 Mary B. Mockler
Mary B. Mockler Melissa J. Conroy
Melissa J. Conroy Joanne Lysaght
Joanne Lysaght- Department of Surgery, Trinity Centre for Health Sciences, St. James’s Hospital, Trinity College Dublin, Dublin, Ireland
The immune system has a key role to play in controlling cancer initiation and progression. T cell activation, which is central to anti-tumor immune responses, coincides with changes in cellular metabolism. Naïve T cells predominantly require an ATP generating metabolic profile, whereas proliferating effector T cells require anabolic metabolic profiles that promote rapid growth and proliferation. Furthermore, specific T cell subsets require distinct energetic and biosynthetic pathways to match their functional requirements. The often hostile tumor microenvironment can affect T cell immune responses by altering the resulting cellular metabolism. Tailoring immune responses by manipulating cellular metabolic pathways may provide an exciting new option for cancer immunotherapy. T cell responses might also be skewed via metabolic manipulation to treat the complications of obesity-associated inflammation, which is a rapidly growing global health problem and a major risk factor for many malignancies. In this review, the diverse metabolic requirements of T cells in anti-tumor immunity are discussed, as well as the profound influence of the tumor microenvironment and the possible avenues for manipulation to enhance anti-tumor immunity.
Introduction
A unique bioenergetic challenge is faced by cells of the immune system when they recognize mutated or over expressed tumor antigens presented by antigen presenting cells. They must rapidly proliferate with a specific phenotype and function to eradicate tumor cells (1). The ability of a T cell to transition from a naïve, to effector, to a memory phenotype is determined by metabolism and the metabolic program varies to match the T cell subset in order to enable cell survival and function (2). Activated T cells switch from naïve T cells, mainly requiring generation of ATP and replacement biosynthesis (3) to effector T cells (TEFF), which must rapidly increase cell mass and elicit effector functions through cytokine production and cytotoxicity (4). This switch from primarily oxidative phosphorylation (OxPhos) to glycolysis supports the demands of rapid cell growth. Primary fuels and amino acids, such as glucose and glutamine must be available to support these distinct metabolic pathways (5, 6). Unavailability of these fuels occurs during malnutrition and can lead to immunosuppression (7), while excess fuel in diet-induced obesity is associated with a constant low level state of inflammation (8). We have previously identified T cells as key players in adipose tissue inflammation in obesity-associated cancer and here, we suggest that their metabolism might be targeted to control chronic inflammation and lessen the clinical manifestations of obesity (9). In comparison, the long lifespan of memory T cells (TMEM) poses a different metabolic demand and to facilitate this need, TMEM cells utilize fatty acid oxidation (FAO) as their main energy source (10). The balance between different T cell subsets can be altered by multiple environmental factors in the tumor resulting in the activation of metabolic pathways and the establishment of inappropriate T cell responses. Direct manipulation of T cell metabolism has the potential to provide a new avenue for cancer immunotherapy and obesity-associated inflammation and this will be discussed.
How T Cell Metabolism Affects Function
At each stage of a T cell’s development, its metabolism must support the function of the cell through the provision of energy and biosynthetic precursors (11) (Figure 1). It is vital that T cells undergo the appropriate activation and differentiation to ensure that homeostasis is maintained (12).
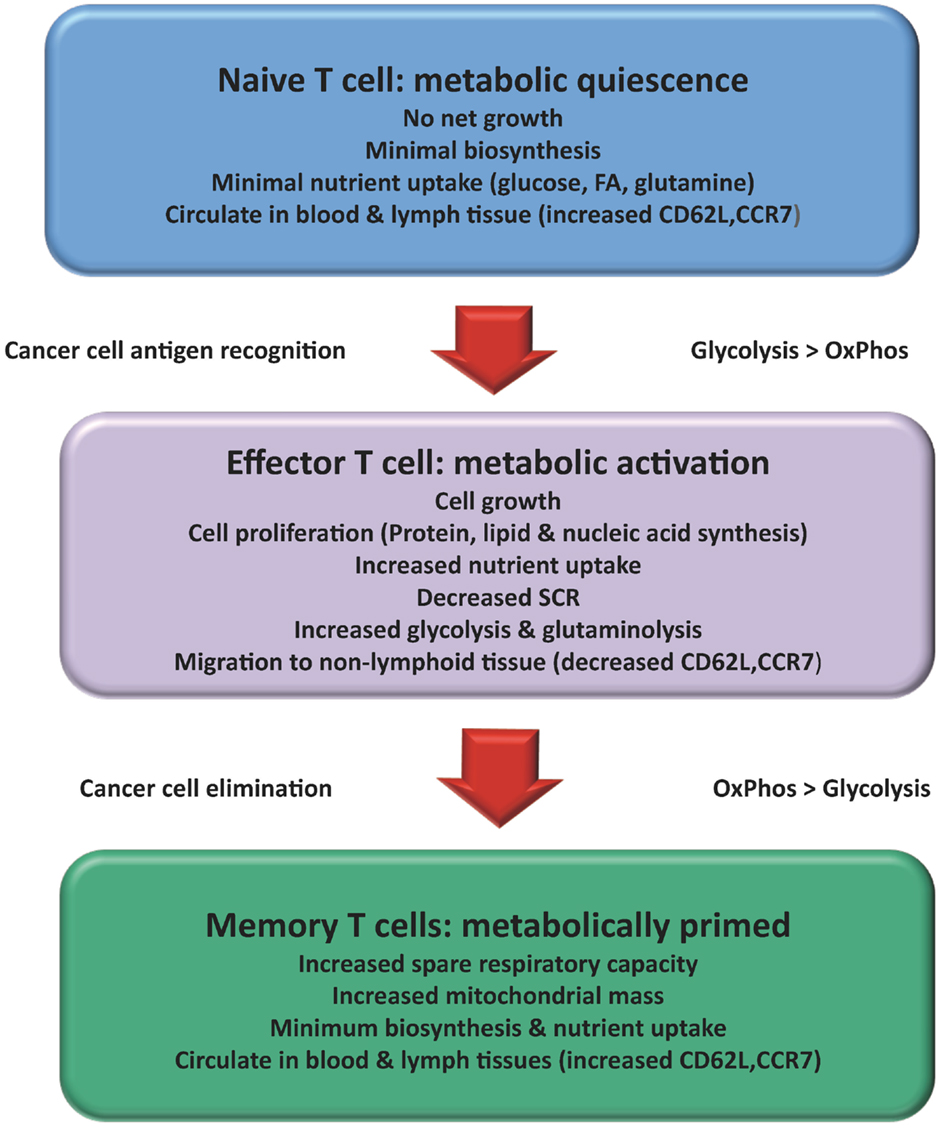
Figure 1. Changes in T cell metabolism are related to function: Over the course of an immune response the metabolism of a T cell changes. Naïve T cells mainly used a mixed fuel OxPhos to produce ATP. Once activated the T cell adopts a metabolic profile that is similar to many cancer cells (Warburg effect). Consumption of glucose and glutamine increases to support increased cell growth and proliferation. A subset of activated T cells survives to become TMEM reverting back to lipid oxidation with increased capacity for efficient energy generation and survival. Activation of T cells results in changes in migration patterns to ensure they can successfully migrate to areas of inflammation and cancer.
