska et al., 2001). Indeed, in either T (Zamo et al., 2002; Zhang et al., 2002; Amin et al., 2004) or B (Momose et al., 2009) ALK+ lymphoma p-STAT3 species are most commonly expressed and in ALCL, ALK signaling via STAT3 represses STAT5 expression (Zhang et al., 2007).
The constitutive activation of STAT3 is achieved through multiple mechanisms, such as NPM–ALK which can directly phosphorylate STAT3 as well as JAK3 that can indirectly contribute, as demonstrated by reduce levels of p-STAT3 after its pharmacological inhibition (Amin et al., 2003). Moreover primary ALCL can display a loss of JAK/STAT pathway negative regulator Shp1 (Honorat et al., 2006) or alternatively demonstrate an overexpression of protein phosphatase 2A, capable of sustaining a STAT3 phosphorylation status (Zhang et al., 2002).
The critical role of STAT3 in ALK+ ALCL models is well established since its knockdown by RNAi invariably leads to first cell cycle arrest (Amin et al., 2004; Piva et al., 2006), followed over time by the execution of irreversible apoptotic program. This phenomenon is associated with well-defined changes in transcription and the reproducible modulation of a large number of genes (∼1500), many of which are physiologically down-regulated in the presence of active p-STAT3. Most of these changes can be observed using anti-ALK inhibitors or anti-ALK RNAi, demonstrating that “oncogenic addiction” of ALK+ ALCL cells requires a STAT3-mediated transcription. Moreover some of these genes can be used as molecular classifiers and may became diagnostic tools for the identification ALCL among primary T-NHL (Chiarle et al., 2008b; Piva et al., 2010). To discover the STAT3 transcriptionally regulated genes, we have recently conducted time kinetics experiments, demonstrating that only a limited number of known and unknown STAT3 genes are directly modulated in ALCL. These, however, can lead to a cascade of downstream changes. Interestingly enough, this STAT3 dependent phenotype requires the concomitant activation of multiple genes, as demonstrated by the individual knockdown by siRNA and/or the activation of a unique miRNA cluster(s). Intriguingly, in ALK+ NSCLC cell lines the STAT3-associated gene expression signature shares a very limited number of targets with that of ALCL, suggesting that STAT3 oncogenic requirements are lineage restricted and/or cell dependent, as shown by the mild phenotype associated with the knockdown of STAT3 in ALK+ NSLCL cells lines (Takezawa et al., 2011).
STAT3 activation controls cell cycle, apoptosis and DNA damage, adhesion and motility, G protein signaling, inflammation and immune response, metabolic pathways, and angiogenesis. Increased expression of many anti-apoptotic molecules, such as Bcl2, Bcl-XL, Survivin, and Mcl-1 proteins, that in association with ALK-regulated genes such as BCL2A1, provide strong anti-apoptotic signals (Amin et al., 2004; Piva et al., 2006). Uncontrolled proliferation is in part regulated through cyclin D3 (Dalton et al., 2005) and c-myc (Amin et al., 2003). Finally several studies have shown how ALK–STAT3 may control angiogenesis (Marzec et al., 2011) and regulate key immune functions (Marzec et al., 2008). Lastly, STAT3 is in part responsible for the null phenotype of ALK+ ALCL, determining T-cell identity and signaling (Zhang et al., 2002; Ambrogio et al., 2009). The strict requirement for STAT3 activation in ALK+ ALCL makes this molecule an ideal target particularly in the context of mutated ALK chimera, resistant to anti-ALK small molecules. Indeed, small selective compounds like S3I-201, niclosamide, and the pyrimethamine show potent apoptotic effects against ALK+ ALCL cells.
Finally, through proteomic studies many molecules were identified and proven to contribute to the neoplastic phenotype protecting from apoptosis (Bassermann et al., 2005), promoting cell cycle, transport, and transcription activities (Sjostrom et al., 2007). Interestingly, this approach has confirmed the discovery of novel ALK chimera and the definition of a new subset of NSCLC tumors (Rikova et al., 2007). Using quantitative proteomic-based approaches coupled with an enrichment strategy that allows the identification specific phosphorylation sites within peptides, we have shown that 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase (ATIC) is phosphorylated at Y104 via ALK and that ALK-mediated ATIC phosphorylation enhances its enzymatic activity, dampening the methotrexate-mediated transformylase activity inhibition (Boccalatte et al., 2009).
Many of the information provided above have been derived using ALK+ ALCL cell lines harboring the NPM–ALK chimera. It is postulated that the ALK-mediated transformation of different fusion proteins may be imposed through analogous mechanisms. Indeed, many of the ALK fusions can elicit similar signals. Although, they share dimerization domains, which allow the constitutive activation of the proteins, different partners display unique features and impose specific intracellular localization to the ALK proteins. This implies that each of them may have unique, yet slightly different properties, whose contribution(s) to the neoplastic phenotype remain yet to be clarified. Toward this end, it has been proven that some fusions display a higher migratory and invasive capacity in vitro and in vivo (Armstrong et al., 2004). The clarification of these features may be critical in the understanding of the oncogenic potential and oncogenic properties of different ALK fusion proteins in human cancers.
Targeted Therapy
The discovery that EML4–ALK fusions can be found in NSCLC has dramatically changed the landscape of ALK, fostering innovative therapies to move very rapidly into the clinical field. Remarkably, the data generated in ALCL and inflammatory myofibroblastic tumors (IMT), although providing the intellectual/scientific bases for the design of drug discovery programs, have not generated sufficient interest from either the pharmaceutical or scientific communities. This perception has definitively changed when (Soda et al., 2007) first described ALK translocations in a set of lung cancers. Since then, we have found that many other human cancers carry ALK lesions in a well-defined subset of patients, making the "ALK story" one of the most attractive and promising of the last decade. Ultimately, the discovery that sporadic as well as familiar neuroblastoma can carry ALK activating mutations has opened additional hopes and new avenues. This has led to the production of different ALK inhibitors that are now in one stage or the other of investigation, with the Crizotinib as the first approved drug for the treatment of lung cancer patients. Although we have witnessed tremendous advances in a very short period of time, many issues remain ahead. These include the design of the most effective chemotherapeutic combinations, the definition of the right combinations for every individual patient, the discovery, early on during treatment, of non-responder patients, and finally new modalities for overpassing the inevitable resistance associated with the usage of anti-ALK inhibitors.
