 Harry R. Haynes
Harry R. Haynes Sandra Camelo-Piragua
Sandra Camelo-Piragua Kathreena M. Kurian
Kathreena M. Kurian- 1Department of Neuropathology, Frenchay Hospital, Bristol, UK
- 2Department of Neuropathology, University of Michigan, Ann Arbor, MI, USA
It is increasingly clear that both adult and pediatric glial tumor entities represent collections of neoplastic lesions, each with individual pathological molecular events and treatment responses. In this review, we discuss the current prognostic biomarkers validated for clinical use or with future clinical validity for gliomas. Accurate prognostication is crucial for managing patients as treatments may be associated with high morbidity and the benefits of high risk interventions must be judged by the treating clinicians. We also review biomarkers with predictive validity, which may become clinically relevant with the development of targeted therapies for adult and pediatric gliomas.
Introduction
At present, histological grading represents the most reliable, accepted overall indicator for the clinical outcome in adult and pediatric glioma patients (1). Gliomas are histologically classified according to the World Health Organization (WHO) classification system, which assigns a malignancy Grade I–IV (2). Traditionally, the higher the grade of gliomas, the worse the prognosis.
Primary glioblastoma (previously referred to as glioblastoma multiforme or GBM) develop de novo. This contrasts with secondary GBM, which undergo progressive malignant transformation from low grade diffuse astrocytoma (WHO Grade II) or anaplastic astrocytoma (WHO Grade III) (3). It has been estimated that 5% of all GBMs are secondary lesions (4). Secondary GBM patients are approximately 15 years younger than those with primary GBM and have a significantly better clinical outcome (5). Although, primary GBM cannot reliably be distinguished histologically from secondary GBM (6), their genetic profiles differ substantially as discussed below. This raises the possibility that they develop from differing neural precursor cells (7).
It is increasingly clear that GBM can be subdivided into tumor groups, which have fundamentally differing molecular drivers and varying treatment responses (8). The Cancer Genome Atlas (TCGA) project aims to establish the genetic and expression profiles for a wide range of tumors (9). Based on this and other profiling, GBM has been divided into four putative subtypes: proneural, neural, classical, and mesenchymal each with a unique biological behavior (10). However, Philips et al. identified alternative prognostic subclasses of high-grade astrocytomas: proneural, proliferative, and mesenchymal (11). The proneural subclass was enriched for neuronal lineage markers, occurred in younger patients and has a better prognosis. The proliferative and mesenchymal markers expressed neural stem cell markers and have worse clinical outcomes. Recurrent gliomas have been observed to shift expression patterns toward the mesenchymal subclass (12). The Philips proneural subtype resembles subsets from the TCGA proneural and neural subtypes, the Philips proliferative group resembles subsets from the TCGA proneural and classical. The mesenchymal subtypes overlap in both studies (13, 14).
The most recent data from the TCGA network has combined copy-number, DNA methylation, protein, mRNA, and microRNA (miRNA) expression profiles of 543 GBMs plus exome DNA sequencing in 291 glioblastomas (15). The key findings of this data will be discussed throughout this review.
It is essential that new tools are developed that better delineate the biological variants of gliomas. Without this more finessed approach, treatment targets may be missed and patients given toxic therapies not sufficiently targeted to their glioma subtype. Such a biomarker approach means that patients with the same histological diagnosis, tumor location, and co-morbidities may receive differing therapy based on the molecular characteristics of their tumors (16).
A prognostic biomarker is a tumor-specific trait that predicts clinical outcome regardless of treatment given. Conversely, a predictive biomarker predicts clinical response to a specific treatment or drug class (17). Using such biomarkers will allow personalized prognostic estimates to be given to patients with a subsequent customized treatment regime, maximizing effectiveness, and minimizing toxicity. Prognostic biomarkers can also be used for patient stratification in clinical trial design, to ensure balance in the arms of randomized control trials of novel glioma therapies (18).
In this review (see Figure 1 and Table 1), we will consider recent advances in our understanding of adult and pediatric gliomas, highlighting both the prospectively validated prognostic and predictive biomarkers (other than tumor grade and morphology) in current use. Additionally, putative biomarkers will be discussed with relevance to both future clinical utility and our current understanding of gliomagenesis.
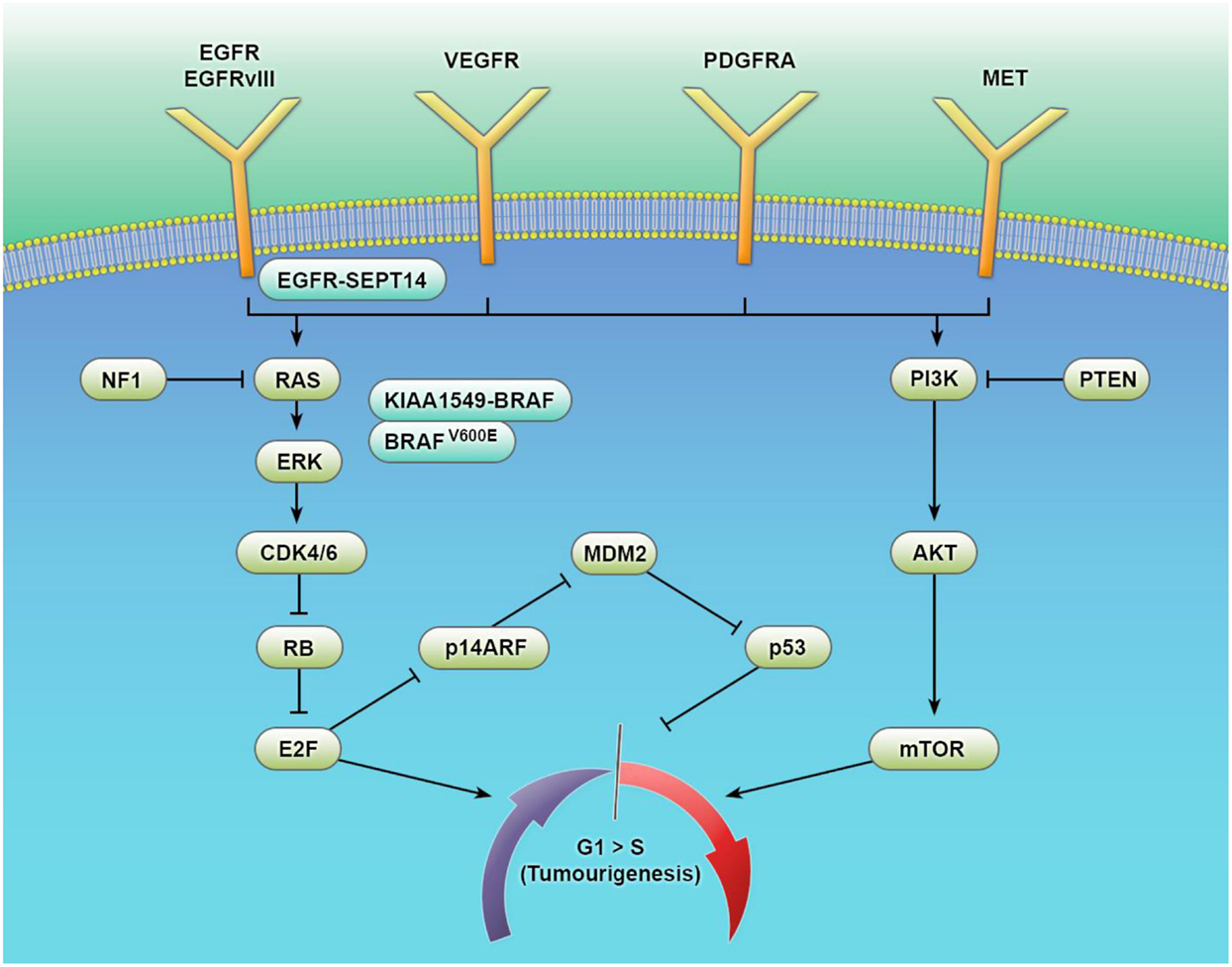
Figure 1. Key signaling pathways in the tumorigenesis of gliomas. The biological behavior of glial tumors is traditionally based on histological typing and grading. Histology is increasingly being supplemented using molecular information on genes, epigenetic markers, transcriptional regulators, and complex gene signatures. This multitude of events converges on a number of common signaling pathways with considerable cross talk. Increased understanding of these pathways and their interactions is facilitating a biomarker-driven approach to glial tumor biology – improving diagnosis, prognostic estimation, and the development of targeted therapies.