Naïve T Cells
The primary function of naïve T cells is antigenic surveillance, which requires relatively small amounts of ATP to support processes such as ion homeostasis, membrane integrity, and rearrangement of the actin cytoskeleton for movement (13). Thus naïve T cells have a catabolic metabolic profile that favors energy production over biosynthesis (14). It is estimated that 96% of ATP in naïve T cells is generated through OxPhos, with the remaining 4% being generated by glycolysis (3). It is an oxygen dependent process and produces 36 molecules of ATP per glucose molecule (1). Naïve T cells require extrinsic signals to maintain this basal metabolism. Interleukin-7 (IL-7) regulates glucose uptake via the cell surface trafficking of Glut1, which is modulated by the PI3K/Akt/mTOR pathway (15). This regulation of glucose uptake by IL-7 is critical to maintain homeostasis and to prevent apoptosis by the pro-apoptotic protein Bim (16).
Activated T Cells
In contrast, TEFF cells adopt an anabolic metabolic profile to meet the increased energy requirements associated with cell growth, proliferation, differentiation, and effector functions (4). TEFF cells utilize aerobic glycolysis even though it is less efficient than OxPhos yielding only four moles of ATP per glucose molecule (17). Aerobic glycolysis provides advantages over OxPhos in rapidly dividing cells; firstly, metabolic intermediates needed for cell growth and proliferation are generated by aerobic glycolysis (11). Secondly, increased OxPhos results in increased production of reactive oxygen species (ROS), which can lead to apoptosis of T cells (18). Thirdly, aerobic glycolysis produces ATP faster than OxPhos. Therefore, once glucose is not a limiting factor, the pace of glycolytic flux will produce enough ATP to meet the energy demand of the TEFF cells (19). Finally glycolysis is not oxygen dependent and can accommodate T cell survival in a hypoxic environment, a common feature of many solid malignancies (20). OxPhos is still utilized in TEFF cells however aerobic glycolysis is the main pathway for glucose metabolism (21). Activated T cells must engage distinct transcriptional programs to both establish their differentiation status and elicit the effector functions required (22). These changes are similar to those observed in rapidly dividing tumor cells with many of the same metabolic regulators involved, including mTOR (23). The main transcription factors up-regulated by mTOR are c-Myc and hypoxia-inducible factor-1α (HIF-1α) and it appears that only c-Myc is necessary for the glycolytic switch that occurs during early T cell growth and proliferation. mTOR dependent up-regulation of c-Myc in the first 24 h following T cell activation is crucial in the transition from a naïve T cell to a TEFF cell (21). Effector CD4+ T cells can be distinguished from effector CD8+ T cells due to their ability to increase OxPhos by up to two-fold after activation, whereas activated CD8+ T cells do not increase OxPhos above that of resting cells (24). This results in activated CD4+ T cells having a survival advantage over CD8+ T cells when pro-glycolytic signals are limited (25). CD8+ T cells execute the majority of immune-mediated tumor killing through the release of cytolytic factors, such as perforin and granzymes (26). Depending on the polarizing signals in the tumor microenvironment the different CD4+ T cell subsets can promote or suppress a defensive host response to cancer (27). Each CD4+ T cell subset has a unique metabolic profile, which is adapted to its function with Th1, Th2, and Th17 cells displaying a glycolytic metabolic phenotype (28). In contrast to Th1 and Th17 cells, Th2 cells readily develop in the absence of mTORC1 but fail to develop in the absence of mTORC2 (29). TREG cells unlike other TEFF cells mainly use lipid metabolism and have elevated levels of AMPK activation (28). The utilization of lipid oxidation by TREG cells might play a central role in their survival advantage over TEFF cells and in the maintenance of a stable pool of pro-tumor immune cells (30).
One area that currently warrants further investigation is how metabolism affects, and is affected by migration of activated T cells. Migration from nutrient and oxygen rich regions of the peripheral blood and lymphatics to the often hypoxic and acidic tumor microenvironment can have a major impact on T cell metabolism and their resulting effector function (31). Naïve and TMEM cells circulate continuously among the blood vessels, lymph vessels, and lymphatic tissue, whereas activated T cells must suspend migration in the lymphoid tissue to proliferate and then regain their motility to migrate to the tumor (32). Naïve and TMEM cells express CD62L, CCR7, and sphingosine-1-phosphate (S1P), which allows homing to and retention in secondary lymphoid organs (33) (Figure 1). Activation of the PI3K-Akt-mTOR pathway down regulates CD62L, CCR7, and S1P (34). Activation of this pathway also leads to up-regulated expression of pro-inflammatory adhesion molecules such as very late antigen-4 (VLA-4) and chemokine receptors such as CXCR3 (32). These changes allow TEFF cells to migrate to areas of inflammation and might influence their prevalence in visceral adipose tissue (VAT), where they play a pathological role in driving obesity-associated inflammation (9, 33).
Memory T Cells
Following resolution of an immune response most TEFF cells undergo apoptosis while some remain as TMEM cells, responsible for enhanced immunity against future re-exposure to invading pathogens or tumors (35). While TEFF cells proliferate rapidly and are short-lived, TMEM cells have lower turnover rates. This long life span of TMEM cells poses a unique metabolic demand on these cells. The production of a stable pool of TMEM cells is reliant on a corresponding decrease in mTOR signaling and a shift to fatty acid oxidation (FAO) (36). TMEM cells unlike naïve and TEFF cells have a greater spare respiratory capacity (SRC) in their mitochondria (37) (Figure 1). This allows for rapid ATP production, which gives a bioenergetic advantage upon secondary exposure to its antigen (38). IL-15 increases SRC by enhancing mitochondrial biogenesis and the expression of carnitine palmitoyltransferase I (CPTI). CPTI is a key enzyme involved in FAO. Since FAO takes place in the mitochondria, TMEM cells can increase their use of fatty acids for energy via OxPhos, with a consequential decrease in dependence on glycolysis. This results in TMEM cells having a survival advantage over TEFF cells in the absence of pro-glycolysis signals, when the initial priming antigen has been eliminated and cytokines such as IL-2 dissipate (37).