We believe that many of these issues could be solved once a systematic molecular stratification is employed in all cancer patients. The recent application of Next Generation Sequencing in lung and colon cancer has not only showed that novel translocations (Lipson et al., 2012) can be discovered among these tumors but they might provide the basis for identifying the molecular fingerprints responsible for the clinical behavior of individual cancers. This will only be possible when the cost of these analyses are affordable and when the data obtained by friendly bioinformatic modules are immediately translated into daily clinical practice.
These and many other questions have been further discussed in the accompanying manuscripts.
Here, we would like to address specific topics and discuss a few alternatives.
Potential Challenges
Indeed, we need to carry forward the progress that we have gained in the NSCLC into the field of ALCL, IMT, and neuroblastoma; despite the fact that these diseases represent only a relatively small market and are rare orphan entities. Nevertheless, many of them occur in children and we have very little therapeutic options for some of them. The usage of anti-ALK inhibitors may provide a unique opportunity reaching relevant clinical benefits in the absence of substantial side effects. This is critical in young individuals who may benefit the most. Anti-ALK inhibitors and innovative approaches are required for refractory or relapsed ALK+ ALCL after conventional treatment. The currently recommended protocol for these patients is an autologous stem cell transplantation (HDC/ASCT) preceded by high-dose chemotherapy in first remission. Alternatively, a few experimental studies currently open are investigating the efficacy of Brentuximab vedotin (SGN-35) alone (NCT01352520, NCT0043084, NCT00866047, etc.) or in combination with conventional drugs (NCT00649584).
There are also clinical studies investigating the usage of Crizotinib (CT00939770) or AP26113 (NCT01449461) ALK inhibitors in these settings.
Finally, since kinase inhibitors are invariably associated to drug resistance and they are incapable of eradicating stem cancer cells, new approaches need to be contemplated. Although ALK signaling is considered to be critical in many ALK+ ALCLs, we have seen that co-synergizing signals may contribute to maintain the neoplastic phenotype (in ALCL IGFR and in NSLCL EGFR, c-Met etc.). These signals often converge on critical downstream molecules, which could be targeted to improve the therapeutic responses. For examples the concomitant treatment with ALK and MEK inhibitors has been shown to increase apoptosis of ALK+ NSCLC cell line in vitro providing a rational approach for clinical interventions (Tanizaki et al., 2012). Notably, the application of geldanamycin-based HSP90 inhibitors has also been proven to be efficacious leading to the forced degradation of the wt or mutated ALK fusion proteins (Chen et al., 2010; Katayama et al., 2011). These and other findings (Bonvini et al., 2004) support clinical trails in ALCL patients NCT00117988).
Moreover, since ALK represents a unique onco-antigen, being required for tumor maintenance, expressed in limited tissue compartments and capable of eliciting relevant ALK-specific B and T-cell responses in lymphoma patients (Pulford et al., 2000; Passoni and Gambacorti-Passerini, 2003; Ait-Tahar et al., 2006, 2007; Chiarle et al., 2008a), this provides the basis for the design and implementation of immune-based therapies, which in association with drug-based approaches could lead to the complete control of the disease. Finally, immunological strategies [i.e., anti-CD25 (NCT00801918) anti-CD30 (Ansell et al., 2007; Forero-Torres et al., 2009) and/or CD26 (Ho et al., 2001) antibodies], in combination with conventional or small molecule approaches, are currently considered to enhance anti-tumor responses or to gain the complete eradication of cancer cells.
Future Developments
Since ALK oncogenic properties rely on the constitutive activation of multiple pathways, it is plausible that combinational therapies may be designed including not only conventional drugs but also small compounds capable of targeting key effectors within ALK signaling pathways (Figure 2). Toward this end, the usage of compounds already in clinics for other applications, but recently discovered to have anti-STAT3 activities, are particularly appealing. These include for example niclosamide and pyrimethamine currently used as anthelmintic or anti-parasitic compounds (Takakura et al., 2011), which display promising therapeutic efficacies in ALK+ ALCL models. Analogously, it is also plausible to consider a set of compounds known to down-regulate members of the Ras-Erk pathway. The most promising are represented by the Mek inhibitors (i.e., AZD6244, PD-325901, GDC-0973 etc.), which can lead to ALCL cell cycle arrest and in combination with anthracycline can become powerful treatment modalities. The inhibitor of PI3K and mTOR (Turturro et al., 2002; Marzec et al., 2007; Staber et al., 2007) have also been proposed and in some cases anti-ALK, HSP90 or mTOR inhibitors have proven to be quite efficacious (Chen et al., 2010; Lovly et al., 2011). Finally, since ALK+ ALCL display a strong expression of BCL2A-138, it is reasonable to propose the usage of BH3 mimetic drug as single or in combination with conventional compounds as previously reported for the treatment of B-cell NHL (Paoluzzi et al., 2008a,b; Stolz et al., 2008).
Thus, the armamentarium that we have in our end has become very extensive requiring a systematic analysis of all possible combinations. This implies the set up of novel in vitro as well in vivo models, which faithfully recapitulate the human clinical scenario. More importantly we need to develop a battery of biomarkers capable of stratifying patients and predicting their clinical outcome. Toward this end, the usage of humanized mouse models in which tumor as well human host cells are reconstituting the host represent the new frontier for drug discovery and personalized oncology modalities (Shultz et al., 2007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC Special Program in Clinical Molecular Oncology 5 × 1000, No. 10007, Milan, Italy), Fondazione Guido Berlucchi; Ministero dell’Università e Ricerca Scientifica; Regione Piemonte; Compagnia di San Paolo, Torino (Progetto Oncologia).
