 Kevin Y. Urayama1,2*
Kevin Y. Urayama1,2* Pamela D. Thompson3
Pamela D. Thompson3 Malcolm Taylor4
Malcolm Taylor4 Elizabeth A. Trachtenberg5
Elizabeth A. Trachtenberg5 Anand P. Chokkalingam1
Anand P. Chokkalingam1
- 1School of Public Health, University of California, Berkeley, CA, USA
- 2Center for Clinical Epidemiology, St. Luke’s Life Science Institute, Tokyo, Japan
- 3Cancer Immunogenetics, St. Mary’s Hospital, University of Manchester, Manchester, UK
- 4Handforth, Cheshire, UK
- 5Center for Genetics, Children’s Hospital Oakland Research Institute, Oakland, CA, USA
The enduring suspicion that infections and immunologic response may play a role in the etiology of childhood leukemia, particularly acute lymphoblastic leukemia (ALL), is now supported, albeit still indirectly, by numerous epidemiological studies. The cumulative evidence includes, for example, descriptive observations of a peculiar peak incidence at age 2–5 years for ALL in economically developed countries, clustering of cases in situations of population mixing associated with unusual patterns of personal contacts, associations with various proxy measures for immune modulatory exposures early in life, and genetic susceptibility conferred by variation in genes involved in the immune system. In this review, our focus is the extended major histocompatibility complex (MHC), an approximately 7.6 Mb region that is well-known for its high-density of expressed genes, extensive polymorphisms exhibiting complex linkage disequilibrium patterns, and its disproportionately large number of immune-related genes, including human leukocyte antigen (HLA). First discovered through the role they play in transplant rejection, the classical HLA class I (HLA-A, -B, and -C) and class II (HLA-DR, HLA-DQ, and HLA-DP) molecules reside at the epicenter of the immune response pathways and are now the targets of many disease susceptibility studies, including those for childhood leukemia. The genes encoding the HLA molecules are only a minority of the over 250 expressed genes in the xMHC, and a growing number of studies are beginning to evaluate other loci through targeted investigations or utilizing a mapping approach with a comprehensive screen of the entire region. Here, we review the current epidemiologic evidence available to date regarding genetic variation contained within this highly unique region of the genome and its relationship with childhood ALL risk.
Introduction
Leukemia, a cancer of the hematopoietic system, is the most common malignancy affecting children less than 15 years of age. Acute lymphoblastic leukemia (ALL) comprises nearly 80% of childhood leukemia diagnoses in developed countries, followed by acute myeloid leukemia (16%) and the very rare chronic sub-types (1, 2). Pediatric patients usually exhibit characteristic chromosomal aberrations such as hyperdiploidy and/or translocations [e.g., t(12;21) TEL-AML1, 11q23MLL-AF4, t(8;21) AML1-ETO, t(15;17) PML-RARA, inv(16) CPFB-MYH11] (3), most of which have been shown to originate prenatally due to unknown causes (2). These prenatal chromosomal aberrations are likely initiating genetic events, occurring during fetal hematopoiesis and operating within a minimal two-hit disease model. Current evidence indicates that transition to overt disease will occur in only a small proportion of children carrying this pre-leukemic clone, after a sufficient second genetic event which likely occurs postnatally as a consequence of a mutagenic exposure (2, 4).
One potential post-natal exposure long thought to modulate this second “hit” is exposure to infectious agents, a premise based on early studies demonstrating the ability of certain viruses to cause leukemia in animals (5, 6) and observing an age distribution that closely mimics the incidence of common childhood infectious diseases (7). In the absence of direct evidence for the involvement of a specific infection (8), this immunologic hypothesis describing a role of infections in childhood leukemia, particularly B-cell precursor (BCP) ALL, has persisted as a result of accumulating epidemiological evidence largely driven by two related infectious hypotheses proposed in the late 1980s. Kinlen proposed the “population mixing” hypothesis in response to childhood leukemia clusters occurring in Seascale and Thurso (UK), and explained that the rise in childhood leukemia incidence may have resulted from children experiencing an abnormal immune response to specific infections introduced during an influx of infected persons into an area populated with non-immune, thus susceptible individuals (9). Greaves posited his “delayed infection” hypothesis in the context of the two-hit disease model, describing that the second genetic hit leading to overt disease in a small fraction (∼1%) of pre-leukemia carriers may be caused by an adverse immune response to infections as a result of insufficient priming of the immune system in early life (10). Abundant epidemiological data are currently available in support of both the population mixing (11) and delayed infection hypotheses (12). Furthermore, new evidence has emerged suggesting that immune dysregulation caused by an infectious exposure in the first months of life may lead to an increased risk of ALL in a genetically susceptible subset of children (2, 13).
Findings from genetic susceptibility studies targeting immune-related genetic loci have also fueled an ongoing suspicion of a role for immunologic response involving infections. Foremost among the candidate loci are those residing within the major histocompatibility complex (MHC), a genomic region well-known for its high-density of genes, complex linkage disequilibrium (LD), and highly polymorphic nature. The human MHC region spans approximately 4 Mb on the short arm of chromosome 6 (6p21.3) and contains about 250 gene loci (Figure 1), with a disproportionately large number encoding genes that play a role in immune function and regulation (14). This region first received attention following the discovery of cell surface molecules called the human leukocyte antigens (HLA) involved in tissue graft and organ transplant rejection. HLA is now well-understood to have a primary functional role in the protection against pathogens through selective binding and presentation, in allele selective fashion, of processed peptides to T lymphocytes critical to both cellular and humoral immune responses.
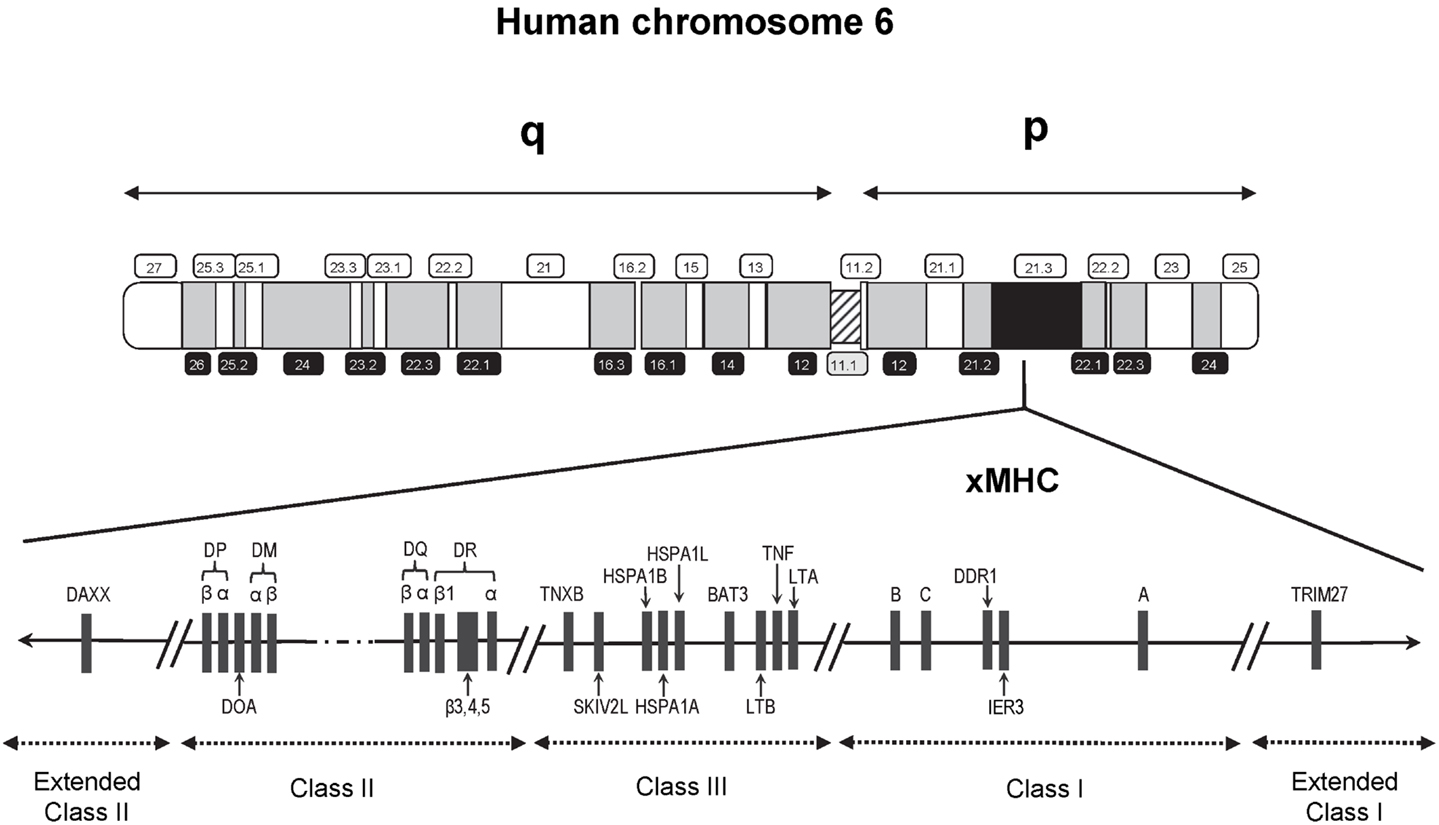
Figure 1. Schematic map (not to scale) of the 7.6 Mb extended human MHC (xMHC) on the short (p) arm of human chromosome 6. The HLA class I gene cluster includes the three expressed classical class I genes, HLA-A, HLA-B, and HLA-C, and is located telomeric of the class III region. The HLA class II gene cluster is located centromeric of the HLA class III region. Of the over 250 known expressed loci of the xMHC, the relative locations of only those that have appeared in previous studies of childhood ALL are indicated.