How the Tumor Microenvironment Can Affect T Cell Metabolism
Hypoxia
Tumor growth tends to outgrow the developing blood supply resulting in a hypoxic microenvironment (39). This hypoxic environment can have multiple effects on T cell responses. Roman et al. demonstrated that under hypoxic conditions the secretion of effector CD4+ cytokines such as IFN-α was increased resulting in an increased immune response (40). In contrast, other studies have found that hypoxia results in an impaired immune response as a result of reduced IL-2 (41). Hypoxia can affect T cell metabolism through the increased production of lactate by tumor cells within the tumor microenvironment. An increase in lactate in the tumor microenvironment results in decreased T cell proliferation and cytokine production, due to decreased glycolysis (42). HIF-1α is a master transcriptional regulator in the response to low O2 levels and is also involved in neoplastic pathways, including angiogenesis, cell survival, and treatment resistance (43). Levels of HIF-1α increase in activated T cells and can also be induced by other stimuli under normoxic conditions such as mTOR signaling (44, 45). HIF-1α is responsible for the glycolytic response that occurs downstream of mTORc1 activation (46). With the exception of Th17 cells, the role of HIF-1α in T cell subset differentiation within the tumor microenvironment has not been clearly defined in the literature and focused studies examining the role of HIF-1α on specific T cell subsets are warranted. Some studies examining T cells in general have shown that it can decrease activated T cell numbers through increased apoptosis (47). Other studies have demonstrated opposing results, showing that HIF-1α prevents apoptosis by up-regulating adrenomedullin (48). A few studies have shown that HIF-1α plays an important role in the differentiation of Th17 cells, which are highly glycolytic and are believed to rely more on glycolysis than other T cell subsets (49). Induction of both Th17 and TREG responses are closely related as they share a common requirement for transforming growth factor (TGF)-β. The expression of HIF-1α plays an important role in regulating the balance of Th17/TREG responses as it inhibits differentiation of TREG cells by targeting Foxp3 for proteasomal degradation (50). It also directly regulates Th17 differentiation by activating transcription of RORγt and collaborates with RORγt to activate Th17 signature genes, such as the IL-17 gene during development (50). The up-regulation of HIF-1α results in high levels of glycolysis in Th17 cells by increasing the expression of Glut1 and enzymes such as pyruvate kinase muscle (PKM) (49). Together with the downregulation of mitochondrial oxygen consumption, this prevents the entry of pyruvate into the TCA cycle (51). It is therefore likely that the constant state of low level inflammation present in many tumors may be due, at least in part, to HIF-1α directed Th17 cell differentiation and decreased TREG cell differentiation (50). A Th17 driven immune response has been linked to infection induced colon cancer in mouse models (52) and Helicobacter Pylori infection which can induce gastric cancer (53). Research has recently discovered that not all Th17 cells are pathogenic and drive autoimmune tissue injury (54). Development of pathogenic Th17 cells is dependent on exposure to IL 23, which diminishes the production of the anti-inflammatory cytokine IL-10 (55).
Nutrient Deprivation
Reduction of nutrients present in the microenvironment is associated with an impaired anti-tumor immune response (56). Nutrient deprivation inhibits mTOR activity which is vital for T cell metabolism (57). Glucose is essential for TEFF cell survival and proliferation (5), IFN-γ production (58), and cytolytic activity via production of granzyme and perforin (59). T cell proliferation is inhibited in the absence of glucose even when other metabolic substrates such as fatty acids and glutamine are present (58). T cell activation is also dependent on extracellular glutamine (6). Glutamine is converted to glutamate and subsequently to α-ketoglutarate, which enters into the TCA cycle to generate citrate and pyruvate. This process is known as anaplerosis. It replaces the metabolites that are removed from the TCA cycle for the biosynthesis of fatty acids, nucleotides, and proteins allowing the TEFF cells to maintain the integrity of the TCA cycle function (60). Chang et al. demonstrated that lymphoma cells can impose nutrient deprivation on T cells by depleting glucose and glutamine resources. This can lead to decreased release of cytokines, such as IFN-γ, from TEFF cells (61). Arginine is an example of another amino acid which is vital for many T cell functions such as proliferation (62). Research carried out by Rodriguez et al. demonstrated that myeloid derived suppressor cells in the tumor microenvironment express high levels of arginase-1. The resulting lower levels of arginine led to inhibition of T cell receptor expression and antigen specific T cell responses (63). Sequestration of cysteine by myeloid derived suppressor cells is another way in which amino acid deprivation occurs and subsequently results in the inhibition of T cell activation (64). Tumor cells and non-malignant stromal cells can elicit immunosuppressive effects through the expression of amino acid catabolic enzymes, such as indoleamine 2,3-dioxygenase (IDO) which catalyzes the degradation of tryptophan (65). In fact, IDO expression by tumor cells has been shown to correlate with a poor clinical prognosis in several cancers including ovarian (66) and endometrial cancer (67). Elevated IDO expression causes both the depletion of tryptophan and the production of immunosuppressive tryptophan metabolites (68). Such metabolites can impair T cell function (69) and lead to T cell apoptosis (70), thus resulting in less effective anti-tumor T cell responses. Nutrient limitation can also induce autophagy in TEFF cells, as a survival mechanism to generate an intracellular source of nutrients (71). Reduced levels of amino acid or decreased ATP/AMP ratios result in AMPK activation, which phosphorylates the protein kinase unc-51-like kinase 1/2 (Ulk1/2). Activation of Ulk1/2 then initiates autophagy (72). In addition to autophagy, increased metabolic stress due to nutrient deprivation can ultimately lead to T cell apoptosis (73).