Footnote
# ^ The European T-Cell Lymphoma Study Group: Italy: Riccardo Bruna, Daniele Corino, Diego Cortese, Ramona Crescenzo, Giuditta Cuccuru, Filomena di Giacomo, Alessandro Fioravanti, Marco Ladetto, Indira Landra, Katia Messana, Rodolfo Machiorlatti, Barbara Martinoglio, Enzo Medico, Marta Mossino, Elisa Pellegrino, and Maria Todaro (University of Torino); Paola Campisi, Luigi Chiusa Annalisa Chiappella, Domenico Novero and Umberto Vitolo (ASO Molinette, and San Luigi Gonzaga Torino); Francesco Abate, Andrea Acquaviva and Elisa Ficarra (Politecnico of Torino); Roberto Freilone (Medicina Trasfusionale ed Ematologia, Ciriè e Chivasso); Marco Chilosi and Alberto Zamó (University of Verona); Fabio Facchetti and Silvia Lonardi (University of Brescia); Anna De Chiara and Franco Fulciniti (National Cancer Institute, Napoli); Claudio Doglioni and Maurilio Ponzoni (San Raffaele Institute, Milano); Luca Agnelli, Antonino Neri and Katia Todoerti, (University of Milan), Claudio Agostinelli, Pier Paolo Piccaluga and Stefano Pileri (University of Bologna); Brunangelo Falini and Enrico Tiacci (University of Perugia). Belgium: Peter Van Loo, Thomas Tousseyn, and Christiane De Wolf-Peeters (University of Leuven). Germany: Eva Geissinger, Hans Konrad Muller-Hermelink and Andreas Rosenwald, (University of Wuerzburg). Spain: Miguel Angel Pirisand Maria E. Rodriguez (Spanish National Cancer Research Centre, CNIO). Switzerland: Francesco Bertoni, Michela Boi, and Ivo Kwee.
References
Adam, P., Katzenberger, T., Seeberger, H., Gattenlohner, S., Wolf, J., Steinlein, C., Schmid, M., Muller-Hermelink, H. K., and Ott, G. (2003). A case of a diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am. J. Surg. Pathol. 27, 1473–1476.
Ait-Tahar, K., Barnardo, M. C., and Pulford, K. (2007). CD4 T-helper responses to the anaplastic lymphoma kinase (ALK) protein in patients with ALK-positive anaplastic large-cell lymphoma. Cancer Res. 67, 1898–1901.
Ait-Tahar, K., Cerundolo, V., Banham, A. H., Hatton, C., Blanchard, T., Kusec, R., Becker, M., Smith, G. L., and Pulford, K. (2006). B and CTL responses to the ALK protein in patients with ALK-positive ALCL. Int. J. Cancer 118, 688–695.
Ambrogio, C., Martinengo, C., Voena, C., Tondat, F., Riera, L., Di Celle, P. F., Inghirami, G., and Chiarle, R. (2009). NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 69, 8611–8619.
Ambrogio, C., Voena, C., Manazza, A. D., Martinengo, C., Costa, C., Kirchhausen, T., Hirsch, E., Inghirami, G., and Chiarle, R. (2008). The anaplastic lymphoma kinase controls cell shape and growth of anaplastic large cell lymphoma through Cdc42 activation. Cancer Res. 68, 8899–8907.
Amin, H. M., Mcdonnell, T. J., Ma, Y., Lin, Q., Fujio, Y., Kunisada, K., Leventaki, V., Das, P., Rassidakis, G. Z., Cutler, C., Medeiros, L. J., and Lai, R. (2004). Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 23, 5426–5434.
Amin, H. M., Medeiros, L. J., Ma, Y., Feretzaki, M., Das, P., Leventaki, V., Rassidakis, G. Z., O’Connor, S. L., Mcdonnell, T. J., and Lai, R. (2003). Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene 22, 5399–5407.
Ansell, S. M., Horwitz, S. M., Engert, A., Khan, K. D., Lin, T., Strair, R., Keler, T., Graziano, R., Blanset, D., Yellin, M., Fischkoff, S., Assad, A., and Borchmann, P. (2007). Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J. Clin. Oncol. 25, 2764–2769.
Armstrong, F., Duplantier, M. M., Trempat, P., Hieblot, C., Lamant, L., Espinos, E., Racaud-Sultan, C., Allouche, M., Campo, E., Delsol, G., and Touriol, C. (2004). Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene 23, 6071–6082.
Bai, R. Y., Dieter, P., Peschel, C., Morris, S. W., and Duyster, J. (1998). Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol. Cell. Biol. 18, 6951–6961.
Bai, R. Y., Ouyang, T., Miething, C., Morris, S. W., Peschel, C., and Duyster, J. (2000). Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 96, 4319–4327.
Bassermann, F., Von Klitzing, C., Munch, S., Bai, R. Y., Kawaguchi, H., Morris, S. W., Peschel, C., and Duyster, J. (2005). NIPA defines an SCF-type mammalian E3 ligase that regulates mitotic entry. Cell 122, 45–57.
Bedwell, C., Rowe, D., Moulton, D., Jones, G., Bown, N., and Bacon, C. M. (2011). Cytogenetically complex SEC31A-ALK fusions are recurrent in ALK-positive large B-cell lymphomas. Haematologica 96, 343–346.
Benharroch, D., Meguerian-Bedoyan, Z., Lamant, L., Amin, C., Brugieres, L., Terrier-Lacombe, M. J., Haralambieva, E., Pulford, K., Pileri, S., Morris, S. W., Mason, D. Y., and Delsol, G. (1998). ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 91, 2076–2084.
Boccalatte, F. E., Voena, C., Riganti, C., Bosia, A., D’amico, L., Riera, L., Cheng, M., Ruggeri, B., Jensen, O. N., Goss, V. L., Lee, K., Nardone, J., Rush, J., Polakiewicz, R. D., Comb, M. J., Chiarle, R., and Inghirami, G. (2009). The enzymatic activity of 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase is enhanced by NPM-ALK: new insights in ALK-mediated pathogenesis and the treatment of ALCL. Blood 113, 2776–2790.
Bonvini, P., Dalla Rosa, H., Vignes, N., and Rosolen, A. (2004). Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 64, 3256–3264.
