
- Division of Cancer, Department of Surgery and Cancer, Imperial College, Hammersmith Hospital, London, UK
S6 kinase 2 (S6K2) is a member of the AGC kinases super-family. Its closest homolog, S6K1, has been extensively studied along the years. However, due to the belief in the community that the high degree of identity between these two isoforms would translate in essentially identical biological functions, S6K2 has been largely neglected. Nevertheless, recent research has clearly highlighted that these two proteins significantly differ in their roles in vitro as well as in vivo. These findings are significant to our understanding of S6 kinase signaling and the development of therapeutic strategies for several diseases including cancer. Here, we will focus on S6K2 and review the protein–protein interactions and specific substrates that determine the selective functions of this kinase.
Introduction
The ribosomal protein S6 kinases constitute a super-family of proteins initially discovered based on their ability to phosphorylate a 40S ribosomal subunit component, the ribosomal S6 protein. The p90 ribosomal S6 kinases (RSKs), comprising RSK1–4 (1), were first identified followed by the p70 ribosomal S6 kinase, S6K1 (2, 3). It took an additional 10 years for the p70 ribosomal S6 kinase homolog, S6K2, to be discovered (4–6). The high degree of homology between S6K1 and S6K2 has for many years led researchers to assume that these were redundant kinases with essentially overlapping functions. This introduced a bias toward S6K1-oriented research, as this isoform came to be considered the prototypical S6K. However, more recent research clearly indicates that these two isoforms also have distinct biological functions the understanding of which may have implications for therapeutic intervention. Therefore, while other publications exist that review S6Ks and their upstream pathways (7, 8), here we will focus specifically on S6K2 and highlight the distinct biological function of this isoform.
Structure of S6K2
Human S6K2 is encoded by the 15 exons of the RPS6KB2 gene on chromosome 11 (11q13). The S6K2 mRNA (ID ENST00000312629) gives rise to two protein products through the use of alternative translational start sites: a long form (p56 S6K2) and a short form (p54 S6K2) that differ by the presence or absence of an N-terminal 13 amino acid segment. The overall structure of S6K2 is very close to that of S6K1 (Figure 1A). The kinase domain of S6K2 shares 83% amino acid identity with that of S6K1, a fact that has long justified the lack of interest in finding isoform-specific substrates for these proteins. The kinase domain is followed toward the C-terminus by a kinase extension domain and a pseudo-substrate inhibitory region. The greatest degree of divergence between S6K1 and S6K2 lies in the C-and N-terminus, a fact that has enabled the development of S6K2-specific antibodies (9). The presence in the C-terminus of S6K2 of a nuclear localization sequence (NLS) means that this isoform is predominantly localized to the nuclei of quiescent cells (10). In addition, the long form of S6K2 contains in its 13 amino acid extension an additional putative NLS. This results in the different cellular distribution of these two isoforms as the two NLS motifs in p56 S6K2 confers constitutive nuclear localization to this variant, while p54 S6K2 shuttles between the nucleus and the cytoplasm in response to growth factor signaling. The C-terminus of S6K2 also contains a proline-rich region which has been proposed to promote interaction with SH3 and WW domains putatively present in its binding partners (4). While shorter isoforms of S6K1 have been shown to be generated by alternate mRNA splicing (11), no such variants have yet been reported for S6K2. However, the high degree of conservation between the two proteins raises the possibility that similar regulation may take place for the RPS6KB2 gene. Indeed, eight transcripts have been reported for S6K2 with a corresponding protein found for only one (ID ENST00000312629) of the four protein coding transcripts (ID ENST00000539188, ENST00000524934, ENST00000524814, ENST00000312629). This may have important functional consequences as, unlike its full length counterpart, an S6K1 splice variant, p31S6K1, was shown to have oncogenic potential (12).
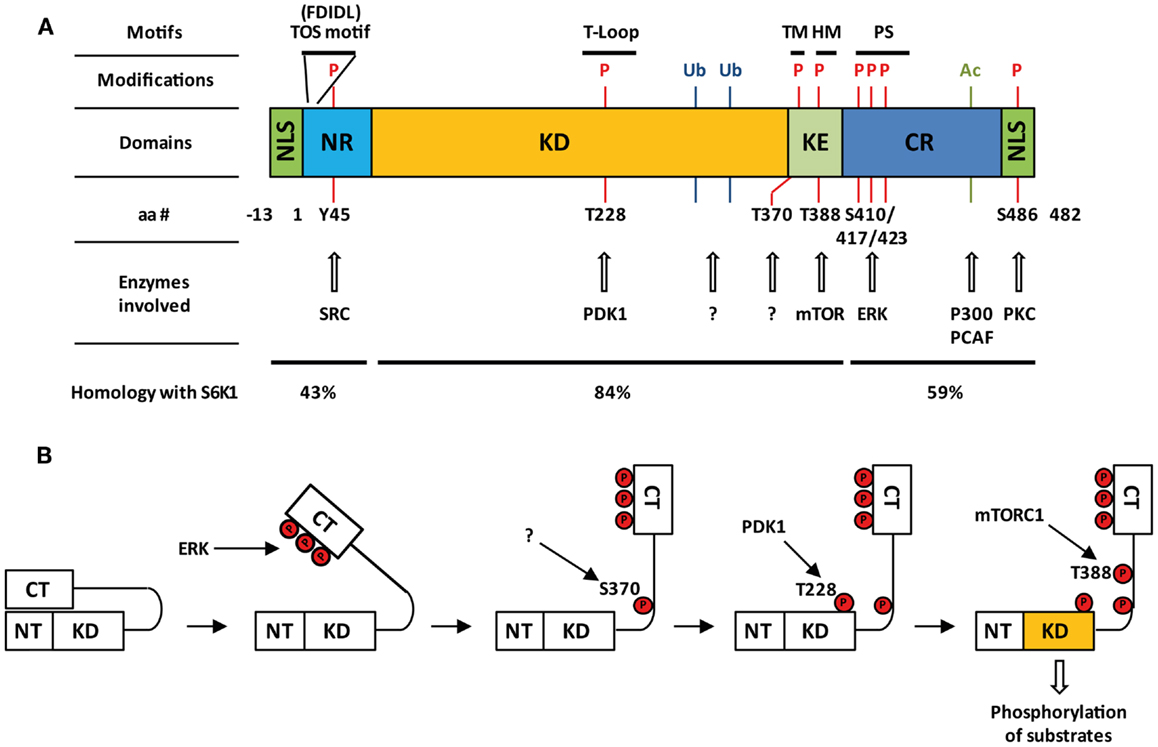
Figure 1. Structure and activation of S6K2. (A) Domain organization of S6K2, post-translational modifications together with involved enzymes, and percentage homology with S6K1. Nuclear localizations sequences (NLS); N-terminal regulatory region (NR); kinase domain (KD); kinase extension region (KE); C-terminal regulatory region (CR); pseudo-substrate domain (PS); turn motif (TM); hydrophobic motif (HM); pseudo-substrate region (PS); phosphorylation (P), ubiquitination (Ub); acetylation (Ac). (B) Step-wise model of activation of S6K2.
S6K2 Activation and Post-Translational Modification
S6K2 Activation
Many of the residues that are required for kinase activation are common between S6K1 and S6K2 as seven of the eight serine/threonine phosphorylation sites present on S6K1 are conserved in S6K2 (Thr-228, Ser-370, Thr-388, Ser-403, Ser-410, Ser-417, and Ser-423 on p54 S6K2) (4, 6, 10) (Figure 1A). The activation of S6K2 occurs in a step-wise manner (Figure 1B). An initial barrier to overcome is the repression exerted by the C-terminal autoinhibitory pseudo-substrate domain. This is dealt with by phosphorylation of the three proline-directed serines in the autoinhibitory domain, Ser-410, Ser-417, and Ser-423 downstream of MEK/ERK signaling. We and others have found this first step to be crucial for S6K2 activation in various cell types (13, 14), as this domain exerts a far more repressive role on S6K2 activity than it’s equivalent for S6K1 (15, 16). This event is presumed to open the kinase conformation, exposing additional phosphorylation sites to activating kinases. In agreement with this, deletion of the autoinhibitory region increases basal activity of S6K2 and sensitizes the kinase to activation by various agonists (15). Subsequent phosphorylation of Ser-370 then enables phosphorylation of Thr-388 by the mTORC1 complex followed by that of Thr-228 by PDK1 (17). The T388 site lies within a conserved sequence of the kinase extension domain (F-X-X-F/Y-S/T-F/Y) known as the hydrophobic motif, a region found in many AGC kinases. Phosphorylation of this site by mTOR is achieved following the binding of the mTORC1 complex component Raptor to the TOR signaling (TOS) motif present in both S6K1 and 2 (18, 19). Interestingly, despite the conservation of the hydrophobic motif, substitution of Thr-388 by a glutamic acid (T388E) renders S6K2, but not S6K1, constitutively active. However, phosphorylation of both the Ser-370 and Thr-228 is crucial for S6K2 activity. Indeed, substitution of the latter site for alanine renders the T388E mutant inactive while that of the first prevents Thr-388 phosphorylation. As S6K2 is mainly a nuclear protein and mTOR shuttles between the cytoplasm and the nucleus, it was shown that S6K2 activity was increased by targeting mTOR expression to the nucleus (20).