Chronic T Cell Activation
Chronic T cell activation occurs due to constant antigen exposure and can induce a state of T cell non-responsiveness termed exhaustion. T cell exhaustion is defined by poor effector function, continued expression of inhibitory receptors, and a gene expression profile distinct from TEFF or TMEM cells (74). The tumor microenvironment establishes an immunosuppressive environment in which T cells respond in a similar manner to exhausted T cells in chronic viral infections (75). This may partially explain why tumors continue to grow despite the presence of tumor specific T cells (76). Baitsch et al. examined T cells from metastases in patients with advanced stage III–IV melanoma and found that these T cells exhibited an exhaustion profile and produced insufficient levels of IFN-γ. The T cell exhaustion at the metastatic tumor sites was induced by continuous antigen exposure coupled with inhibitory signals from tumor cells and non-malignant stromal cells (77). The programed death receptor 1 (PD-1) is an immune-inhibitory receptor, which is expressed on chronically activated T cells. Ahmadzadeh et al. found increased expression of this receptor on tumor infiltrating T cells compared with T cells in peripheral tissue and the blood of patients with metastatic melanoma. The augmented PD-1 expression was associated with impaired effector function in these T cells, decreased proliferation, and reduced ability to differentiate into TMEM cells (76). Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is another negative regulator of T cells, which is up-regulated following T cell activation and during T cell exhaustion (78). Higher levels of CTLA-4 expression on T cells out competes CD28 for binding to CD80 and CD86 on the antigen presenting cell, thus preventing co-stimulation of the T cell (79). CTLA-4 and PD-1 also participate in the regulation of cellular metabolism by blocking CD28-mediated metabolic changes following T cell activation. Ligations of CTLA-4 and PD-1 have been shown to inhibit PI3K/Akt/mTOR signaling resulting in diminished glucose uptake and glycolytic rate (80). Binding of CTLA-4 expressed on TREG cells to CD80 and CD86 on dendritic cells is believed to suppress T cell responses through the up-regulation of IDO expression, thus revealing another mechanism through which CTLA-4 can contribute to tumor antigen tolerance.
Targeting T Cell Metabolism; A New Avenue for Cancer Immunotherapy
Dysregulation of T cell subsets can result in many immune-mediated disorders, due to a disruption in the balance between protective immunity and suppression of pathological inflammation (1). Modulation of immune cell metabolism has been investigated in conditions where there is a chronic overreaction of the immune system, such as asthma. Chronically activated CD4+ T cells isolated from asthma patients have been shown to overexpress pyruvate dehydrogenase kinase-1 (PDK-1), which results in the production of high levels of lactate which promotes aerobic glycolysis. Treatment with the PDK-1 inhibitor dichloroacetate (DCA) inhibited the proliferation of T cells and reduced inflammatory cytokine production by switching the metabolism back to OxPhos. In mouse models this resulted in reduced airway inflammation (81). This study, together with others, has attempted to dampen excessive inflammatory responses through attenuation of TEFF cell responses and enhancement of TREG cell responses by targeting metabolic pathways. Treatment of T cells isolated from a mouse model of asthma with metformin induced the generation of TREG cells through the activation of AMPK and therefore, conferred protective immunity without inappropriate inflammation (28). Metformin is an oral anti-diabetic drug, which reduces hepatic glucose production and increases insulin sensitivity and glucose uptake by muscles and adipocytes, resulting in decreased insulinemia and insulin sensitivity (82). Zhou et al. demonstrated that following inhibition of complex I of the mitochondrial respiratory chain, metformin induces a decrease in ATP synthesis and increases the AMP/ATP ratio, thus stimulating AMPK (83). Studies targeting T cell metabolism have already been conducted in mouse models of arthritis, lupus, uveitis, and asthma and similar studies in obesity-associated cancer might elucidate a mechanism by which the T cell inflammatory responses can be abrogated in adipose tissue to attenuate tumor-promoting chronic inflammation (84–87). Targeting T cell metabolism for cancer immunotherapy must take a different approach by augmenting the beneficial anti-tumor responses of TEFF cells initially, leading to TMEM cell generation and by attenuating the responses of TREG cells. A detailed understanding of the exact metabolic needs of different T cell subsets will allow therapies to be designed in order to achieve this (27).
Decreasing TREG Cell Proliferation
Increased numbers of TREG cells within the tumor microenvironment have been reported in patients with cancer (88) and are associated with a poorer outcome (89). One theory proposed is that tumor cells both secrete and induce the secretion of TGF-β/IL-10 from immature dendritic cells and results in the generation of TREG cells from CD4+CD25− naïve T cells (90). It has been demonstrated in mouse models of cancer that depletion of TREG cells improves endogenous immune-mediated tumor rejection (91). Clinical trials have shown the feasibility and relative safety of managing TREG cells in human cancer, although treatment effects have been modest. Ontak is a monovalent human IL-2 fusion toxin licensed to treat human CD25+ cutaneous T cell lymphoma (92). Due to the phenotypic similarity of CD4+CD25+ cutaneous T cell lymphoma cells and CD4+CD25+ TREG cells, Ontak was tested in many cancers. Some studies demonstrated depletion in TREG cell numbers and improved immunity in patients with metastatic ovarian carcinoma (93), while other studies showed no benefit in melanoma patients (94). Modulation of TREG cell metabolism to suppress their differentiation is a novel approach being investigated in vivo. TREG cells require lipid oxidation for differentiation and treatment of these cells with etomoxir, a CPT1A blocker, has been successful in suppressing TREG cell generation (Figure 2). This study is very promising for cancer therapy due to the fact that TREG cell generation was decreased but the development of IFN-γ secreting Th1 cells was not affected (28).
