 Marko Jakopovic1 Anish Thomas2 Sanjeeve Balasubramaniam2 David Schrump3 Giuseppe Giaccone4 Susan E. Bates2*
Marko Jakopovic1 Anish Thomas2 Sanjeeve Balasubramaniam2 David Schrump3 Giuseppe Giaccone4 Susan E. Bates2*- 1University of Zagreb, School of Medicine, Department for Respiratory Diseases Jordanovac, University Hospital Center Zagreb, Zagreb, Croatia
- 2Medical Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
- 3Surgical Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
- 4Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC, USA
Epigenetic aberrations offer dynamic and reversible targets for cancer therapy; increasingly, alteration via overexpression, mutation, or rearrangement is found in genes that control the epigenome. Such alterations suggest a fundamental role in carcinogenesis. Here, we consider three epigenetic mechanisms: DNA methylation, histone tail modification and non-coding, microRNA regulation. Evidence for each of these in lung cancer origin or progression has been gathered, along with evidence that epigenetic alterations might be useful in early detection. DNA hypermethylation of tumor suppressor promoters has been observed, along with global hypomethylation and hypoacetylation, suggesting an important role for tumor suppressor gene silencing. These features have been linked as prognostic markers with poor outcome in lung cancer. Several lines of evidence have also suggested a role for miRNA in carcinogenesis and in outcome. Cigarette smoke downregulates miR-487b, which targets both RAS and MYC; RAS is also a target of miR-let-7, again downregulated in lung cancer. Together the evidence implicates epigenetic aberration in lung cancer and suggests that targeting these aberrations should be carefully explored. To date, DNA methyltransferase and histone deacetylase inhibitors have had minimal clinical activity. Explanations include the possibility that the agents are not sufficiently potent to invoke epigenetic reversion to a more normal state; that insufficient time elapses in most clinical trials to observe true epigenetic reversion; and that doses often used may provoke off-target effects such as DNA damage that prevent epigenetic reversion. Combinations of epigenetic therapies may address those problems. When epigenetic agents are used in combination with chemotherapy or targeted therapy it is hoped that downstream biological effects will provoke synergistic cytotoxicity. This review evaluates the challenges of exploiting the epigenome in the treatment of lung cancer.
Introduction
Lung cancer is the second most common cancer and leading cause of cancer-related death in both males and females in the United States (1). Traditionally, lung cancer histology allowed it to be divided into two morphologic groups that also demonstrate distinct clinical features: non-small cell lung cancer (NSCLC), which represents around 85% of all cases, and small-cell lung cancer (SCLC). About 80–90% of NSCLCs are directly related to tobacco smoke, while nearly all small cell lung cancers are associated with smoking (2). The vast majority of lung cancers present as advanced and incurable disease at the time of diagnosis (SEER Cancer Statistics Review, 1975–2008); in these patients platinum-based chemotherapy remains a standard treatment for the majority of patients with NSCLC and SCLC, with overall survival (OS) less than 12 months (3, 4). Tailoring chemotherapy according to histologic subset has been shown to improve efficacy in patients with NSCLC (5).
Further progress has been made more recently with improved understanding of the molecular mechanisms involved in oncogenesis (6). These advances enabled the development of drugs that target cancer cell specific gene alterations. These targeted drugs significantly improved response rates and progression-free survival (PFS) in patients with specific genetic alterations (7–9). Despite these advances, the prognosis of patients with both advanced NSCLC and advanced SCLC, remains poor.
Gene expression is regulated by epigenetic mechanisms; epigenetic alterations play important roles in many physiological and pathophysiological conditions, including carcinogenesis, without changes in DNA sequence (10). In addition to genetic alterations in DNA sequence, cancers harbor numerous epigenetic alterations, which regulate gene expression and signaling pathways in the malignant cell. Moreover, these alterations can outnumber genetic alterations and usually occur early in carcinogenesis (11). Numerous data suggest that epigenetics together with genetics intersect to promote carcinogenesis at all stages of cancer development. Increasingly, genomic sequencing of human tumors has identified mutations in genes that encode proteins regulating the epigenome. Epigenetic alterations are in many cases dynamic and reversible, thus representing interesting targets for cancer therapy (12). This review will focus on epigenetic mechanisms of gene regulation in lung cancer and discuss the therapeutic implication of these alterations.
Types of Epigenetic Changes
Epigenetic mechanisms of gene expression involve various reversible alterations in chromatin structure without changes in nucleotide sequence. Chromatin is the macromolecular complex of DNA and histone proteins that allows packing the entire genome in a single cell. The basic functional unit of chromatin is the nucleosome, an octamer containing two each of the histones H2A, H2B, H3, and H4, around which 146 bp of DNA are wrapped (13). Consecutive nucleosomes are separated by linker DNA, usually 20 and 50 bp in length (14). Classically, nucleosomal DNA is less accessible than linker DNA; the degree of compaction of nucleosomes strongly influences the ability of proteins to target sequences within DNA, modulating transcription, repair, and replication of genes. There are three main types of epigenetic mechanisms: DNA methylation, histone tail modification and non-coding, micro RNA regulation (15).
DNA Methylation
The longest studied epigenetic mechanism of gene expression regulation is the methylation of cytosine residues in CpG sites in the 5′ region of genes. Both DNA hypomethylation and hypermethylation are commonly described in human cancer cells (10). Hypermethylation generally leads to gene silencing and inactivation in tumor suppressor genes, whereas hypomethylation leads to genomic instability and active transcription (15, 16). Global DNA hypermethylation occurs early in the carcinogenesis of lung cancer. Liu et al. showed that hypomethylation of DNA repeats and upregulation of imprinted alleles precede hypermethylation and epigenetic silencing of tumor suppressor genes in epithelial cells exposed to tobacco smoke (17). DNA methylation is mediated via three DNA methyltransferases (DNMT): DNMT1 – 3a and 3b. DNMT1 binds to hemimethylated DNA to maintain methylation patterns after DNA replication (18). On the other hand, DNMTs 3a and 3b bind to unmethylated or hemimethylated DNA to mediate de novo DNA methylation (19). Pre-clinical studies have suggested that aberrant expression of DNMTs is involved in carcinogenesis of lung cancer via tumor suppressor gene silencing (20). For example, DNMT1 and DNMT 3b overexpression in lung cancer cells has been correlated with promotor hypermethylation and silencing of the tumor suppressor gene p16 in lung cancer cells (21). Simultaneous overexpression of all three DNMTs and hypermethylation of several tumors suppressor genes including p16, FHIT, and RAR-β was reported by Lin and colleagues (22). Multiple reports have suggested that epigenetic silencing of tumors suppressor genes is involved in the initiation and progression of lung cancer (23–26).
Modification of Histone Tails
Lysine-rich tails of core histones (H2A, H2B, H3, and H4) protrude from the nucleosome providing sites for reversible modifications that alter chromatin structure and modulate gene expression (27). These modifications include methylation, acetylation, phosphorylation, sumoylation, and ubiquitination – some of these modifications mark active and some inactive chromatin states (15). The most extensively studied modifications are histone lysine acetylation/deacetylation and methylation/demethylation (27).
Acetylation of histone tails is mediated by a number of histone acetyltransferases (HAT) including GNAT, MYST, and p300 families (27, 28). On the other hand, histone deacetylation is mediated by the histone deacetylase enzymes (HDAC), which are classified in four subfamilies (29). Histone acetylation leads to chromatin relaxation and gene expression, whereas deacetylation leads to gene silencing (30). Non-histone proteins also undergo changes in acetylation state mediated by HATs and HDACs (31).
Numerous histone methyltransferases (KMT) mediate mono, di-, or trimethylation of lysine residues (27). Histone lysine demethylation, on the other hand, is mediated by histone dimethyltransferases (KDMT) (32). Histone methylation may either activate or inhibit gene transcription, depending on the site of action. For example, methylation of lysine 4 on H3 (H3K4) is strongly associated with transcription activation, whereas methylation of lysine 27 on H3 is frequently associated with gene silencing (15). Like histone acetylation, many non-histone proteins such as p53, E2F1, and NFB can be targets of KMT and KDMT (27).