Despite S6K1 and S6K2 both lying downstream of mTOR (Figure 2), there is evidence to indicate that they may be regulated through different pools of this upstream kinase. Indeed, both S6K isoforms react differently to nutrient deprivation, a known modulator of mTOR activity. For instance inhibition of protein synthesis by leucine deprivation in myotubes, results in dephosphorylation of S6K1, without affecting S6K2 activity (21). The existence of two separate pools of mTOR regulating the two S6K isoforms is further suggested by the differential sensitivity of S6K1 and 2 kinase activity to the mTOR inhibitor, rapamycin. Indeed, the involvement of mTORC1 in the activation of S6K2, led several researchers to report the sensitivity of S6K2 to this inhibitor (14, 17, 22). However, the majority of reports suggesting equivalent sensitivity of S6K1 and 2 to rapamycin used concentrations of this drug that non-selectively inhibit the MEK/ERK pathway, therefore indirectly targeting S6K2 independently of its effect on mTOR (13). Hence, when used at the minimal concentrations that fully inhibit S6K1 activity, rapamycin often fails to significantly alter S6K2 activity in several cell systems [(13) and unpublished data from our lab]. These findings are consistent with the reported existence of a rapamycin-resistant mTORC1 activity pool (23, 24) that can efficiently be targeted by mTOR ATP-competitive inhibitors (23, 25).
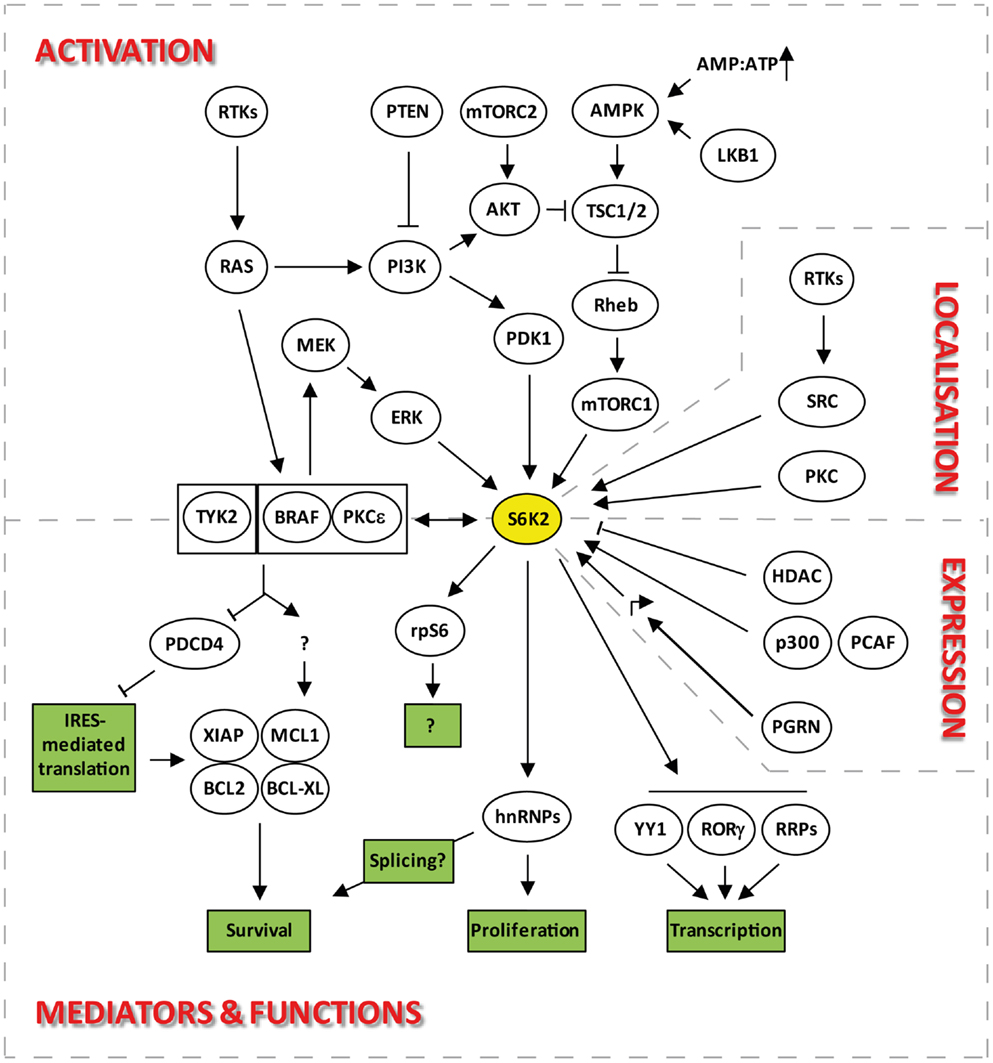
Figure 2. Signaling pathways upstream and downstream of S6K2 that regulate its activation, localization, expression, and functions.
Additional Phosphorylation Events
While expression of an mTOR variant targeted to the nucleus increases activation of S6K2, nuclear localization of S6K2 is not indispensable for activation of this kinase. Indeed, S6K2, but not S6K1, is phosphorylated in vitro as well as in vivo by protein kinase C (PKC) (26). The site of phosphorylation was identified as S486 in p54 S6K2 (S486 in p56 S6K2), located within the C-terminal NLS. While phosphorylation of this site did not affect the activity of S6K2, it impaired the function of the NLS leading to cytoplasmic accumulation of the kinase upon cell stimulation with PKC agonists such as PMA. In contrast, S6K1 sub-cellular localization was not modulated by this treatment, highlighting a specific mechanism regulating S6K2 nucleo-cytoplasmic shuttling. All PKC isoforms were capable of phosphorylating S6K2, with PKCδ appearing to be the most efficient in vitro. However, this specificity seemed to disappear in vivo with all PKCs being equally potent.
In addition to being serine/threonine phosphorylated, S6K2, as well as S6K1, can be tyrosine phosphorylated downstream of receptor tyrosine kinase activation (27). Both S6Ks were found to associate with the PDGFR, HGFR, and CSFR. Upon stimulation of these receptors, N-terminal tyrosine phosphorylation of S6Ks occurred (Y39 on S6K1 and Y45 on S6K2) in a SRC-dependent manner. This event did not result in modulation of S6Ks activity or the gross redistribution of these enzymes, although a fraction of S6K1 was found to relocates to membrane ruffles where the activated RTKs are expected to reside. Although this pathway seems shared between S6K1 and 2, it is worth noting that while SRC family members were equally able to phosphorylate both isoforms in vitro, S6K2, but not S6K1, was tyrosine phosphorylated in response to FYN transgene expression in vivo. This may reflect differential wiring of these isoforms to the SRC family members through alternate cellular multi-protein complexes.
In addition to phosphorylation events, S6K2 is also the target of ubiquitination (28, 29) and of acetylation on a lysine residue close to the C-terminal PDZ binding motif (30). The latter modification does not impact on S6K2 kinase activity or sub-cellular localization but increases the stability of this kinase (see Control of S6K2 Steady-State Levels).