Recent interrogation of the flanking regions led to the discovery of additional MHC-relevant genes that extended beyond the classically defined boundaries (15). This expansion resulted in a region that is now referred to as the extended MHC (xMHC) spanning 7.6 Mb and is divided into five subregions consisting of the extended class I region at the telomeric end, and successively the classical class I, III, and II clusters bounded by an extended class II region at the centromeric end (Figure 1). While the HLA superfamily of genes are among the most highly implicated targets in disease susceptibility, they account for only a small proportion of the estimated 252 expressed xMHC loci (15), including genes involved in other immune response and inflammatory processes (e.g., cytokines and complement factors), leukocyte maturation, stress response, cell-cycle regulation, transcriptional and translational machinery, and other cellular processes. Thus, strong rationale exists supporting the need for comprehensive examinations of the xMHC not limited only to the classical HLA loci, which historically have been given the most attention in disease association studies.
Because childhood ALL is suspected to be caused by an adverse immune response, possibly triggered by infectious exposures, and because the xMHC includes a wide range of genes with potential relevance to leukemogenesis, the xMHC is a highly appropriate candidate region for investigation. Indeed, there is currently a substantial collection of studies that have evaluated a potential involvement of xMHC loci in childhood ALL risk. In this review, we aimed to provide the reader with a synthesis of the current epidemiological evidence available to date regarding the association between genetic variation of the xMHC and risk of childhood ALL.
Classical HLA Genes
Human leukocyte antigen is polygenic and encodes heterodimeric cell surface glycoproteins involved in antigen presentation that selectively interact, in a peptide-binding groove, with short peptide fragments derived from non-self and self-proteins. The classical HLA class I molecules (HLA-A, HLA-B, and HLA-C) present foreign antigens derived predominantly from intracellular infection, and initiate CD8+ T-cell responses, while class II molecules (HLA-DRB1, HLA-DRB3, HLA-DRB4, HLA-DRB5, HLA-DQB1, HLA-DQA1, HLA-DPB1, and HLA-DPA1) present antigens derived from the extracellular space to CD4+ T helper cells, which, in turn, activate macrophage and B-cells. The genes encoding HLA molecules are highly polymorphic with up to many hundreds of alleles segregating at a single locus (16). Because most are confined to the region encoding the peptide-binding groove, the HLA polymorphisms have functional relevance and significance for disease susceptibility. Sequence homology and epitope sharing between alleles and loci in the HLA genes is evident from an inspection of the sequences (17) for current alignments1. Selective pressures for balanced polymorphism act to increase amino-acid diversity of HLA molecules in populations, but stoichiometric constraints influence the amino-acid make-up of an HLA molecule, and both influence the types of peptides that can be bound by the HLA molecule (18).
Scrutiny of the HLA system in leukemia susceptibility in humans began in the 1960s following landmark studies by Lilly and colleagues who demonstrated that leukemogenesis by the Gross leukemia virus in mice is strongly influenced by H-2 (the MHC system in mice) type (19). Utilizing the best available typing methods and knowledge of the HLA system at the time, the earliest studies in humans showed a link with class I loci such as HLA-A2 antigen serotype (20–22), but were limited by relatively small study populations and absence of restriction to age groups (i.e., children or adults) and sub-types. In addition, these studies used low-resolution HLA typing methods based on serological recognition of cell surface antigens, which do not fully capture the allelic diversity of HLA. Over time, with advances in understanding of HLA genetics, development of higher resolution molecular typing techniques, and availability of well-defined study populations, more refined and methodologically rigorous evaluations of HLA in childhood leukemia susceptibility has been possible. Here, we summarize the results of 23 previous studies, the first of which was published in 1980, that specifically examined leukemia in children in relation to genetic variation at the classical class I and class II HLA loci (Table 1).
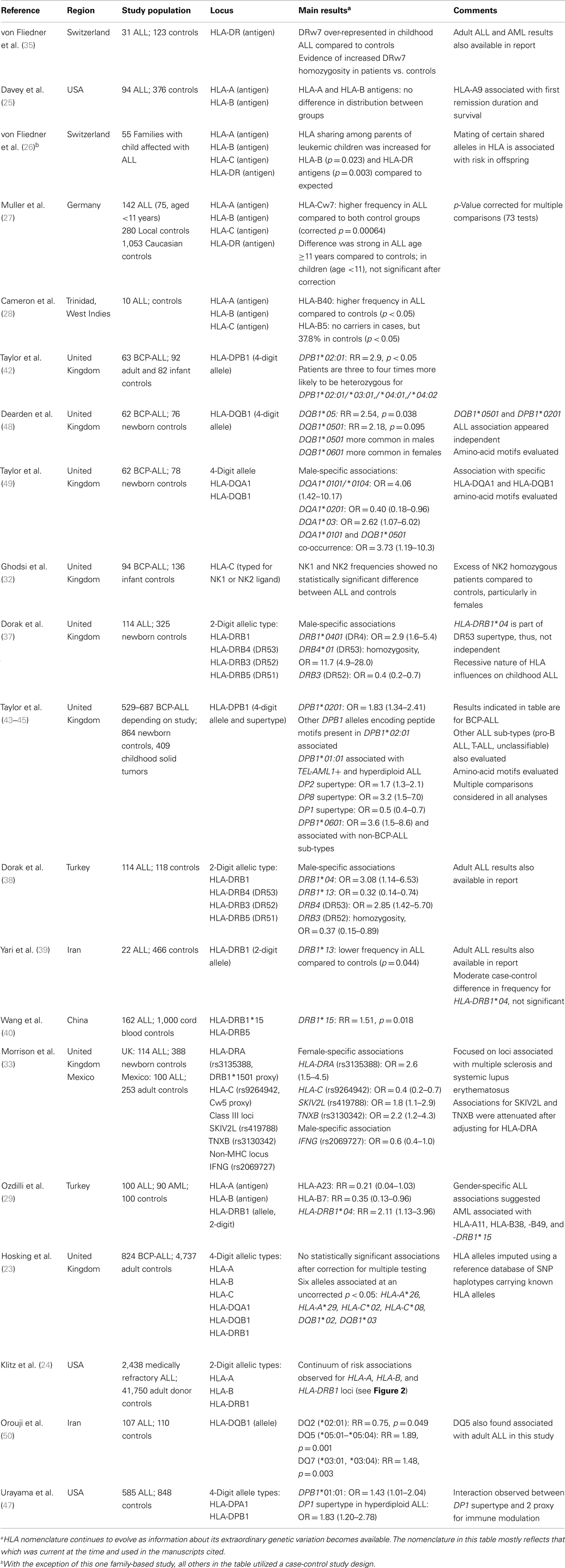
Table 1. Studies evaluating the association between HLA genetic variation and ALL risk specifically in children.
HLA Class I Loci
HLA-A and HLA-B
With the exception of two reports (23, 24), studies of HLA-A, HLA-B, and HLA-C associations with childhood ALL risk have been based on serotypes rather than allelic level classifications. The HLA-A2 antigen association found in the previous generation of studies of ALL (all age groups) was not observed in subsequent studies conducted specifically in children. In one of the earliest studies of HLA and childhood ALL risk, Davey et al. (25) observed no difference in distribution of HLA-A and HLA-B antigens between patients and controls. Among four additional studies that investigated HLA-A antigens in childhood ALL (26–29), only one reported an association with this locus, but with the HLA-A23 serotype (29). An association with the HLA-B locus was found in three of these studies (26, 28, 29), one of which was a family-based study comprising 55 families with a child affected with ALL (26). A comparison made between observed vs. expected numbers of shared antigens among parents with ALL offspring showed increased compatibility for HLA-B antigens, suggesting a role for this locus as a recessive determinant. Using the case-control design, one study reported higher HLA-B40 and lower HLA-B5 antigen frequencies in Trinidadian cases compared to controls (28), and another study conducted in Turkey reported a reduced risk associated with the HLA-B7 antigen (29).