Kim et al showed that increased activity of KMT DOT1L, which mediates methylation of H3K79, supports carcinogenesis of lung cancer cells (33). It is thought that methylation at K79 promotes/inhibits transcriptional elongation, thereby inducing overexpression/underexpression of different cell cycle regulatory genes and different tumor suppressor genes such as HOXA9 and RASSF1A. Overexpression of JARID1B (KDM5B), which demethylates H3K4Me3/Me2, has been observed in both NSCLC and SCLC (34). This overexpression correlated with increased expression of E2F1 and E2F2. Upregulation of LSD1 (KDM1A), which catalyzes demethylation of H3K4Me2/Me1 and possibly H3K9Me2/Me1, was observed in small cell lung cancers relative to normal lung tissues (35). Various alterations in methylation/demethylation and acetylation/deacetylation of core histone in lung cancer cells can be involved in lung carcinogenesis. For example, hyperacetylation of H4K5 and H4K8, hypoacetylation of H4K12/H4K16, and decreased H4K20Me3 levels were observed in lung cancer cells relative to adjacent normal respiratory epithelia (36). It is important to note that these studies report on global changes in expression of specific histone modifications. What is missing is an understanding of which gene promoters are critically affected by these modifications, and which ensuing gene expression changes are involved in carcinogenesis. It is not clear whether changes in histone methylation or acetylation are oncogenic in themselves, or more permissive in allowing the dysregulation of cell growth that is a hallmark of cancer.
MicroRNAs
MicroRNAs (miRNAs) are small regulatory RNAs, approximately 22 nucleotides long, that control gene expression by binding to the 3′ untranslated region of messenger RNA (mRNA), leading to either mRNA degradation or inhibition of protein translation (37). As negative regulatory factors, miRNAs have emerged as key post-transcriptional regulators of gene expression, involved in many physiological and pathological processes, such as proliferation, differentiation, death, and stress resistance, mediated by altering levels of gene expression (16, 37). There are more than 1,000 mature miRNAs in the human genome according to the miRBase (http://www.mirbase.org/) and it is expected that many more miRNAs will be identified in the future, making their interactions even more complex (15). A single miRNA can target many different mRNAs, and a single mRNA can be targeted by multiple miRNAs, thereby creating a complex network of molecular pathways in cells. Altered expression of miRNAs is commonly found in cancer, and is thus thought to be associated with and potentially contributing to the pathogenesis of most malignancies, including lung cancer, with a miRNA having the potential to serve as either oncogene or tumor suppressor gene, depending on its gene target (38, 39).
The role of the let-7 miRNA family in lung carcinogenesis has been extensively studied. One of the main targets of let-7 miRNA is KRAS (40, 41), leading to its down-regulation. Johnson et al showed that let-7 miRNA expression is lower in lung tumors than in normal lung tissue, while RAS protein is significantly higher in lung tumors, providing a possible mechanistic involvement of let-7 miRNA in carcinogenesis in a subset of lung cancers (40). Hayashita et al. observed that the polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation and tumor development (42). A recent study demonstrated that loss of miR-365 might also be involved in lung carcinogenesis via decreased suppression of NKX2-1, a transcription factor also known as TTF-1 that is thought to be involved in lung cancer carcinogenesis (43). Enforced NKX2-1 overexpression significantly increased cell proliferation, overcoming the suppressive effect of miR-365 (43).
Xi et al. observed that cigarette smoke repressed miR-487b in cultured respiratory epithelia and lung cancer cells (44). Subsequent experiments revealed significant repression of miR-487b in primary lung cancers – particularly those from smokers, relative to adjacent normal lung tissues. Repression of miR-487b in cultured cells following cigarette smoke exposure and in primary lung cancers coincided with DNA methylation and recruitment of polycomb repressor proteins to the miR-487b regulatory region. Notably, miR-487b directly targets transcripts encoding the non-canonical Wnt ligand, Wnt5a; polycomb repressor proteins BMI1 and SUZ12; and the oncogenes KRAS and MYC. Repression of miR-487b correlated with overexpression of all five transcripts in primary lung cancers. Collectively, these findings demonstrate links between cigarette smoke and the epigenetic repression of a microRNA regulating the expression of five genes involved in oncogenesis.
Prognostic and Predictive Value of Epigenetic Changes
Early detection of lung cancer could change disease outcome. Until recently, there had not been a screening test that demonstrated a mortality reduction in lung cancer. The National Lung Cancer Screening Trial showed that screening high-risk persons with low-dose CT scanning could significantly reduce lung cancer mortality (45). However, the expense and the potential for harm from even low-dose radiation, raises the question of other approaches. Epigenetic changes develop in smokers and early in lung carcinogenesis, making them potential biomarkers for early detection of lung cancer. Several early but interesting studies have been reported utilizing sputum as a source of tumor cell DNA. Unfortunately, these studies suffer from the problem of “searching under the lamp-post.” As a result there are multiple different epigenetic marks that have been associated with outcome of lung cancer and no comparative analysis that would allow clinical investigators to choose among them for validation studies.
Methylguanine-DNA methyltransferase is a DNA repair enzyme that protects cells from the carcinogenic effects of alkylating agents by removing adducts from the O6 position of guanine; MGMT is frequently inactivated by aberrant promoter methylation in NSCLC (46). Palmisano et al. demonstrated that aberrant methylation of the promoters of the tumor suppressor genes p16 and/or O6-methylguanine-DNA methyltransferase (MGMT) can be detected in DNA from sputum in 100% of patients with squamous cell lung carcinoma up to 3 years before clinical diagnosis (47). Another study confirmed that aberrant promoter hypermethylation of the p16 gene, and to a lesser extent DAP kinase, is detectable in sputum, occurs frequently in smokers and persists after smoking cessation (48).
The melanoma antigen gene (MAGE) is a highly specific tumor marker, and MAGE-A3 expression has been detected in 35% of lung cancer samples and also in pre-cancerous lesions (49, 50). Shin et al. collected sputum from 133 patients with lung diseases (65 lung cancers and 68 benign lung diseases) and showed methylation abnormalities in patients with MAGE-positive sputum (in both malignant and benign diseases). Thus, MAGE expression in the sputum suggests the presence of lung cancer cells or pre-cancerous cells (51).
Interestingly, sputum miRNA profiling using a cluster of five miRNAs (miR-21, miR-143, miR-155, miR-210, and miR-372) detected NSCLC (with 83.3% sensitivity and 100% specificity) in 30 patients. If validated in larger, prospective studies, sputum miRNA profiling could represent a potentially valuable approach for the early detection of NSCLC (52).
Prognostic Markers
Epigenetic changes have been linked to early recurrence in resected NSCLC. Brock and colleagues reported that DNA methylation in the promoter region of four genes (TP16, CDH13, RASSFIA, and APC) was associated with early recurrence in patients with resected stage I NSCLC (53). Barlési et al. classified 138 lung cancer patients into seven groups based on histology, stage, and global expression levels of H3K4Me2, H2AK5Ac, and H3K9Ac. The groups showed significant differences in disease-free and OS. High intratumoral levels of H3K4Me2 showed significant improvement in OS compared to patients with high intratumoral levels of H3K9Ac (147 vs. 10 months) (54). These changes may help in selecting early-stage NSCLC patients for adjuvant treatment.
DNMT1 accumulation and subsequent hypermethylation of the promoter of tumor suppressor genes may lead to tumorigenesis and provide an important link between tobacco smoking and lung cancer (55). Xing et al. examined DMNT1 and DNMT3b, as well as methylated DNA-binding protein 2 (MBD2) expression in 148 resected NSCLC samples. High DNMT1 expression correlated significantly with increased risk of cancer-related death in all patients, whereas increased DNMT3b expression was associated with poor outcome in patients less than 65 years of age. High-level expression of MBD2 correlated with poor survival in male patients and those with squamous cell carcinomas (56). The previously mentioned study conducted by Kim and colleagues demonstrated that overexpression of DNMT1 is correlated with p16 promoter hypermethylation and diminished patient survival (21). High intranuclear DNMT1 levels correlated significantly with smoking status and poor survival in 124 patients with lung cancer (55). Importantly, DNMT1 overexpression in lung cancer patients who smoked continuously correlated with poor prognosis. The key genes beyond p16 that are hypermethylated and lead to a poor prognosis in patients diagnosed with lung cancer are not known.