Cellular Expression and Localization of S6K2
Control of S6K2 Steady-State Levels
There is currently little to no information on the transcriptional or translational regulation of S6K2 expression. However, much more is known about the regulation of S6K2 stability. Steady-state levels of S6K2 and 1 are regulated through the opposing effects of ubiquitination and acetylation. Indeed, degradation of S6K1 and S6K2 is mediated by ubiquitination followed by proteosomal degradation (28, 29). This is promoted by growth factor signaling in cell lines although independently of phosphorylation/activation of these kinases. Conversely, cell stress, such as that induced by UV exposure, stabilizes both proteins. However, unlike for S6K1, the molecular pathway regulating S6K2 ubiquitination is currently unknown. Indeed, while the ROC1 ubiquitin ligase was shown to specifically interact and ubiquitinate S6K1 (31), the corresponding partner for S6K2 has not yet been identified. S6K1/2 degradation is counteracted by the stabilization of this protein through a C-terminal lysine acetylation event. This occurs through interaction with the acetyltransferases p300 and p300/CBP-associated factor (PCAF) (Figure 2). Hence, overexpression of p300 or inhibition of deacetylases leads to an increase in the levels of both kinases (30). Interestingly, overexpression of p300 has been linked to decreased overall survival in patients suffering from a wide variety of malignancies (32–36) while that of PCAF has been associated with drug resistance (37–39). However, the role of S6K2 in these backgrounds still remains to be established. S6K1 seems to be targeted by both HDAC and sirtuins for de-acetylation, but S6K2 seems entirely dependent on HDAC showing differential regulation of these isoforms. The involvement of HDACs in this process may provide a further link between these kinases and the transcriptional machinery (see Regulation of Transcription). Moreover, the stabilization of S6K2 by acetylation together with the pro-survival and drug resistance phenotypes associated with overexpression of this kinase (see Control of Cell Survival) may hinder the efficiency of HDAC inhibitors in the clinic.
Sub-Cellular Localization of S6K2
As mentioned above, S6K2 mainly resides in the nucleus of resting cells. Closer examination reveals that it is the long form of S6K2 that is predominantly nuclear by virtue of its two NLSs. In contrast, the short form of S6K2 shuttles between the nucleus and the cytoplasm in response to growth factor signaling. In addition to its diffuse nuclear localization, a proportion of S6K2, but not S6K1, has been shown to co-localize with CTR453 and γ-tubulin at the level of the centrosome. This localization, demonstrated in multiple cell lines using both immunofluorescence and immunoblotting of purified centrosome (40), was stable throughout the cell cycle. Finally, cytoplasmic S6K2 has been shown to have a speckled distribution (40), although the structural components of these speckles remains undetermined.
Tissue Expression of S6K2 in Health and Disease
Physiological Expression of S6K2
S6K2 is expressed at various levels in different mouse and human tissues, and its expression levels often inversely correlate with those of S6K1 (41). In Humans, S6K2 expression was found in all tissues with the exception of the neuropil, the peripheral nervous system, and adipocytes. However, expression levels between organs vary considerably, with highest levels found in the gastrointestinal tract, the central nervous system and the lung. In contrast, most mesenchymal cells stain weakly for S6K2. Although S6K2 is detected in normal tissues, its expression levels are often very low compared to those found in corresponding tumor samples [(42) and see S6K2 Protein Levels in Cancer and Normal Corresponding Tissues].
Expression of S6K2 in Cancer and its Significance
S6K2 protein levels in cancer and normal corresponding tissues
S6K2 has been shown to be expressed in the overwhelming majority (88%) of cancer samples investigated and the level for this kinase compared between several cancer types and corresponding normal tissue. These studies demonstrated that normal tissue usually express low levels of this kinase as compared to those found in tumor samples (42–45) and that overexpression of S6K2 was more common than that of S6K1 (e.g., 80 versus 25% in breast and 18 versus 8% for endometrial cancer). In addition to changes in the levels of expression of S6K2, investigators also identified changes in the sub-cellular localization of this kinase between normal and malignant tissues. For instance, Filonenko et al. demonstrated that nuclear accumulation of S6K2 was a distinguishing feature of breast cancer cell in situ, whereas this kinase was only found in the cytoplasm of normal breast cells (43). Moreover, presence of S6K2 in the nucleus positively correlated with proliferating cell nuclear antigen (PCNA) and Ki-67 staining in breast cancer tissues, demonstrating a link between the presence of S6K2 in this compartment and cell proliferation (46). It is noteworthy that no such correlation existed in the case of S6K1. Interestingly, nuclear localization of S6K2 was further increased in cells localized at the periphery of the tumor where tumor cells are in contact with healthy tissue. Hence, it is tempting to speculate that the nuclear activity of S6K2 is somehow promoting tumorigenesis. The role of S6K2 in tumorigenesis is further suggested by the fact that expression levels of S6K2 in various tissues seem to influence the role of S6K1 in mediating PTEN haplo-insufficiency-driven tumorigenicity. Indeed, S6K1 downregulation impaired tumor development downstream of mTORC1 hyperactivation in Pten± mice only in tissues where S6K2 expression levels were low (41). Furthermore, in endometrial cancer, increased nuclear localization of S6K2 correlated with tumor grade (44), while in lung cancer, increased expression of S6K2 correlated with drug resistance (42). This would further suggest that S6K2 expression is linked to cancer progression.
Interestingly, phosphorylation of the ribosomal S6 protein was shown not to correlate with expression levels of either S6K1 or S6K2 in endometrial and breast cancer (43, 44). This lack of correlation is in contradiction with the results obtained from animal models showing that S6K2 knockout mice, unlike their S6K1 counterpart, showed a dramatic reduction in the cellular levels of S6 phosphorylation (47). Hence, the lack of correlation found in tissue samples may be an artifact generated by the modalities of tissue processing or the saturation of these phosphorylation events beyond a certain threshold of S6K expression. However, conclusions on this issue are further complicated by results obtained in knock-in mice where the five phosphorylation sites in S6 are replaced by alanine residues. Indeed, these animals show a phenotypic overlap with that of S6K1−/− mice including a cell growth defect associated with reduction in cell size (48). This is somehow surprising considering the lack of impact on S6 phosphorylation of knocking out S6K1 and the previously mentioned privileged link between S6K2 and S6 phosphorylation in mice (47), a finding that our lab corroborated in human cell lines using an siRNA approach (42). However, some reports suggest that reliance of S6 phosphorylation on one or the other S6K isoform may depend on the nature of the mitogen stimulation (49). Also, these discrepancies may be explained by date failure to take into account the sub-cellular localizations of S6K1, S6K2, and S6, variables that may impact considerably on the overall phosphorylation of the latter protein. The change in sub-cellular distribution of S6K2 observed between tumor and healthy tissues would support this hypothesis (43, 44). Finally, it may be postulated that increased expression of S6K1 and S6K2 does not correlate with increased activity of these kinases.
Amplification of S6K2 in cancer
Amplification of the chromosomal region 11q13 where the S6K2 resides is found in 15–20% of breast cancers samples studied, an event implicated in resistance to endocrine therapy (50). Amplification of S6K2 among these samples correlated with increased mRNA levels for this kinase, ER positive status and worse prognosis (45). However, S6K2 copy number gain and nuclear localization of the protein was related to an improved benefit from tamoxifen among patients with ER+/PgR+ tumors, while in the ER+/PgR− subgroup, nuclear S6K2 rather indicated decreased tamoxifen responsiveness. The presence of the S6K2 amplicon correlated significantly with amplification of the 8p12 region, containing the FGFR1, PPAPDC1B, and 4EBP1 genes (50). This latter amplification was associated with increased mRNA levels for all three genes. Of the genes present in the proximal 11q13 region, S6K2 expression alone correlated with that of FGFR1, PPAPDC1B, and 4EBP1. Using univariate analysis, it was found that 8p12 gain/amplification was significantly associated with increased risk of distant recurrence among patients with 11q13 positive tumors. This analysis could be further refined to demonstrate that high level of FGFR1 and 4EBP1 mRNA expression alone predicted a worse outcome in this patient group. Although co-expression of FGFR1 and S6K2 would be logical in view of the link between FGF2-mediated survival and downstream S6K2 activity (42), an associated increased expression of 4EBP1, a reported tumor suppressor, may seem surprising. However, phosphorylated 4EBP1 has been suggested to stimulate mTORC1 activity (51), a process that would in turn increase S6K2 activation. In addition to the case of breast cancer, S6K2, but not S6K1, was found amplified in about 5% of gastric carcinoma patient samples (52). This amplification was associated with a significance decrease in the overall survival of patients with advanced disease.
S6K2 variants and disease
One of the neuropathological hallmarks in Alzheimer’s disease (AD) is the neurofibrillary tangles formed by hyperphosphorylated microtubule-associated Tau protein. Vazquez-Higuera et al. investigated genetic variations in a set of 20 candidates kinases involved in tau phosphorylation at sites correlating with AD (53). They reported that the distribution of the minor allele frequencies for these kinases did not differ significantly between sufferers and control groups, except for S6K2 where variations in intron 2 was increased in patients (50%) versus controls (39%). In addition to correlating with increased risks of developing the disease, this minor allele was also associated with late onset of AD. Genetic variations in the S6K2 gene have also been linked to the risk of developing colon cancer where they are found in tumors with SNPs in PIK3CA, CIMP positivity, and mutated KRAS2 (54). While CIMP positivity in colon cancer generally correlates with poor tumor differentiation and patient prognosis (55, 56), it was also shown to independently predict the survival benefit from 5-FU chemotherapy (57, 58). Hence, the polymorphism in S6K2, and its pathway associations, may have prognostic value and therapeutic significance. However, the true impact of these SNPs to S6K2 biological activity is yet to be experimentally confirmed.