The lack of consistent results for HLA class I loci from these mostly small studies (all less than 100 patients) utilizing low-resolution HLA typing complicates interpretation. Addressing these shortcomings, in the largest study to date evaluating HLA and childhood ALL risk, Klitz et al. (24) utilized HLA allelic data from the National Marrow Donor Program (NMDP) in an analysis of 2,438 medically refractory childhood ALL patients being considered for or undergoing hematopoietic stem cell transplantation and 41,750 controls that were matched on age (2–15 years), sex, and geographical location in the United States. This comprehensive analysis of the HLA-A and HLA-B loci revealed a spectrum of HLA associations conferring predisposition to or protection from refractory disease (Figure 2). Four significantly (p-value <0.01) protective HLA-A alleles (A*33, A*01, A*03, and A*26) and six predisposing-alleles (A*24, A*31, A*23, A*30, A*68, and A*74) were found, with 50% of alleles as a whole either predisposing or protective in this high-risk ALL population (Figure 2A). HLA-B allelic associations revealed six protective alleles (B*14, B*38, B*08, B*07, B*57, and B*44) and eight predisposing (B*15, B*18, B*40, B*51, B*53, B*39, B*48, and a binned category “rare” with <50 copies) (Figure 2B). As with HLA-A, they showed a continuum of odds ratios ranging from 0.57 to 3.20, with more predisposing than protective alleles, and 24.0% of alleles being neutral with respect to disease. These analyses show a spectrum of HLA associations and indicate that studies with larger sample sizes might clarify the predisposing, neutral, or protective status of alleles whose confidence intervals are close to, or include, an odds ratio of 1.0. Further, each allele may be regarded as a marker effect for sequences in LD with the antigen.
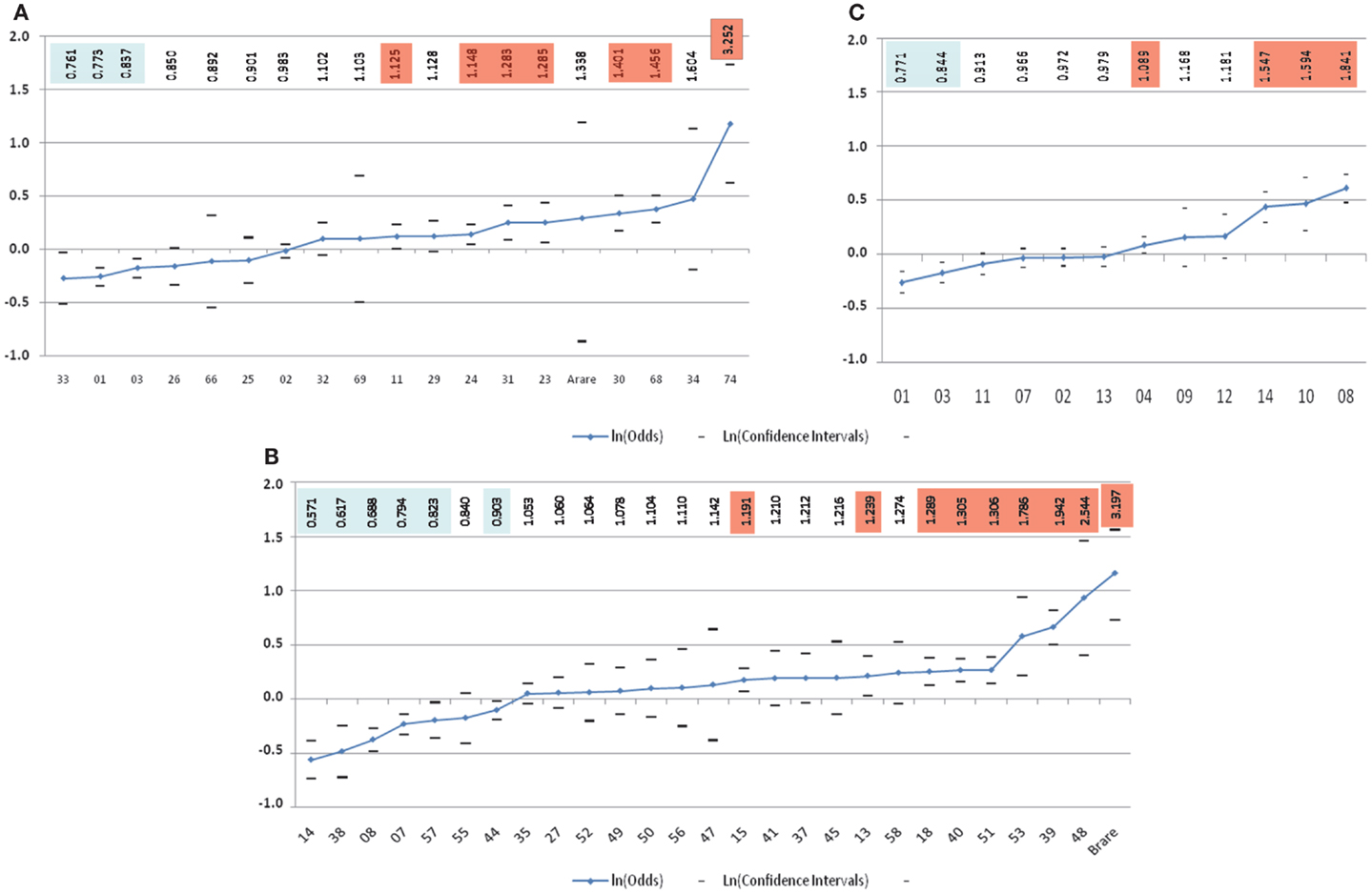
Figure 2. Risk of medically refractory childhood ALL according to 2-digit HLA alleles at HLA-A (A), HLA-B (B), and HLA-DRB1(C) (24). The vertical scale is in ln(Odds) with odds values listed alone the top highlighted in blue for favorable and orange for predisposing-allele.
Next, the authors classified genotypic groups according to allelic composition, with alleles being protective, predisposing, or neutral. Single allele effects for protective-neutral heterozygotes and homozygous protective genotypes both conferred significantly greater protection. In contrast, homozygous were significantly more predisposing than heterozygous predisposing genotypes. Haplotypes were similarly grouped, with homozygous predisposing-allele haplotypes more disposed to medically refractory childhood ALL than heterozygous predisposing-allele haplotypes. Haplotypes bearing favorable alleles revealed a different pattern, with significant protection only from HLA-B haplotypes with protective alleles, while the mixed predisposing/protective haplotypes were neutral, suggesting that these cancel each other out. Overall, these results indicate novel qualities of HLA loci in medically refractory childhood ALL. In these analyses, HLA-A appeared to have the weakest effect on ALL predisposition and protection in these children. HLA-B alleles show substantial protective and predisposing effects, with stronger effects seen in homozygous states. This continuum in disease susceptibility suggests a system in which many alleles take part in disease predisposition based on differences in binding affinity to one or a few peptides of exogenous origin, adding to the numerous studies pointing to a role for an infectious origin in childhood ALL.
HLA-C
Childhood ALL associations with HLA-C have also been reported, but like the other class I loci, the associated antigens have not been consistent across studies. Muller et al. (27) in a German study compared ALL patients (all ages) to two different control groups and found statistically significant associations with HLA-Cw7 even after correction for multiple comparisons. However, the association was less evident when restricted to patients less than aged 11 years. Associations with HLA-Cw3 and HLA-Cw4 have also been noted in studies of ALL conducted among children and adults (30, 31). Since different HLA types of a specific locus may encode proteins with overlapping functional specificities, a situation of having multiple ALL-associated HLA types is a possibility. Ghodsi et al. (32) examined this by distinguishing HLA-Cw types with respect to whether they encode either NK1 or NK2, ligands that interact with receptors on natural killer cells. The results did not support a role for the HLA-C locus in childhood ALL susceptibility. Building on previous observations of an increased risk of ALL in children of women with autoimmune disorders, Morrison et al. (33) specifically examined multiple sclerosis and systemic lupus erythematosus risk markers. A childhood ALL association with the single nucleotide polymorphism (SNP) rs9264942, a marker for HLA-Cw5, was found specifically among females similar to the protective effect observed in multiple sclerosis.
HLA Class II Loci
HLA-DRB
Suggestion of a putative role of the HLA class II DR antigen on ALL risk was first reported in the late 1970s (34) and showed an association with DR7. Additional evidence specifically in childhood ALL was available shortly after based on studies by Von Fliedner et al., one of which confirmed the DR7 association (35) and the other showing a higher than expected sharing of DR antigens among parents of children with leukemia (26).