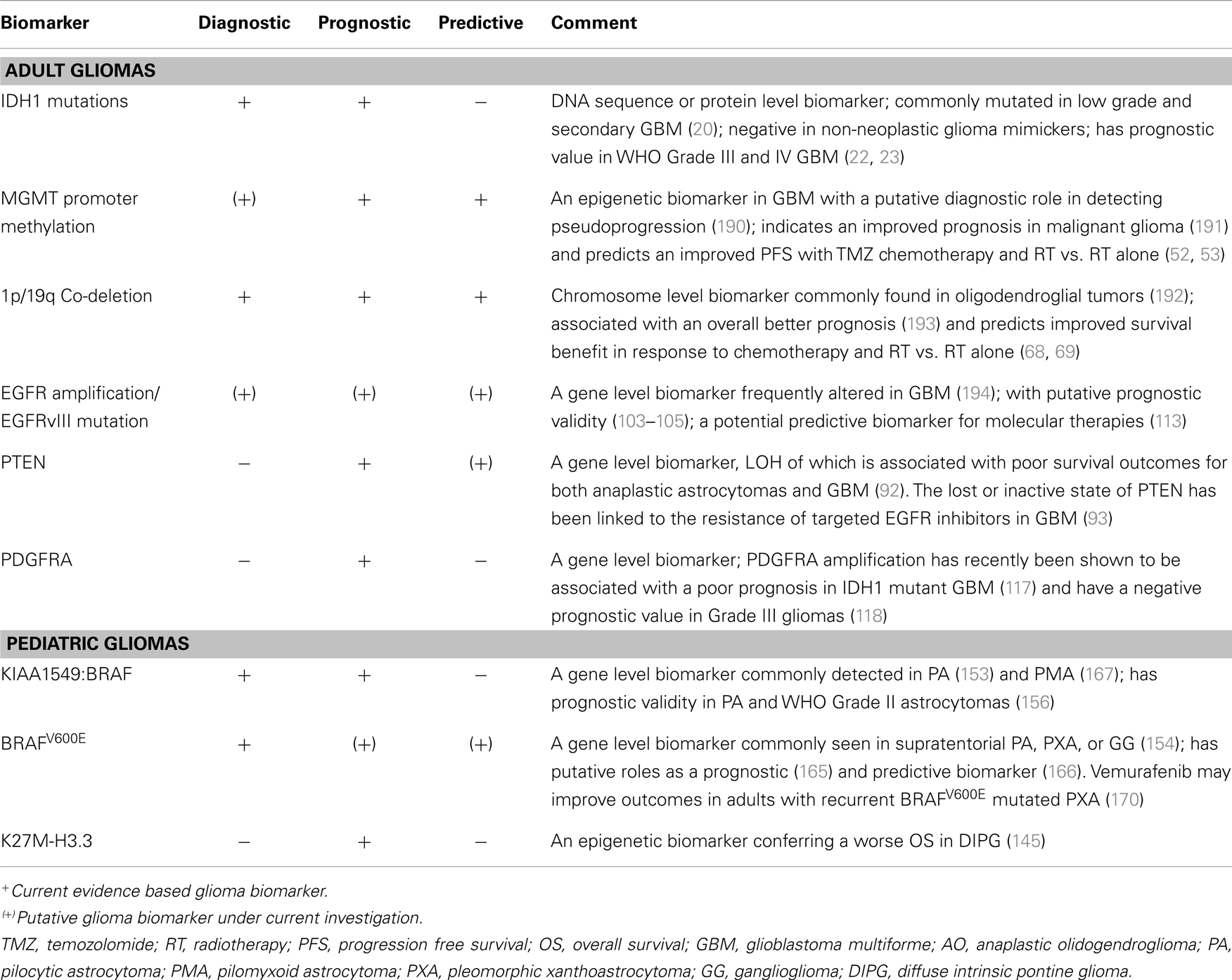
Table 1. Tissue biomarkers with current clinical or promising relevance in malignant glioma.
Biomarkers in Adult Gliomas
Primary vs. Secondary Gliomas and Isocitrate Dehydrogenase (IDH1 and IDH2)
Isocitrate dehydrogenases are members of a group of enzymes involved in energy metabolism that catalyze the decarboxylation of isocitrate (ICT) into α-ketoglutarate (19). Several studies have confirmed that IDH1 is mutated in approximately 60–80% of diffusely infiltrating gliomas (WHO Grade II and III) and in secondary GBMs, which may derive from them (20). However <10% of primary GBM carry this mutation (20, 21).
Although patients with IDH1 mutations are generally younger, a multivariate analysis has confirmed that IDH1 mutation can be considered a favorable independent prognostic marker in Grade III and IV gliomas, but not in WHO Grade II astrocytomas (22, 23). IDH2 (isoenzyme) mutations are less common than IDH1 mutations and confer a similar improved prognosis, but as yet the precise mechanism for this has not been elucidated (20, 24).
Watanabe et al. reported that IDH1 mutations always precede TP53 mutations or 1p/19q loss (see below) in a large cohort of Grade II/III and secondary gliomas (25). Disruption of the p53 pathway disruption is frequent in gliomagenesis [dysregulated in 85.3% of GBM according to the latest TCGA findings (15)] and TP53 mutations play a particularly important role in the development of secondary GBM (26). The prognostic significance of mutated TP53 in GBM is unclear (27, 28). However, it has been shown that mutant TP53 is associated with increased activity of a homologous recombination-mediated mechanism called alternative lengthening of telomeres (ALT). This confers a more benign biology and favorable prognosis (29, 30).
The ALT phenotype has been positively correlated with IDH mutations, TP53 mutations, and mutations of the alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene in astrocytomas but not in the oligodendroglial tumor lineage (31, 32). ATRX encodes a subunit of the chromatin remodeling complex, which is key for H3.3 incorporation into heterochromatin at centromeres and telomeres (33).
In oligodendrogliomas, a high frequency of TERT promotor mutations have been reported (34), occurring concurrently with IDH mutations and total 1p/19q loss [resulting in the loss of the tumor suppressor genes CIC and far upstream element binding protein 1 (FUBP1) – see below] (35). TERT is a catalytic subunit of telomerase and occurs mutually exclusively with ATRX mutations (34). This suggests that ATRX and TERT promotor mutations serve as alternative mechanisms for telomere lengthening.
This recent evidence points to two recurrent genetic signatures in gliomas. The first group progresses along an astrocytic lineage with ALT, alterations in ATRX, TP53, and IDH. The second group has a strong oligodendroglial component and carries IDH mutations and alterations in either CIC and FUBP1 and/or 1p/19q loss. The latter group has the longest median overall survival (OS) (36).
These finding support current theories that IDH1 mutation occurs early in gliomagenesis and may affect a glial proneural cell population that can give rise to both astrocytes (with ATRX and TP53 mutations) and oligodendrocytes (with TERT mutations and 1p/19q co-deletions) (37). Most recently, TERT mutations have been shown to have a significant overlap with IDH1 wild type primary GBM (38), suggesting that alternative progenitor cells give rise to these tumors.
A remarkable study showed that the R132H IDH1 point mutation results in the production of a different functional metabolite called 2-hydroxyglutarate (2-HG) (39). Increased 2-HG is known to inhibit histone demethylation (40), in part because histone demethylases and TET 5-methylcytosine hydroxylases are α-ketoglutarate-dependent dioxygenases involved in epigenetic control (41). Mutated IDH1 and increased 2-HG may therefore pre-dispose glioma cells to DNA hypermethylation.
Extensive DNA hypermethylation can be part of the so-called glioma CpG island methylator phenotype (G-CIMP) (42). A recent study reported a significant association between IDH mutation and methylation status (determined by the H3k9me3 methylation mark) in Grade II but not Grade III (anaplastic) astrocytomas or glioblastomas (43). Furthermore, H3K9me3-positive Grade II oligodendrogliomas showed improved OS when compared with H3K9me3-negative cases (43).
Glioma CpG island methylator phenotype GBM has been shown to be associated with younger patients, with IDH1 mutations and an improved prognosis, clustering in the TCGA proneural subgroup (44). The G-CIMP phenotype therefore appears to be a feature of lower grade gliomas and provides a molecular definition of secondary glioblastoma (45). It may be possible in the future to monitor mutant IDH1 glioma-associated 2-HG using non-invasive MR spectroscopy (46). Furthermore, the early role of mutant IDH in neoplastic transformation has lead to development of mutant IDH specific inhibitors (47). IDH could therefore assume an additional role as a predictive biomarker.