Biological Functions of S6K2
Mouse Models of S6K2 Reveal Little of its Biological Functions
Knockout mice for S6K1 and S6K2, in isolation and combination, have been bred (47). Single-knockout animals were viable and born at the expected Mendelian ratio. S6K1−/− mice were significantly smaller than their wild-type counterpart while S6K2−/− animals tended to be slightly larger. The latter change is thought to result from compensatory mechanisms whereby S6K2 knockout leads to increased S6K1 activity. This possibility is independently supported by cell line-based experiments in which RNAi-mediated silencing or inhibition of S6K2 leads to increased baseline S6K1 activity (13, 42). Taken together, these data provide confirmation for the proposed role of S6K1 in the control of cell size in mammalian cell lines (59). Conversely, S6K1 knockout mice presented with increased S6K2 mRNA levels in all organs tested, a fact that may explain the observed physiological inverse correlation between the tissue expression for S6K1 and S6K2 (see Physiological Expression of S6K2). In contrast to single-knockout animals, mice lacking both S6K1 and S6K2 suffered from perinatal lethality. This was not due to defects in cell cycle progression or 5′-TOP mRNAs translation in these animals. Also, analysis of embryos at 18.5 days of gestation indicated normal Mendelian distribution. Instead, one third of the double-knockout animals were born dead and the majority of those born alive developed signs of cyanosis leading to death shortly after birth. The minority of mice surviving past the first few days following delivery succeeded in reaching adulthood with similar growth rates as the S6K1−/− animals and were fertile. Histopathological analysis performed to understand the reason for perinatal lethality amongst the double-knockout litters revealed no gross anatomical abnormalities. However non-viable animals showed hyperemic internal organs, occasional dilated heart chambers as well as several hemorrhagic sites. In short, none of the phenotypes observed in these animal models could be attributed to some distinct role of S6K2 and further biochemical analysis was required before biological functions of this kinase started to transpire.
Regulation of Transcription
Genome-wide microarray experiments revealed that S6K1 and 2 regulate the general transcriptional program. Indeed, 456 mRNAs were downregulated in the whole-cell extracts from starved S6K1/S6K2 double-knockout mice livers as compared to that of wild-type controls following re-feeding (60). Specifically, S6K2, but not S6K1, has been shown to directly interact in a mitogen-inducible fashion with the general transcription factor Yin Yang 1(YY1), an association that required the C-terminal region of S6K2 (61). YY1 has been involved in a wide range of biological processes through the recruitment of general components of the transcriptional machinery such as RNA polymerase II, ATF and SP1 as well as various transcriptional co-activators and co-repressors including DNA methyltransferases (DNMTs), histone acetyl transferases (p300, CBP, PCAF), histone deacetylases (HDAC 4), protein arginine methyltransferases (PRMTs), histone-lysine N-methyltransferase (Ezh1/2), Sumo-conjugating enzyme (Ubc9), and ubiquitin ligases (Mdm2). The function of YY1 is regulated by post-translational modifications including phosphorylation, sumoylation, acetylation, and ubiquitination so it is possible that interaction with S6K2 results in the phosphorylation and regulation of transcriptional activity of this protein. S6K2, along with S6K1, has also been involved in the transcription of ribosomal proteins (60). Indeed, analysis of the ribosome biogenesis transcriptional program after feeding in liver cells from S6K1−/−/S6K2−/− mice showed that over 75 factors involved in ribosome biogenesis were controlled by S6Ks. Importantly, these changes were also observed in knock-in mice for a non-phosphorylatable mutant of rpS6. However, this was not associated with changes in the recruitment of RNAs into polysomes, revealing a role for S6Ks in the regulation of transcription that is dissociated from the translational program. Interestingly, S6K1 and 2 were functionally redundant in this biological process as overexpression of either isoform into the double-knockout cells rescued the phenotype.
The regulation of transcription by S6K2 also plays an important role in immune cells differentiation. Indeed, the import into the nucleus of RORγ, a critical transcription factor for the differentiation of a sub-class of IL-17-secreting CD4+ T-helper lymphocytes, is dependent on the binding of this protein to S6K2 (Figure 2) (62). This occurs because RORγ, a protein that lacks an NLS, uses S6K2 to piggyback its ways into the nucleus. The interaction of S6K2 with RORγ was resistant to rapamycin treatment. However, the import of RORγ into the nucleus was rapamycin-sensitive, suggesting a role for mTORC1 in S6K2-mediated nuclear import of RORγ.
Regulation of Protein Translation
S6K2, like S6K1, appear not to be involved in the general protein translation program. Indeed, despite the impairment in S6 phosphorylation found in cells from S6K1/2 double-knockout mice, translation of 5′-TOP mRNAs were still promoted by mitogen stimulation (47). This and other data (63, 64) indicated that S6 phosphorylation is dispensable for this process., Furthermore, these results demonstrated that other kinases lying downstream of the MAPK pathway substituted for S6K1 and S6K2 in phosphorylating two serine sites (235 and 236) on S6 in response to mitogen stimuli. This role has since been attributed to members of the RSK family. However, unlike RSKs, S6Ks are capable of catalyzing the ordered phosphorylation of the five sites present in the C-terminus of the S6 protein (S236, S235, S240, S244, S247) (65), although the physiological significance of these events still remains unclear (66). While it is clear that S6K2 does not play a major role in the cap-dependent translation of housekeeping proteins, this kinase is involved in the cap-independent translation of selective mRNAs (Figure 2). These, including the Bcl-XL, MCL1, and XIAP mRNAs, may be selected for regulation by S6K2 through the presence in their 5′UTR of an internal ribosome entry site (IRES) (67, 68), the activity of which S6K2 controls through the phosphorylation of IRES transactivating factors (ITAFs) such as PDCD4 (69) (see Control of Cell Survival).
Cell Cycle Regulation
S6K2 has been suggested to play a role in mitosis, as a pool of this kinase localizes at the centrosome and S6K2 activity peaks at the G2 and M phases of the cell cycle (70). However, it is worth noting that neither MEFs from S6K1/S6K2 double-knockout mice nor their corresponding embryonic stem cells show significant defects in cell proliferation or cell cycle distribution (47). Hence, the role of S6K2 in the cell cycle may be context dependent and limited to situations where mitogenic stimulus is applied or compensatory mechanisms are not available. Indeed, in IL3-dependent immortalized murine bone marrow-derived pro-B-cells and primary mast cells, IL3 stimulation activated S6K2, which contributed to the mitogenic effect of this growth factor (71). In these cells, S6K2 activity was able to shorten the G1 phase of the cell cycle, enabling cells to enter the S-phase at an increased rate. However, S6K2 activity alone was not able to substitute the need for IL3, as cells expressing a kinase-active S6K2 in the absence of IL3 failed to proliferate. This suggests that S6K2 is only playing a facilitating role in this process, a finding that may help explain the non-essential role of this kinase for cell cycle progression in animal models. It is worth noting that our recent collaborative research has highlighted specific S6K2 partners that mediate the effect of this kinase on the cell cycle. We showed that S6K2 binds several heterogenous ribonuclear proteins (hnRNPs) in a mitogen-inducible manner. One such RNP, hnRNPF was required for cell proliferation driven by S6K2 (72). Indeed, silencing of hnRNPF thwarted the proliferative effects of S6K2 while overexpression of this RNP increased cell proliferation in a rapamycin-sensitive manner. Consistent with the latter result, mitogen stimulation led to the recruitment of preformed S6K2-hnRNPF complexes to mTORC1.
The contradictory findings published on the role of S6K2 in the regulation of the cell cycle may also be explained by the extensive rewiring of signaling pathways occurring during tumorigenesis together with accompanying overexpression of S6K2. These may significantly change the contribution of this kinase to the promotion of mitosis, a possibility supported by the correlation between Ki-67 staining, a marker for cell proliferation, and S6K2 overexpression found in tumor samples (44). In addition, the increased nuclear localization of S6K2 in cancer (43, 44) may also impact on its pro-mitotic function. A possible increased involvement of S6K2 in the regulation of cell cycle in malignant tumors as compared to normal tissue could provide a therapeutic window for the targeting of this kinase in the treatment of cancer patients.