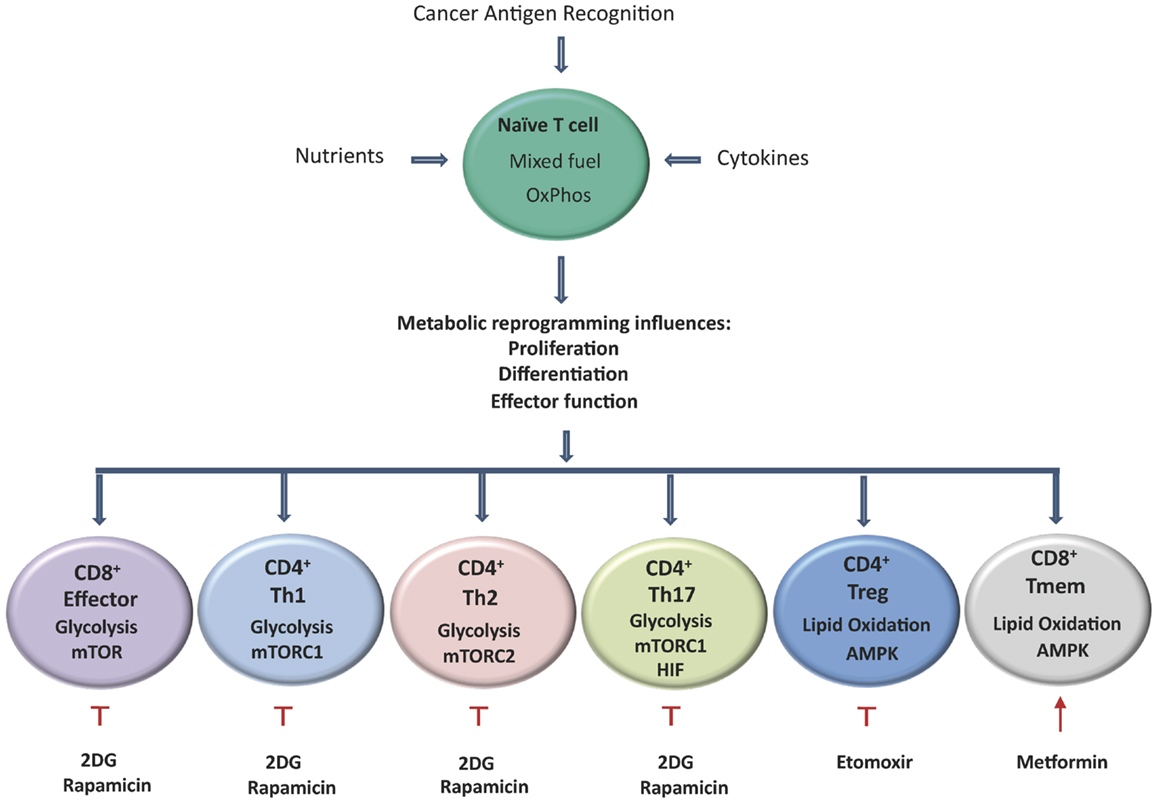
Figure 2. Activation of T cells results in metabolic reprograming, which influences their proliferation, differentiation, and effector functions. Aerobic glycolysis becomes the predominant metabolic pathway used by CD8+ effector cells, CD4+ Th1, Th2, and Th17 cells, by contrast TREG and TMEM rely on lipid oxidation. T cells that differentiate into Th17 cells primarily rely on glycolysis controlled by HIF-1α. TMEM and TREG rely on lipid oxidation under the control of AMPK. Tumor specific TMEM cell prevalence can be enhanced through the inhibition of glycolysis following treatment with the hexokinase-2 inhibitor 2DG. In addition, inhibition of mTOR by rapamycin and activation of AMPK by the mitochondrial complex I inhibitor metformin can also led to increased survival of TMEM cells. Etomoxir, a specific inhibitor of CPT1A results in the reduction of TREG prevalence and has also shown promising results in delaying tumor development.
Increasing TEFF Cell Proliferation
Perhaps the most beneficial approach to boost anti-tumor immunity is to increase activated tumor specific TEFF cell numbers (Figure 2). The development of IDO inhibitors is one avenue currently being explored for reactivation of T cells which have become exhausted or tolerant to tumor antigens. One of the most widely studied IDO inhibitors is 1-methyl-tryptophan. In combination with other chemotherapeutic agents such as cyclophosphamide, it led to the regression of established tumors in a mouse model of HER-2 breast cancer (95). Another novel IDO inhibitor: INCB024360 inhibited tumor growth in cell and mouse models due to increased T cell proliferation and IFN-γ production (96). Reactivating TEFF cells by increasing tryptophan levels is advantageous over reactivation by increasing glycolysis, due to the similar glycolytic metabolic pathways used by both T cells and tumor cells for cell survival and growth (97). Doedens et al. have demonstrated that genetically activating the HIF-1α pathway in a B16 mouse melanoma tumor cell line can prevent TEFF cell exhaustion by sustaining the effector functions of T cells despite persistent antigen exposure (98). As previously discussed, the HIF-1α pathway can serve as both a positive and negative regulator of TEFF cells. Further research is required to elucidate the net effect of using HIF-1α activators to boost TEFF cell-mediated responses to persistent tumor antigen challenge. Blockade of CTLA-4 by antibodies prevents the inhibitory signal to the T cell resulting in an enhanced T cell response against tumor cells and improved anti-tumor activity (79). Ipilimumab is a fully humanized IgG1 monoclonal antibody against the extracellular domain of CTLA-4 and has been approved for the treatment of metastatic melanoma (99). Ipilimumab binding to CTLA-4 allows CD28 on the T cell to bind to the co-stimulatory molecules CD80 and CD86 on the antigen presenting cell. This is cited as the mechanism of action of ipilimumab (99). CTLA-4 also inhibits increased glucose metabolism following T cell activation which is highly relevant as increased glucose metabolism is vital to sustain the increased metabolic demands that occur in the transition of a naïve T cell to a TEFF cell (80, 100). Therefore, it can be concluded from this evidence that the success of ipilimumab in the treatment of malignant melanoma (101) might be in part due to its negative effects on glucose metabolism. Some drug therapies currently available affect T cell metabolism as well as their intended therapeutic targets. Imatinib is a tyrosine kinase inhibitor originally designed to target Bcr-Abl in patients with chronic myeloid leukemia (102). However, it has been shown to have opposing effects on T cell metabolism. Balachandran et al. found that imatinib can activate CD8+ T cells and induce TREG cell apoptosis in gastrointestinal tumors through downregulation of IDO, thus resulting in augmented anti-tumor activity (103). Others found that imatinib inhibits both normal and leukemic T cell response through the inhibition of Lck-mediated T cell receptor signaling (104, 105). T cell receptor signaling is necessary for maximum glucose uptake following TCR stimulation, through elevated expression and cell surface trafficking of the glucose transporter Glut1. Without correct T cell receptor signaling, T cells cannot correctly adjust their metabolic profile to become TEFF cells and in this manner, imatinib might be detrimental to T cell mediated anti-tumor immune responses (58).