Increased HDAC1 mRNA levels appear to be more common in advanced stages of disease in lung cancer patients, thus suggesting a role of HDAC in more aggressive tumors (57). Minamyia et al demonstrated that lung cancer patients with high intratumor levels of HDAC3 had significantly shorter disease-free survivals than patients whose tumors exhibited low HDAC3 expression. They also stated that HDAC3 overexpression was an independent prognostic factor for poor survival in patients with adenocarcinomas, but not in those with squamous cell carcinomas (58). Given that 94 patients were included in this retrospective analysis, confirmatory studies of the role of HDAC3 in NSCLC are needed.
Cellular levels of both H3K4me2 and H3K18ac were reported to predict clinical outcome in lung cancer patients, with lower levels predicting significantly poorer survival (59). Lower global levels of histone modifications are generally predictive of a more aggressive cancer phenotype, revealing a surprising commonality in prognostic epigenetic patterns of adenocarcinomas of different tissue origins, including lung cancer (59).
In a study that analyzed NSCLC and neighboring normal lung tissues, high levels of miR-155 and low miR-let-7a-2 expression were found to correlate with poor survival in lung adenocarcinomas (39). In another study, low let-7 miRNA expression was also significantly associated with shorter survival in 143 resected lung cancer patients (60). Further attempts in addition to sputum analysis, have attempted to find valid biomarkers to detect lung cancer. Interestingly, three separate studies reported that miR-21 overexpression correlated with the aggressiveness of the disease and high levels of miR-21 in serum or plasma were strongly associated with lymph node metastasis, advanced clinical stage, and poor survival in patients with NSCLC (61–63).
Predictive Markers
A predictive marker is one that may aid in choice among therapeutic options; there are only limited data for predictive epigenetic aberrations in lung cancer patients. Molecular mechanisms of drug resistance are not completely understood and believed to be multifactorial, involving host factors, numerous molecular events, and genetic and epigenetic changes (64). In addition, chemotherapeutics induce epigenetic changes in the promoter area of specific genes, altering expression and possibly underlying resistance in many tumor types (65). One possible reason for the development of chemoresistance in NSCLC might be the epigenetic inactivation of certain tumor suppressor genes as a consequence of chemotherapy treatment (66). Ibanez de Caceres and colleagues reported that a loss of insulin-like growth factor binding protein-3 (IGFBP-3) expression, mediated by promoter hypermethylation, resulted in a reduction of tumor cell sensitivity to cisplatin in NSCLC. Authors suggested that basal methylation status of IGFBP-3 before treatment may be a clinical biomarker and a predictor of chemotherapy outcome, helping to identify patients who are most likely to benefit from platinum-based chemotherapy therapy alone or in combination with epigenetic treatment (66). Expression levels of a miRNA described above as associated with poor outcome, miR-21, were evaluated in tumor tissue and plasma by Gao et al. in 58 patients with resected NSCLC (67). The investigators found increased levels of miR-21 expression in patients with chemotherapy-resistant NSCLC, and concluded that miR-21 may be useful as a biomarker to predict adjuvant platinum-based chemotherapy response and disease-free survival in patients with NSCLC. Thus, it may serve as a novel therapeutic target to modulate platinum-based chemotherapy.
Targeting the Epigenome
Unlike oncogenic mutations, which are fixed, epigenetic alterations are potentially reversible. The reversibility of epigenetic alterations provides the foundation for targeting them therapeutically. To date, histone deacetylation and DNA methylation have been successfully targeted in the clinic. Several epigenetic modifiers have received FDA approval: DNMT inhibitors, decitabine, and 5-azacitidine are approved for treatment of myelodysplastic syndromes (68, 69) and HDAC inhibitors romidepsin and vorinostat are approved for T-cell lymphoma (70–72).
The adverse effect profiles of these epigenetic drugs are well known from their use for these approved indications. Peripheral cytopenias are among the most common adverse effects of DNMT inhibitors whereas gastrointestinal adverse effects and injection-site reactions are among the most common non-hematological adverse effects. The major adverse effects of HDAC inhibitors are fatigue, nausea, and vomiting. Most of the agents cause thrombocytopenia, lymphopenia, neutropenia, and electrocardiographic changes including ST and T wave flattening and QT prolongation (72, 73). Selective HDAC inhibition may provide greater efficacy and a wider therapeutic window by reducing adverse effects.
Epigenetic Therapy in NSCLC
Table 1 shows the main classes of epigenetic therapies in lung cancer. Table 2 summarizes clinical trials of epigenetic therapies in lung cancer (74–87). Unlike hematological malignancies and CTCL, monotherapy with epigenetic therapies have not proven particularly efficacious in lung cancer, although in vitro both romidepsin and vorinostat alone induce apoptosis in NSCLC and SCLC (Figure 1). Available pre-clinical and clinical data suggests that most HDAC inhibitors will be optimally used in combination: with chemotherapies, targeted therapies, radiation, or other epigenetic modifiers, rather than as single agents (88–94).
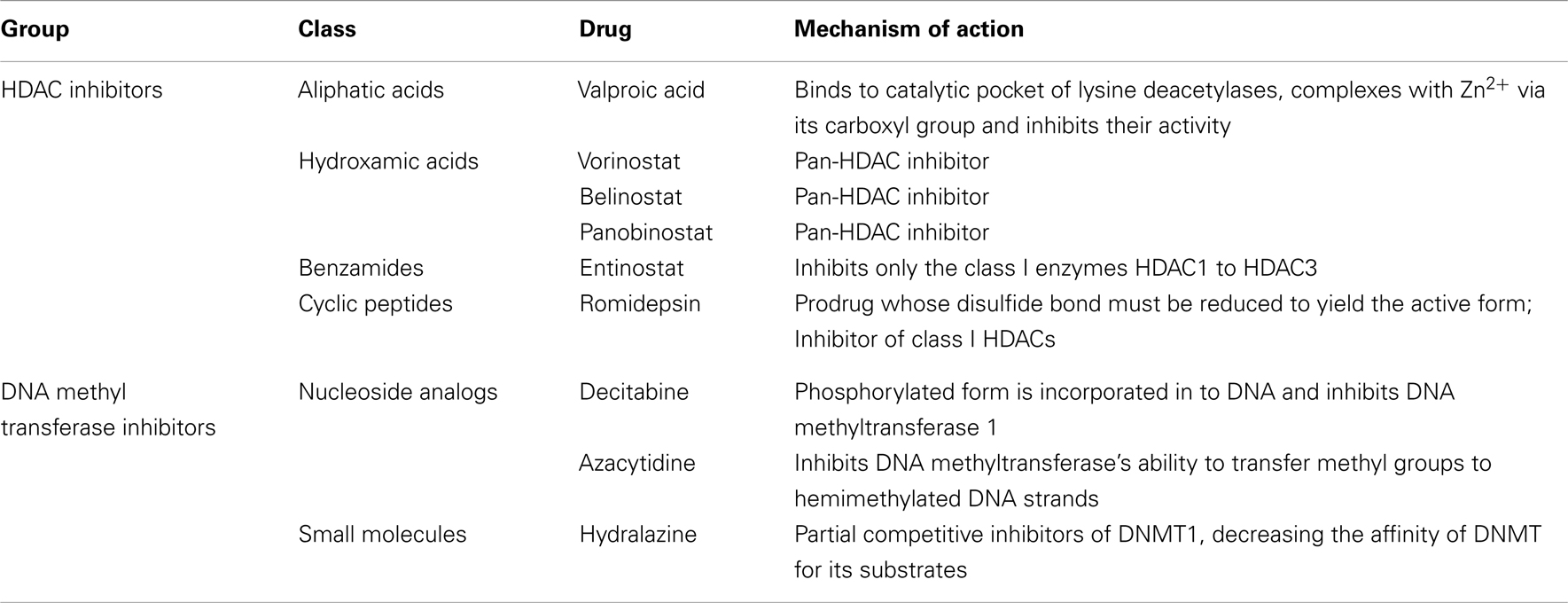
Table 1. Selected epigenetic drugs which are undergoing clinical evaluation in lung cancer.