Control of Cell Survival
The role of S6K2 in the regulation of apoptotic cell death was first demonstrated by research from our lab (42). The serum levels for basic fibroblast growth factor (FGF2) are often elevated in patients with a variety of malignancies and are a poor prognostic factor on uni- and multi-variate analysis (73–78). We found that treatment of lung cancer cells with FGF2, used at concentrations commonly found in the serum of carcinoma patients, promoted cell survival, and drug resistance through translational up-regulation of anti-apoptotic proteins such as Bcl-XL and XIAP (79, 80). The mRNAs for these proteins are characterized by the presence in their 5′UTR of an IRES, a three-dimensional structure that represses the efficiency of their translation. Hence, increased expression of these proteins requires the unwinding of these structures and the recruitment of ITAFs that de-represses their translation. Investigation into the signaling involved in this response revealed that S6K2, but not S6K1, was required for the increased translation of these anti-apoptotic proteins in the absence of de novo mRNA synthesis (42, 80). In support of this, silencing of S6K2 using siRNAs prevented FGF2-induced drug resistance as well as up-regulation of Bcl-XL and XIAP. Moreover, in un-stimulated cells, S6K2 downregulation was accompanied by a decrease in the steady-state levels of both anti-apoptotic proteins, suggesting that this kinase is not only involved in promoting their production downstream of pro-survival signaling but also participates to their baseline translation. In contrast, silencing S6K1 had no impact on either protein whether in the presence or absence of mitogen stimulation. Conversely, overexpression of a kinase-active mutant for S6K2, but not S6K1, increased the translation of Bcl-XL and XIAP, promoted baseline cell survival and induced drug resistance in the absence of FGF2 stimulation (42). The anti-apoptotic function of S6K2 was dependent on the FGF2-inducible formation of a multi-protein complex comprising S6K2, BRAF, and PKCε (Figure 2). Indeed, disruption of this complex through the silencing of BRAF or PKCε prevented the pro-survival activity of S6K2. The composition of this multi-protein interaction was selective as it did not include other PKC or RAF isoforms. Similarly, S6K1 was unable to form a complex with these S6K2 partners. Tandem affinity purification using S6K2 as bait in the presence and absence of FGF2 stimulation in HEK93 cells enabled the identification of downstream mediators that regulate the translation of S6K2’s anti-apoptotic targets. One such interactor, the tumor suppressor programed cell death 4 (PDCD4) that normally binds to the IRES of Bcl-XL and XIAP to repress their translation, is phosphorylated by S6K2 (69). This post-translational modification leads to the degradation of PDCD4 and subsequent derepression of Bcl-XL and XIAP translation. It is also noteworthy that hnRNPF and hnRNPH, two ribonucleoproteins previously found to regulate the differential splicing of BCL-X into the anti-apoptotic protein BCL-XL or the pro-apoptotic proteins Bcl-XS (81) were found to associate with S6K2 in a mitogen-inducible manner (72). Hence, in addition to being able to regulate the translation of this protein, S6K2 may also be able to promote the preferential splicing of BCL-X toward BcL-XL.
Further work in U2OS osteosarcoma cells demonstrated that the Janus kinase TYK2 participated to the initially identified S6K2/BRAF/PKCε complex downstream of FGF2 signaling (82). In this cell system, TYK2 was required for the induction of the anti-apoptotic proteins BCL2 and MCL1 and for the promotion of cell survival in response to this growth factor. Whether this finding extends to lung, or other, cancer cells in which FGF2 signaling is relevant to the development of chemoresistance is at this point unknown. However, it suggests that inhibition of TYK2 may represent a new therapeutic strategy to target drug resistance downstream of FGF2 signaling.
The role of S6K2 in the control of cell survival may extend to neurodegenerative disorders. Frontotemporal dementia (FTD) is a form of pre-senile dementia associated with focal atrophy of the frontal or temporal lobes accompanied by deficits in cognition, behavior, and language. Mutations in progranulin (PGRN), a protein involved in cell growth and survival (83–85), are a common cause of FTD (86). Human neurons obtained from FTD patients with mutant PGRN were shown to have reduced cell viability that correlated with a downregulation of S6K2 transcription (87). All these changes were rescued by expression of wild-type PGRN, directly linking this factor with expression of S6K2. This link may have far wider relevance as serum levels of PGRN are a clinically significant predictive marker for recurrence in patients with HR-positive breast cancer during adjuvant tamoxifen therapy (88). Also, high PGRN expression levels correlate with an aggressive phenotype in cancer cell lines and clinical specimens from gliomas, breast, ovarian, and renal cancers (83). However, the link between PGRN and S6K2 in these settings is yet to be established.
S6K2 and Cognition
Behavioral analysis of S6K knockout mice highlighted non-overlapping cognitive functions for S6K1 and 2. S6K1 and S6K2-deficient mice were tested for contextual fear memory, conditioned taste aversion, Morris water maze acquisition, exploratory behavior, and long-term potentiation (89). Deficit in individual S6K isoforms resulted in distinct patterns of behavioral modifications with S6K1 being associated with the most pronounced phenotype. While both isoforms participated to contextual fear memory, conditioned taste aversion, and early-phase long-term potentiation, S6K2 deficit impacted particularly on long-term contextual fear memory and reduced latent inhibition of conditioned taste aversion. However, S6K2−/− mice displayed normal spatial learning in the Morris water maze.
S6K2 as a Therapeutic Target for Cancer
Important Considerations in the Designs of S6K2 Targeting Therapy
Since knockout mice for S6K2 have shown that this kinase was dispensable for normal development and homeostasis, this kinase may represent an excellent therapeutic target for cancer. However, several lines of evidence suggest that S6K2 targeting should be selective and not impinge on S6K1 activity. Indeed, as indicated above, unlike S6K2 single-knockout animals, S6K1−/−/S6K2−/− mice displayed perinatal lethality (47), suggesting that acute inhibition of both isoforms simultaneously in the absence of compensatory mechanisms may be deleterious to normal homeostasis. This is confirmed by work done in Drosophila where disruption of the unique S6K gene, dS6K, results in the death of the majority of flies at the larval stage or early pupation (90). Moreover, the S6Ks have been involved in negative feedback loops that regulate the PI3K and mTOR pathways. First, it was found that S6K1 and S6K2 phosphorylate IRS-1 on serine 302, a site adjacent to its PTB domain. This phosphorylation event inhibits the binding of IRS-1 to the insulin receptor, preventing further stimulation of the PI3K pathway by insulin (91, 92). Conversely, silencing of S6K1 using siRNAs was shown to decrease IRS-1 phosphorylation on several serine residues leading to an increase in PI3K/AKT signaling (93). In addition to targeting IRS-1, it has recently been revealed that S6K1, but not S6K2, modulates the activation of AKT by phosphorylating the mTORC2 complex component RICTOR on T1135 (49, 94, 95). This phosphorylation event does not seem to directly modulate the kinase activity of the mTORC2 complex. However, expression of a phospho-deficient mutant version of RICTOR promoted phosphorylation of AKT on S473, a site associated with activation of this kinase (49). Hence, considering the well-documented function of AKT in tumorigenesis (96) and the lethality of S6K double-knockout animals, a therapeutic strategy targeting both S6K isoforms in cancer patient may not be advisable. This hypothesis is further supported by results obtained using the pan-S6K inhibitor, LYS6K2, which increases basal and mitogen-induced AKT phosphorylation in treated cell lines (97). Conversely, the use of rapamycin analogs to inhibit the mTOR pathway, an approach that has been the subject of numerous cancer clinical trials (98), would not be appropriate in targeting the biological effects of S6K2 as some of the functions of this kinase are resistant to this inhibitor (13, 42, 79). Hence, it would be highly desirable to develop S6K2-specific compounds. However, at present, no drug discovery project has attempted to specifically target S6K2.
Small-Molecule ATP Competitors
While S6K kinase inhibitors do exist, these are either pan-S6K compounds that also target other AGC kinases [e.g., Ref. (99, 100)] or compounds that show relative selectivity for S6K1 (101, 102). It was initially thought that the high degree of identity between the kinase domains of S6K1 and S6K2 would prevent the development of S6K2-selective kinase inhibitors. However, the existence of “selective” S6K1 inhibitors (101, 102) together with data acquired through 3-D modeling suggest that the development of such compounds would be possible. Indeed, comparison of the crystal structure of the kinase domain of S6K1 bound to Staurosporine (103) with the 3-D model of the same region in S6K2 reveals that the two kinases may differ in their contact with this inhibitor. This divergence occurs at the level of cysteine 150, a residue within the hinge region of S6K2 and may be exploitable to tweak selectivity of pan-S6K compounds toward this isoform. In support for this idea, the modulation of interaction of chemical compounds with the hinge region has previously been exploited to introduce selectivity among kinase inhibitors (104). Nevertheless, the high level of homology between the kinase domains of AGC kinases family members will always render the broader selectivity of these compounds hard to secure, especially in vivo where their intracellular concentration cannot be easily controlled. Hence, other approaches for the selective inhibition of S6K2 should also be considered.