Bridge, J. A., Kanamori, M., Ma, Z., Pickering, D., Hill, D. A., Lydiatt, W., Lui, M. Y., Colleoni, G. W., Antonescu, C. R., Ladanyi, M., and Morris, S. W. (2001). Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am. J. Pathol. 159, 411–415.
Chen, Z., Sasaki, T., Tan, X., Carretero, J., Shimamura, T., Li, D., Xu, C., Wang, Y., Adelmant, G. O., Capelletti, M., Lee, H. J., Rodig, S. J., Borgman, C., Park, S. I., Kim, H. R., Padera, R., Marto, J. A., Gray, N. S., Kung, A. L., Shapiro, G. I., Janne, P. A., and Wong, K. K. (2010). Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 70, 9827–9836.
Cheng, L. Y., Bailey, A. P., Leevers, S. J., Ragan, T. J., Driscoll, P. C., and Gould, A. P. (2011). Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila. Cell 146, 435–447.
Cheng, M., Quail, M. R., Gingrich, D. E., Ott, G. R., Lu, L., Wan, W., Albom, M. S., Angeles, T. S., Aimone, L. D., Cristofani, F., Machiorlatti, R., Abele, C., Ator, M. A., Dorsey, B. D., Inghirami, G., and Ruggeri, B. A. (2012). CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 11, 670–679.
Chiarle, R., Gong, J. Z., Guasparri, I., Pesci, A., Cai, J., Liu, J., Simmons, W. J., Dhall, G., Howes, J., Piva, R., and Inghirami, G. (2003). NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 101, 1919–1927.
Chiarle, R., Martinengo, C., Mastini, C., Ambrogio, C., D’escamard, V., Forni, G., and Inghirami, G. (2008a). The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat. Med. 14, 676–680.
Chiarle, R., Voena, C., Ambrogio, C., Piva, R., and Inghirami, G. (2008b). The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 8, 11–23.
Chiarle, R., Simmons, W. J., Cai, H., Dhall, G., Zamo, A., Raz, R., Karras, J. G., Levy, D. E., and Inghirami, G. (2005). Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 11, 623–629.
Christensen, J. G., Zou, H. Y., Arango, M. E., Li, Q., Lee, J. H., Mcdonnell, S. R., Yamazaki, S., Alton, G. R., Mroczkowski, B., and Los, G. (2007). Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 6, 3314–3322.
Colleoni, G. W., Bridge, J. A., Garicochea, B., Liu, J., Filippa, D. A., and Ladanyi, M. (2000). ATIC-ALK: a novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am. J. Pathol. 156, 781–789.
Cools, J., Wlodarska, I., Somers, R., Mentens, N., Pedeutour, F., Maes, B., De Wolf-Peeters, C., Pauwels, P., Hagemeijer, A., and Marynen, P. (2002). Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 34, 354–362.
Cussac, D., Greenland, C., Roche, S., Bai, R. Y., Duyster, J., Morris, S. W., Delsol, G., Allouche, M., and Payrastre, B. (2004). Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood 103, 1464–1471.
Dalton, R. R., Rassidakis, G. Z., Atwell, C., Wang, S., Oyarzo, M. P., and Medeiros, L. J. (2005). Differential expression of cyclin D3 in ALK+ and ALK- anaplastic large cell lymphoma. Hum. Pathol. 36, 806–811.
De Paepe, P., Baens, M., Van Krieken, H., Verhasselt, B., Stul, M., Simons, A., Poppe, B., Laureys, G., Brons, P., Vandenberghe, P., Speleman, F., Praet, M., De Wolf-Peeters, C., Marynen, P., and Wlodarska, I. (2003). ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood 102, 2638–2641.
Debelenko, L. V., Arthur, D. C., Pack, S. D., Helman, L. J., Schrump, D. S., and Tsokos, M. (2003). Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab. Invest. 83, 1255–1265.
Debelenko, L. V., Raimondi, S. C., Daw, N., Shivakumar, B. R., Huang, D., Nelson, M., and Bridge, J. A. (2011). Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod. Pathol. 24, 430–442.
Debiec-Rychter, M., Marynen, P., Hagemeijer, A., and Pauwels, P. (2003). ALK-ATIC fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 38, 187–190.
Delsol, G., Jaffe, E. S., Falini, B., Gascoyne, R. D., Muller-Hermelink, H. K., Stein, H., Campo, E., and Kinney, M. C. (2008). “Anaplastic large cell lymphoma (ALCL), ALK-positive,” in WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 312–316.
Du, X. L., Hu, H., Lin, D. C., Xia, S. H., Shen, X. M., Zhang, Y., Luo, M. L., Feng, Y. B., Cai, Y., Xu, X., Han, Y. L., Zhan, Q. M., and Wang, M. R. (2007). Proteomic profiling of proteins dysregulated in Chinese esophageal squamous cell carcinoma. J. Mol. Med. 85, 863–875.
Falini, B., Pileri, S., Zinzani, P. L., Carbone, A., Zagonel, V., Wolf-Peeters, C., Verhoef, G., Menestrina, F., Todeschini, G., Paulli, M., Lazzarino, M., Giardini, R., Aiello, A., Foss, H. D., Araujo, I., Fizzotti, M., Pelicci, P. G., Flenghi, L., Martelli, M. F., and Santucci, A. (1999). ALK+ lymphoma: clinico-pathological findings and outcome. Blood 93, 2697–2706.
Forero-Torres, A., Leonard, J. P., Younes, A., Rosenblatt, J. D., Brice, P., Bartlett, N. L., Bosly, A., Pinter-Brown, L., Kennedy, D., Sievers, E. L., and Gopal, A. K. (2009). A Phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br. J. Haematol. 146, 171–179.
Fujimoto, J., Shiota, M., Iwahara, T., Seki, N., Satoh, H., Mori, S., and Yamamoto, T. (1996). Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc. Natl. Acad. Sci. U.S.A. 93, 4181–4186.
Galkin, A. V., Melnick, J. S., Kim, S., Hood, T. L., Li, N., Li, L., Xia, G., Steensma, R., Chopiuk, G., Jiang, J., Wan, Y., Ding, P., Liu, Y., Sun, F., Schultz, P. G., Gray, N. S., and Warmuth, M. (2007). Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. U.S.A. 104, 270–275.