Epigenetic Modifiers of Glioblastoma – MGMT Promoter Methylation and miRNAs
Recently, epigenetic modifiers, such as hypermethylation have been implicated in the significant differences in glioma response to treatment (48). Of particular importance in glioblastoma is the DNA repair protein O6-methlyguanine methyl transferase – MGMT – (10q26). Active MGMT is able to remove alkyl groups from DNA, thereby reducing the efficacy and promoting cancer cell resistance to alkylating chemotherapeutic agents such as temozolomide (TMZ) (49).
Following on from the work of Esteller et al. (50), a landmark paper by Hegi et al. showed that MGMT promoter methylation silences gene expression in 45% cases of GBM. (51). This results in a significantly better prognosis for patients.
Following this discovery, the EORTC-NCIC trial and other studies have shown that MGMT promoter methylation increases progression free survival (PFS) in cases of GBM treated with TMZ and radiotherapy (RT) vs. RT alone (52, 53). Variability in methylation load at individual CpG sites also seems to affect PFS and OS in patients receiving TMZ (54).
In addition, the prospective randomized NOA-08 (55) and Nordic (56) trials showed that MGMT promoter methylation in elderly patients (who traditionally receive RT alone for malignant astrocytoma) correlates with longer event free survival and OS (respectively) when treated with TMZ. The latest TCGA findings suggest that MGMT promoter methylation may only have such predictive validity in classical subtype GBM (15). A multivariable analysis has additionally shown that the MGMT promoter methylation (MGMT-STP27) status and G-CIMP phenotype have a significant prognostic role in anaplastic oligodendrogliomas/oligoastrocytomas and are predictive of OS outcomes when treated with adjuvant procarbazine/CCNU/vincristine (PCV) (see below) chemotherapy (57).
Further epigenetic regulation is imposed on gliomas by miRNAs exerting complex effects on cancer gene networks (12). miRNAs may exert tumor suppressive or oncogenic functions through the post-transcriptional regulation of gene expression. miR-21 was the first miRNA to be investigated in gliomas (58) and is elevated in gliomas compared to healthy cortex (59). miR-21 acts as an oncogenic miRNA, inhibiting matrix metalloproteinase regulators and promoting glioma cell migration (60). miR-181a/b/c were originally found to be downregulated in glioblastoma (61). Of note, downregulation of miR-181b and miR-181c has been associated with clinical response to RT and TMZ when compared to patients with progressive disease (62). miR-181d has been shown to exert a suppressive effect on MGMT expression with a corresponding inverse association with TMZ response in GBM (63). Most recently, this has been reported to occur due to the effect of miR-181d on MGMT protein translation, downregulating MGMT expression independently of promoter methylation (64). miRNA data has also identified GBM subclasses within the TCGA, with significant clinical differences (65). Interestingly, miRNAs (such as miR-9) were shown to regulate subclass-specific gene expression and thus characterize and contribute to the phenotypic diversity of glioblastoma subclasses. Full discussion of miRNAs implicated in glioma initiation and progression are out with the scope of this review. Nevertheless, miRNAs present exciting opportunities for further biomarker research with direct application for patient stratification in clinical trials or as future therapeutic agents (58).
Co-Deletion of Chromosomes 1p/19q in Oligodendrogliomas and Gross Chromosomal Alterations in GBM
Loss of the short arm of chromosome 1 and the long arm of chromosome 19 (co-deletion of 1p/19q) is seen in >60% of Grade II/III oligodendroglial tumors (66, 67).
The recent findings of the major randomized clinical trial RTOG 9402 showed an OS benefit in response to PCV chemotherapy plus RT vs. RT alone in patients with 1p/19q co-deletions and both anaplastic oligodendroglioma and anaplastic oligoastrocytoma (68). A recent update of the similar EORTC 26951 trial confirmed that patients with 1p/19q co-deleted anaplastic oligodendroglial tumors showed an improved OS and PFS when treated with adjuvant PCV (69).
The cause for this survival benefit is still unclear. However, recently two candidate tumor suppressor genes have recently been discovered within the areas of chromosomal loss. FUBP1 and CIC (the homolog of the Drosophila capicua gene) have been identified on 1p and 19q, respectively (70, 71). FUBP1 is thought to be a dual (activator/inhibitor) transcriptional modulator of MYC (72). CIC is thought to act as a transcriptional repressor, regulating target gene expression of activated epidermal growth factor receptor (EGFR), Ras/Raf, and MAPK cancer pathways (73).
In previous studies, the combination of 1p and 19q loss in GBM is rare, and numbers are too small to reliably determine any survival benefit (28). However, loss of 1p alone has been observed in both primary and secondary GBM (74). Interestingly, a multivariate analysis found that LOH 1p alone was associated with longer GBM survival (75). Detailed studies have shown that 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumors and homozygously deleted in a subset of glioblastomas (76). It has been also been suggested that 19q LOH alone may confer a longer OS in patients with GBM (77).
The short arm of chromosome 9 contains the tumor suppressor genes CDKN2A and CDKN2B, which encode p14ARF, p16INK4A, and p15INK4B. These proteins have key roles in the p53 and RB pathways and hence, the G1 cell cycle checkpoint. LOH 9p is frequently seen in GBM samples (78, 79) and has been associated with a shorter OS (77) although other studies have not confirmed this finding (80). Interestingly, the absence of CDKN2A or CDKN2B deletion and a frequent loss of 17p (which includes p53) differentiate diffuse intrinsic pontine glioma from non-brainstem high-grade pediatric glioma (see later) (81).
Most recently, a small subset of GBMs has been reported as carrying chromosomal translocations fusing the tyrosine kinase domains of FGFR genes (FGFR1 or 3) to TACC1 or 3, respectively (82). The resulting fusion protein localizes to metaphase spindle poles, inducing aneuploidy. An FGFR inhibitor (83) was shown to prolong survival in FGFR3-TACC3-initiated glioma mice models. This gives promise to FGFR-TACC rearrangements as putative predictive biomarkers for FGFR inhibitor response (84).
EGFR Activation, EGFRvIII Mutation, and Downstream Mediators of GBM Tumorigenesis
It is well known up to 65% of so-called primary glioblastomas show epidermal growth factor receptor (7p12) amplification, overexpression, and/or mutations of this pathway (10). Such glioblastomas are part of the “classic” expression subtype (10) occurring mutually exclusively with IDH-mutated secondary GBM (20). EGFR is a receptor tyrosine kinase. Ligand binding by EGF promotes receptor dimerization and autophosphorylation of the cytoplasmic domain (85). Such EGFR activation is thought to promote cellular proliferation via activation of the MAPK and PI3K-Akt pathways (86).
The small molecule inhibitors gefitinib and erlotinib compete with ATP to prevent phosphorylation and therefore, ameliorate oncogenic downstream signaling (87). However, this mitogenic signaling network shows significant modularity in GBM (88). Gliomas are therefore able to escape the need for receptor function by activating alternative oncogenic pathways when challenged by receptor targeted-agents (89), dramatically revealing the complexity of tyrosine kinase signaling in these tumors (85). This means that treatments targeted to multiple signaling hubs and inhibitors of compensatory signaling paths are needed to inhibit tumor growth and recurrence.
Previous trials of EGFR kinase inhibitor therapy have been unsuccessful (90), most likely due to the role of downstream mediators of the pathway, which can undergo mutation (91). For example, PTEN is a tumor suppressor gene located at 10q23. It functions to suppress the PI3K-Akt signaling pathway. LOH at 10q is associated with poor survival outcomes for both anaplastic astrocytomas and GBM (92) and the lost or inactive state of PTEN has been linked to the resistance of targeted EGFR inhibitors in GBM (93). However, in vitro studies have shown that PTEN deficient GBM cells show significant responses to combined mTOR/EGFR kinase inhibitor therapy (94, 95).
Further predictive response stratification may be possible through baseline measurements of downstream mTOR activators such as s6 kinase (96). Similarly in GBM patients, levels of phosphorylated Akt have been shown to be predictive of clinical response to erlotinib in EGFR amplified tumors (97, 98). This highlights the importance of carefully selected molecular determinants for rational trials of EGFR inhibitors in GBM. Downstream pathway inhibitors such as BMK120 – a pan PI3K inhibitor currently undergoing phase II trials in patients stratified by PTEN status (99) – may also have efficacy when used in combination with EGFR inhibitors. Furthermore, gain-of-function mutations in PI3KCA have been found in 15% of glioma samples (100) and pre-clinical studies have shown that dual PI3KCA/mTOR inhibitors augment the antiproliferative effects of EGFR inhibition (101). Such combination therapy could be used in concert with traditional chemo- and radiotherapy to abrogate alternative survival pathway activation and enhance cytotoxicity (102).