Protein–Protein Interaction Inhibitors
We propose that the development of protein–protein interaction inhibitors would be a more appropriate strategy to specifically target S6K2. Indeed, drug discovery efforts into protein–protein interaction inhibitors have multiplied in the last few years [e.g., Ref. (105–109) to only cite a few] and some have already yielded compounds that entered the clinic (105). The heightened interest in this approach has been fueled by the relative lack of success of small-molecule kinase inhibitors in the clinic, together with the development of novel bioinformatics tools to predict disruption of protein–protein interactions (110, 111). We and others have performed co-purification of S6K2 with its interacting partners downstream of mitogen stimulation in various cell lines [(69, 72) and unpublished], an effort that resulted in the identification of S6K2-specific interactors that regulate the biological functions of this kinase (42, 69, 72). For instance, interaction of S6K2 with BRAF and PKCε was shown to regulate the anti-apoptotic function of this kinase while that of S6K2 and hnRNPF modulated its effect on the cell cycle. Hence, one could envision drug discovery efforts aimed at targeting these identified interactions.
Allosteric S6K2 Inhibitors
Two more characteristics of S6K2 may be potentially exploitable for the development of selective compounds. The first is its C-terminal domain which differs significantly from the same region in S6K1 and is predicted through 3-D modeling to be fairly unstructured and exposed. This region could be used to fish out interacting compounds that may, through binding, alter the 3-D conformation of S6K2. As this region also contains the NLS for S6K2, interacting compounds may also interfere with its sub-cellular localization. The second S6K2 characteristic is its higher reliance for activation on the MEK/ERK-mediated derepression from the pseudo-substrate domain. Compounds that would bind to the kinase extension domain or hinder access to the proline-directed phosphorylation sites may prevent S6K2 from exposing the serine/threonine sites responsible for its activation to upstream kinases.
Conclusion
It has now been 15 years since the cloning of S6K2. However, many aspects of the regulation and biological functions of this enzyme are still a mystery. Indeed, the high degree of homology between S6K1 and S6K2 led us to bias most of our research toward the first of these two isoforms, as these were thought to have identical roles. Instead, it is now becoming clear that these enzymes have distinct biological functions mediated by their distinct repertoires of substrates and interactors. Our limited knowledge of S6K2 suggests that this isoform may play a particularly important role in the pathobiology of cancer and targeting this isoform could provide a therapeutic benefit in patients. However, this will most probably require the development of S6K2 isoform-selective compounds that exploit its known specific protein–protein interactions and downstream substrates that mediate its functions. The time has come to embrace S6K2, recognize the biological diversity that this enzyme introduces into the S6 kinase pathway and exploit this information for new therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Michael J. Seckl is supported by the Department of Health and Cancer Research UK funded Imperial College NIHR/Biomedical Research Center and Imperial Experimental Cancer Medicine Center. Olivier E. Pardo and Michael J. Seckl are funded by the European Union FP7 as part of the LungTarget consortium.
References
1. Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol (2008) 9:747–58. doi:10.1038/nrm2509
2. Jeno P, Ballou LM, Novak-Hofer I, Thomas G. Identification and characterization of a mitogen-activated S6 kinase. Proc Natl Acad Sci U S A (1988) 85:406–10.
3. Nemenoff RA, Price DJ, Mendelsohn MJ, Carter EA, Avruch J. An S6 kinase activated during liver regeneration is related to the insulin-stimulated S6 kinase in H4 hepatoma cells. J Biol Chem (1988) 263:19455–60.
4. Gout I, Minami T, Hara K, Tsujishita Y, Filonenko V, Waterfield MD, et al. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase beta containing a proline-rich region. J Biol Chem (1998) 273:30061–4.
5. Saitoh M, Ten Dijke P, Miyazono K, Ichijo H. Cloning and characterization of p70(S6K beta) defines a novel family of p70 S6 kinases. Biochem Biophys Res Commun (1998) 253:470–6. doi:10.1006/bbrc.1998.9784
6. Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene (1999) 18:5108–14.
7. Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol (2011) 43:47–59. doi:10.1016/j.biocel.2010.09.018
8. Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J (2012) 441:1–21. doi:10.1042/BJ20110892
9. Savinska L, Skorokhod O, Klipa O, Gout I, Filonenko V. Development of monoclonal antibodies specific to ribosomal protein S6 kinase 2. Hybridoma (Larchmt) (2012) 31:289–94. doi:10.1089/hyb.2012.0032
10. Koh H, Jee K, Lee B, Kim J, Kim D, Yun YH, et al. Cloning and characterization of a nuclear S6 kinase, S6 kinase-related kinase (SRK); a novel nuclear target of Akt. Oncogene (1999) 18:5115–9.
11. Karni R, De Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol (2007) 14:185–93.
12. Ben-Hur V, Denichenko P, Siegfried Z, Maimon A, Krainer A, Davidson B, et al. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep (2013) 3:103–15. doi:10.1016/j.celrep.2012.11.020
13. Pardo OE, Arcaro A, Salerno G, Tetley TD, Valovka T, Gout I, et al. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene (2001) 20:7658–67.
14. Wang L, Gout I, Proud CG. Cross-talk between the ERK and p70 S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in cardiomyocytes. J Biol Chem (2001) 276:32670–7. doi:10.1074/jbc.M102776200
15. Martin KA, Schalm SS, Richardson C, Romanelli A, Keon KL, Blenis J. Regulation of ribosomal S6 kinase 2 by effectors of the phosphoinositide 3-kinase pathway. J Biol Chem (2001) 276:7884–91.
16. Martin KA, Schalm SS, Romanelli A, Keon KL, Blenis J. Ribosomal S6 kinase 2 inhibition by a potent C-terminal repressor domain is relieved by mitogen-activated protein-extracellular signal-regulated kinase kinase-regulated phosphorylation. J Biol Chem (2001) 276:7892–8.
17. Phin S, Kupferwasser D, Lam J, Lee-Fruman KK. Mutational analysis of ribosomal S6 kinase 2 shows differential regulation of its kinase activity from that of ribosomal S6 kinase 1. Biochem J (2003) 373:583–91.
18. Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol (2002) 12:632–9. doi:10.1016/S0960-9822(02)00762-5
19. Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem (2003) 278:15461–4.
20. Park IH, Bachmann R, Shirazi H, Chen J. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J Biol Chem (2002) 277:31423–9.
21. Talvas J, Obled A, Fafournoux P, Mordier S. Regulation of protein synthesis by leucine starvation involves distinct mechanisms in mouse C2C12 myoblasts and myotubes. J Nutr (2006) 136:1466–71.
22. Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J (1998) 17:6649–59. doi:10.1093/emboj/17.22.6649
23. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem (2009) 284:8023–32. doi:10.1074/jbc.M900301200
24. Moorman NJ, Shenk T. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J Virol (2010) 84:5260–9. doi:10.1128/JVI.02733-09
25. Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol (2009) 7::e38. doi:10.1371/journal.pbio.1000038
26. Valovka T, Verdier F, Cramer R, Zhyvoloup A, Fenton T, Rebholz H, et al. Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol Cell Biol (2003) 23:852–63. doi:10.1128/MCB.23.3.852-863.2003
27. Rebholz H, Panasyuk G, Fenton T, Nemazanyy I, Valovka T, Flajolet M, et al. Receptor association and tyrosine phosphorylation of S6 kinases. FEBS J (2006) 273:2023–36.
28. Wang ML, Panasyuk G, Gwalter J, Nemazanyy I, Fenton T, Filonenko V, et al. Regulation of ribosomal protein S6 kinases by ubiquitination. Biochem Biophys Res Commun (2008) 369:382–7. doi:10.1016/j.bbrc.2008.02.032
29. Gwalter J, Wang ML, Gout I. The ubiquitination of ribosomal S6 kinases is independent from the mitogen-induced phosphorylation/activation of the kinase. Int J Biochem Cell Biol (2009) 41:828–33.