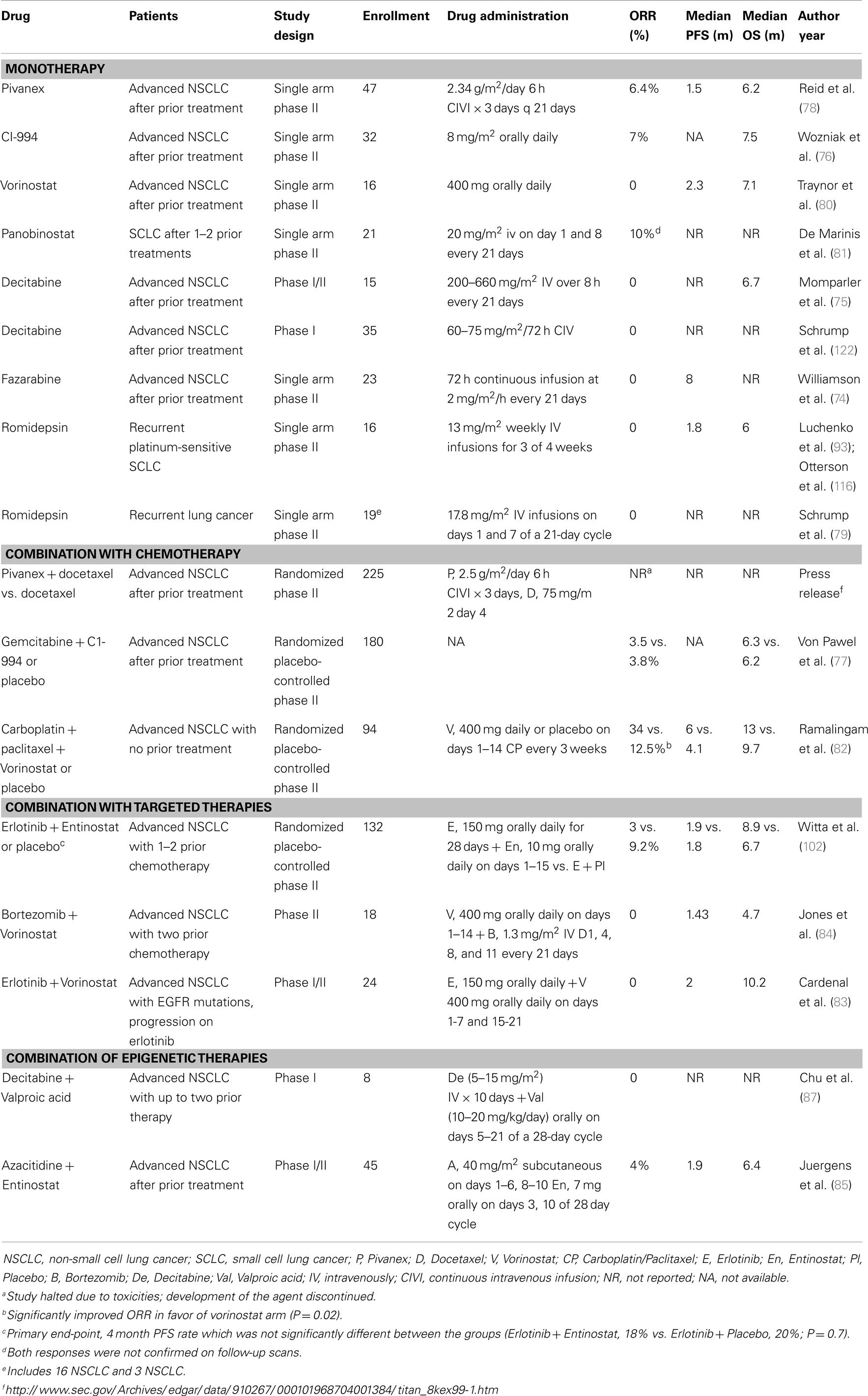
Table 2. Selected clinical trials of epigenetic therapies in lung cancer.
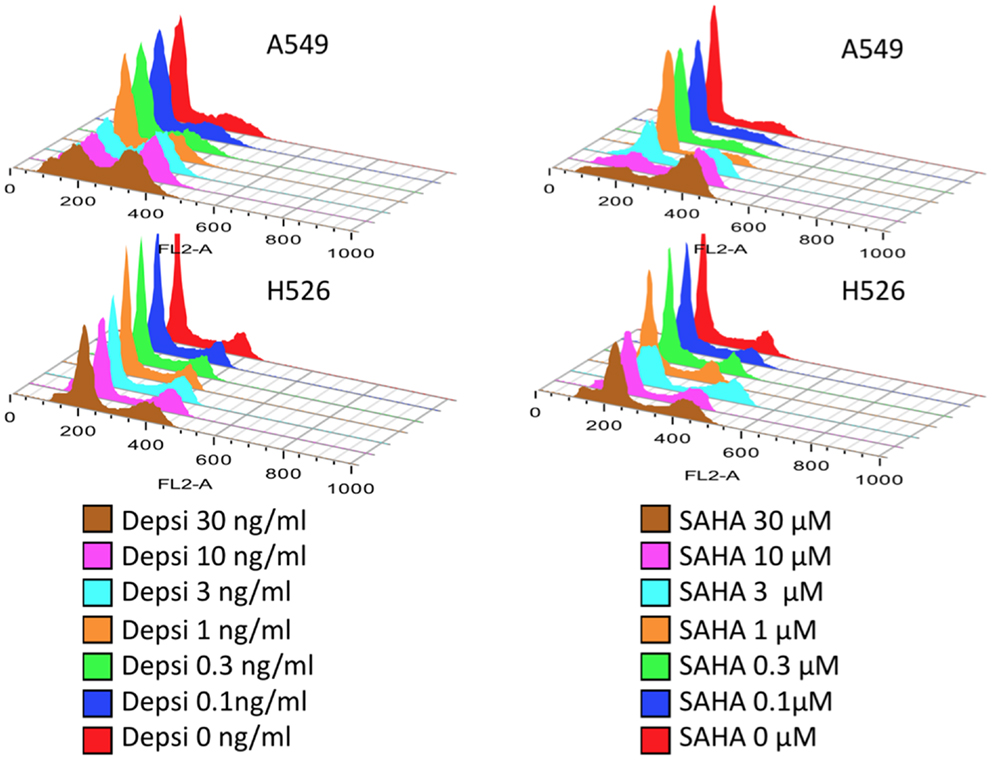
Figure 1. Lung carcinoma cells treated with HDAC inhibitors. Dose response studies of cell cycle following 24 h treatment with either romidepsin (DEPSI) or vorinostat (SAHA) are show in the histograms. Concentrations are shown in the legend and represent equipotent concentrations for growth inhibition (Luchenko et al., manuscript in preparation). The NSCLC cell line responds to the HDAC inhibitor with G2 arrest and loss of the G1 peak, while the SCLC line H526 responds with both G1 and G2 arrests and apoptosis.
Combination with Chemotherapy and Radiation
Combinations with chemotherapy or radiation therapy and epigenetic agents have been based on the rationale that alterations in gene expression induced by the epigenetic agent may allow increased sensitivity and reverse resistance to treatment. For example, increased topoisomerase II gene expression following HDAC inhibition may increase sensitivity to etoposide (94). Alternately, increased accessibility of DNA due to the “relaxed chromatin” following HDAC inhibition may result in increased platinum binding and increased efficacy. In yet another hypothesis, altered chromatin states resulting from epigenetic alterations have also been identified in small populations of cells that acquire a drug-tolerant phenotype (95). This drug-tolerant subpopulation may be selectively ablated by epigenetic therapies.