Increasing TMEM Cell Prevalence
Increasing the number of long-lived tumor specific TMEM cells can lead to sustained anti-tumor activity due to their increased survival and proliferative capacities. In a study by Sukumar et al., the glucose analog 2-deoxyglucose (2DG), an inhibitor of hexokinase-2 (Hk2), was used to block glycolysis (Figure 2). This resulted in the increased activity of signaling pathways and transcription factors that enhance TMEM cell development, such as AMPK which negatively regulated mTOR and Foxo1, and resulted in enhanced anti-tumor activity of CD8+ T cells (106). Other methods to increase the generation of TMEM cells by the modulation of fatty acid metabolism have also been investigated. Treatment with metformin to activate AMPK promoted TMEM cell generation and was able to significantly improve the efficacy of an experimental anti-cancer vaccine in vivo (10) (Figure 2). Metformin has already been shown to inhibit tumor formation due to its direct action on the tumor, but its use as a potentiator of TMEM cell activity still requires further investigation (107). Many mechanisms of action have been proposed for the anti-cancer properties of metformin, including its actions on mTOR (108) and novel actions such as up-regulation of miR33a resulting in down regulated c-Myc expression (109). Rapamycin, through the inhibition of mTOR (specifically complex 1) reduces glycolysis and increases lipid oxidation and can also enhance the formation of TMEM cells (36) (Figure 2). Other modes of action investigated for rapamycin-induced development of TMEM cells include the inhibition of the Th1 promoting transcription factor T-bet and the stabilization of the expression of the related transcription factor eomesodermin (110). Treatment of mice with rapamycin following viral infection not only enhanced frequencies but also accelerated the transition from TEFF cells to virus specific TMEM cells without inhibiting the ability of the infected animal to clear the antigen (111). Due to the multiple actions of mTOR, rapamycin can potentially exert multiple effects on T cell metabolism, particularly its rapid induction of autophagy, which can be both a pro- and anti-survival process in T cells (112). Like metformin, rapamycin can directly target and inhibit tumor cell growth in many ways such as induction of apoptosis (113) and inhibiting angiogenesis (114). Unlike metformin however, rapamycin has immunosuppressive properties and is used as anti-rejection therapy due to its anti-proliferative actions (115). Although to date, the anti-cancer effects of rapamycin and its derivatives are more prominent than the immunosuppressant effects, further research is warranted to ensure this is also the case in the long term (116). Another important role for any drugs developed to enhance the formation of TMEM cells is to enhance the efficacy of cancer vaccines. The main goal of any vaccination is to induce an effective TMEM cell response (117), many of the experiments targeting TMEM cell generation have a better recall response and are more protective following re-challenge (111).
Targeting T Cell Metabolism to Prevent and Treat Obesity-Associated Cancer
Obesity induces a state of tumor-promoting chronic inflammation, marked by elevated levels of free fatty acids, abnormal cytokine production, and activation of inflammatory signaling pathways (118, 119). The expansion of VAT in obesity, results in increased blood supply, inflammatory immune cell infiltration, and enhanced pro-inflammatory cytokine release (9, 120–122, 126). Our group have previously shown that activated IFN-γ producing CD4+ and CD8+ T cells are significantly enriched in obese VAT, while others have found that frequencies of TREG cells are decreased (9, 123, 124). Therefore, obese VAT is characterized by an increased prevalence of pro-inflammatory T cells and reduced numbers of anti-inflammatory T cells, which is likely to contribute to adipose tissue inflammation and the clinical manifestations of obesity, including cancer. Developing effective immunotherapies for obesity-associated malignancies represents a unique challenge; to attenuate chronic systemic inflammation while simultaneously augmenting specific immunity at tumor sites. As pro-inflammatory Th1 cells appear to play a crucial role in obesity-associated inflammation, the alteration of the metabolic profile of T cells in the VAT may present a favorable therapeutic option. There is currently a paucity of studies investigating the efficacy of targeting immunometabolism as a mechanism to control tumor-promoting systemic inflammation associated with obesity. However, there is some precedence for targeting metabolism to control systemic inflammation in autoimmune diseases. For instance, inhibition of both the mTOR and MAPK (ERK) pathways using rapamycin and the MEK1/2 inhibitor PD325901 was identified as a means of robustly blocking effector CD4+ T cell proliferation in a mouse model of arthritis (84). This study found that inhibition of both pathways was more effective than inhibiting either mTOR or ERK alone and resulted in greatly decreased disease severity in a mouse model of arthritis. Since our group has previously identified TEFF cells as key players in adipose tissue inflammation, ERK and mTOR inhibitors might elicit a similar anti-inflammatory effect in obesity (9). Others have found that beauvericin-mediated inhibition of T cell activation in colitis was achieved by targeting P13K and AKT (86). Treatment with beauvericin reduced pro-inflammatory cytokine production, weight loss, diarrhea, and mortality in this mouse model of colitis (86). In addition, pro-inflammatory cytokine production and clinical symptoms in experimental autoimmune uveitis (EAU) were reduced through the inhibition of PI3K/AKT by a novel anti-angiogenic peptide H-RN, thus presenting another potential therapeutic option for attenuating obesity-associated inflammation (87). The small molecule inhibitor IPI-145 has also shown promising therapeutic value in the blockade of PI3K in collagen-induced arthritis, ovalbumin-induced asthma, and systemic lupus suggesting that this molecule might be applicable and effective in autoimmune and multiple inflammatory disorders, including obesity (85). The inhibitor elicited its effects through blockade of both PI3K-δ and PI3K-γ and subsequent suppression of both innate and adaptive immune responses. This resulted in reduced pro-inflammatory cytokine production and disease severity in all mouse models tested (85). From these studies, it appears that anti-inflammatory agents targeting PI3K, AKT, and mTOR could be a favorable option in the treatment of obesity-associated inflammation and cancer. However, metabolic targets for the treatment of obesity-associated cancer and disease should first be identified through definitive studies that will elucidate the metabolic profiles of T cells in VAT and the corresponding tumor. A therapy targeting specific metabolic pathways of VAT T cells could be used alone as a prophylactic in obese individuals who are at risk of developing malignancy or in combination with standard therapies in patients who have already developed an obesity-associated malignancy. Focused studies are currently lacking but clearly warranted in this area.
Conclusion
It is clear that T cell survival, function, and differentiation are intimately linked to T cell metabolism. The increase in glycolysis and glutaminolysis facilitate the rapid proliferation of T cells required during activation to allow fast clearance of pathogens or tumor cells. TREG and TMEM cells favor FAO, which allows these cells to survive long term, when glucose and growth factors are limited. This ensures the control of inappropriate immune responses and long term immunity. Extensive metabolic reprograming not only fulfils the extra biosynthetic demands of activated T cells, but it is also integrated with signaling pathways that determine T cell fate, thus enhancing our understanding of distinct T cell subset function and regulation. The different metabolic pathways may also provide a new opportunity for modulating the immune response during tumor development and progression. One of the hallmarks of cancer is evasion of the immune system (125) and in the past few years our understanding of how this occurs has greatly improved. One area which holds promise for cancer immunotherapy is the manipulation of immune responses, such that TEFF and TMEM cell-mediated responses are enhanced, while those of TREG cells are decreased, ultimately providing a therapy that might “switch” back on the immune system to target the tumor cell. Reactivating exhausted T cells by adjusting their metabolism also holds potential as a treatment for cancer, however more research needs to be carried out to fully elucidate the role of cellular metabolism in T cell exhaustion. Due to the similarities between tumor cells and activated T cell’s metabolic programing, any therapies designed to modulate tumor cell metabolism must take into account their effects on T cell metabolism in the immune response to cancer.