30. Fenton TR, Gwalter J, Ericsson J, Gout IT. Histone acetyltransferases interact with and acetylate p70 ribosomal S6 kinases in vitro and in vivo. Int J Biochem Cell Biol (2010) 42:359–66. doi:10.1016/j.biocel.2009.11.022
31. Panasyuk G, Nemazanyy I, Filonenko V, Gout I. Ribosomal protein S6 kinase 1 interacts with and is ubiquitinated by ubiquitin ligase ROC1. Biochem Biophys Res Commun (2008) 369:339–43. doi:10.1016/j.bbrc.2008.02.016
32. Xiao XS, Cai MY, Chen JW, Guan XY, Kung HF, Zeng YX, et al. High expression of p300 in human breast cancer correlates with tumor recurrence and predicts adverse prognosis. Chin J Cancer Res (2011) 23:201–7. doi:10.1007/s11670-011-0201-5
33. Hou X, Li Y, Luo RZ, Fu JH, He JH, Zhang LJ, et al. High expression of the transcriptional co-activator p300 predicts poor survival in resectable non-small cell lung cancers. Eur J Surg Oncol (2012) 38:523–30.
34. Liao ZW, Zhou TC, Tan XJ, Song XL, Liu Y, Shi XY, et al. High expression of p300 is linked to aggressive features and poor prognosis of nasopharyngeal carcinoma. J Transl Med (2012) 10::110. doi:10.1186/1479-5876-10-110
35. Li Y, Yang HX, Luo RZ, Zhang Y, Li M, Wang X, et al. High expression of p300 has an unfavorable impact on survival in resectable esophageal squamous cell carcinoma. Ann Thorac Surg (2011) 91:1531–8. doi:10.1016/j.athoracsur.2010.12.012
36. Yokomizo C, Yamaguchi K, Itoh Y, Nishimura T, Umemura A, Minami M, et al. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer Lett (2011) 310:140–7. doi:10.1016/j.canlet.2011.06.030
37. Hirano G, Izumi H, Kidani A, Yasuniwa Y, Han B, Kusaba H, et al. Enhanced expression of PCAF endows apoptosis resistance in cisplatin-resistant cells. Mol Cancer Res (2010) 8:864–72. doi:10.1158/1541-7786.MCR-09-0458
38. Shiota M, Yokomizo A, Tada Y, Uchiumi T, Inokuchi J, Tatsugami K, et al. P300/CBP-associated factor regulates Y-box binding protein-1 expression and promotes cancer cell growth, cancer invasion and drug resistance. Cancer Sci (2010) 101:1797–806. doi:10.1111/j.1349-7006.2010.01598.x
39. Toth M, Boros IM, Balint E. Elevated level of lysine 9-acetylated histone H3 at the MDR1 promoter in multidrug-resistant cells. Cancer Sci (2012) 103:659–69. doi:10.1111/j.1349-7006.2012.02215.x
40. Rossi R, Pester JM, Mcdowell M, Soza S, Montecucco A, Lee-Fruman KK. Identification of S6K2 as a centrosome-located kinase. FEBS Lett (2007) 581:4058–64.
41. Nardella C, Lunardi A, Fedele G, Clohessy JG, Alimonti A, Kozma SC, et al. Differential expression of S6K2 dictates tissue-specific requirement for S6K1 in mediating aberrant mTORC1 signaling and tumorigenesis. Cancer Res (2011) 71:3669–75.
42. Pardo OE, Wellbrock C, Khanzada UK, Aubert M, Arozarena I, Davidson S, et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J (2006) 25:3078–88.
43. Filonenko VV, Tytarenko R, Azatjan SK, Savinska LO, Gaydar YA, Gout IT, et al. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp Oncol (2004) 26:294–9.
44. Lyzogubov VV, Lytvyn DI, Dudchenko TM, Lubchenko NV, Pogrybniy PV, Nespryadko SV, et al. Immunohistochemical analysis of S6K1 and S6K2 expression in endometrial adenocarcinomas. Exp Oncol (2004) 26:287–93.
45. Perez-Tenorio G, Karlsson E, Waltersson MA, Olsson B, Holmlund B, Nordenskjold B, et al. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res Treat (2011) 128:713–23. doi:10.1007/s10549-010-1058-x
46. Lyzogubov V, Khozhaenko Y, Usenko V, Antonjuk S, Ovcharenko G, Tikhonkova I, et al. Immunohistochemical analysis of Ki-67, PCNA and S6K1/2 expression in human breast cancer. Exp Oncol (2005) 27:141–4.
47. Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol (2004) 24:3112–24.
48. Ruvinsky I, Katz M, Dreazen A, Gielchinsky Y, Saada A, Freedman N, et al. Mice deficient in ribosomal protein S6 phosphorylation suffer from muscle weakness that reflects a growth defect and energy deficit. PLoS One (2009) 4::e5618. doi:10.1371/journal.pone.0005618
49. Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol (2009) 29:5657–70. doi:10.1128/MCB.00735-09
50. Karlsson E, Waltersson MA, Bostner J, Perez-Tenorio G, Olsson B, Hallbeck AL, et al. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer (2011) 50:775–87. doi:10.1002/gcc.20900
51. Wang L, Rhodes CJ, Lawrence JC Jr. Activation of mammalian target of rapamycin (mTOR) by insulin is associated with stimulation of 4EBP1 binding to dimeric mTOR complex 1. J Biol Chem (2006) 281:24293–303. doi:10.1074/jbc.M603566200
52. Yoshida S, Matsumoto K, Arao T, Taniguchi H, Goto I, Hanafusa T, et al. Gene amplification of ribosomal protein S6 kinase-1 and -2 in gastric cancer. Anticancer Res (2013) 33:469–75.
53. Vazquez-Higuera JL, Mateo I, Sanchez-Juan P, Rodriguez-Rodriguez E, Pozueta A, Calero M, et al. Genetic variation in the tau kinases pathway may modify the risk and age at onset of Alzheimer’s disease. J Alzheimers Dis (2011) 27:291–7. doi:10.3233/JAD-2011-110794
54. Slattery ML, Lundgreen A, Herrick JS, Wolff RK. Genetic variation in RPS6KA1, RPS6KA2, RPS6KB1, RPS6KB2, and PDK1 and risk of colon or rectal cancer. Mutat Res (2011) 706:13–20. doi:10.1016/j.mrfmmm.2010.10.005
55. Kim JH, Shin SH, Kwon HJ, Cho NY, Kang GH. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch (2009) 455:485–94. doi:10.1007/s00428-009-0857-0
56. Bae JM, Kim MJ, Kim JH, Koh JM, Cho NY, Kim TY, et al. Differential clinicopathological features in microsatellite instability-positive colorectal cancers depending on CIMP status. Virchows Arch (2011) 459:55–63. doi:10.1007/s00428-011-1080-3
57. Van Rijnsoever M, Elsaleh H, Joseph D, Mccaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res (2003) 9:2898–903.
58. Iacopetta B, Kawakami K, Watanabe T. Predicting clinical outcome of 5-fluorouracil-based chemotherapy for colon cancer patients: is the CpG island methylator phenotype the 5-fluorouracil-responsive subgroup? Int J Clin Oncol (2008) 13:498–503. doi:10.1007/s10147-008-0854-3
59. Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev (2002) 16:1472–87. doi:10.1101/gad.995802
60. Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene (2013). [Epub ahead of print]. doi:10.1038/onc.2012.606
61. Ismail HM, Myronova O, Tsuchiya Y, Niewiarowski A, Tsaneva I, Gout I. Identification of the general transcription factor Yin Yang 1 as a novel and specific binding partner for S6 Kinase 2. Cell Signal (2013) 25:1054–63. doi:10.1016/j.cellsig.2013.02.002
62. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep (2012) 1:360–73. doi:10.1016/j.celrep.2012.02.007
63. Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev (2005) 19:2199–211.
64. Patursky-Polischuk I, Stolovich-Rain M, Hausner-Hanochi M, Kasir J, Cybulski N, Avruch J, et al. The TSC-mTOR pathway mediates translational activation of TOP mRNAs by insulin largely in a raptor- or rictor-independent manner. Mol Cell Biol (2009) 29:640–9. doi:10.1128/MCB.00980-08
65. Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J Biol Chem (1991) 266:22770–5.
66. Meyuhas O. Physiological roles of ribosomal protein S6: one of its kind. Int Rev Cell Mol Biol (2008) 268:1–37.
67. Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta (2009) 1789:518–28. doi:10.1016/j.bbagrm.2009.07.004
68. Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle (2011) 10:229–40.