Early clinical observations in NSCLC have not supported the pre-clinical findings of synergy between chemotherapy and epigenetic therapy. A randomized placebo-controlled phase II trial of previously untreated NSCLC patients showed improved response rates when vorinostat was added to first-line carboplatin and paclitaxel (34% with vorinostat vs. 12.5% with placebo; P = 0.02) (82). However, a phase III randomized trial was prematurely terminated because of no anticipated improvement in response rates, PFS, or OS (96).
These early failures at combining epigenetic therapy with conventional DNA damaging agents may relate to a lack of understanding of the mechanisms underlying the in vitro synergy and the conflicting data regarding the optimal schedule for the combination. Whereas some studies suggest that pretreatment with HDAC inhibitors sensitizes cancer cells to chemotherapeutic agents, presumably because sequential treatment may facilitate access to DNA (88–90, 92, 94); other studies are not in agreement with this observation (97, 98). We found that in SCLC cell lines, simultaneous but not sequential treatment with HDAC inhibitors enhanced double-stranded DNA breaks by cisplatin and etoposide. In fact, pretreatment with HDAC inhibitors mitigated the cytotoxicity of DNA damaging agents without increasing their access to DNA (93).
Histone deacetylase enzymes inhibitors have been shown to sensitize NSCLC to the cytotoxic effects of radiation through persistence of DNA double-strand breaks and apoptotic cell death both in vitro and in vivo (99). The enhanced cytotoxic effects of radiation co-administered with HDAC inhibitors are thought to be due to the effects of HDAC inhibitors decreasing expression or function (via acetylation) of DNA repair proteins (100). Although in theory, HDAC inhibitor induced tumor cell cycle arrest could provide an efficient period for an enhanced response to radiation-induced cell injury, many tumor cells lack the ability to arrest in G1 and by that means to repair their DNA.
Combination with Targeted Therapy
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI), erlotinib, and gefitinib are standard treatments for NSCLC and have striking activity against tumors with EGFR mutations (101). However, resistance to the EGFR TKI is observed, and often correlates with markers of epithelial-mesenchymal transition status. Higher levels of E-cadherin, an epithelial marker, indicate sensitivity, whereas higher levels of vimentin and ZEB-1, both mesenchymal markers, indicate resistance. Pre-clinical evidence suggests that HDAC inhibitors can delay as well as reverse EGFR-TKI resistance by inhibiting epigenetic modifications leading to drug tolerance as well as reverting the EMT phenotype (102, 103). A BIM (BCL2L11) deletion polymorphism that results in the generation of alternatively spliced isoforms of BIM that lack the crucial BH3 domain and confers an EGFR TKI resistant phenotype in NSCLC cell lines (104) can be epigenetically restored restored by HDAC inhibition. BIM function is required for apoptosis induction by EGFR-TKIs in EGFR mutant NSCLC (105).
A randomized phase II study that evaluated erlotinib with and without entinostat in previously treated patients with advanced NSCLC and no prior EGFR-TKIs did not find any difference in the primary end-point, 4 month PFS rate between the two groups (Table 2) (102). However, in the subset of patients with high E-cadherin levels, OS was longer in the patients who received erlotinib with entinostat (9.4 vs. 5.4 months; hazard ratio, 0.35; 95% CI, 0.13–0.92; P = 0.03). Interim results of a phase I/II study of concurrent administration of vorinostat and erlotinib in patients with advanced NSCLC with EGFR mutations who had prior disease progression on erlotinib showed no objective responses (83). Whether the subset analysis of the erlotinib/entinostat study will be sufficient to move the combination forward with a selection strategy to enroll patients with evidence of EMT in their tumors remains to be determined. Given the in vitro evidence we would view the ability to revert EMT as an important question to resolve.
Many kinases, such as EGFR, rely on heat shock protein 90 (Hsp90) chaperone function for conformational maturation and function. HDAC6 deacetylates Hsp90; HDAC6 inhibition results in Hsp90 acetylation, which impairs its chaperone function. This leads to degradation of client proteins. In NSCLC cell lines, HDAC inhibition leads to Hsp90 acetylation, depletion of EGFR, and other key survival signaling proteins, and triggers apoptosis only in lung cancer cells harboring EGFR mutations (103). This mechanism should also provoke synergy with EGFR inhibitors by reducing the level of mutant EGFR that requires inhibition. As noted above, to date few clinical trials have explored this mechanism of synergy, and most unsuccessfully.
Synergistic anti-proliferative effects were seen in pre-clinical models with histone acetylation and proteasome inhibition. HDAC6, a cytoplasmic, microtubule-associated member of the class II family of HDACs plays an essential role in aggresomal protein degradation, a pathway that is upregulated in the setting of proteasome inhibition. Cells that lack HDAC6 do not form aggresomes properly and fail to clear misfolded protein aggregates, which by themselves are toxic (106). HDAC6 inhibition causes the same failure of protein aggregates to traffic to the aggresome. In some cell types, inhibition of proteasome-dependent pathways with bortezomib and the aggresome pathway with HDAC inhibitors leads to a greater accumulation of polyubiquitinated proteins with a resultant increase in cell stress and apoptosis (107). Despite a strong pre-clinical rationale, a phase II study of bortezomib and vorinostat showed no evidence of clinical activity in NSCLC (84).
Combinations of Epigenetic Modifiers
DNA methyltransferases inhibitors and HDAC inhibitors in combination have shown synergistic growth inhibition in NSCLC cell lines (108, 109). Clinical studies in patients with hematologic malignancies suggest that sequential administration of DNMT inhibitors and HDAC inhibitors may reverse the silencing of a subset of tumor suppressor genes by a combination of CpG hypermethylation and histone hypoacetylation (110). Initial clinical studies in NSCLC suggest that such combinations may increase clinical efficacy without unacceptable toxicity. A phase I/II trial combined azacitidine and entinostat, in patients with metastatic NSCLC (n = 45) (85). One patient had a complete response that lasted 14 months. A second patient had a partial response that lasted 8 months. In the intent-to-treat population, the median PFS was 7.4 weeks (95% CI, 7.0–8.0 weeks) and median OS was 6.4 months (95% CI, 3.8–9.2 months). Median survival among patients who completed at least one cycle of epigenetic therapy was 8.6 months (95% CI, 5.5–12.2 months). One observation in the post-study follow-up of these patients was a major objective response in four out of the 19 patients who received subsequent chemotherapy (21%), which the authors suggested could indicate stable changes in gene expression resulting from epigenetic therapy that altered the cancer cell sensitivity to subsequent cytotoxic therapy.
Epigenetic Therapy in SCLC
Unlike NSCLC, there is a paucity of data on therapeutic targeting of epigenetic alterations in SCLC. In pre-clinical in vitro studies, SCLC has proven sensitive to a number of HDAC inhibitors including panobinostat (111), romidepsin (112), trichostatin (113), and valproic acid (114, 115). Proposed mechanisms of activity of HDAC inhibitors in SCLC include activation of caspases, down-regulation of antiapoptotic factors such as Bcl-2 and Bcl-XL, and upregulation of p21 (111). However, three phase II studies involving monotherapy with two HDAC inhibitors, panobinostat and romidepsin, yielded no objective tumor responses (79, 81, 116).
Several pre-clinical studies of SCLC have shown additive effects of combining epigenetic therapy with chemotherapy (111, 117), and other epigenetic modifiers (118, 119) as well as noting the importance of sequence of administration (93). In an ongoing phase I study, we are evaluating the safety of the combination of belinostat with cisplatin and etoposide in SCLC (NCT00926640). Histone acetylation in peripheral blood mononuclear cells will be determined following belinostat exposure to determine the duration of sustained global acetylation.