Gu, T. L., Tothova, Z., Scheijen, B., Griffin, J. D., Gilliland, D. G., and Sternberg, D. W. (2004). NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood 103, 4622–4629.
Hernandez, L., Bea, S., Bellosillo, B., Pinyol, M., Falini, B., Carbone, A., Ott, G., Rosenwald, A., Fernandez, A., Pulford, K., Mason, D., Morris, S. W., Santos, E., and Campo, E. (2002). Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am. J. Pathol. 160, 1487–1494.
Hernandez, L., Pinyol, M., Hernandez, S., Bea, S., Pulford, K., Rosenwald, A., Lamant, L., Falini, B., Ott, G., Mason, D. Y., Delsol, G., and Campo, E. (1999). TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 94, 3265–3268.
Ho, L., Aytac, U., Stephens, L. C., Ohnuma, K., Mills, G. B., Mckee, K. S., Neumann, C., Lapushin, R., Cabanillas, F., Abbruzzese, J. L., Morimoto, C., and Dang, N. H. (2001). In vitro and in vivo antitumor effect of the anti-CD26 monoclonal antibody 1F7 on human CD30+ anaplastic large cell T-cell lymphoma Karpas 299. Clin. Cancer Res. 7, 2031–2040.
Honorat, J. F., Ragab, A., Lamant, L., Delsol, G., and Ragab-Thomas, J. (2006). SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood 107, 4130–4138.
Hsu, F. Y., Johnston, P. B., Burke, K. A., and Zhao, Y. (2006). The expression of CD30 in anaplastic large cell lymphoma is regulated by nucleophosmin-anaplastic lymphoma kinase-mediated JunB level in a cell type-specific manner. Cancer Res. 66, 9002–9008.
Jaffe, E. S. (2001). Anaplastic large cell lymphoma: the shifting sands of diagnostic hematopathology. Mod. Pathol. 14, 219–228.
Jazii, F. R., Najafi, Z., Malekzadeh, R., Conrads, T. P., Ziaee, A. A., Abnet, C., Yazdznbod, M., Karkhane, A. A., and Salekdeh, G. H. (2006). Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer. World J. Gastroenterol. 12, 7104–7112.
Jung, Y., Kim, P., Jung, Y., Keum, J., Kim, S. N., Choi, Y. S., Do, I. G., Lee, J., Choi, S. J., Kim, S., Lee, J. E., Kim, J., Lee, S., and Kim, J. (2012). Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosomes Cancer 51, 590–597.
Katayama, R., Khan, T. M., Benes, C., Lifshits, E., Ebi, H., Rivera, V. M., Shakespeare, W. C., Iafrate, A. J., Engelman, J. A., and Shaw, A. T. (2011). Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. U.S.A. 108, 7535–7540.
Koivunen, J. P., Mermel, C., Zejnullahu, K., Murphy, C., Lifshits, E., Holmes, A. J., Choi, H. G., Kim, J., Chiang, D., Thomas, R., Lee, J., Richards, W. G., Sugarbaker, D. J., Ducko, C., Lindeman, N., Marcoux, J. P., Engelman, J. A., Gray, N. S., Lee, C., Meyerson, M., and Janne, P. A. (2008). EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 14, 4275–4283.
Kuefer, M. U., Look, A. T., Pulford, K., Behm, F. G., Pattengale, P. K., Mason, D. Y., and Morris, S. W. (1997). Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood 90, 2901–2910.
Lamant, L., Dastugue, N., Pulford, K., Delsol, G., and Mariame, B. (1999). A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 93, 3088–3095.
Lamant, L., Gascoyne, R. D., Duplantier, M. M., Armstrong, F., Raghab, A., Chhanabhai, M., Rajcan-Separovic, E., Raghab, J., Delsol, G., and Espinos, E. (2003). Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer 37, 427–432.
Lawrence, B., Perez-Atayde, A., Hibbard, M. K., Rubin, B. P., Dal Cin, P., Pinkus, J. L., Pinkus, G. S., Xiao, S., Yi, E. S., Fletcher, C. D., and Fletcher, J. A. (2000). TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am. J. Pathol. 157, 377–384.
Levy, D. E., and Inghirami, G. (2006). STAT3: a multifaceted oncogene. Proc. Natl. Acad. Sci. U.S.A. 103, 10151–10152.
Lin, E., Li, L., Guan, Y., Soriano, R., Rivers, C. S., Mohan, S., Pandita, A., Tang, J., and Modrusan, Z. (2009). Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res. 7, 1466–1476.
Lipson, D., Capelletti, M., Yelensky, R., Otto, G., Parker, A., Jarosz, M., Curran, J. A., Balasubramanian, S., Bloom, T., Brennan, K. W., Donahue, A., Downing, S. R., Frampton, G. M., Garcia, L., Juhn, F., Mitchell, K. C., White, E., White, J., Zwirko, Z., Peretz, T., Nechushtan, H., Soussan-Gutman, L., Kim, J., Sasaki, H., Kim, H. R., Park, S. I., Ercan, D., Sheehan, C. E., Ross, J. S., Cronin, M. T., Janne, P. A., and Stephens, P. J. (2012). Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 18, 382–384.
Lovly, C. M., Heuckmann, J. M., De Stanchina, E., Chen, H., Thomas, R. K., Liang, C., and Pao, W. (2011). Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 71, 4920–4931.
Ma, Z., Cools, J., Marynen, P., Cui, X., Siebert, R., Gesk, S., Schlegelberger, B., Peeters, B., De Wolf-Peeters, C., Wlodarska, I., and Morris, S. W. (2000). Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood 95, 2144–2149.
Ma, Z., Hill, D. A., Collins, M. H., Morris, S. W., Sumegi, J., Zhou, M., Zuppan, C., and Bridge, J. A. (2003). Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 37, 98–105.
Marino-Enriquez, A., Wang, W. L., Roy, A., Lopez-Terrada, D., Lazar, A. J., Fletcher, C. D., Coffin, C. M., and Hornick, J. L. (2011). Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am. J. Surg. Pathol. 35, 135–144.