Subsequent inspections at the allelic and supertype level of HLA-DRB1, together with the expressed DR beta chain paralogs (HLA-DRB3, -DRB4, and -DRB5) have yielded some of the most consistent evidence to date of an HLA link with childhood ALL risk. Dorak et al. (36) reported a strong male-specific childhood ALL association with homozygosity for the HLA DR53 supertypic antigen (encoded by HLA-DRB4), consistent with effects seen in other major forms of leukemia such as chronic lymphocytic and myeloid leukemia and acute myeloid leukemia (36). A few years later, using an expanded series of cases and controls, these findings were confirmed in a comprehensive examination of all DR beta chain genes (37). An effect specifically in males was found for DRB1*04, a common DRB1 allelic group, and part of the DRB4 haplotype expressing the DR53 antigen. Examination of DRB3, DRB4, and DRB5 (encoding the DR52, DR53, and DR51 supertypes, respectively) showed considerably higher DRB4*01 (DR53) homozygosity rates in patients compared to controls that was specific to males. In addition, a decreased DRB3 frequency was observed in patients compared to controls, again in males only. These findings were subsequently replicated in an independent series of high-risk and relapse childhood ALL patients and controls and additionally noted marked protective effects for DRB1*12 and DRB1*13, alleles linked to the DRB3 (DR52) haplotypic group (38).
Additional evidence for a DRB1 association comes from three other studies. Two of these, an Iranian (39) and a Turkish (29) study, both showed protective and predisposing effects of DRB1*13 and DRB1*04, respectively. The third was the large NMDP study of European Americans described earlier (24). Similar to their findings for the class I HLA-A and HLA-B loci, Klitz et al. described a spectrum of associations at DRB1 with odds ratios ranging from 0.77 to 1.84, and observed an imbalance toward predisposing effects including DRB1*04, DRB1*14, DRB1*10, and DRB1*08 (Figure 2C).
Finally, DRB1*15, a risk allele for multiple sclerosis, has also been implicated in the susceptibility to childhood ALL. At least two recent studies have reported an association, one in a Chinese population comprising 162 childhood ALL cases and 1,000 controls (40), and another in a UK population using the SNP rs3135388 as a proxy for DRB1*15:01 (33). The latter study examined the effects by gender and observed the association only in females, which is consistent with a female-specific effect of the allele also seen in multiple sclerosis (41).
HLA-DP
The first study to identify an association of HLA-DPB1 types with childhood ALL was a preliminary study by Taylor et al. (42), based on comparison of typing data from childhood common ALL cases and two sets of controls (42). The study found that the allele DPB1*02:01 was more than twice as common among children with ALL as in either set of controls. These findings were confirmed and extended in 2002 (43) in the United Kingdom Childhood Cancer Study (UKCCS) demonstrating that DPB1*02:01 was significantly more common among a large series of 875 childhood ALL patients than in either of two control groups (corrected p-value <0.05). A negative association of DPB1*01:01 with childhood ALL was also noted. Further, additional analyses generated a global p-value <10−6 for a difference between HLA-DPB1 phenotype frequencies between common ALL and controls and indicated that alleles other than DPB1*02:01 also contribute to susceptibility. The authors examined the distribution of key peptide-binding residues in the pocket domains of these alleles, which indicated that there was increased sharing of these between HLA molecules predicted to contribute to disease risk, compared to those encoded by other alleles. Examination of the phenotype frequencies of specific peptide-binding pocket residues demonstrated that significant associations with childhood ALL susceptibility were almost exclusively restricted to those peptide motifs present in DPB1*02:01; with significantly different associations for total leukemias, total ALL, common ALL, TEL-AML1+ ALL, and high-hyperdiploid ALL, compared to controls.
In an extension of this work, Taylor et al. (44) defined six HLA-DPB1 supertypes, based on amino-acid dimorphisms at positions 11 (G/L), 69 (E/K), and 84 (G/D) (Table 2), consisting of unique peptide haplotypes encoded by the hypervariable second exon of HLA-DPB1, which group alleles predicted to demonstrate functional similarities with regard to antigen binding. Among the UK childhood leukemia cases and controls, of the six supertypes examined, three (DP2, DP6, and DP8) showed evidence of increased frequencies in cases, although this was only significant in DP2 and DP8 after correction for multiple testing. These two supertypes were also significantly increased in BCP-ALL after correction, whereas DP6 appeared to be more common among non-BCP leukemia cases (p-value = 0.007). Moreover, the DP1 supertype frequency was much lower among BCP-ALL cases (6.4%) compared to controls (11.1%; p-value <10−5), a finding which did not extend to non-BCP leukemia. Stratification of cases by age at diagnosis revealed a significant association of the DP2 supertype with cases occurring at the typical peak age for diagnosis for BCP-ALL (3–6 years; p-value = 10−4), but not among cases diagnosed before 3 years or after 6 years of age. By contrast, the decreased frequency of the DP1 supertype was apparent in all groups of age at diagnosis. In a separate analysis of supertype distributions in ALL sub-types (44), a paucity of another DP1 supertype (GKD) was found among cases with TEL-AML1+ and high-hyperdiploid karyotypes relative to controls.
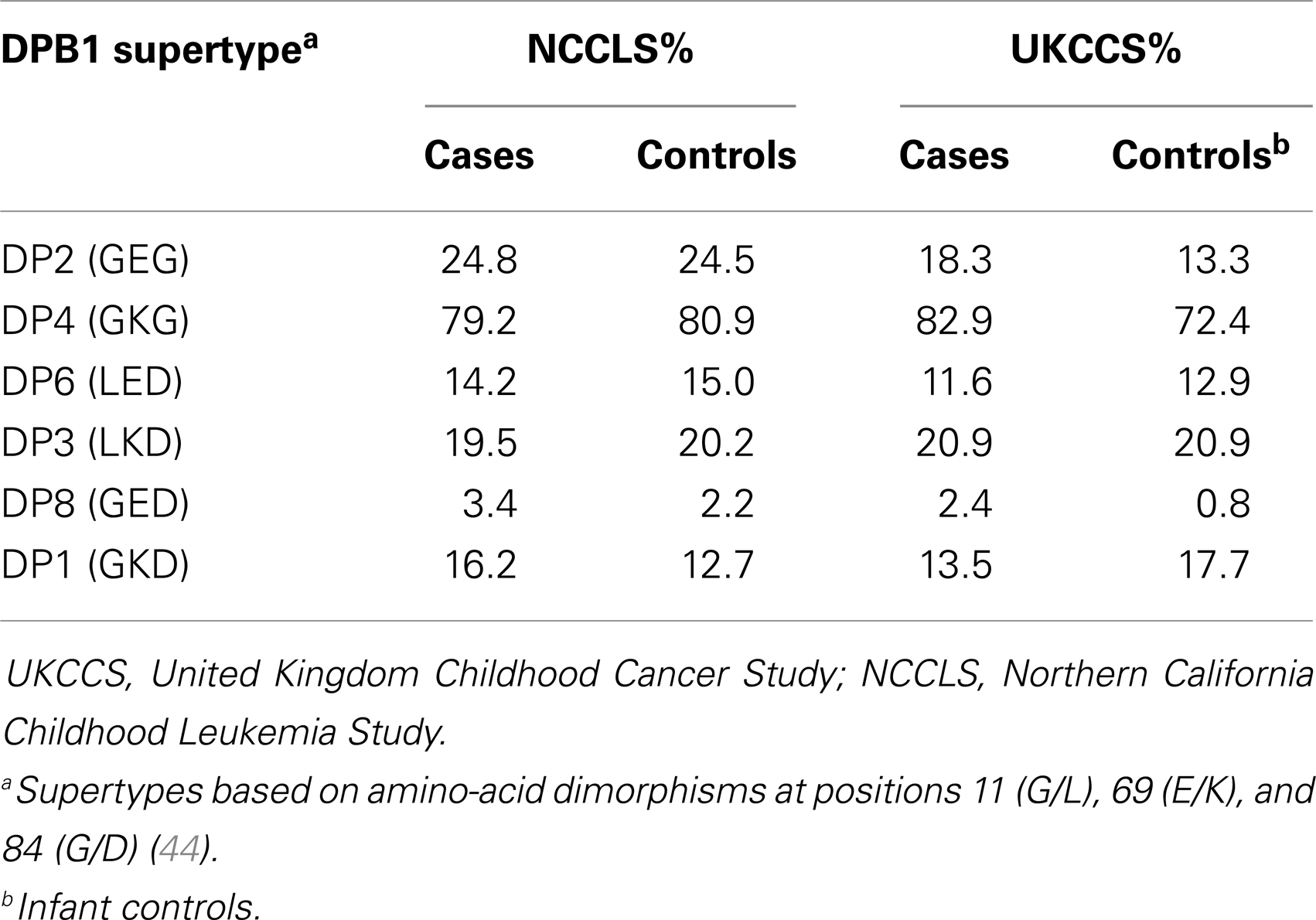
Table 2. HLA class II DPB1 supertype distributions (percentage of individuals carrying) reported in the UKCCS and NCCLS studies.