Challenges and Future Directions
Clinical application of epigenetic therapies in lung cancer, and in other tumor types, is still in its early days. Other epigenetic targets such as histone methylation and miRNAs are still in pre-clinical evaluation. There is more to learn about the mechanisms most critical for cancer cell death caused by epigenetic drugs: what are the genes that when demethylated and reexpressed, result in differentiation or cell death? HDAC inhibitors cause global epigenetic changes and some, as we have alluded to above, may act in opposition to promotion of cell death. Further, in lung cancer cell lines and animal models, HDAC inhibition has been shown to enhance cell migration and metastasis through induction of multiple protein kinases and downstream pathways (120). This may diminish therapeutic efficacy, leading to unfavorable outcomes. There is also a lack of understanding of resistance to epigenetic drugs in lung cancer. Although in vitro studies have suggested several mechanisms of resistance, they do not typically reflect the insensitivity of solid tumors in the clinic (121). The dual goals of overcoming resistance and the inherent potential of epigenetic drugs to reactivate tumor suppressor genes, point toward the need for rational drug combinations. Although early clinical evidence suggests efficacy of combination therapies in NSCLC, the optimal combinations, sequence, and doses need further study.
The experience with epigenetic drugs in the hematological malignancy setting and the limited experience with solid tumors suggest a delayed response to treatment possibly due to delay between epigenetic effects and ensuing differentiation and cytotoxic effects. For example, the two responses to combination epigenetic therapy in the phase I/II trial that combined azacitidine and entinostat were gradual and maximal response was achieved after only 6–8 months (81). Schrump et al. observed progressive increases in NY-ESO-1 and MAGE-A3 expression in sequential biopsies of an endobronchial tumor from a lung cancer patient treated with Decitabine over a 12-month period (122). NY-ESO-1 and MAGE-A3 protein expression in these biopsies, and serum antibodies recognizing these cancer-testis antigens, were detectable on and after the 6-month timepoint. These as well as other observations from that trial provide molecular evidence that prolonged exposures are necessary to mediate epigenetic alterations in lung cancer cells by DNA demethylating agents. Thus, the conventional strategy of testing drugs in the advanced setting may preclude detection of any clinical benefit from epigenetic drugs in a rapidly growing tumor such as lung cancer. Such delayed benefits may be best detected in the adjuvant setting; a randomized phase II trial comparing 3 year PFS following adjuvant combined epigenetic therapy with azacytidine and entinostat vs. standard care in resected stage I NSCLC (NCT01207726) recently closed. It can also be argued that effectiveness of epigenetic therapy will depend upon its ability to revert the abnormal cancer-related epigenetic changes rather than on direct or indirect cytotoxicity. Hence, the traditional strategy of finding the highest dose that is deemed safe, i.e., the maximum tolerated dose, may not necessarily identify the dose with optimal biological effect. Lastly, identification of lung cancer specific predictive biomarkers is important to identify the appropriate subgroup that may respond to treatment. As noted earlier, multiple biomarkers have been identified and need further evaluation.
Emerging data from large-scale sequencing studies underscore the major role of epigenetics in human cancer and point to a much closer collaboration between genetic and epigenetic events in carcinogenesis (123). Coupled with results of ongoing efforts to map human epigenomes in great detail, this understanding will significantly expand the implications of epigenome in lung cancer detection, prevention, and treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Victoria Luchenko for providing representative histograms of the effect of HDAC inhibitors on cell cycle in lung cancer cell lines. The authors would also like to thank Robert Robey for editorial assistance. Intramural Program, National Cancer Institute, National Institutes of Health.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi: 10.3322/caac.21166
2. Khuder SA. Effect of cigarette smoking on major histological types of lung cancer: a meta-analysis. Lung Cancer (2001) 31(2–3):139–48. doi:10.1016/S0169-5002(00)00181-1
3. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, et al. Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med (2002) 346(2):92–8. doi:10.1056/NEJMoa011954
4. van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet (2011) 378(9804):1741–55. doi:10.1016/S0140-6736(11)60165-7
5. Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol (2008) 26(21):3543–51. doi:10.1200/JCO.2007.15.0375
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
7. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi:10.1056/NEJMoa0810699
8. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi:10.1016/S1470-2045(11)70393-X
9. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol (2012) 13(10):1011–9. doi:10.1016/S1470-2045(12)70344-3
10. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer (2004) 4(2):143–53. doi:10.1038/nrc1279
11. Geutjes EJ, Bajpe PK, Bernards R. Targeting the epigenome for treatment of cancer. Oncogene (2012) 31(34):3827–44. doi:10.1038/onc.2011.552
12. Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA (2008) 299(24):2877–83. doi:10.1001/jama.299.24.2877
13. Dawson MA, Kouzarides T, Huntly BJP. Targeting epigenetic readers in cancer. N Engl J Med (2012) 367:647–57. doi:10.1056/NEJMra1112635
14. Segal E, Widom J. What controls nucleosome positions? Trends Genet (2009) 25:335–43. doi:10.1016/j.tig.2009.06.002
15. Yang IV, Schwartz DA. Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med (2011) 183(10):1295–301. doi:10.1164/rccm.201010-1579PP
16. Gomperts BN, Spira A, Massion PP, Walser TC, Wistuba II, Minna JD, et al. Evolving concepts in lung carcinogenesis. Semin Respir Crit Care Med (2011) 32(1):32–43. doi:10.1055/s-0031-1272867
17. Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene (2010) 29(25):3650–6. doi:10.1038/onc.2010.129
18. Cheng X, Blumenthal RM. Mammalian DNA methyltransferases: a structural perspective. Structure (2008) 16(3):341–50. doi:10.1016/j.str.2008.01.004
19. Wienholz BL, Kareta MS, Moarefi AH, Gordon CA, Ginno PA, Chédin F. DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLoS Genet (2010) 6(9):e1001106. doi:10.1371/journal.pgen.1001106
20. Damiani LA, Yingling CM, Leng S, Romo PE, Nakamura J, Belinsky SA. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res (2008) 68(21):9005–14. doi:10.1158/0008-5472.CAN-08-1276
21. Kim H, Kwon YM, Kim JS, Han J, Shim YM, Park J, et al. Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factors in primary non small cell lung cancer. Cancer (2006) 107:1042–9. doi:10.1002/cncr.22087
22. Hsu HS, Chen TP, Hung CH, Wen CK, Lin RK, Lee HC, et al. Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer (2007) 110(9):2019–26. doi:10.1002/cncr.23001
23. Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A (1998) 95(20):11891–6. doi:10.1073/pnas.95.20.11891
24. Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res (2001) 61(11):4556–60.
25. Toyooka S, Toyooka KO, Maruyama R, Virmani AK, Girard L, Miyajima K, et al. DNA methylation profiles of lung tumors. Mol Cancer Ther (2001) 1(1):61–7.
26. Machida EO, Brock MV, Hooker CM, Nakayama J, Ishida A, Amano J, et al. Hypermethylation of ASC/TMS1 is a sputum marker for late-stage lung cancer. Cancer Res (2006) 66(12):6210–8. doi:10.1158/0008-5472.CAN-05-4447
27. Schrump DS. Targeting epigenetic mediators of gene expression in thoracic maligancies. Biochim Biophys Acta (2012) 1819(7):836–45. doi:10.1016/j.bbagrm.2012.03.009
28. Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol (2008) 18(6):682–9. doi:10.1016/j.sbi.2008.11.004
29. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol (2010) 6(3):238–43. doi:10.1038/nchembio.313
30. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21(3):381–95. doi:10.1038/cr.2011.22
31. Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol (2009) 41(1):185–98. doi:10.1016/j.biocel.2008.08.027
32. Nottke A, Colaiácovo MP, Shi Y. Developmental roles of the histone lysine demethylases. Development (2009) 136(6):879–89. doi:10.1242/dev.020966
33. Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem (2012) 287(8):5588–99. doi:10.1074/jbc.M111.328138
34. Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T, et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer (2010) 9:59. doi:10.1186/1476-4598-9-59
35. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer (2011) 128(3):574–86. doi:10.1002/ijc.25349
36. Van Den Broeck A, Brambilla E, Moro-Sibilot D, Lantuejoul S, Brambilla C, Eymin B, et al. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin Cancer Res (2008) 14(22):7237–45. doi:10.1158/1078-0432.CCR-08-0869
37. Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet (2008) 9(11):831–42. doi:10.1038/nrg2455
38. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet (2009) 10(10):704–14. doi:10.1038/nrg2634
39. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell (2006) 9(3):189–98. doi:10.1016/j.ccr.2006.01.025
40. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell (2005) 120(5):635–47. doi:10.1016/j.cell.2005.01.014
41. Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A (2008) 105(10):3903–8. doi:10.1073/pnas.0712321105
42. Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res (2005) 65(21):9628–32. doi:10.1158/0008-5472.CAN-05-2352
43. Kang SM, Lee HJ, Cho JY. MicroRNA-365 regulates NKX2-1, a key mediator of lung cancer. Cancer Lett (2013) 335(2):487–94. doi:10.1016/j.canlet.2013.03.006.