The evidence for EGFR amplification as an independent predictor for survival in GBM varies between studies (103–105). However, higher expression of the down stream mediator phosphorylated MAPK has been reported to independently confirm a lower OS in newly diagnosed GBM (106).
Of current interest is the EGFRvIII mutation, which is the most common EGFR mutation resulting from an in-frame deletion of 801bp spanning exons 2–7 (107). This leads to a constitutively active EGFR (107). This EGFRvIII mutation occurs in 20–30% of GBM patients and is detected exclusively in cases with EGFR amplification (108).
Interestingly, in vitro models have suggested that GBM cells expressing EGFRvIII are resistant to small molecule tyrosine kinase inhibitors (109). EGFRvIII signals to a mTOR complex 2 induced mechanism (110), thereby differing from the EGFR-mTOR complex 1 signaling axis, which may contribute to such therapy resistance. Nevertheless, co-expression of EGFRvIII and wild type PTEN has previously been shown to be associated with response to erlotinib in GBM patients (111). Conversely, erlotinib has been suggested to act synergistically with a c-Met inhibitor against in vitro models lacking PTEN (112). An ongoing trial of the EGFRvIII vaccine (Rindopepimut) CDX-110 has shown longer OS in patients treated after GBM resection (113).
Recently, an analysis of in-frame gene fusions in GBM has identified EGFR-SEPT14 fusions, which tend to cluster in the TCGA classical and mesenchymal subgroups, constitutively activate STAT3 signaling and confer mitogen-independent growth (114). EGFR-SEPT14-positive GBM xenograft cells additionally showed a significant response to lapatinib (114), indicating a predictive role for EGFR fusions in EGFR inhibitor clinical trials. The latest whole genome sequencing work from the TCGA has identified further complex rearrangements of the EGFR gene or structural variants of genes surrounding its locus (15). The prognostic and predictive effects of these rearrangements have yet to be elucidated.
In GBM, platelet-derived growth factor receptor alpha (PDGFRA) is the most commonly altered receptor tyrosine kinase after EGFR (10, 115). GBM with amplified PDGFRA have been shown to be associated with either amplified EGFR or amplified MET (the hepatocyte growth factor receptor) (116). PDGFRA amplification has recently been shown to be associated with a poor prognosis in IDH1 mutant GBM (117) and have a negative prognostic value in Grade III gliomas (118). In PDGFRA amplified gliomas two genetic rearrangements have been described – a gene fusion between kinase insert domain receptor (KDR) and the PDGRFA gene and PDGFRA (Δ8, 9), an intragenic deletion rearrangement (115). Both PDGFRA mutants behave as transforming oncogenes with elevated tyrosine kinase activity. Unfortunately, early phase clinical trials of dual tyrosine kinase and PDGFR inhibitors have not been successful (119), indicating that a better understanding of this signaling pathway is needed prior to further clinical testing.
Vascular Endothelial Growth Factor and Its Receptors
Vascular endothelial growth factor (VEGF) plays a key role in the malignant angiogenesis seen in GBM. Activated vascular endothelial growth factor and its receptors 2 (VEGFR2) activates the Ras-Raf and PI3K-Akt transduction pathway (120). Although >60% of GBM express VEGF by immunohistochemistry, VEGF expression does not correlate with survival in GBM (121). However, shorter PFS has been associated with increased VEGFR2 immunohistochemical expression at tumor recurrence (122).
Bevacizumab (Avastin) is a humanized monoclonal antibody against VEGF (123). The RTOG-0825 (US) and AVAGlio (Europe) trials are large randomized phase III trials using bevacizumab in addition to standard radio- and chemotherapy in newly diagnosed GBM (124). These trials showed no improvement in OS with bevacizumab (125, 126). However, subgroup and molecular analysis may still reveal positive results (127) as could prospective randomized trials of combination therapies that include bevacizumab (128).
The tyrosine kinase inhibitor sorafenib is currently being trialed in combination with mTOR inhibitors in patients with recurrent GBM (129) and with standard chemotherapy and RT in untreated GBM (130). Sorafenib has multi-target activity against VEGFR2, VEGFR3, PDGFRB, c-Kit, BRAF, and Raf (131). The clinical response outcomes have so far been disappointing in recurrent GBM (132). Sunitinib is an alternative multi-target inhibitor against VEGFR1-3, PDGFRA/B, FLT-3, c-Kit, and RET (133, 134). Unfortunately, results so far indicate no significant improvement in PFS in recurrent GBM (135).
Biomarkers in Pediatric Gliomas
Pontine, Hemispheric, and Thalamic Gliomas
Although pediatric GBM are morphologically indistinguishable from adult GBM, they have a unique biological signature (136). For example, while TP53 and PIK3CA mutations occur in all high grade gliomas (137), <10% of childhood GBM harbor EGFR amplifications or PTEN mutations (138, 139). Similarly, IDH mutations are found in <5% of pediatric GBM although become more common in adolescents (140).
K27M and G34V/R gain-of-function mutations in H3F3A, which encodes histone H3.3, have recently been studied in pediatric and young adult GBM (141, 142). H3.3 is known to modulate gene expression during brain development (143). H3.3 mutations were shown to be prevalent in pediatric cohorts and mutually exclusive with IDH mutations (142, 144). K27M mutations have been shown to occur in over 70% of pediatric diffuse intrinsic pontine gliomas (DIPG) and confer a worse OS when compared to patients with wild type H3.3 (145). Additionally, 80% of pediatric thalamic GBM carry this mutation (145). Indeed, K27M-H3.3 mutations characterize pediatric midline GBM (136). In cohorts of childhood brainstem and thalamic tumors, this mutation occurs commonly in the younger (median 10 years) age group (142, 145), overlaps significantly with TP53 but only co-occurs with ATRX mutations in approximately half of all cases (145). The K27M mutation has also been found in the related histone H3.1 in 18% of DIPG (141).
G34V/R H3.3 mutations have been noted to cluster in childhood hemispheric GBM (144, 145). Such supratentorial tumors occur in an older population (median 18 years) and almost always overlap with TP53 and ATRX mutations (142). Recently, it has been reported that loss-of-function mutations in the H3K36 trimethyltransferase SETD2 also occur in supratentorial high grade gliomas of older children and young adults and are mutually exclusive with H3.3 mutations (146).
How H3.3 mutations promote tumorigenesis through interference with chromatin function has not been fully elucidated. However, the K27M mutation has been shown to inhibit K27 methylation (associated with polycomb-mediated gene repression) (147) and K27 acetylation (which is present on active gene promoters) (148). Both H3.3 mutations may additionally alter the expression of neural development genes (149). Furthermore, future work on the role of H3K36 methylation in tumorigenesis and the effects of differing histone mutations may reveal roles as predictive biomarkers (146).
Pilocytic Astrocytomas: KIAA1549:BRAF and BRAFV600E
Pilocytic astrocytomas are the most common childhood brain tumor, accounting for approximately 20% of brain tumors under the age of 20 (150). Pilocytic astrocytomas are slow-growing, non-infiltrating tumors with WHO malignancy Grade I (2). They may occur throughout the central nervous system but are found most frequently in the posterior fossa and the hypothalamic/chiasmatic region. Gross surgical resection frequently results in a cure and over 90% of patients survive more than 10 years (151). However, local recurrence occurs in 10–20% of cases and the primary lesion, its recurrence and the subsequent treatment can cause considerable morbidity as well as neurocognitive and endocrine dysfunction (152).
Jones et al. (153) were the first to describe a tandem duplication at 7q34 in a high proportion of pilocytic astrocytomas. This rearrangement creates a fusion gene (KIAA1549:BRAF) with constitutive BRAF kinase activity and putative abnormal activation of MAPK/ERK pathway. KIAA1549 is an as yet uncharacterized gene.
Multiple exonic fusion combinations between KIAA1549 and BRAF have now been reported, all of which result in loss of the BRAF autoregulatory N-terminal domain while the C-terminal kinase domain is retained, resulting in constitutive activation of the oncogenic MAPK pathway (154). The frequency of this fusion gene in pilocytic astrocytomas reported in the literature varies from 50 to 100%, depending on the patient demographic (154). The genetic mechanism resulting in this fusion is the subject of ongoing research (155).
It was originally reported that there was no significant difference in survival at follow up of fusion positive vs. fusion negative pilocytic astrocytomas (153). Hawkins et al. later found that the KIAA1549:BRAF fusion was an independent prognostic marker for significantly improved 5 year PFS for pilocytic astrocytomas as well as Grade II diffuse and pilomyxoid astrocytomas (156).