Further investigation of the association of the DP6 supertype with leukemia (45) found that, of the seven alleles included in this supertype, only DPB1*06:01 was significantly associated with childhood leukemia (corrected p-value = 0.02), including total ALL, AML, BCP-ALL, Pro-B ALL, and T-ALL. Further analyses indicated that, of all DPB1 alleles and supertypes, DPB1*06:01 was the most strongly associated with leukemia. The genotype DPB1*04:01/*06:01 is particularly strongly associated with ALL (OR, 95% CI; 10.6, 2.4–47.4). Transmission disequilibrium tests indicated that this allele was over-transmitted to children who go on to develop leukemia (76.9%; p-value = 0.03). Subsequent extended analysis of DPB1 allele transmission revealed no evidence of over- or under-transmission of DP1 or DP2 supertypes among UK leukemia cases, although there was a suggestion of under-transmission of other rare alleles (46).
More recently, Urayama et al. (47) investigated HLA-DP in relation to childhood leukemia in the Northern California Childhood Leukemia Study (NCCLS), which includes an ethnically heterogeneous sample consisting of two main populations; non-Hispanic white and Hispanic. This study found no gross differences in HLA-DPA1 allele frequencies or DPA1-DPB1 haplotypes between cases and controls. However, analyses conditioned on age, sex, and ethnicity identified a statistically significant association with DPB1*01:01. Using the supertype classification of Taylor et al. (44), this translated as an association of DP1, with increased ORs in both non-Hispanic whites and Hispanics, significant for high-hyperdiploid ALL (p-value = 0.005). In contrast, Taylor et al. found the DP1 supertype was associated with decreased risk. Importantly, as an extension beyond previous studies of HLA-DPB1 and childhood leukemia, the California study also examined interactions between proxies for delayed infections in childhood (daycare attendance, ear infections, and breastfeeding) with the observed DP1 association. Interactions were detected between DP1 and two proxies for early immune modulation, older sibling (p-value = 0.036), and breastfeeding (p-value = 0.094) in the risk of childhood ALL. Although the power to detect interactions between DP1 and daycare/ear infections in the first year was limited, the point estimates were in the expected direction should the association be related to delayed infection in childhood ALL, as hypothesized by Greaves (4).
The observation of opposing effects of the DP1 supertype on childhood ALL susceptibility in two apparently ethnically similar populations is intriguing, especially as both studies narrowed the significant associations to the high-hyperdiploid subtype of ALL. Comparison of supertype frequencies suggests heterogeneity between the two populations (Table 2). The DP1 supertype appears to be less common in the NCCLS non-Hispanic white controls (12.7%) compared with those employed in the UKCCS (17.7%). Conversely, the frequency of DP2 is lower among UKCCS controls (13.3%) compared to the NCCLS (24.5%) (Table 2). These underlying differences may indicate differences in selective pressure acting on the two geographically distinct populations, or alternatively be a consequence of other processes, such as genetic drift.
HLA-DQ
In comparison to the other class II loci, there are relatively few studies examining HLA-DQ in relation to childhood ALL risk. Two of the three previous studies were based on the same study population with an initial report by Dearden et al. (48) describing for the first time an ALL association with the DQB1*05:01 allele that was independent of their previously identified DPB1*02:01 effect. In their subsequent report, Taylor et al. (49) conducted a more detailed examination of HLA-DQ that included the tightly linked HLA-DQA1 locus. Their findings showed an increased risk associated with DQA1*0101/*0104 exclusively in males and a protective effect associated with HLA-DQA1*0201. A joint analysis of DQA1 and DQB1 indicated a strong effect associated with carriers of both DQA1*0101/*0104 and DQB1*0501 and was seen only in males. Interestingly, in a third DQB1 study conducted in Iranian childhood ALL patients and controls by Orouji et al. (50), a consistent predisposing effect was seen for DQB1*05 (DQ5), and additionally a protective and predisposing effect for DQB1*02:01 (DQ2) and DQ7 (DQB1*03:01, *03:04), respectively.
Candidate Gene Studies of Non-HLA Loci in xMHC
The vast majority of genes in the xMHC that have been studied with respect to childhood leukemia are HLA; however, a number of non-HLA genes in the xMHC region have also been examined. While in general these genes have not been studied in multiple populations, they highlight the potential importance of genetic variation in the xMHC in the etiology of childhood leukemia.
Han et al. (51) conducted a case-control study of the role of immune response genes in total childhood leukemia in Korea. Among the studied loci were two genes in or near the xMHC region, TNF and NFKBIE (6p21.1). Using gene-wise analytical methods to adjust for examination of multiple SNPs in each gene, the authors observed no significant associations for any of the genes included. This study included a relatively small sample size (63 cases and 148 controls) and examined total childhood leukemia, including ALL (59%) and AML (26%). Results for individual leukemia sub-types were not presented.
Similarly, in a study of xenobiotic transport and metabolism genes, Chokkalingam et al. (52) examined whether GPX6/GPXP3 (6p22.1) and TPMT (6p22.3) were associated with childhood ALL risk among 377 cases and 448 controls. The authors found no significant associations for the studied SNPs in these genes either individually or in haplotype analyses.
In a study of genes involved in p53 signaling, Do et al. (53) examined a number of loci within the xMHC region. Among 114 ALL cases and 414 controls, they found significant associations with SNPs in BAT3/BAG6, a molecular chaperone involved in transcriptional control of the p53 tumor suppressor. However, no associations were observed for other genes within the xMHC, including DAXX, LTA, DDR1, and IER3.
The human hemochromatosis gene (HFE) modulates serum iron levels, which have been linked to risk of several cancers. Interestingly, evidence linking risk of childhood ALL to high birthweight has been reasonably consistent (54), and it is possible that this observation may reflect or be modulated by serum iron levels. In a study of 163 ALL cases and 995 controls, Dorak et al. (55) explored this hypothesis and found that just one of 15 SNPs in HFE was significantly associated with childhood ALL risk. This SNP, rs9366637, in intron 1 of HFE, was also associated with birthweight. However, among the 14 other studied SNPs were two well-known functional SNPs, rs1800562, and rs1799945, which were not associated with risk of childhood ALL.
Heat shock proteins are molecular chaperones that may play a role in both adaptive and innate immune responses to tumors and virus-infected cells. Ucisik-Akkaya et al. (56) examined functional polymorphisms in three HSP genes that reside in the class III region: HSPA1L, HSPA1A, and HSPA1B. Among 114 ALL cases and 414 controls from the UK, they found that rs1061581, in HSPA1B, was significantly associated with childhood ALL risk. The association was replicated in a separate set of 100 ALL cases and 253 controls from Mexico (56). The authors noted that although this SNP is in LD with HLA-DRB3, which was associated with childhood ALL in the UK dataset, the HSPA1B association was independent of HLA-DRB3. SNPs in HSPA1L and HSPA1A showed no significant associations with childhood ALL risk. Using the same UK and Mexican populations, Morrison et al. (33) examined the role of multiple sclerosis susceptibility factors in childhood ALL risk. Included were two non-HLA genes in the xMHC region: SKIV2L and TNXB, which were not found to have significant single SNP associations with childhood ALL risk.
In summary, reports of childhood ALL risk associated with non-HLA loci within the xMHC provide limited evidence that the MHC may have relevance to childhood leukemia etiology above and beyond the HLA. The reports described above include a somewhat arbitrary and incomplete representation of the non-HLA genes in the xMHC. Furthermore, with the exception of the Morrison et al. and Ucisik-Akkaya et al. reports, which included independent replication populations, none of the genes described in this section were examined in more than one study population, and none were described in more than one report.
xMHC Association Mapping Studies
Hosking et al. (23) used data generated in the UKCCS genome-wide association study (GWAS) to conduct an analysis of BCP-ALL association across a 4.5-Mb xMHC region. They performed single SNP comparisons, alongside a sliding window haplotype analysis and imputation of HLA genotypes at the major class I (HLA-A, -B, and -C) and class II (HLA-DQA1, -DQB1, and -DRB1) loci, with data derived from UK population samples as controls (824 cases and 4,737 controls). No associations, significant after correction for multiple testing were observed; however, the strongest single SNP-based association was with rs3135034 (p-value = 0.0017; not significant after adjustment for multiple testing), which is approximately 92 kb telomeric of HLA-DPB1, between the BRD2 and HLA-DOA loci. In addition, after correction for multiple testing, no significant associations were observed with imputed HLA alleles. The strongest (non-significant) associations were found for A*26:01, A*29:02, B*14:02, C*02:02, and C*08:02 class I alleles and DQB1*02:01, DQB1*03:02, DQB1*03:03, and DRB1*13:02 class II alleles.