Martinelli, P., Bonetti, P., Sironi, C., Pruneri, G., Fumagalli, C., Raviele, P. R., Volorio, S., Pileri, S., Chiarle, R., Mcduff, F. K., Tusi, B. K., Turner, S. D., Inghirami, G., Pelicci, P. G., and Colombo, E. (2011). The lymphoma-associated NPM-ALK oncogene elicits a p16INK4a/pRb-dependent tumor-suppressive pathway. Blood 117, 6617–6626.
Marzec, M., Kasprzycka, M., Liu, X., Raghunath, P. N., Wlodarski, P., and Wasik, M. A. (2007). Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene 26, 813–821.
Marzec, M., Liu, X., Wong, W., Yang, Y., Pasha, T., Kantekure, K., Zhang, P., Woetmann, A., Cheng, M., Odum, N., and Wasik, M. A. (2011). Oncogenic kinase NPM/ALK induces expression of HIF1alpha mRNA. Oncogene 30, 1372–1378.
Marzec, M., Zhang, Q., Goradia, A., Raghunath, P. N., Liu, X., Paessler, M., Wang, H. Y., Wysocka, M., Cheng, M., Ruggeri, B. A., and Wasik, M. A. (2008). Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. U.S.A. 105, 20852–20857.
Mcdonnell, S. R., Hwang, S. R., Basrur, V., Conlon, K. P., Fermin, D., Wey, E., Murga-Zamalloa, C., Zeng, Z., Zu, Y., Elenitoba-Johnson, K. S., and Lim, M. S. (2011). NPM-ALK signals through glycogen synthase kinase 3beta to promote oncogenesis. Oncogene. doi: 10.1038/onc.2011.542
Meech, S. J., Mcgavran, L., Odom, L. F., Liang, X., Meltesen, L., Gump, J., Wei, Q., Carlsen, S., and Hunger, S. P. (2001). Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4 – anaplastic lymphoma kinase gene fusion. Blood 98, 1209–1216.
Mi, R., Chen, W., and Hoke, A. (2007). Pleiotrophin is a neurotrophic factor for spinal motor neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 4664–4669.
Momose, S., Tamaru, J., Kishi, H., Mikata, I., Mori, M., Toyozumi, Y., and Itoyama, S. (2009). Hyperactivated STAT3 in ALK-positive diffuse large B-cell lymphoma with clathrin-ALK fusion. Hum. Pathol. 40, 75–82.
Morris, S. W., Kirstein, M. N., Valentine, M. B., Dittmer, K. G., Shapiro, D. N., Saltman, D. L., and Look, A. T. (1994). Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263, 1281–1284.
Nieborowska-Skorska, M., Slupianek, A., Xue, L., Zhang, Q., Raghunath, P. N., Hoser, G., Wasik, M. A., Morris, S. W., and Skorski, T. (2001). Role of signal transducer and activator of transcription 5 in nucleophosmin/anaplastic lymphoma kinase-mediated malignant transformation of lymphoid cells. Cancer Res. 61, 6517–6523.
Okuwaki, M. (2008). The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 143, 441–448.
Onciu, M., Behm, F. G., Downing, J. R., Shurtleff, S. A., Raimondi, S. C., Ma, Z., Morris, S. W., Kennedy, W., Jones, S. C., and Sandlund, J. T. (2003). ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood 102, 2642–2644.
Ott, G., Katzenberger, T., Siebert, R., Decoteau, J. F., Fletcher, J. A., Knoll, J. H., Kalla, J., Rosenwald, A., Ott, M. M., Weber-Matthiesen, K., Kadin, M. E., and Muller-Hermelink, H. K. (1998). Chromosomal abnormalities in nodal and extranodal CD30+ anaplastic large cell lymphomas: infrequent detection of the t(2;5) in extranodal lymphomas. Genes Chromosomes Cancer 22, 114–121.
Palmer, R. H., Vernersson, E., Grabbe, C., and Hallberg, B. (2009). Anaplastic lymphoma kinase: signalling in development and disease. Biochem. J. 420, 345–361.
Panagopoulos, I., Nilsson, T., Domanski, H. A., Isaksson, M., Lindblom, P., Mertens, F., and Mandahl, N. (2006). Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int. J. Cancer 118, 1181–1186.
Paoluzzi, L., Gonen, M., Bhagat, G., Furman, R. R., Gardner, J. R., Scotto, L., Gueorguiev, V. D., Heaney, M. L., Manova, K., and O’Connor, O. A. (2008a). The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood 112, 2906–2916.
Paoluzzi, L., Gonen, M., Gardner, J. R., Mastrella, J., Yang, D., Holmlund, J., Sorensen, M., Leopold, L., Manova, K., Marcucci, G., Heaney, M. L., and O’Connor, O. A. (2008b). Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 111, 5350–5358.
Passoni, L., and Gambacorti-Passerini, C. (2003). ALK a novel lymphoma-associated tumor antigen for vaccination strategies. Leuk. Lymphoma 44, 1675–1681.
Patel, A. S., Murphy, K. M., Hawkins, A. L., Cohen, J. S., Long, P. P., Perlman, E. J., and Griffin, C. A. (2007). RANBP2 and CLTC are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet. Cytogenet. 176, 107–114.
Pearson, J. D., Lee, J. K., Bacani, J. T., Lai, R., and Ingham, R. J. (2011). NPM-ALK and the JunB transcription factor regulate the expression of cytotoxic molecules in ALK-positive, anaplastic large cell lymphoma. Int. J. Clin. Exp. Pathol. 4, 124–133.
Perez-Pinera, P., Zhang, W., Chang, Y., Vega, J. A., and Deuel, T. F. (2007). Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. J. Biol. Chem. 282, 28683–28690.
Piva, R., Agnelli, L., Pellegrino, E., Todoerti, K., Grosso, V., Tamagno, I., Fornari, A., Martinoglio, B., Medico, E., Zamo, A., Facchetti, F., Ponzoni, M., Geissinger, E., Rosenwald, A., Muller-Hermelink, H. K., De Wolf-Peeters, C., Piccaluga, P. P., Pileri, S., Neri, A., and Inghirami, G. (2010). Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J. Clin. Oncol. 28, 1583–1590.