The KIAA1549:BRAF fusion gene was initially thought to be highly specific for pilocytic astrocytomas (154) thereby suggesting a useful diagnostic biomarker for a tumor with varied and challenging histology. However, Lin et al. recently identified KIAA1549:BRAF in 36% of glioneuronal tumors studied as well as 33% of pilomyxoid astrocytomas (157). In addition, KIAA1549:BRAF has been identified in 9% of diffuse adult gliomas (pre-dominantly oligodendroglial neoplasms) (158). In a large proportion of these cases, KIAA1549:BRAF and IDH mutations were present as independent molecular events. This suggests that in a small proportion of adult glial tumors 7q34 rearrangements may contribute to Ras-RAF-ERK signaling dysregulation. This has important implications for the design of novel therapies.
The above discussion highlights the single pathway nature of pilocytic astrocytomas – a disease driven by abnormal MAPK/ERK pathway activation. Indeed, recent evidence has highlighted further oncogenic “hits” driving this pathway. Two new fusion genes involving the kinase domain of the known oncogene NTRK2 and point mutations within the kinase domain of FGFR1 have been identified (159). All FGFR1-mutant tumors are extracerebellar and may be further modified by co-expression of the mutated phosphatase gene PTPN11 to upregulate phosphorylated ERK (159). NTRK2, FGFR1, and PTPN11 are likely to act as upstream drivers of MAPK/ERK pathway activation and may be responsible for the paradoxical pathway activation seen in KIAA1549:BRAF tumors treated with BRAF inhibitors (160). These genetic changes could therefore become key as predictive biomarkers for stratifying patients in ongoing trials of combination FGFR, NTRK2, and/or MAPK/ERK kinase inhibitors (161, 162).
Infratentorial posterior fossa pilocytic astrocytomas tend to display a high frequency of the KIAA1549:BRAF fusion (163). KIAA1549:BRAF fusion positive pilocytic astrocytomas also occur less frequently with increasing age (164). Supratentorial tumors are less frequently fusion positive but have an increased frequency of the oncogenic BRAFV600E mutation (154), a putative pediatric low grade prognostic biomarker (165), and promising predictive biomarker (166).
Additionally, BRAF V600E is associated with 60–80% of pleomorphic xanthoastrocytomas (PXA, WHO Grade II) across all age groups (167). These tumors do not have concomitant 7q34 BRAF duplications (168), indicating a potential diagnostic role for the BRAFV600E mutation. A BRAFV600E mutation specific antibody is available (169) and a recent small study suggested that vemurafenib (a BRAF inhibitor with significant activity against BRAF mutated metastatic melanoma) may improve outcomes in adults with recurrent BRAFV600E mutated PXA (170). BRAFV600E is also reported in 20–25% of pediatric and adult gangliogliomas (171) [or higher (172)] and may have a role as a negative prognostic biomarker (173). BRAFV600E mutated gangliogliomas have been shown to be associated with concomitant mTOR pathway activation, which may aid the development of targeted treatments for this tumor (174).
Gliomas, Intratumoural Heterogeneity, and Brain Tumor Stem Cells
The discussion above highlights our developing understanding of glial tumors as entities with individual pathological molecular events and treatment responses. It is increasingly recognized, however, that individual tumors contain distinct spatial regions with differing molecular profiles. For example, microdissected glioma tissue specimens have been shown to harbor unique molecular profiles in central vs. peripheral invasive regions (175). Receptor kinases (EGFR, MET, PDGFRA) have also been found to be amplified in single tumors in different cells in a mutually exclusive fashion (176). Such intratumoural heterogeneity results in divergent subclones of neoplastic cells within the same tumor and is likely to drive treatment failure, treatment resistance, and subsequent recurrence of the malignancy (177, 178). Indeed, it has recently been shown that different samples from the same GBM can be classified into different GBM subtypes (179). It follows that the impact of sampling bias must be considered when stratifying patients in future clinical trials using molecular criteria.
Furthermore, tumor recurrence post-treatment may consist of multiple neoplastic clones coexisting in the same lesion. Analysis of such patterns of heterogeneity could enable patients to receive targeted multimodal therapies for recurrent tumors (12, 180). This aim will additionally require a greater understanding of how brain cancer stem cells (also termed brain tumor initiating cells) drive such divergent neoplastic clones and cause tumor relapse (181–183). It has been shown both in vivo and in vitro that brain tumor initiating cells can be epigenetically differentiated into mature neuronal type cells (184, 185). Such cells can also be directed into non-neuronal cell types with a resultant suppression of malignant cellular behavior (186). It is hoped that such epigenetic manipulation could 1 day lead to further, more refined, personalized treatment (187, 188).
Conclusion
Over the last decade, there has been an unprecedented surge in our understanding of what drives neoplastic growth in glial tumors. Further molecular characterization of these tumors in the future will accelerate biomarker discovery and facilitate the creation of new diagnostic categories for gliomas (189). Only IDH mutation status (prognostic) and MGMT methylation status and 1p/19q co-deletion (predictive) are currently routinely used for evaluation of glioma patients by clinicians in the US and UK. However, the ongoing development of targeted therapies as mono and combination treatments necessitates the discovery of optimal molecular predictive biomarkers, which will further our understanding of these tumors.
Additionally, biomarker analysis will become a major factor in glioma clinical trials, with rapid identification of putative biomarkers in early stage trials with sufficient statistical design to validate predictive associations in phase III trials. Care will therefore be required to distinguish biomarkers that provide prognostic information from those that have predictive validity. This approach will allow us to determine future personalized therapeutic choices with minimal toxicity and improve clinical outcomes for patients for whom the diagnosis of a malignant glioma still portends a dismal outlook.
Author Contributions
Harry R. Haynes: manuscript research and writing; Sandra Camelo-Piragua: manuscript review and revision; Kathreena M. Kurian: manuscript research and writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Mr. Ben Kirk, University of Bristol, for Figure 1.
References
1. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol (2005) 109:93–108. doi:10.1007/s00401-005-0991-y
2. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4th ed. Geneva: IARC (2007).
3. Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res (2013) 19:764–72. doi:10.1158/1078-0432.CCR-12-3002
4. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre P-L, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res (2004) 64:6892–9. doi:10.1158/0008-5472.CAN-04-1337
5. Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol (1996) 6:217–23; discussion 23–4. doi:10.1111/j.1750-3639.1996.tb00848.x
6. McDonald KL, Aw G, Kleihues P. Role of biomarkers in the clinical management of glioblastomas: what are the barriers and how can we overcome them? Front Neurol (2012) 3:188. doi:10.3389/fneur.2012.00188
7. Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol (2011) 29:4482–90. doi:10.1200/JCO.2010.33.8715
8. Bartek J, Ng K, Fischer W, Carter B, Chen CC. Key concepts in glioblastoma therapy. J Neurol Neurosurg Psychiatry (2012) 83:753–60. doi:10.1136/jnnp-2011-300709
9. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi:10.1038/nature07385
10. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17:98–110. doi:10.1016/j.ccr.2009.12.020
11. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell (2006) 9:157–73. doi:10.1016/j.ccr.2006.02.019
12. Riemenschneider MJ, Louis DN, Weller M, Hau P. Refined brain tumor diagnostics and stratified therapies: the requirement for a multi- disciplinary approach. Acta Neuropathol (2013) 126:21–37. doi:10.1007/s00401-013-1127-4
13. Zheng S, Chheda MG, Verhaak RG. Studying a complex tumor: potential and pitfalls. Cancer J (2012) 18:107–14. doi:10.1097/PPO.0b013e3182431c57
14. Olar A, Aldape KD. Biomarkers classification and therapeutic decision-making for malignant gliomas. Curr Treat Options Oncol (2012) 13:417–36. doi:10.1007/s11864-012-0210-8
15. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155:462–77. doi:10.1016/j.cell.2013.09.034
16. Johnson DR, Galanis E. Incorporation of prognostic and predictive factors into glioma clinical trials. Curr Oncol Rep (2013) 15:56–63. doi:10.1007/s11912-012-0279-z
17. Galanis E, Wu W, Sarkaria J, Chang SM, Colman H, Sargent D, et al. Incorporation of biomarker assessment in novel clinical trial designs: personalizing brain tumor treatments. Curr Oncol Rep (2011) 13:42–9. doi:10.1007/s11912-010-0144-x
18. Conley BA, Taube SE. Prognostic and predictive markers in cancer. Dis Markers (2004) 20:35–43. doi:10.1155/2004/202031
19. Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol (2011) 12:83–91. doi:10.1016/S1470-2045(10)70053-X
20. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360:765–73. doi:10.1056/NEJMoa0808710
21. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol (2008) 116:597–602. doi:10.1007/s00401-008-0455-2
22. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol (2009) 27:4150–4. doi:10.1200/JCO.2009.21.9832
23. Ahmadi R, Stockhammer F, Becker N, Hohlen K, Misch M, Christians A, et al. No prognostic value of IDH1 mutations in a series of 100 WHO grade II astrocytomas. J Neurooncol (2012) 109:15–22. doi:10.1007/s11060-012-0863-y
24. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol (2009) 118:469–74. doi:10.1007/s00401-009-0561-9
25. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol (2009) 174:1149–53. doi:10.2353/ajpath.2009.080958
26. Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest (2012) 30:48–56. doi:10.3109/07357907.2011.630050
27. Rich JN, Hans C, Jones B, Iversen ES, McLendon RE, Rasheed BK, et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res (2005) 65:4051–8. doi:10.1158/0008-5472.CAN-04-3936
28. Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain (2007) 130:2596–606. doi:10.1093/brain/awm204
29. Chen Y-J, Hakin-Smith V, Teo M, Xinarianos GE, Jellinek DA, Carroll T, et al. Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res (2006) 66:6473–6. doi:10.1158/0008-5472.CAN-06-0910
30. Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet (2003) 361:836–8. doi:10.1016/S0140-6736(03)12681-5
31. Liu X-Y, Gerges N, Korshunov A, Sabha N, Khuong-Quang D-A, Fontebasso AM, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol (2012) 124(5):615–25. doi:10.1007/s00401-012-1031-3
32. Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget (2012) 3:1194–203.