Recently a high-density SNP analysis of the MHC region in the NCCLS was conducted (57). In single SNP analyses this study identified one locus, rs9296068, as significantly associated with BCP-ALL risk after correction for multiple testing (OR = 1.40, 95% CI = 1.19–1.66, corrected p-value = 0.036). Of interest in the context of the UKCCS MHC study (23), this SNP lies only approximately 37 kb from rs3135034, the most strongly associated UK xMHC SNP, upstream of the HLA-DOA transcription start site. Sliding window haplotype analysis was also undertaken as part of this study, yielding two significantly associated MHC haplotypes, the first close to the TRIM27 gene in the extended class I region (rs1237485-rs3118361-rs2032502-rs7747023; nominal global p-value = 2.7 × 10−5) and the second including rs9296068 and flanking HLA-DOA (rs423639-rs7754316-rs9296068; nominal global p-value = 9.2 × 10−5). The haplotype showed little evidence of correlation with the DP1 supertype, previously found to be associated with ALL susceptibility in the same group of patients, and analysis adjusting for carriers of the DP1 supertype indicated an independent effect for rs9296068 (p-value = 3.0 × 10−4). In contrast to the Hosking et al. study, this investigation did not extend to the imputation of additional SNPs in the MHC region or the prediction of classical HLA types. This fact may have contributed to the identification of significant associations not apparent in the UK data; these may have been missed in the UK study as a result of the additional burden of correction for multiple testing.
Although the two SNPs identified in the UK and California BCP-ALL MHC association studies were physically close to one another, neither SNP showed any indication of association in the reciprocal studies. However, the two SNPs flank a strong meiotic recombination hotspot, known as DNA3 (58). It is established that commonly used methods of genetic association mapping would not be expected to identify associations under recombination hotspots, since they rely on positive LD, which hotspots lack by definition. Hence, it is possible that these two SNPs tag a HLA-DOA association in both populations, partially masked by the strong DNA3 hotspot.
This hypothesis led to another study, consisting of a detailed investigation of haplotypes and recombination rates in this region of the MHC in UK childhood ALL cases and controls (Thompson et al., submitted). Using the program PHASE (59) to predict haplotypes and recombination rates among UKCCS ALL patients and UK population controls from data generated in the UKCCS GWAS (60), this study found evidence for an association with recombination rates at DNA3 and childhood ALL. Moreover, a novel analysis using LDsplit (61) showed that loci on both sides of the hotspot, including rs9296068 and rs3135034, contribute to recombination at DNA3. Using data from an Icelandic study (62) the authors demonstrated that recombination at DNA3 is strongly affected by PRDM9 genotype, the relevance of which is underlined by a recent publication demonstrating a marked increase in the prevalence of rare PRDM9 alleles among parents of childhood B-cell ALL patients, and altered recombination patterns in patients, in two North American populations (63).
Evidence from these analyses indicates that there are two physically close, but weakly linked risk loci for childhood ALL at HLA-DOA and HLA-DPB1 in the MHC class II region, both substantiated by results from two separate studies in the UK and California. However, data from these studies are inconsistent in terms of the nature of the HLA-DPB1 allelic associations with risk of ALL and in the precise location of the association in the vicinity of HLA-DOA. The former is complicated by apparent underlying population differences in frequencies of HLA-DP, but may be attributable to geographic variations in exposures to antigens such as infectious agents. The weak but significant associations in both studies with loci flanking the DNA3 meiotic recombination hotspot are suggestive of an association with HLA-DOA, a molecule critical for HLA class II antigen processing, masked by the breakdown of LD in this region. The increased rates of recombination at DNA3 may further exacerbate the heterogeneity of genotypes at this locus. Previously identified functionally important variants within recombination hotspots have been associated with recombination mediated sequence alterations (64, 65); it remains to be determined whether this applies to HLA-DOA in the context of childhood ALL.
Conclusion and Future Directions
Results based on mostly low-resolution genotypes and HLA technology that has evolved over time, compounded by small study sizes and conducted independently across populations of different ancestral backgrounds, present challenges to fully interpreting the collective HLA evidence. Thus far, there appear to be no striking childhood ALL genetic susceptibility loci in the xMHC that are similar in nature and magnitude to those seen for autoimmune and infectious diseases. However, mounting evidence does suggest that more modestly associated susceptibility loci showing population and subtype specificity may exist. In particular, this is exemplified by multiple previous studies showing male-specific effects for DR53 and evidence of HLA-DPB1 BCP-ALL associations with potentially more detectable effects when in the presence of certain environmental exposures. A role for HLA-DPB1 or a gene localized to that region is also supported by two independent mapping studies of the xMHC demonstrating a need for targeted fine-mapping efforts. Further, a highly powered study of children with medically refractory ALL, those being considered for or going to stem cell transplantation, has revealed highly significant associations to both HLA class I (HLA-A and -B) and class II (HLA-DRB1), results which may shed light on the role of HLA in the less severe disease form of pediatric ALL. While this is a noteworthy study due to its methodological rigor that is unique over previous studies, validation in comparable and appropriately large study populations is still required.
There are several opportunities for improvement in future studies given our currently expanded knowledge of the genetic complexities of the xMHC (14, 66, 67), technological advances in genomic characterization and high-resolution HLA genotyping (68), and increased recognition of the importance of international collaboration and independent validation (69). The genomic complexities of the xMHC, such as long-range LD that gives rise to extended haplotypes and extensive genetic variation of the region, are now well-characterized and facilitate the initial localization of genomic regions, but complicate the effort to fine-map causal genetic loci to disease risk (70). The classical HLA genes are the most widely studied of the xMHC loci and have been associated with dozens of diseases, but in many cases, including childhood ALL, it is not clear whether the associations can be unambiguously linked to the HLA gene or instead indicate an association with an adjacent causal locus in LD. To address this, comprehensive examination of genetic variability of the entire xMHC, including high-resolution HLA allelic typing, is needed and is currently achievable using recently developed technologies. High-density MHC SNP genotyping studies, two of which were described in this review (23, 57), are a step in this direction, but they lacked direct allelic typing of all classical HLA loci. The UKCCS applied previously derived methods (71, 72) to impute HLA alleles based on GWAS-derived SNPs for six of the nine classical HLA loci. While this is a reasonable alternative to a costly and intensive HLA allelic typing effort of all loci, whether such imputation methods can replace classical typing is still questionable and should be considered on an individual study basis depending on sample size, ancestral composition of the study population, and availability of SNP and HLA data from a suitable reference panel (73).
Methods for routine next-generation DNA sequencing-based HLA typing have recently been developed (68) and provide the highest quality and resolution of allelic types without the need for time-consuming efforts to resolve allelic ambiguities in heterozygotes. Full implementation of these methods has recently been applied to a case-control study of follicular lymphoma, demonstrating their feasibility for use in disease association studies (74). With rapidly falling costs, next-generation sequencing of the xMHC appears to be a feasible and efficient strategy for future comprehensive examination of the region in childhood ALL.
It should be noted, however, this ability to test a large number of markers simultaneously, including up to hundreds of high-resolution allelic types for a single HLA locus presents a major concern regarding increased probabilities of false-positive findings due to the multiple tests being conducted. A few available methods for addressing multiple testing include Sidak and Bonferroni corrections, controlling the false-discovery rate, and permutation-based methods (75, 76), but these criteria for defining a statistical significance threshold, in return, put a greater strain on statistical power and sample size requirements. Careful consideration of these requirements, particularly for a rare disease like childhood ALL that appears to benefit from gender-specific and gene-environment interaction evaluation, and the importance of independent replication, suggests the need for coordinated studies and collaboration to fully characterize the role of the xMHC in childhood ALL risk. The Childhood Leukemia International Consortium (CLIC2), which currently includes 22 epidemiological studies of childhood leukemia representing 12 countries, was formed in 2005 to facilitate collaborative efforts such as those needed for genetic studies (69). Furthermore, because the extensive LD patterns across the xMHC show population-specificity (70, 77) that vary in the breakdown of conserved sequences, the comparison of multiple homogenous populations of different ancestries can aid in the fine-mapping effort of disease loci (78).
Although the xMHC was one of the first candidate regions examined in childhood ALL over three decades ago, only during these last several years have sufficient advances in understanding of the xMHC been made to permit comprehensive and definitive studies in evaluating its role in childhood ALL. Given the persistent epidemiological support for an infection-related cause, the inconsistent but suggestive evidence of the xMHC and childhood ALL literature, and clear opportunities for application of the most recent knowledge and technologies, we believe that future studies are warranted and have tremendous potential for elucidating the role of this complex region in the genetic susceptibility of childhood ALL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the U.S. National Institute of Environmental Health Sciences (R01ES09137) (Kevin Y. Urayama, Anand P. Chokkalingam), the Children with Cancer UK (Pamela D. Thompson), U.S. National Institutes of Health (NIH 2 PO1 CA111412-06) (Elizabeth A. Trachtenberg), and the My Brother Joey Foundation (Elizabeth A. Trachtenberg).
Footnotes
References
1. Smith M, Ries L, Gurney J, Ross J. Leukemia. In: Ries L, Smith M, Gurney J, Linet M, Tamra T, Young J, editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute, SEER Program (1999). p. 17–34.