Piva, R., Pellegrino, E., Mattioli, M., Agnelli, L., Lombardi, L., Boccalatte, F., Costa, G., Ruggeri, B. A., Cheng, M., Chiarle, R., Palestro, G., Neri, A., and Inghirami, G. (2006). Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Invest. 116, 3171–3182.
Pulford, K., Falini, B., Banham, A. H., Codrington, D., Roberton, H., Hatton, C., and Mason, D. Y. (2000). Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood 96, 1605–1607.
Pulford, K., Morris, S. W., and Mason, D. Y. (2001). Anaplastic lymphoma kinase proteins and malignancy. Curr. Opin. Hematol. 8, 231–236.
Riera, L., Lasorsa, E., Ambrogio, C., Surrenti, N., Voena, C., and Chiarle, R. (2010). Involvement of Grb2 adaptor protein in nucleophosmin-anaplastic lymphoma kinase (NPM-ALK)-mediated signaling and anaplastic large cell lymphoma growth. J. Biol. Chem. 285, 26441–26450.
Rikova, K., Guo, A., Zeng, Q., Possemato, A., Yu, J., Haack, H., Nardone, J., Lee, K., Reeves, C., Li, Y., Hu, Y., Tan, Z., Stokes, M., Sullivan, L., Mitchell, J., Wetzel, R., Macneill, J., Ren, J. M., Yuan, J., Bakalarski, C. E., Villen, J., Kornhauser, J. M., Smith, B., Li, D., Zhou, X., Gygi, S. P., Gu, T. L., Polakiewicz, R. D., Rush, J., and Comb, M. J. (2007). Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203.
Salaverria, I., Bea, S., Lopez-Guillermo, A., Lespinet, V., Pinyol, M., Burkhardt, B., Lamant, L., Zettl, A., Horsman, D., Gascoyne, R., Ott, G., Siebert, R., Delsol, G., and Campo, E. (2008). Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br. J. Haematol. 140, 516–526.
Savage, K. J., Harris, N. L., Vose, J. M., Ullrich, F., Jaffe, E. S., Connors, J. M., Rimsza, L., Pileri, S. A., Chhanabhai, M., Gascoyne, R. D., Armitage, J. O., and Weisenburger, D. D. (2008). ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood 111, 5496–5504.
Shi, P., Lai, R., Lin, Q., Iqbal, A. S., Young, L. C., Kwak, L. W., Ford, R. J., and Amin, H. M. (2009). IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood 114, 360–370.
Shiota, M., Fujimoto, J., Takenaga, M., Satoh, H., Ichinohasama, R., Abe, M., Nakano, M., Yamamoto, T., and Mori, S. (1994). Diagnosis of t(2;5)(p23;q35)-associated Ki-1 lymphoma with immunohistochemistry. Blood 84, 3648–3652.
Shultz, L. D., Ishikawa, F., and Greiner, D. L. (2007). Humanized mice in translational biomedical research. Nat. Rev. Immunol. 7, 118–130.
Siebert, R., Gesk, S., Harder, L., Steinemann, D., Grote, W., Schlegelberger, B., Tiemann, M., Wlodarska, I., and Schemmel, V. (1999). Complex variant translocation t(1;2) with TPM3-ALK fusion due to cryptic ALK gene rearrangement in anaplastic large-cell lymphoma. Blood 94, 3614–3617.
Sjostrom, C., Seiler, C., Crockett, D. K., Tripp, S. R., Elenitoba Johnson, K. S., and Lim, M. S. (2007). Global proteome profiling of NPM/ALK-positive anaplastic large cell lymphoma. Exp. Hematol. 35, 1240–1248.
Slupianek, A., Nieborowska-Skorska, M., Hoser, G., Morrione, A., Majewski, M., Xue, L., Morris, S. W., Wasik, M. A., and Skorski, T. (2001). Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 61, 2194–2199.
Soda, M., Choi, Y. L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K., Hatanaka, H., Bando, M., Ohno, S., Ishikawa, Y., Aburatani, H., Niki, T., Sohara, Y., Sugiyama, Y., and Mano, H. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566.
Soda, M., Takada, S., Takeuchi, K., Choi, Y. L., Enomoto, M., Ueno, T., Haruta, H., Hamada, T., Yamashita, Y., Ishikawa, Y., Sugiyama, Y., and Mano, H. (2008). A mouse model for EML4-ALK-positive lung cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 19893–19897.
Staber, P. B., Vesely, P., Haq, N., Ott, R. G., Funato, K., Bambach, I., Fuchs, C., Schauer, S., Linkesch, W., Hrzenjak, A., Dirks, W. G., Sexl, V., Bergler, H., Kadin, M. E., Sternberg, D. W., Kenner, L., and Hoefler, G. (2007). The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 110, 3374–3383.
Stein, H., Foss, H. D., Durkop, H., Marafioti, T., Delsol, G., Pulford, K., Pileri, S., and Falini, B. (2000). CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 96, 3681–3695.
Stolz, C., Hess, G., Hahnel, P. S., Grabellus, F., Hoffarth, S., Schmid, K. W., and Schuler, M. (2008). Targeting Bcl-2 family proteins modulates the sensitivity of B-cell lymphoma to rituximab-induced apoptosis. Blood 112, 3312–3321.
Takakura, A., Nelson, E. A., Haque, N., Humphreys, B. D., Zandi-Nejad, K., Frank, D. A., and Zhou, J. (2011). Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum. Mol. Genet. 20, 4143–4154.
Takeuchi, K., Choi, Y. L., Togashi, Y., Soda, M., Hatano, S., Inamura, K., Takada, S., Ueno, T., Yamashita, Y., Satoh, Y., Okumura, S., Nakagawa, K., Ishikawa, Y., and Mano, H. (2009). KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res. 15, 3143–3149.