33. De La Fuente R, Baumann C, Viveiros MM. Role of ATRX in chromatin structure and function: implications for chromosome instability and human disease. Reproduction (2011) 142:221–34. doi:10.1530/REP-10-0380
34. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A (2013) 110:6021–6. doi:10.1073/pnas.1303607110
35. Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A, et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol (2013) 126:267–76. doi:10.1007/s00401-013-1141-6
36. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget (2012) 3:709–22.
37. Ohgaki H, Kleihues P. Genetic profile of astrocytic and oligodendroglial gli- omas. Brain Tumor Pathol (2011) 28:177–83. doi:10.1007/s10014-011-0029-1
38. Nonoguchi N, Ohta T, Oh J-E, Kim Y-H, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol (2013) 126(6):931–7. doi:10.1007/s00401-013-1163-0
39. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462:739–44. doi:10.1038/nature08617
40. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi:10.1038/nature10860
41. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol (2012) 108:11–27. doi:10.1007/s11060-011-0793-0
42. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi:10.1038/nature10866
43. Venneti S, Felicella MM, Coyne T, Phillips JJ, Gorovets D, Huse JT, et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J Neuropathol Exp Neurol (2013) 72:298–306. doi:10.1097/NEN.0b013e3182898113
44. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell (2010) 17:510–22. doi:10.1016/j.ccr.2010.03.017
45. Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol (2014) 232:165–77. doi:10.1002/path.4282
46. Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, et al. 2-Hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med (2012) 18:624–9. doi:10.1038/nm.2682
47. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (2013) 340:626–30. doi:10.1126/science.1236062
48. Malzkorn B, Wolter M, Riemenschneider MJ, Reifenberger G. Unraveling the glioma epigenome: from molecular mechanisms to novel biomarkers and therapeutic targets. Brain Pathol (2011) 21:619–32. doi:10.1111/j.1750-3639.2011.00536.x
49. Pegg AE, Dolan ME, Moschel RC. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog Nucleic Acid Res Mol Biol (1995) 51:167–223. doi:10.1016/S0079-6603(08)60879-X
50. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med (2000) 343:1350–4. doi:10.1056/NEJM200011093431901
51. Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med (2005) 352:997–1003. doi:10.1056/NEJMoa043331
52. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi:10.1016/S1470-2045(09)70025-7
53. Olson RA, Brastianos PK, Palma DA. Prognostic and predictive value of epigenetic silencing of MGMT in patients with high grade gliomas: a systematic review and meta-analysis. J Neurooncol (2011) 105:325–35. doi:10.1007/s11060-011-0594-5
54. Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer (2009) 101:124–31. doi:10.1038/sj.bjc.6605127
55. Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol (2012) 13:707–15. doi:10.1016/S1470-2045(12)70164-X
56. Malmström A, Grønberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol (2012) 13:916–26. doi:10.1016/S1470-2045(12)70265-6
57. van den Bent MJ, Erdem-Eraslan L, Idbaih A, de Rooi J, Eilers PH, Spliet WG, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res (2013) 19:5513–22. doi:10.1158/1078-0432.CCR-13-1157
58. Zhang Y, Dutta A, Abounader R. The role of microRNAs in glioma initiation and progression. Front Biosci (Landmark Ed) (2012) 17:700–12. doi:10.2741/3952
59. Conti A, Aguennouz M, La Torre D, Tomasello C, Cardali S, Angileri FF, et al. miR-21 and 221 upregulation and miR-181b downregulation in human grade II-IV astrocytic tumors. J Neurooncol (2009) 93:325–32. doi:10.1007/s11060-009-9797-4
60. Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol (2008) 28:5369–80. doi:10.1128/MCB.00479-08
61. Ciafrè SA, Galardi S, Mangiola A, Ferracin M, Liu C-G, Sabatino G, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun (2005) 334:1351–8. doi:10.1016/j.bbrc.2005.07.030
62. Slaby O, Lakomy R, Fadrus P, Hrstka R, Kren L, Lzicarova E, et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma (2010) 57:264–9. doi:10.4149/neo_2010_03_264
63. Zhang W, Zhang J, Hoadley K, Kushwaha D, Ramakrishnan V, Li S, et al. miR-181d: a predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol (2012) 14:712–9. doi:10.1093/neuonc/nos089
64. Kreth S, Limbeck E, Hinske LC, Schütz SV, Thon N, Hoefig K, et al. In human glioblastomas transcript elongation by alternative polyadenylation and miRNA targeting is a potent mechanism of MGMT silencing. Acta Neuropathol (2013) 125:671–81. doi:10.1007/s00401-013-1081-1
65. Kim T-M, Huang W, Park R, Park PJ, Johnson MD. A developmental taxonomy of glioblastoma defined and maintained by MicroRNAs. Cancer Res (2011) 71:3387–99. doi:10.1158/0008-5472.CAN-10-4117
66. Cairncross G, Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer J (2008) 14:352–7. doi:10.1097/PPO.0b013e31818d8178
67. Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol (2000) 18:636–45.
68. Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, et al. Chemotherapy plus radiotherapy (CT-RT) versus RT alone for patients with anaplastic oligodendroglioma: long-term results of the RTOG 9402 phase III study. J Clin Oncol (2012) 30:2008b.
69. van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre J-Y, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol (2012) 31(3):344–50. doi:10.1200/JCO.2012.43.2229
70. Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science (2011) 333:1453–5. doi:10.1126/science.1210557
71. Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol (2012) 226:7–16. doi:10.1002/path.2995
72. Hsiao H-H, Nath A, Lin C-Y, Folta-Stogniew EJ, Rhoades E, Braddock DT. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry (2010) 49:4620–34. doi:10.1021/bi9021445
73. Roch F, Jiménez G, Casanova J. EGFR signalling inhibits capicua-dependent repression during specification of Drosophila wing veins. Development (2002) 129:993–1002.
74. Nakamura M, Yang F, Fujisawa H, Yonekawa Y, Kleihues P, Ohgaki H. Loss of heterozygosity on chromosome 19 in secondary glioblastomas. J Neuropathol Exp Neurol (2000) 59:539–43.
75. Homma T, Fukushima T, Vaccarella S, Yonekawa Y, Di Patre PL, Franceschi S, et al. Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol (2006) 65:846–54. doi:10.1097/01.jnen.0000235118.75182.94
76. Ichimura K, Vogazianou AP, Liu L, Pearson DM, Bäcklund LM, Plant K, et al. 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene (2008) 27:2097–108. doi:10.1038/sj.onc.1210848
77. Burton EC, Lamborn KR, Feuerstein BG, Prados M, Scott J, Forsyth P, et al. Genetic aberrations defined by comparative genomic hybridization distinguish long-term from typical survivors of glioblastoma. Cancer Res (2002) 62:6205–10.