2. Wiemels J. Perspectives on the causes of childhood leukemia. Chem Biol Interact (2012) 196:59–67. doi:10.1016/j.cbi.2012.01.007
3. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol (2011) 29:551–65. doi:10.1200/JCO.2010.30.7405
4. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer (2006) 6:193–203. doi:10.1038/nrc1816
5. Gross L. “Spontaneous” leukemia developing in C3H mice following inoculation in infancy, with AK-leukemic extracts, or AK-embryos. Proc Soc Exp Biol Med (1951) 76:27–32. doi:10.3181/00379727-76-18379
7. Cooke JV. The incidence of acute leukemia in children. JAMA (1942) 119:547–50. doi:10.1001/jama.1942.02830240011004
8. MacKenzie J, Greaves MF, Eden TO, Clayton RA, Perry J, Wilson KS, et al. The putative role of transforming viruses in childhood acute lymphoblastic leukemia. Haematologica (2006) 91:240–3.
9. Kinlen L. Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet (1988) 2:1323–7. doi:10.1016/S0140-6736(88)90867-7
10. Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia (1988) 2:120–5.
11. Kinlen LJ. An examination, with a meta-analysis, of studies of childhood leukaemia in relation to population mixing. Br J Cancer (2012) 107:1163–8. doi:10.1038/bjc.2012.402
12. McNally RJ, Eden TO. An infectious aetiology for childhood acute leukaemia: a review of the evidence. Br J Haematol (2004) 127:243–63. doi:10.1111/j.1365-2141.2004.05166.x
13. Chang JS, Tsai CR, Tsai YW, Wiemels JL. Medically diagnosed infections and risk of childhood leukaemia: a population-based case-control study. Int J Epidemiol (2012) 41:1050–9. doi:10.1093/ije/dys113
14. The MHC Sequencing Consortium. Complete sequence and gene map of a human major histocompatibility complex. Nature (1999) 401:921–3. doi:10.1038/44853
15. Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, et al. Gene map of the extended human MHC. Nat Rev Genet (2004) 5:889–99. doi:10.1038/nrg1489
16. Robinson J, Waller MJ, Parham P, De Groot N, Bontrop R, Kennedy LJ, et al. IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Res (2003) 31:311–4. doi:10.1093/nar/gkg070
17. Parham P, Lomen CE, Lawlor DA, Ways JP, Holmes N, Coppin HL, et al. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc Natl Acad Sci U S A (1988) 85:4005–9. doi:10.1073/pnas.85.11.4005
18. Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, et al. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature (1994) 368:711–8. doi:10.1038/368711a0
19. Lilly F, Boyse EA, Old LJ. Genetic basis of susceptibility to viral leukaemogenesis. Lancet (1964) 2:1207–9. doi:10.1016/S0140-6736(64)91043-8
20. Walford RL, Finkelstein S, Neerhout R, Konrad P, Shanbrom E. Acute childhood leukaemia in relation to the HL-A human transplantation genes. Nature (1970) 225:461–2. doi:10.1038/225461a0
21. Harris R, Lawler SD, Oliver RT. The HLA system in acute leukaemia and Hodgkin’s disease. Br Med Bull (1978) 34:301–4.
22. Dausset J. The major histocompatibility complex in man. Science (1981) 213:1469–74. doi:10.1126/science.6792704
23. Hosking FJ, Leslie S, Dilthey A, Moutsianas L, Wang Y, Dobbins SE, et al. MHC variation and risk of childhood B-cell precursor acute lymphoblastic leukemia. Blood (2010) 117:1633–40. doi:10.1182/blood-2010-08-301598
24. Klitz W, Gragert L, Trachtenberg E. Spectrum of HLA associations: the case of medically refractory pediatric acute lymphoblastic leukemia. Immunogenetics (2012) 64:409–19. doi:10.1007/s00251-012-0605-5
25. Davey FR, Lachant NA, Dock NL, Hubbell C, Stockman JA III, Henry JB. HLA antigens and childhood acute lymphocytic leukaemia. Br J Haematol (1981) 47:211–20. doi:10.1111/j.1365-2141.1981.tb02781.x
26. von Fliedner VE, Merica H, Jeannet M, Barras C, Feldges A, Imbach P, et al. Evidence for HLA-linked susceptibility factors in childhood leukemia. Hum Immunol (1983) 8:183–93. doi:10.1016/0198-8859(83)90036-8
27. Muller CA, Hasmann R, Grosse-Wilde H, Vogeler U, Bei-Jun C, Dopfer R, et al. Significant association of acute lymphoblastic leukemia with HLA-Cw7. Genet Epidemiol (1988) 5:453–61. doi:10.1002/gepi.1370050609
28. Cameron ES, Labastide WB, Wilson V, Charles W, Holder Y, Reid HF. Immunophenotypic and HLA studies in childhood acute lymphoblastic leukemia in Trinidad, West Indies. Leukemia (1990) 4:615–9.
29. Ozdilli K, Oguz FS, Anak S, Kekik C, Carin M, Gedikoglu G. The frequency of HLA class I and II alleles in Turkish childhood acute leukaemia patients. J Int Med Res (2010) 38:1835–44. doi:10.1177/147323001003800531
30. D’Amaro J, Bach FH, Van Rood JJ, Rimm AA, Bortin MM. HLA C associations with acute leukaemia. Lancet (1984) 2:1176–8. doi:10.1016/S0140-6736(84)92741-7
31. Bortin MM, D’amaro J, Bach FH, Rimm AA, Van Rood JJ. HLA associations with leukemia. Blood (1987) 70:227–32.
32. Ghodsi K, Taylor GM, Gokhale DA, Dearden S, Stevens RF, Birch JM, et al. Lack of association between childhood common acute lymphoblastic leukaemia and an HLA-C locus dimorphism influencing the specificity of natural killer cells. Br J Haematol (1998) 102:1279–83. doi:10.1046/j.1365-2141.1998.00919.x
33. Morrison BA, Ucisik-Akkaya E, Flores H, Alaez C, Gorodezky C, Dorak MT. Multiple sclerosis risk markers in HLA-DRA, HLA-C, and IFNG genes are associated with sex-specific childhood leukemia risk. Autoimmunity (2010) 43:690–7. doi:10.3109/08916930903567492
34. de Moerloose P, Chardonnens X, Vassalli P, Jeannet M. HL-A D antigens from B-lymphocytes and susceptibility to certain diseases. Schweiz Med Wochenschr (1977) 107:1461.
35. von Fliedner VE, Sultan-Khan Z, Jeannet M. HLA-DRw antigens associated with acute leukemia. Tissue Antigens (1980) 16:399–404. doi:10.1111/j.1399-0039.1980.tb00321.x
36. Dorak MT, Owen G, Galbraith I, Henderson N, Webb D, Mills KI, et al. Nature of HLA-associated predisposition to childhood acute lymphoblastic leukemia. Leukemia (1995) 9:875–8.
37. Dorak MT, Lawson T, Machulla HK, Darke C, Mills KI, Burnett AK. Unravelling an HLA-DR association in childhood acute lymphoblastic leukemia. Blood (1999) 94:694–700.
38. Dorak MT, Oguz FS, Yalman N, Diler AS, Kalayoglu S, Anak S, et al. A male-specific increase in the HLA-DRB4 (DR53) frequency in high-risk and relapsed childhood ALL. Leuk Res (2002) 26:651–6. doi:10.1016/S0145-2126(01)00189-8
39. Yari F, Sobhani M, Sabaghi F, Zaman-Vaziri M, Bagheri N, Talebian A. Frequencies of HLA-DRB1 in Iranian normal population and in patients with acute lymphoblastic leukemia. Arch Med Res (2008) 39:205–8. doi:10.1016/j.arcmed.2007.09.009
40. Wang XJ, Ai XF, Sun HY, Wang F, Xu SC, Gui FM, et al. Relation of HLA-DRB1*15 with pathogenesis in 162 childhood cases of acute lymphoblastic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2009) 17:1507–10.
41. Hensiek AE, Sawcer SJ, Feakes R, Deans J, Mander A, Akesson E, et al. HLA-DR 15 is associated with female sex and younger age at diagnosis in multiple sclerosis. J Neurol Neurosurg Psychiatry (2002) 72:184–7. doi:10.1136/jnnp.72.2.184
42. Taylor GM, Robinson MD, Binchy A, Birch JM, Stevens RF, Jones PM, et al. Preliminary evidence of an association between HLA-DPB1*0201 and childhood common acute lymphoblastic leukaemia supports an infectious aetiology. Leukemia (1995) 9:440–3.