Takeuchi, K., Soda, M., Togashi, Y., Ota, Y., Sekiguchi, Y., Hatano, S., Asaka, R., Noguchi, M., and Mano, H. (2011). Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica 96, 464–467.
Takeuchi, K., Soda, M., Togashi, Y., Suzuki, R., Sakata, S., Hatano, S., Asaka, R., Hamanaka, W., Ninomiya, H., Uehara, H., Lim Choi, Y., Satoh, Y., Okumura, S., Nakagawa, K., Mano, H., and Ishikawa, Y. (2012). RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 18, 378–381.
Takezawa, K., Okamoto, I., Nishio, K., Janne, P. A., and Nakagawa, K. (2011). Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin. Cancer Res. 17, 2140–2148.
Tanizaki, J., Okamoto, I., Takezawa, K., Sakai, K., Azuma, K., Kuwata, K., Yamaguchi, H., Hatashita, E., Nishio, K., Janne, P. A., and Nakagawa, K. (2012). Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br. J. Cancer 106, 763–767.
Tilly, H., Gaulard, P., Lepage, E., Dumontet, C., Diebold, J., Plantier, I., Berger, F., Symann, M., Petrella, T., Lederlin, P., and Briere, J. (1997). Primary anaplastic large-cell lymphoma in adults: clinical presentation, immunophenotype, and outcome. Blood 90, 3727–3734.
Togashi, Y., Soda, M., Sakata, S., Sugawara, E., Hatano, S., Asaka, R., Nakajima, T., Mano, H., and Takeuchi, K. (2012). KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 7, e31323. doi:10.1371/journal.pone.0031323
Tort, F., Campo, E., Pohlman, B., and Hsi, E. (2004). Heterogeneity of genomic breakpoints in MSN-ALK translocations in anaplastic large cell lymphoma. Hum. Pathol. 35, 1038–1041.
Tort, F., Pinyol, M., Pulford, K., Roncador, G., Hernandez, L., Nayach, I., Kluin-Nelemans, H. C., Kluin, P., Touriol, C., Delsol, G., Mason, D., and Campo, E. (2001). Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab. Invest. 81, 419–426.
Touriol, C., Greenland, C., Lamant, L., Pulford, K., Bernard, F., Rousset, T., Mason, D. Y., and Delsol, G. (2000). Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 95, 3204–3207.
Trinei, M., Lanfrancone, L., Campo, E., Pulford, K., Mason, D. Y., Pelicci, P. G., and Falini, B. (2000). A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res. 60, 793–798.
Turner, S. D., and Alexander, D. R. (2005). What have we learnt from mouse models of NPM-ALK-induced lymphomagenesis? Leukemia 19, 1128–1134.
Turturro, F., Arnold, M. D., Frist, A. Y., and Pulford, K. (2002). Model of inhibition of the NPM-ALK kinase activity by herbimycin A. Clin. Cancer Res. 8, 240–245.
Van Roosbroeck, K., Cools, J., Dierickx, D., Thomas, J., Vandenberghe, P., Stul, M., Delabie, J., De Wolf-Peeters, C., Marynen, P., and Wlodarska, I. (2010). ALK-positive large B-cell lymphomas with cryptic SEC31A-ALK and NPM1-ALK fusions. Haematologica 95, 509–513.
Vose, J., Armitage, J., and Weisenburger, D. (2008). International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J. Clin. Oncol. 26, 4124–4130.
Wan, W., Albom, M. S., Lu, L., Quail, M. R., Becknell, N. C., Weinberg, L. R., Reddy, D. R., Holskin, B. P., Angeles, T. S., Underiner, T. L., Meyer, S. L., Hudkins, R. L., Dorsey, B. D., Ator, M. A., Ruggeri, B. A., and Cheng, M. (2006). Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood 107, 1617–1623.
Watanabe, M., Sasaki, M., Itoh, K., Higashihara, M., Umezawa, K., Kadin, M. E., Abraham, L. J., Watanabe, T., and Horie, R. (2005). JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and Reed-Sternberg cells of Hodgkin lymphoma. Cancer Res. 65, 7628–7634.
Weiss, J. B., Xue, C., Benice, T., Xue, L., Morris, S. W., and Raber, J. (2012). Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol. Biochem. Behav. 100, 566–574.
Wong, D. W., Leung, E. L., Wong, S. K., Tin, V. P., Sihoe, A. D., Cheng, L. C., Au, J. S., Chung, L. P., and Wong, M. P. (2011). A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer 117, 2709–2718.
Zamo, A., Chiarle, R., Piva, R., Howes, J., Fan, Y., Chilosi, M., Levy, D. E., and Inghirami, G. (2002). Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 21, 1038–1047.
Zettl, A., Rudiger, T., Konrad, M. A., Chott, A., Simonitsch-Klupp, I., Sonnen, R., Muller-Hermelink, H. K., and Ott, G. (2004). Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am. J. Pathol. 164, 1837–1848.
Zhang, Q., Raghunath, P. N., Xue, L., Majewski, M., Carpentieri, D. F., Odum, N., Morris, S., Skorski, T., and Wasik, M. A. (2002). Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 168, 466–474.
Keywords: anaplastic large cell lymphoma, chimeric fusion proteins, signaling pathways, molecular targeted therapy, anaplastic lymphoma kinase
Citation: Tabbó F, Barreca A, Piva R, Inghirami G and The European T-Cell Lymphoma Study Group (2012) ALK signaling and target therapy in anaplastic large cell lymphoma. Front. Oncol. 2:41. doi: 10.3389/fonc.2012.00041
Received: 04 January 2012; Accepted: 10 April 2012; Published online: 11 May 2012.
Edited by:
Yisong Wang, National Cancer Institute at the National Institutes of Health, USAReviewed by:
Min Hee Kang, School of Medicine TTUHSC, USALokesh Jain, Food and Drug Administration, USA
Copyright: © 2012 Tabbó, Barreca, Piva, Inghirami and The European T-Cell Lymphoma Study Group. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Giorgio Inghirami, Department of Pathology, Center for Experimental Research and Medical Studies, University of Torino, Via Santena 7, Torino 10126, Italy. e-mail: giorgio.inghirami@unito.it