44. Xi S, Xu H, Shan J, Tao Y, Hong JA, Inchauste S, et al. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J Clin Invest (2013) 123(3):1241–61. doi:10.1172/JCI61271
45. National Lung Screening Trial Research TeamAberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med (2011) 365(5):395–409. doi:10.1056/NEJMoa1102873
46. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res (1999) 59(4):793–7.
47. Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res (2000) 60(21):5954–8.
48. Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res (2002) 62(8):2370–7.
49. Sienel W, Varwerk C, Linder A, Kaiser D, Teschner M, Delire M, et al. Melanoma associated antigen (MAGE)-A3 expression in stages I and II non-small cell lung cancer: results of a multi-center study. Eur J Cardiothorac Surg (2004) 25(1):131–4. doi:10.1016/j.ejcts.2003.09.015
50. Jang SJ, Soria JC, Wang L, Hassan KA, Morice RC, Walsh GL, et al. Activation of melanoma antigen tumor antigens occurs early in lung carcinogenesis. Cancer Res (2001) 61(21):7959–63.
51. Shin KC, Lee KH, Lee CH, Shin IH, Suh HS, Jeon CH. MAGE A1-A6 RT-PCR and MAGE A3 and p16 methylation analysis in induced sputum from patients with lung cancer and non-malignant lung diseases. Oncol Rep (2012) 27(4):911–6. doi:10.3892/or.2011.1566
52. Roa WH, Kim JO, Razzak R, Du H, Guo L, Singh R, et al. Sputum microRNA profiling: a novel approach for the early detection of non-small cell lung cancer. Clin Invest Med (2012) 35(5):E271.
53. Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med (2008) 358(11):1118–28. doi:10.1056/NEJMoa0706550
54. Barlési F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol (2007) 25(28):4358–64. doi:10.1200/JCO.2007.11.2599
55. Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, et al. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest (2010) 120(2):521–32. doi:10.1172/JCI40706
56. Xing J, Stewart DJ, Gu J, Lu C, Spitz MR, Wu X. Expression of methylation-related genes is associated with overall survival in patients with non-small cell lung cancer. Br J Cancer (2008) 98(10):1716–22. doi:10.1038/sj.bjc.6604343
57. Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Kiriyama M, Fukai I, et al. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer (2004) 46(2):171–8. doi:10.1016/j.lungcan.2004.03.021
58. Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Motoyama S, et al. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumour Biol (2010) 31(5):533–9. doi:10.1007/s13277-010-0066-0
59. Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, et al. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol (2009) 174(5):1619–28. doi:10.2353/ajpath.2009.080874
60. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res (2004) 64(11):3753–6. doi:10.1158/0008-5472.CAN-04-0637
61. Liu XG, Zhu WY, Huang YY, Ma LN, Zhou SQ, Wang YK, et al. High expression of serum miR-21 and tumor miR-200c associated with poor prognosis in patients with lung cancer. Med Oncol (2012) 29(2):618–26. doi:10.1007/s12032-011-9923-y
62. Zandberga E, Kozirovskis V, Abols A, Andrejeva D, Purkalne G, Line A. Cell-free microRNAs as diagnostic, prognostic, and predictive biomarkers for lung cancer. Genes Chromosomes Cancer (2013) 52(4):356–69. doi:10.1002/gcc.22032
63. Wang ZX, Bian HB, Wang JR, Cheng ZX, Wang KM, De W. Prognostic significance of serum miRNA-21 expression in human non-small cell lung cancer. J Surg Oncol (2011) 104(7):847–51. doi:10.1002/jso.22008
64. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med (2002) 53:615–27. doi:10.1146/annurev.med.53.082901.103929
65. Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene (2005) 24(54):8061–75. doi:10.1038/sj.onc.1208955
66. Ibanez de Caceres I, Cortes-Sempere M, Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, Manguán-García C, et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene (2010) 29(11):1681–90. doi:10.1038/onc.2009.454
67. Gao W, Lu X, Liu L, Xu J, Feng D, Shu Y. MiRNA-21: a biomarker predictive for platinum-based adjuvant chemotherapy response in patients with non-small cell lung cancer. Cancer Biol Ther (2012) 13(5):330–40. doi:10.4161/cbt.19073
68. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol (2009) 10(3):223–32. doi:10.1016/S1470-2045(09)70003-8
69. Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol (2011) 29(15):1987–96. doi:10.1200/JCO.2010.30.9245
70. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol (2007) 25(21):3109–15. doi:10.1200/JCO.2006.10.2434
71. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol (2009) 27(32):5410–7. doi:10.1200/JCO.2008.21.6150
72. Harrison SJ, Bishton M, Bates SE, Grant S, Piekarz RL, Johnstone RW, et al. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax(®)). Epigenomics (2012) 4(5):571–89. doi:10.2217/epi.12.52
73. Noonan AM, Eisch RA, Liewehr DJ, Sissung TM, Venzon DJ, Flagg TP, et al. Electrocardiographic studies of romidepsin demonstrate its safety and identify a potential role for KATP channel. Clin Cancer Res (2013) 19(11):3095–104. doi:10.1158/1078-0432.CCR-13-0109
74. Williamson SK, Crowley JJ, Livingston RB, Panella TJ, Goodwin JW. Phase II trial and cost analysis of fazarabine in advanced non-small cell carcinoma of the lung: a Southwest Oncology Group study. Invest New Drugs (1995) 13(1):67–71. doi:10.1007/BF00872867
75. Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs (1997) 8(4):358–68. doi:10.1097/00001813-199704000-00008
76. Wozniak A, O’Shaughnessy J, Fiorica J, Grove W. Phase II trial of CI- 994 in patients (pts) with advanced nonsmall cell lung cancer (NSCLC). Proc Am Soc Clin Oncol (1999) 18:487a.[abstract 1878],
77. Von Pawel J, Shepherd F, Gatzmeier U, Natale RB, O’Brien MER, Otterson GA, et al. Randomized phase 2 study of the oral histone deacetylase inhibitor CI-994 plus gemcitabine (Gem) versus placebo (PBO) plus Gem in second-line nonsmall cell lung cancer (NSCLC). Proc Am Soc Clin Oncol (2002) 21:310a.(Abstract 1239),
78. Reid T, Valone F, Lipera W, Irwin D, Paroly W, Natale R, et al. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer (2004) 45(3):381–6. doi:10.1016/j.lungcan.2004.03.002
79. Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res (2008) 14(1):188–98. doi:10.1158/1078-0432.CCR-07-0135
80. Traynor AM, Dubey S, Eickhoff JC, Kolesar JM, Schell K, Huie MS, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol (2009) 4(4):522–6. doi:10.1097/JTO.0b013e3181952478
81. De Marinis F, Atmaca A, Tiseo M, Ciuffreda L, Gridelli C, Gebbia V, et al. Deacetylase inhibitor (DACI) panobinostat in relapsed small cell lung cancer (SCLC) patients: results of a multicenter phase II trial. J Clin Oncol (2010) 28:2010.suppl; abstr e17521,
82. Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(1):56–62. doi:10.1200/JCO.2009.24.9094
83. Cardenal F, Reguart N, Morán T, Insa A, Isla L, Magern M, et al. Phase I/II Trial of Vorinostat (V) in Combination with Erlotinib (E) in Advanced Non-Small Cell Lung Cancer (NSCLC) Patients (pts) With EGFR Mutations After Erlotinib Progression - The TARZO Trial. Stockholm: The European Multidisciplinary Cancer Congress (2011).