69. Liwak U, Thakor N, Jordan LE, Roy R, Lewis SM, Pardo O, et al. Tumour suppressor PDCD4 represses IRES-mediated translation of anti-apoptotic proteins and is regulated by S6 kinase 2. Mol Cell Biol (2012) 32(10):1818–29. doi:10.1128/MCB.06317-11
70. Boyer D, Quintanilla R, Lee-Fruman KK. Regulation of catalytic activity of S6 kinase 2 during cell cycle. Mol Cell Biochem (2008) 307:59–64. doi:10.1007/s11010-007-9584-5
71. Cruz R, Hedden L, Boyer D, Kharas MG, Fruman DA, Lee-Fruman KK. S6 kinase 2 potentiates interleukin-3-driven cell proliferation. J Leukoc Biol (2005) 78:1378–85. doi:10.1189/jlb.0405225
72. Goh ET, Pardo OE, Michael N, Niewiarowski A, Totty N, Volkova D, et al. Involvement of heterogeneous ribonucleoprotein F in the regulation of cell proliferation via the mammalian target of rapamycin/S6 kinase 2 pathway. J Biol Chem (2010) 285:17065–76. doi:10.1074/jbc.M109.078782
73. Fujimoto K, Ichimori Y, Kakizoe T, Okajima E, Sakamoto H, Sugimura T, et al. Increased serum levels of basic fibroblast growth factor in patients with renal cell carcinoma. Biochem Biophys Res Commun (1991) 180:386–92.
74. Linder C, Linder S, Munck W, Strander H. Independent expression of serum vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) in patients with carcinoma and sarcoma. Anticancer Res (1998) 18:2063–8.
75. Graeven U, Andre N, Achilles E, Zornig C, Schmiegel W. Serum levels of vascular endothelial growth factor and basic fibroblast growth factor in patients with soft-tissue sarcoma. J Cancer Res Clin Oncol (1999) 125:577–81.
76. Salven P, Teerenhovi L, Joensuu H. A high pretreatment serum basic fibroblast growth factor concentration is an independent predictor of poor prognosis in non-Hodgkin’s lymphoma. Blood (1999) 94:3334–9.
77. Joensuu H, Anttonen A, Eriksson M, Makitaro R, Alfthan H, Kinnula V, et al. Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res (2002) 62:5210–7.
78. Ruotsalainen T, Joensuu H, Mattson K, Salven P. High pretreatment serum concentration of basic fibroblast growth factor is a predictor of poor prognosis in small cell lung cancer. Cancer Epidemiol Biomarkers Prev (2002) 11:1492–5.
79. Pardo OE, Arcaro A, Salerno G, Raguz S, Downward J, Seckl MJ. Fibroblast growth factor-2 induces translational regulation of Bcl-XL and Bcl-2 via a MEK-dependent pathway: correlation with resistance to etoposide-induced apoptosis. J Biol Chem (2002) 277:12040–6.
80. Pardo OE, Lesay A, Arcaro A, Lopes R, Ng BL, Warne PH, et al. Fibroblast growth factor 2-mediated translational control of IAPs blocks mitochondrial release of Smac/DIABLO and apoptosis in small cell lung cancer cells. Mol Cell Biol (2003) 23:7600–10.
81. Garneau D, Revil T, Fisette JF, Chabot B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem (2005) 280:22641–50. doi:10.1074/jbc.M501070200
82. Carmo CR, Lyons-Lewis J, Seckl MJ, Costa-Pereira AP. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS One (2011) 6::e19861. doi:10.1371/journal.pone.0019861
83. He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (2003) 81:600–12.
84. Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med (2010) 207:117–28. doi:10.1084/jem.20091568
85. Tapia L, Milnerwood A, Guo A, Mills F, Yoshida E, Vasuta C, et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci (2011) 31:11126–32. doi:10.1523/JNEUROSCI.6244-10.2011
86. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature (2006) 442:916–9. doi:10.1038/nature05016
87. Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, et al. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep (2012) 2:789–98. doi:10.1016/j.celrep.2012.09.007
88. Koo DH, Park CY, Lee ES, Ro J, Oh SW. Progranulin as a prognostic biomarker for breast cancer recurrence in patients who had hormone receptor-positive tumors: a cohort study. PLoS One (2012) 7::e39880. doi:10.1371/journal.pone.0039880
89. Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, et al. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem (2008) 15:29–38. doi:10.1101/lm.661908
90. Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science (1999) 285:2126–9.
91. Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol (2004) 166:213–23. doi:10.1083/jcb.200403069
92. Zick Y. Uncoupling insulin signalling by serine/threonine phosphorylation: a molecular basis for insulin resistance. Biochem Soc Trans (2004) 32:812–6. doi:10.1042/BST0320812
93. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature (2004) 431:200–5.
94. Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol (2010) 30:908–21.
95. Treins C, Warne PH, Magnuson MA, Pende M, Downward J. Rictor is a novel target of p70 S6 kinase-1. Oncogene (2010) 29:1003–16. doi:10.1038/onc.2009.401
96. Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog (2012) 17:69–95. doi:10.1615/CritRevOncog.v17.i1.60
97. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A (2010) 107:3441–6. doi:10.1073/pnas.0914798107
98. Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol (2009) 2::45. doi:10.1186/1756-8722-2-45
99. Grimshaw KM, Hunter LJ, Yap TA, Heaton SP, Walton MI, Woodhead SJ, et al. AT7867 is a potent and oral inhibitor of AKT and p70 S6 kinase that induces pharmacodynamic changes and inhibits human tumor xenograft growth. Mol Cancer Ther (2010) 9:1100–10. doi:10.1158/1535-7163.MCT-09-0986
100. Bussenius J, Anand NK, Blazey CM, Bowles OJ, Bannen LC, Chan DS, et al. Design and evaluation of a series of pyrazolopyrimidines as p70S6K inhibitors. Bioorg Med Chem Lett (2012) 22:2283–6. doi:10.1016/j.bmcl.2012.01.105
101. Bilanges B, Vanhaesebroeck B. A new tool to dissect the function of p70 S6 kinase. Biochem J (2010) 431e1–3. doi:10.1042/BJ20101445
102. Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J (2010) 431:245–55.
103. Sunami T, Byrne N, Diehl RE, Funabashi K, Hall DL, Ikuta M, et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation. J Biol Chem (2010) 285:4587–94. doi:10.1074/jbc.M109.040667
104. Akritopoulou-Zanze I, Hajduk PJ. Kinase-targeted libraries: the design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discov Today (2009) 14:291–7. doi:10.1016/j.drudis.2008.12.002
105. Wendt MD. Discovery of ABT-263, a Bcl-family protein inhibitor: observations on targeting a large protein-protein interaction. Expert Opin Drug Discov (2008) 3:1123–43. doi:10.1517/17460441.3.9.1123
106. Clark RC, Lee SY, Searcey M, Boger DL. The isolation, total synthesis and structure elucidation of chlorofusin, a natural product inhibitor of the p53-mDM2 protein-protein interaction. Nat Prod Rep (2009) 26:465–77. doi:10.1039/b821676b
107. Zhao J, Du Y, Horton JR, Upadhyay AK, Lou B, Bai Y, et al. Discovery and structural characterization of a small molecule 14-3-3 protein-protein interaction inhibitor. Proc Natl Acad Sci U S A (2011) 108:16212–6. doi:10.1073/pnas.1100012108
108. Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg Med Chem Lett (2013) 23:3039–43. doi:10.1016/j.bmcl.2013.03.013
109. Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, et al. PFI-1 – a highly selective protein interaction inhibitor targeting BET bromodomains. Cancer Res (2013) 73(11):3336–46. doi:10.1158/0008-5472.CAN-12-3292
110. Koes DR, Camacho CJ. PocketQuery: protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res (2012) 40:W387–92. doi:10.1093/nar/gks336
Keywords: S6K2, S6 kinase, selectivity, specificity, function, RPS6KB2, cancer
Citation: Pardo OE and Seckl MJ (2013) S6K2: the neglected S6 kinase family member. Front. Oncol. 3:191. doi: 10.3389/fonc.2013.00191
Received: 22 May 2013; Paper pending published: 11 June 2013;
Accepted: 08 July 2013; Published online: 24 July 2013.
Edited by:
Alexandre Arcaro, University of Bern, SwitzerlandReviewed by:
Enrico Vittorio Avvedimento, University Naples Federico II, ItalyShailender Singh Kanwar, University of Michigan, USA
Copyright: © 2013 Pardo and Seckl. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Olivier E. Pardo and Michael J. Seckl, Division of Cancer, Department of Surgery and Cancer, Imperial College, 1st Floor ICTEM Building, Hammersmith Hospital, Du Cane Road, London W12 0NN, UK e-mail: o.pardo@imperial.ac.uk; m.seckl@imperial.ac.uk