78. Solomon DA, Kim J-S, Jean W, Waldman T. Conspirators in a capital crime: co-deletion of p18INK4c and p16INK4a/p14ARF/p15INK4b in glioblastoma multiforme. Cancer Res (2008) 68:8657–60. doi:10.1158/0008-5472.CAN-08-2084
79. Rao SK, Edwards J, Joshi AD, Siu I-M, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol (2010) 96:169–79. doi:10.1007/s11060-009-9959-4
80. Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol (2009) 27:5743–50. doi:10.1200/JCO.2009.23.0805
81. Kim J-H, Huse JT, Huang Y, Lyden D, Greenfield JP. Molecular diagnostics in paediatric glial tumours. Lancet Oncol (2013) 14:e19–27. doi:10.1016/S1470-2045(12)70577-6
82. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science (2012) 337:1231–5. doi:10.1126/science.1220834
83. Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res (2012) 72:2045–56. doi:10.1158/0008-5472.CAN-11-3034
84. Guagnano V, Kauffmann A, Wöhrle S, Stamm C, Ito M, Barys L, et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov (2012) 2:1118–33. doi:10.1158/2159-8290.CD-12-0210
85. Hegi ME, Rajakannu P, Weller M. Epidermal growth factor receptor: a re-emerging target in glioblastoma. Curr Opin Neurol (2012) 25:774–9. doi:10.1097/WCO.0b013e328359b0bc
86. Patel R, Leung HY. Targeting the EGFR-family for therapy: biological challenges and clinical perspective. Curr Pharm Des (2012) 18:2672–9. doi:10.2174/138161212800626148
87. Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene (2000) 19:6550–65. doi:10.1038/sj.onc.1204082
88. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science (2007) 318:287–90. doi:10.1126/science.1142946
89. Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets (2012) 12:197–209. doi:10.2174/156800912799277557
90. Lassman AB, Rossi MR, Raizer JJ, Razier JR, Abrey LE, Lieberman FS, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res (2005) 11:7841–50. doi:10.1158/1078-0432.CCR-05-0421
91. Mawrin C, Diete S, Treuheit T, Kropf S, Vorwerk CK, Boltze C, et al. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int J Oncol (2003) 23:641–8.
92. Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther (2008) 7:1321–5. doi:10.4161/cbt.7.9.6954
93. Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci U S A (2010) 107:6459–64. doi:10.1073/pnas.0911188107
94. Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res (2006) 66:7864–9. doi:10.1158/0008-5472.CAN-04-4392
95. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med (2008) 5:e8. doi:10.1371/journal.pmed.0050008
96. Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol (2005) 23:5294–304. doi:10.1200/JCO.2005.23.622
97. Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst (2005) 97:880–7. doi:10.1093/jnci/dji161
98. van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol (2009) 27:1268–74. doi:10.1200/JCO.2008.17.5984
99. Available from: http://clinicaltrials.gov/show/NCT01339052
100. Gallia GL, Rand V, Siu I-M, Eberhart CG, James CD, Marie SK, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res (2006) 4:709–14. doi:10.1158/1541-7786.MCR-06-0172
101. Fan Q-W, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res (2007) 67:7960–5. doi:10.1158/0008-5472.CAN-07-2154
102. Fan Q-W, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol (2010) 347:279–96. doi:10.1007/82_2010_67
103. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res (2003) 63:6962–70.
104. Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst (2001) 93:1246–56. doi:10.1093/jnci/93.16.1246
105. Hobbs J, Nikiforova MN, Fardo DW, Bortoluzzi S, Cieply K, Hamilton RL, et al. Paradoxical relationship between the degree of EGFR amplification and outcome in glioblastomas. Am J Surg Pathol (2012) 36:1186–93. doi:10.1097/PAS.0b013e3182518e12
106. Pelloski CE, Lin E, Zhang L, Yung WK, Colman H, Liu J-L, et al. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res (2006) 12:3935–41. doi:10.1158/1078-0432.CCR-05-2202
107. Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene (1994) 9:2313–20.
108. Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res (2005) 11:1462–6. doi:10.1158/1078-0432.CCR-04-1737
109. Huang PH, Cavenee WK, Furnari FB, White FM. Uncovering therapeutic targets for glioblastoma: a systems biology approach. Cell Cycle (2007) 6:2750–4. doi:10.4161/cc.6.22.4922
110. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B, et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov (2011) 1:524–38. doi:10.1158/2159-8290.CD-11-0124
111. Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med (2005) 353:2012–24. doi:10.1056/NEJMoa051918
112. Lal B, Goodwin CR, Sang Y, Foss CA, Cornet K, Muzamil S, et al. EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol Cancer Ther (2009) 8:1751–60. doi:10.1158/1535-7163.MCT-09-0188
113. Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther (2009) 9:1087–98. doi:10.1517/14712590903124346
114. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet (2013) 45:1141–9. doi:10.1038/ng.2734
115. Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev (2010) 24:2205–18. doi:10.1101/gad.1972310
116. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A (2012) 109:3041–6. doi:10.1073/pnas.1114033109
117. Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol (2013) 23(5):565–73. doi:10.1111/bpa.12043
118. Alentorn A, Marie Y, Carpentier C, Boisselier B, Giry M, Labussière M, et al. Prevalence, clinico-pathological value, and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro Oncol (2012) 14:1393–403. doi:10.1093/neuonc/nos217
119. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol (2013) 10:14–26. doi:10.1038/nrclinonc.2012.204
120. Knizetova P, Ehrmann J, Hlobilkova A, Vancova I, Kalita O, Kolar Z, et al. Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF-VEGFR2 (KDR) interplay. Cell Cycle (2008) 7:2553–61. doi:10.4161/cc.7.16.6442
121. Oehring RD, Miletic M, Valter MM, Pietsch T, Neumann J, Fimmers R, et al. Vascular endothelial growth factor (VEGF) in astrocytic gliomas – a prognostic factor? J Neurooncol (1999) 45:117–25. doi:10.1023/A:1006333005563
122. Kuczynski EA, Patten SG, Coomber BL. VEGFR2 expression and TGF-β signaling in initial and recurrent high-grade human glioma. Oncology (2011) 81:126–34. doi:10.1159/000332849
123. Specenier P. Bevacizumab in glioblastoma multiforme. Expert Rev Anticancer Ther (2012) 12:9–18. doi:10.1586/era.11.179
124. Chinot OL, de La Motte Rouge T, Moore N, Zeaiter A, Das A, Phillips H, et al. AVAglio: phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv Ther (2011) 28:334–40. doi:10.1007/s12325-011-0007-3
125. Gilbert MR, Dignam J, Won M, Blumenthal DT, Vogelbaum MA, Aldape KD, et al. RTOG 0825: phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM). J Clin Oncol (2013) 31:2013.
126. Wick W, Cloughesy TF, Nishikawa R, Mason W, Saran F, Henriksson R, et al. Tumor response based on adapted Macdonald criteria and assessment of pseudoprogression (PsPD) in the phase III AVAglio trial of bevacizumab (Bv) plus temozolomide (T) plus radiotherapy (RT) in newly diagnosed glioblastoma (GBM). J Clin Oncol (2013) 4:11.
127. Weller M, Yung WK. Angiogenesis inhibition for glioblastoma at the edge: beyond AVAGlio and RTOG 0825. Neuro Oncol (2013) 15:971. doi:10.1093/neuonc/not106
128. Rahmathulla G, Hovey EJ, Hashemi-Sadraei N, Ahluwalia MS. Bevacizumab in high-grade gliomas: a review of its uses, toxicity assessment, and future treatment challenges. Onco Targets Ther (2013) 6:371–89. doi:10.2147/OTT.S38628
129. US National Library of Medicine. Clinicaltrials.gov (2012). Available from: http://www.clinicaltrials.gov/ct2/show/NCT00329719?term=nct00329719&rank=1
130. US National Library of Medicine. Clinicaltrails.gov (2012). Available from: http://www.clinicaltrials.gov/ct2/show/NCT00734526?term=nct00734526&rank=1
131. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov (2006) 5:835–44. doi:10.1038/nrd2130
132. Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol (2012) 14:1511–8. doi:10.1093/neuonc/nos264
133. Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol (2011) 103:491–501. doi:10.1007/s11060-010-0402-7
134. Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, et al. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol (2012) 110:111–8. doi:10.1007/s11060-012-0943-z
135. Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, et al. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol (2013) 111:41–8. doi:10.1007/s11060-012-0988-z
136. Fontebasso AM, Liu X-Y, Sturm D, Jabado N. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol (2013) 23:210–6. doi:10.1111/bpa.12023
137. Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med (2002) 346:420–7. doi:10.1056/NEJMoa012224
138. Pollack IF, Hamilton RL, Burger PC, Brat DJ, Rosenblum MK, Murdoch GH, et al. Akt activation is a common event in pediatric malignant gliomas and a potential adverse prognostic marker: a report from the Children’s Oncology Group. J Neurooncol (2010) 99:155–63. doi:10.1007/s11060-010-0297-3
139. Pollack IF, Hamilton RL, James CD, Finkelstein SD, Burnham J, Yates AJ, et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg (2006) 105:418–24.
140. Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst (2011) 27:87–94. doi:10.1007/s00381-010-1264-1
141. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet (2012) 44:251–3. doi:10.1038/ng.1102
142. Schwartzentruber J, Korshunov A, Liu X-Y, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482:226–31. doi:10.1038/nature10833
143. Bosch A, Suau P. Changes in core histone variant composition in differentiating neurons: the roles of differential turnover and synthesis rates. Eur J Cell Biol (1995) 68:220–5.
144. Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell (2012) 22:425–37. doi:10.1016/j.ccr.2012.08.024
145. Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu X-Y, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol (2012) 124:439–47. doi:10.1007/s00401-012-0998-0
146. Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, Liu X-Y, Sturm D, Korshunov A, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol (2013) 125:659–69. doi:10.1007/s00401-013-1095-8
147. Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol (2009) 10:697–708. doi:10.1038/nrm2763
148. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet (2011) 12:7–18. doi:10.1038/nrg2905
149. Rheinbay E, Louis DN, Bernstein BE, Suvà ML. A tell-tail sign of chromatin: histone mutations drive pediatric glioblastoma. Cancer Cell (2012) 21:329–31. doi:10.1016/j.ccr.2012.03.001
150. Pfister S, Witt O. Pediatric gliomas. Recent Results Cancer Res (2009) 171:67–81. doi:10.1007/978-3-540-31206-2_4
151. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol (2005) 64:479–89.
152. Armstrong GT, Conklin HM, Huang S, Srivastava D, Sanford R, Ellison DW, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol (2011) 13:223–34. doi:10.1093/neuonc/noq178
153. Jones DT, Kocialkowski S, Liu L, Pearson DM, Bäcklund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68:8673–7. doi:10.1158/0008-5472.CAN-08-2097
154. Jones DT, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci (2012) 69:1799–811. doi:10.1007/s00018-011-0898-9
155. Lawson AR, Hindley GF, Forshew T, Tatevossian RG, Jamie GA, Kelly GP, et al. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res (2011) 21:505–14. doi:10.1101/gr.115782.110
156. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res (2011) 17:4790–8. doi:10.1158/1078-0432.CCR-11-0034
157. Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol (2012) 71:66–72. doi:10.1097/NEN.0b013e31823f2cb0
158. Badiali M, Gleize V, Paris S, Moi L, Elhouadani S, Arcella A, et al. KIAA1549-BRAF fusions and IDH mutations can coexist in diffuse gliomas of adults. Brain Pathol (2012) 22(6):841–7. doi:10.1111/j.1750-3639.2012.00603.x
159. Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet (2013) 45:927–32. doi:10.1038/ng.2682
160. Sievert AJ, Lang S-S, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A (2013) 110:5957–62. doi:10.1073/pnas.1219232110
161. Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov (2013) 3:264–79. doi:10.1158/2159-8290.CD-12-0362
162. Rusconi P, Caiola E, Broggini M. RAS/RAF/MEK inhibitors in oncology. Curr Med Chem (2012) 19:1164–76. doi:10.2174/092986712799320510
163. Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, et al. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer (2009) 101:722–33. doi:10.1038/sj.bjc.6605179
164. Hasselblatt M, Riesmeier B, Lechtape B, Brentrup A, Stummer W, Albert FK, et al. BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol (2011) 37:803–6. doi:10.1111/j.1365-2990.2011.01193.x
165. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol (2012) 14:777–89. doi:10.1093/neuonc/nos077
166. Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res (2011) 17:7595–604. doi:10.1158/1078-0432.CCR-11-1456
167. Horbinski C. To BRAF or not to BRAF: is that even a question anymore? J Neuropathol Exp Neurol (2013) 72:2–7. doi:10.1097/NEN.0b013e318279f3db
168. Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One (2011) 6:e17948. doi:10.1371/journal.pone.0017948
169. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol (2011) 122:11–9. doi:10.1007/s00401-011-0841-z
170. Chamberlain MC. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol (2013) 114:237–40. doi:10.1007/s11060-013-1176-5
171. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol (2011) 121:397–405. doi:10.1007/s00401-011-0802-6
172. Koelsche C, Wöhrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, et al. Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol (2013) 125:891–900. doi:10.1007/s00401-013-1100-2
173. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol (2013) 125:901–10. doi:10.1007/s00401-013-1120-y
174. Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schouten-van Meeteren AY, Spliet WG, et al. BRAF V600E mutation is associated with mTOR signaling activation in glioneuronal tumors. Brain Pathol (2014) 24:52–66. doi:10.1111/bpa.12081
175. Delic S, Lottmann N, Jetschke K, Reifenberger G, Riemenschneider MJ. Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol Appl Neurobiol (2012) 38:201–12. doi:10.1111/j.1365-2990.2011.01207.x
176. Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell (2011) 20:810–7. doi:10.1016/j.ccr.2011.11.005
177. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer (2012) 12:487–93. doi:10.1038/nrc3298
178. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer (2012) 12:323–34. doi:10.1038/nrc3261
179. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A (2013) 110:4009–14. doi:10.1073/pnas.1219747110
180. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta (2010) 1805:105–17. doi:10.1016/j.bbcan.2009.11.002
181. Kurian KM. The impact of neural stem cell biology on CNS carcinogenesis and tumor types. Patholog Res Int (2011) 2011:685271. doi:10.4061/2011/685271
182. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res (2004) 64:7011–21. doi:10.1158/0008-5472.CAN-04-1364
183. Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A (2003) 100:15178–83. doi:10.1073/pnas.2036535100
184. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature (2006) 444:761–5. doi:10.1038/nature05349
185. Nakano I, Saigusa K, Kornblum HI. BMPing off glioma stem cells. Cancer Cell (2008) 13:3–4. doi:10.1016/j.ccr.2007.12.018
186. Stricker SH, Feber A, Engström PG, Carén H, Kurian KM, Takashima Y, et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev (2013) 27:654–69. doi:10.1101/gad.212662.112
187. Yin C, Lv S, Chen X, Guo H. The role of glioma stem cells in glioma tumorigenesis. Front Biosci (Landmark Ed) (2014) 19:818–24. doi:10.2741/4249
188. Cruceru ML, Neagu M, Demoulin J-B, Constantinescu SN. Therapy targets in glioblastoma and cancer stem cells: lessons from haematopoietic neoplasms. J Cell Mol Med (2013) 17:1218–35. doi:10.1111/jcmm.12122
189. Louis DN. The next step in brain tumor classification: “let us now praise famous men”… or molecules? Acta Neuropathol (2012) 124:761–2. doi:10.1007/s00401-012-1067-4
190. Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol (2008) 26:2192–7. doi:10.1200/JCO.2007.14.8163
191. Lalezari S, Chou AP, Tran A, Solis OE, Khanlou N, Chen W, et al. Combined analysis of O6-methylguanine-DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro Oncol (2013) 15:370–81. doi:10.1093/neuonc/nos308
192. Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol (1994) 145:1175–90.
193. Aldape K, Burger PC, Perry A. Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med (2007) 131:242–51.
Keywords: gliomas, prognostic biomarkers, predictive biomarkers
Citation: Haynes HR, Camelo-Piragua S and Kurian KM (2014) Prognostic and predictive biomarkers in adult and pediatric gliomas: toward personalized treatment. Front. Oncol. 4:47. doi: 10.3389/fonc.2014.00047
Received: 26 January 2014; Accepted: 27 February 2014;
Published online: 24 March 2014.
Edited by:
Andreas Hoeflich, Leibniz-Institute for Farm Animal Biology, GermanyReviewed by:
Federica Barbieri, University of Genova, ItalyPeter F. Ambros, Children’s Cancer Research Institute, Austria
Carl Friedrich Classen, University Children’s Hospital, Rostock, Germany
Copyright: © 2014 Haynes, Camelo-Piragua and Kurian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathreena M. Kurian, Brain Tumor Research Group, Institute of Clinical Neurosciences, Frenchay Hospital, Frenchay Park Road, Bristol BS16 1LE, UK e-mail: kathreena.kurian@nbt.nhs.uk