84. Jones MW, Zhang C, Oettel KR, Blank JH, Robinson EG, Ahuja HG, et al. Vorinostat (V) and bortezomib (B) as third-line treatment in patients with advanced non-small cell lung cancer (NSCLC): a Wisconsin oncology network phase II study. J Clin Oncol (2011) 29:2011. doi:10.1200/JCO.2010.33.5091 suppl; abstr 7567,
85. Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov (2011) 1(7):598–607. doi:10.1158/2159-8290.CD-11-0214
86. Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res (2006) 66(2):944–50. doi:10.1158/0008-5472.CAN-05-1988
87. Chu BF, Karpenko MJ, Liu Z, Aimiuwu J, Villalona-Calero MA, Chan KK, et al. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol (2013) 71(1):115–21. doi:10.1007/s00280-012-1986-8
88. Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther (2003) 2(10):971–84.
89. Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res (2003) 63(21):7291–300.
90. Zhang Y, Jung M, Dritschilo A, Jung M. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiat Res (2004) 161(6):667–74. doi:10.1667/RR3192
91. Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol (2007) 25(26):4051–6. doi:10.1200/JCO.2007.11.6202
92. Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche TJ, et al. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst (2007) 99(14):1107–19. doi:10.1093/jnci/djm044
93. Luchenko VL, Salcido CD, Zhang Y, Agama K, Komlodi-Pasztor E, Murphy RF, et al. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle (2011) 10(18):3119–28. doi:10.4161/cc.10.18.17190
94. Kurz EU, Wilson SE, Leader KB, Sampey BP, Allan WP, Yalowich JC, et al. The histone deacetylase inhibitor sodium butyrate induces DNA topoisomerase II alpha expression and confers hypersensitivity to etoposide in human leukemic cell lines. Mol Cancer Ther (2001) 1(2):121–31.
95. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell (2010) 141(1):69–80. doi:10.1016/j.cell.2010.02.027
96. Belani C, Ramalingam S, Kalemkerian G, Mok T, Rosell R, Ahn MJ, et al. Randomized, double-blind phase II/III study of first-line paclitaxel (P) plus carboplatin (C) in combination with vorinostat or placebo in patients with advanced non-small-cell lung cancer (NSCLC). Eur J Cancer (2009) 7(Suppl):507. doi:10.1016/S1359-6349(09)71720-3 abstr O-9007,
97. Bevins RL, Zimmer SG. It’s about time: scheduling alters effect of histone deacetylase inhibitors on camptothecin-treated cells. Cancer Res (2005) 65(15):6957–66. doi:10.1158/0008-5472.CAN-05-0836
98. Sato T, Suzuki M, Sato Y, Echigo S, Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int J Oncol (2006) 28(5):1233–41.
99. Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs (2007) 18(7):793–800. doi:10.1097/CAD.0b013e328011fdab
100. Robert C, Rassool FV. HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res (2012) 116:87–129. doi:10.1016/B978-0-12-394387-3.00003-3
101. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer – Molecular and clinical predictors of outcome. N Eng J Med (2005) 353(2):133–44. doi:10.1056/NEJMoa050736
102. Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, et al. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol (2012) 30(18):2248–55. doi:10.1200/JCO.2011.38.9411
103. Edwards A, Li J, Atadja P, Bhalla K, Haura EB. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol Cancer Ther (2007) 6(9):2515–24. doi:10.1158/1535-7163.MCT-06-0761
104. Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med (2012) 18(4):521–8. doi:10.1038/nm.2713
105. Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, Nanjo S, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented by in combination with HDAC inhibition. Cancer Res (2013) 73(8):2428–34. doi:10.1158/0008-5472.CAN-12-3479
106. Shimamura T, Shapiro GI. Heat shock protein 90 inhibition in lung cancer. J Thorac Oncol (2008) 3(6):S152–9. doi:10.1097/JTO.0b013e318160c0e7
107. Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res (2004) 10(11):3839–52. doi:10.1158/1078-0432.CCR-03-0561
108. Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, et al. Sequential 5-Aza-2 deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother (2001) 24(2):151–61. doi:10.1097/00002371-200103000-00010
109. Chai G, Li L, Zhou W, Wu L, Zhao Y, Wang D, et al. HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS ONE (2008) 3(6):e2445. doi:10.1371/journal.pone.0002445
110. Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood (2009) 114(13):2764–73. doi:10.1182/blood-2009-02-203547
111. Crisanti MC, Wallace AF, Kapoor V, Vandermeers F, Dowling ML, Pereira LP, et al. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther (2009) 8(8):2221–31. doi:10.1158/1535-7163.MCT-09-0138
112. Doi S, Soda H, Oka M, Tsurutani J, Kitazaki T, Nakamura Y, et al. The histone deacetylase inhibitor FR901228 induces caspase-dependent apoptosis via the mitochondrial pathway in small cell lung cancer cells. Mol Cancer Ther (2004) 3(11):1397–402.
113. Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. J Surg Res (2007) 142(2):219–26. doi:10.1016/j.jss.2006.12.555
114. Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J Surg Res (2008) 148(1):31–7. doi:10.1016/j.jss.2008.03.008
115. Hubaux R, Vandermeers F, Crisanti MC, Kapoor V, Burny A, Mascaux C, et al. Preclinical evidence for a beneficial impact of valproate on the response of small cell lung cancer to first-line chemotherapy. Eur J Cancer (2010) 46(9):1724–34. doi:10.1016/j.ejca.2010.03.021
116. Otterson GA, Hodgson L, Pang H, Vokes EE, Cancer and Leukemia Group B. Phase II study of the histone deacetylase inhibitor romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304). J Thorac Oncol (2010) 5(10):1644–8. doi:10.1097/JTO.0b013e3181ec1713
117. Gray J, Cubitt CL, Zhang S, Chiappori A. Combination of HDAC and topoisomerase inhibitors in small cell lung cancer. Cancer Biol Ther (2012) 13(8):614–22. doi:10.4161/cbt.19848
118. Luszczek W, Cheriyath V, Mekhail TM, Borden EC. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: correlation of resistance with IFN- stimulated gene expression. Mol Cancer Ther (2010) 9(8):2309–21. doi:10.1158/1535-7163.MCT-10-0309
119. Kaminskyy VO, Surova OV, Vaculova A, Zhivotovsky B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis (2011) 32(10):1450–8. doi:10.1093/carcin/bgr135
120. Lin KT, Wang YW, Chen CT, Ho CM, Su WH, Jou YS. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin Cancer Res (2012) 18(17):4691–701. doi:10.1158/1078-0432.CCR-12-0633
121. Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, et al. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm (2011) 8(6):2021–31. doi:10.1021/mp200329f
122. Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, et al. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res (2006) 12(19):5777–85. doi:10.1158/1078-0432.CCR-06-0669
Keywords: epigenetics, non-small cell lung cancer, small-cell lung cancer, DNA methylation, histone modification, microRNA
Citation: Jakopovic M, Thomas A, Balasubramaniam S, Schrump D, Giaccone G and Bates SE (2013) Targeting the epigenome in lung cancer: expanding approaches to epigenetic therapy. Front. Oncol. 3:261. doi: 10.3389/fonc.2013.00261
Received: 15 July 2013; Accepted: 18 September 2013;
Published online: 09 October 2013.
Edited by:
Markus Joerger, Kantonsspital St. Gallen, SwitzerlandReviewed by:
Marina Chiara Garassino, Istituto Nazionale dei Tumori, ItalyShahab Babakoohi, Medstar Good Samaritan Hospital, USA
Copyright: © 2013 Jakopovic, Thomas, Balasubramaniam, Schrump, Giaccone and Bates. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan E. Bates, National Cancer Institute, Room 12N226, 10 Center Drive, Bethesda, MD 20892, USA e-mail: batess@helix.nih.gov