43. Taylor GM, Dearden S, Ravetto P, Ayres M, Watson P, Hussain A, et al. Genetic susceptibility to childhood common acute lymphoblastic leukaemia is associated with polymorphic peptide-binding pocket profiles in HLA-DPB1*0201. Hum Mol Genet (2002) 11:1585–97. doi:10.1093/hmg/11.14.1585
44. Taylor GM, Hussain A, Lightfoot TJ, Birch JM, Eden TO, Greaves MF. HLA-associated susceptibility to childhood B-cell precursor ALL: definition and role of HLA-DPB1 supertypes. Br J Cancer (2008) 98:1125–31. doi:10.1038/sj.bjc.6604257
45. Taylor GM, Hussain A, Verhage V, Thompson PD, Fergusson WD, Watkins G, et al. Strong association of the HLA-DP6 supertype with childhood leukaemia is due to a single allele, DPB1*0601. Leukemia (2009) 23:863–9. doi:10.1038/leu.2008.374
46. Taylor M, Bergemann TL, Hussain A, Thompson PD, Spector L. Transmission of HLA-DP variants from parents to children with B-cell precursor acute lymphoblastic leukemia: log-linear analysis using the case parent design. Hum Immunol (2011) 72:897–903. doi:10.1016/j.humimm.2011.05.011
47. Urayama KY, Chokkalingam AP, Metayer C, Ma X, Selvin S, Barcellos LF, et al. HLA-DP genetic variation, proxies for early life immune modulation and childhood acute lymphoblastic leukemia risk. Blood (2012) 120:3039–47. doi:10.1182/blood-2012-01-404723
48. Dearden SP, Taylor GM, Gokhale DA, Robinson MD, Thompson W, Ollier W, et al. Molecular analysis of HLA-DQB1 alleles in childhood common acute lymphoblastic leukaemia. Br J Cancer (1996) 73:603–9. doi:10.1038/bjc.1996.104
49. Taylor GM, Dearden S, Payne N, Ayres M, Gokhale DA, Birch JM, et al. Evidence that an HLA-DQA1-DQB1 haplotype influences susceptibility to childhood common acute lymphoblastic leukaemia in boys provides further support for an infection-related aetiology. Br J Cancer (1998) 78:561–5. doi:10.1038/bjc.1998.540
50. Orouji E, Tavakkol Afshari J, Badiee Z, Shirdel A, Alipour A. Association between HLA-DQB1 gene and patients with acute lymphoblastic leukemia (ALL). Int J Hematol (2012) 95:551–5. doi:10.1007/s12185-012-1051-8
51. Han S, Koo HH, Lan Q, Lee KM, Park AK, Park SK, et al. Common variation in genes related to immune response and risk of childhood leukemia. Hum Immunol (2012) 73:316–9. doi:10.1016/j.humimm.2011.12.018
52. Chokkalingam AP, Metayer C, Scelo GA, Chang JS, Urayama KY, Aldrich MC, et al. Variation in xenobiotic transport and metabolism genes, household chemical exposures, and risk of childhood acute lymphoblastic leukemia. Cancer Causes Control (2012) 23:1367–75. doi:10.1007/s10552-012-9947-4
53. Do TN, Ucisik-Akkaya E, Davis CF, Morrison BA, Dorak MT. TP53 R72P and MDM2 SNP309 polymorphisms in modification of childhood acute lymphoblastic leukemia susceptibility. Cancer Genet Cytogenet (2009) 195:31–6. doi:10.1016/j.cancergencyto.2009.05.009
54. Caughey RW, Michels KB. Birth weight and childhood leukemia: a meta-analysis and review of the current evidence. Int J Cancer (2009) 124:2658–70. doi:10.1002/ijc.24225
55. Dorak MT, Mackay RK, Relton CL, Worwood M, Parker L, Hall AG. Hereditary hemochromatosis gene (HFE) variants are associated with birth weight and childhood leukemia risk. Pediatr Blood Cancer (2009) 53:1242–8. doi:10.1002/pbc.22236
56. Ucisik-Akkaya E, Davis CF, Gorodezky C, Alaez C, Dorak MT. HLA complex-linked heat shock protein genes and childhood acute lymphoblastic leukemia susceptibility. Cell Stress Chaperones (2010) 15:475–85. doi:10.1007/s12192-009-0161-6
57. Urayama KY, Chokkalingam AP, Metayer C, Hansen H, May S, Ramsay P, et al. SNP association mapping across the extended major histocompatibility complex and risk of B-cell precursor acute lymphoblastic leukemia in children. PLoS One (2013) 8:e72557. doi:10.1371/journal.pone.0072557
58. Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet (2001) 29:217–22. doi:10.1038/ng1001-217
59. Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics (2003) 165:2213–33.
60. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet (2009) 41:1006–10. doi:10.1038/ng.430
61. Zheng J, Khil PP, Camerini-Otero RD, Przytycka TM. Detecting sequence polymorphisms associated with meiotic recombination hotspots in the human genome. Genome Biol (2010) 11:R103. doi:10.1186/gb-2010-11-10-r103
62. Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature (2010) 467:1099–103. doi:10.1038/nature09525
63. Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, et al. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res (2013) 23:419–30. doi:10.1101/gr.144188.112
64. Ng SK, Lo WS, Pun FW, Zhao C, Yu Z, Chen J, et al. A recombination hotspot in a schizophrenia-associated region of GABRB2. PLoS One (2010) 5:e9547. doi:10.1371/journal.pone.0009547
65. Tan IK, Mackin L, Wang N, Papenfuss AT, Elso CM, Ashton MP, et al. A recombination hotspot leads to sequence variability within a novel gene (AK005651) and contributes to type 1 diabetes susceptibility. Genome Res (2010) 20:1629–38. doi:10.1101/gr.101881.109
66. Allcock RJ, Atrazhev AM, Beck S, De Jong PJ, Elliott JF, Forbes S, et al. The MHC haplotype project: a resource for HLA-linked association studies. Tissue Antigens (2002) 59:520–1. doi:10.1034/j.1399-0039.2002.590609.x
67. Abecasis GR, Auton A, Brooks LD, Depristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature (2012) 491:56–65. doi:10.1038/nature11632
68. Bentley G, Higuchi R, Hoglund B, Goodridge D, Sayer D, Trachtenberg EA, et al. High-resolution, high-throughput HLA genotyping by next-generation sequencing. Tissue Antigens (2009) 74:393–403. doi:10.1111/j.1399-0039.2009.01345.x
69. Metayer C, Milne E, Clavel J, Infante-Rivard C, Petridou E, Taylor M, et al. The childhood leukemia international consortium. Cancer Epidemiol (2013) 37:336–47. doi:10.1016/j.canep.2012.12.011
70. de Bakker PI, Mcvean G, Sabeti PC, Miretti MM, Green T, Marchini J, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet (2006) 38:1166–72. doi:10.1038/ng1885
71. Leslie S, Donnelly P, Mcvean G. A statistical method for predicting classical HLA alleles from SNP data. Am J Hum Genet (2008) 82:48–56. doi:10.1016/j.ajhg.2007.09.001
72. de Bakker PI, Raychaudhuri S. Interrogating the major histocompatibility complex with high-throughput genomics. Hum Mol Genet (2012) 21:R29–36. doi:10.1093/hmg/dds384
73. Huang L, Li Y, Singleton AB, Hardy JA, Abecasis G, Rosenberg NA, et al. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet (2009) 84:235–50. doi:10.1016/j.ajhg.2009.01.013
74. Skibola CF, Akers NK, Conde L, Ladner M, Hawbecker SK, Cohen F, et al. Multi-locus HLA class I and II allele and haplotype associations with follicular lymphoma. Tissue Antigens (2012) 79:279–86. doi:10.1111/j.1399-0039.2012.01845.x
75. Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol (2000) 279:R1–8.
76. Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol (2008) 32:361–9. doi:10.1002/gepi.20310
77. Miretti MM, Walsh EC, Ke X, Delgado M, Griffiths M, Hunt S, et al. A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am J Hum Genet (2005) 76:634–46. doi:10.1086/429393
Keywords: childhood leukemia, epidemiology, genetic susceptibility, human leukocyte antigen, major histocompatibility complex
Citation: Urayama KY, Thompson PD, Taylor M, Trachtenberg EA and Chokkalingam AP (2013) Genetic variation in the extended major histocompatibility complex and susceptibility to childhood acute lymphoblastic leukemia: a review of the evidence. Front. Oncol. 3:300. doi: 10.3389/fonc.2013.00300
Received: 06 August 2013; Paper pending published: 18 October 2013;
Accepted: 26 November 2013; Published online: 12 December 2013.
Edited by:
Sophia S. Wang, City of Hope, USAReviewed by:
Sophia S. Wang, City of Hope, USAQian Li, Icahn School of Medicine at Mount Sinai, USA
Copyright: © 2013 Urayama, Thompson, Taylor, Trachtenberg and Chokkalingam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin Y. Urayama, School of Public Health, University of California, 1995 University Avenue, Suite 460, Berkeley, CA 94704, USA e-mail: kurayama@luke.or.jp