Since the WHO now estimates that obesity is directly attributable for as many as 41% of malignancies and with the rates of obesity continuing to increase dramatically, it is clear that novel therapeutic options are urgently required. Since activated inflammatory TEFF cells are found to be enriched in the VAT of cancer patients, it is likely that they play pathological roles in driving obesity-associated inflammation. Attenuation of this inflammation might be achieved via metabolic manipulation of inflammatory TEFF cell differentiation and activation. Suppression of excessive pro-inflammatory T cell responses through inhibition of metabolic pathways has already been demonstrated across a spectrum of chronic inflammatory disorders in mice and could represent a viable therapeutic target for obesity-associated cancers.
The possibilities of modulating anti-tumor immune responses through targeting T cell metabolic pathways hold huge promise and clearly warrant further investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Health Research Board of Ireland Research Award HRA_POR/2011/91 and Mary Mockler is currently enrolled on the M.Sc. in Translational Oncology in Trinity College Dublin.
References
1. Annunziato F, Romagnani S. Heterogeneity of human effector CD4+ T cells. Arthritis Res Ther (2009) 11(6):257. doi: 10.1186/ar2843
2. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi:10.1126/science.1242454
3. Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem (1993) 212(1):95–9. doi:10.1111/j.1432-1033.1993.tb17637.x
4. van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, et al., Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol (2003) 4(4):361–5. doi:10.1038/ni912
5. Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem (1994) 269(50):31484–90.
6. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al., Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185(2):1037–44. doi:10.4049/jimmunol.0903586
7. Chandra RK. Nutrition and the immune system: an introduction. Am J Clin Nutr (1997) 66(2):460S–3S.
8. Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev (2012) 249(1):218–38. doi:10.1111/j.1600-065X.2012.01151.x
9. Lysaght J, Allott EH, Donohoe CL, Howard JM, Pidgeon GP, Reynolds JV. T lymphocyte activation in visceral adipose tissue of patients with oesophageal adenocarcinoma. Br J Surg (2011) 98(7):964–74. doi:10.1002/bjs.7498
10. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al., Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
11. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5(11):844–52. doi:10.1038/nri1710
12. Kronenberg M, Rudensky A. Regulation of immunity by self-reactive T cells. Nature (2005) 435(7042):598–604. doi:10.1038/nature03725
13. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol (2004) 172(8):4661–5. doi:10.4049/jimmunol.172.8.4661
14. Bental M, Deutsch C. Metabolic changes in activated T cells: an NMR study of human peripheral blood lymphocytes. Magn Reson Med (1993) 29(3):317–26. doi:10.1002/mrm.1910290307
15. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111(4):2101–11. doi:10.1182/blood-2007-06-096297
16. Ruppert SM, Li W, Zhang G, Carlson AL, Limaye A, Durum SK, et al., The major isoforms of Bim contribute to distinct biological activities that govern the processes of autophagy and apoptosis in interleukin-7 dependent lymphocytes. Biochim Biophys Acta (2012) 1823(10):1877–93. doi:10.1016/j.bbamcr.2012.06.017
17. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
18. Fleury C, Mignotte B, Vayssière J-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie (2002) 84(2–3):131–41. doi:10.1016/S0300-9084(02)01369-X
19. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27(2):173–8. doi:10.1016/j.immuni.2007.10.014
20. Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res (2012) 18(5):1207–13. doi:10.1158/1078-0432.CCR-11-1591
21. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi:10.1016/j.immuni.2011.09.021
22. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
23. Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol (1999) 162(5):2775–84.
24. Wahl DR, Byersdorfer CA, Ferrara JL, Opipari AW Jr., Glick GD. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol Rev (2012) 249(1):104–15. doi:10.1111/j.1600-065X.2012.01148.x
25. De Boer RJ, Homann D, Perelson AS. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol (2003) 171(8):3928–35. doi:10.4049/jimmunol.171.8.3928
26. Titu LV, Monson JR, Greenman J. The role of CD8(+) T cells in immune responses to colorectal cancer. Cancer Immunol Immunother (2002) 51(5):235–47. doi:10.1007/s00262-002-0276-4
27. Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev (2012) 249(1):176–94. doi:10.1111/j.1600-065X.2012.01141.x
28. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al., Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
29. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al., The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi:10.1038/ni.2005
30. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al., Stability of the regulatory T cell lineage in vivo. Science (2010) 329(5999):1667–71. doi:10.1126/science.1191996
31. Marelli-Berg FM, Fu H, Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology (2012) 136(4):363–9. doi:10.1111/j.1365-2567.2012.03583.x
32. Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol (2011) 11(2):109–17. doi:10.1038/nri2888
33. Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al., Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol (2008) 9(5):513–21. doi:10.1038/ni.1603
34. Waugh C, Sinclair L, Finlay D, Bayascas JR, Cantrell D. Phosphoinositide (3,4,5)-triphosphate binding to phosphoinositide-dependent kinase 1 regulates a protein kinase B/Akt signaling threshold that dictates T-cell migration, not proliferation. Mol Cell Biol (2009) 29(21):5952–62. doi:10.1128/MCB.00585-09
35. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol (2008) 8(2):107–19. doi:10.1038/nri2251
36. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
37. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al., Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi:10.1016/j.immuni.2011.12.007
38. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al., CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110(35):14336–41. doi:10.1073/pnas.1221740110
39. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med (1997) 3(2):177–82. doi:10.1038/nm0297-177
40. Roman J, Rangasamy T, Guo J, Sugunan S, Meednu N, Packirisamy G, et al., T-cell activation under hypoxic conditions enhances IFN-gamma secretion. Am J Respir Cell Mol Biol (2010) 42(1):123–8. doi:10.1165/rcmb.2008-0139OC
41. Zuckerberg AL, Goldberg LI, Lederman HM. Effects of hypoxia on interleukin-2 mRNA expression by T lymphocytes. Crit Care Med (1994) 22(2):197–203. doi:10.1097/00003246-199401000-00352
42. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al., Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
43. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123(9):3664–71. doi:10.1172/JCI67230
44. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al., NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature (2008) 453(7196):807–11. doi:10.1038/nature06905
45. Lukashev D, Caldwell C, Ohta A, Chen P, Sitkovsky M. Differential regulation of two alternatively spliced isoforms of hypoxia-inducible factor-1 alpha in activated T lymphocytes. J Biol Chem (2001) 276(52):48754–63. doi:10.1074/jbc.M104782200
46. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al., Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39(2):171–83. doi:10.1016/j.molcel.2010.06.022
47. Carraro F, Pucci A, Pellegrini M, Pelicci PG, Baldari CT, Naldini A. p66Shc is involved in promoting HIF-1alpha accumulation and cell death in hypoxic T cells. J Cell Physiol (2007) 211(2):439–47. doi:10.1002/jcp.20951
48. Makino Y, Nakamura H, Ikeda E, Ohnuma K, Yamauchi K, Yabe Y, et al., Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol (2003) 171(12):6534–40. doi:10.4049/jimmunol.171.12.6534
49. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al., HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
50. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al., Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
51. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3(3):187–97. doi:10.1016/j.cmet.2006.01.012
52. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al., A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15(9):1016–22. doi:10.1038/nm.2015
53. Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, Rizzo A, et al., IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol (2008) 38(2):470–8. doi:10.1002/eji.200737635
54. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al., Induction and molecular signature of pathogenic TH17 cells. Nat Immunol (2012) 13(10):991–9. doi:10.1038/ni.2416
55. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al., TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi:10.1038/ni1539
56. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8(1):74–80. doi:10.1038/nri2233
57. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al., mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell (2002) 110(2):163–75. doi:10.1016/S0092-8674(02)00808-5
58. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al., Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180(7):4476–86. doi:10.4049/jimmunol.180.7.4476
59. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol (2008) 38(9):2438–50. doi:10.1002/eji.200838289
60. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem (2002) 277(34):30409–12. doi:10.1074/jbc.R200006200
61. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al., Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
62. Ochoa JB, Strange J, Kearney P, Gellin G, Endean E, Fitzpatrick E. Effects of L-arginine on the proliferation of T lymphocyte subpopulations. JPEN J Parenter Enteral Nutr (2001) 25(1):23–9. doi:10.1177/014860710102500123
63. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al., Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
64. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi:10.1158/0008-5472.CAN-09-2587
65. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al., Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi:10.1038/nm934
66. Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, et al., Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res (2005) 11(16):6030–9. doi:10.1158/1078-0432.CCR-04-2671
67. Ino K, Yoshida N, Kajiyama H, Shibata K, Yamamoto E, Kidokoro K, et al., Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer (2006) 95(11):1555–61. doi:10.1038/sj.bjc.6603477
68. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117(5):1147–54. doi:10.1172/JCI31178
69. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med (2002) 196(4):459–68. doi:10.1084/jem.20020121
70. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al., T cell apoptosis by tryptophan catabolism. Cell Death Differ (2002) 9(10):1069–77. doi:10.1038/sj.cdd.4401073
71. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330(6009):1344–8. doi:10.1126/science.1200403
72. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al., Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331(6016):456–61. doi:10.1126/science.1196371
73. Mason EF, Rathmell JC. Cell metabolism: an essential link between cell growth and apoptosis. Biochim Biophys Acta (2011) 1813(4):645–54. doi:10.1016/j.bbamcr.2010.08.011
75. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
76. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al., Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi:10.1182/blood-2008-12-195792
77. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al., Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest (2011) 121(6):2350–60. doi:10.1172/JCI46102
78. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol (2001) 19:565–94. doi:10.1146/annurev.immunol.19.1.565
79. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
80. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al., CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
81. Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q, et al., The role of low-level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol (2012) 302(3):L300–7. doi:10.1152/ajplung.00221.2011
82. Rosilio C, Ben-Sahra I, Bost F, Peyron JF. Metformin: a metabolic disruptor and anti-diabetic drug to target human leukemia. Cancer Lett (2014) 346(2):188–96. doi:10.1016/j.canlet.2014.01.006
83. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al., Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108(8):1167–74. doi:10.1172/JCI200113505
84. Lin JT, Stein EA, Wong MT, Kalpathy KJ, Su LL, Utz PJ, et al., Differential mTOR and ERK pathway utilization by effector CD4 T cells suggests combinatorial drug therapy of arthritis. Clin Immunol (2012) 142(2):127–38. doi:10.1016/j.clim.2011.09.008
85. Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, et al., PI3K-delta and PI3K-gamma inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol (2013) 20(11):1364–74. doi:10.1016/j.chembiol.2013.09.017
86. Wu XF, Xu R, Ouyang ZJ, Qian C, Shen Y, Wu XD, et al., Beauvericin ameliorates experimental colitis by inhibiting activated T cells via downregulation of the PI3K/Akt signaling pathway. PLoS One (2013) 8(12):e83013. doi:10.1371/journal.pone.0083013
87. Wang L, Xu Y, Yu Q, Sun Q, Xu Y, Gu Q, et al., H-RN, a novel antiangiogenic peptide derived from hepatocyte growth factor inhibits inflammation in vitro and in vivo through PI3K/AKT/IKK/NF-kappaB signal pathway. Biochem Pharmacol (2014) 89(2):255–65. doi:10.1016/j.bcp.2014.02.026
88. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res (2003) 9(2):606–12.
89. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al., Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi:10.1038/nm1093
90. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al., Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202(7):919–29. doi:10.1084/jem.20051511
91. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol (1999) 163(10):5211–8.
92. Foss FM. DAB(389)IL-2 (ONTAK): a novel fusion toxin therapy for lymphoma. Clin Lymphoma (2000) 1(2):110–6. doi:10.3816/CLM.2000.n.009
93. Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol (2005) 54(6):369–77. doi:10.1111/j.1600-0897.2005.00330.x
94. Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (denileukin diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother (2005) 28(6):582–92. doi:10.1097/01.cji.0000175468.19742.10
95. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med (2005) 11(3):312–9. doi:10.1038/nm1196
96. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al., Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood (2010) 115(17):3520–30. doi:10.1182/blood-2009-09-246124
97. Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab (2013) 1(1):5. doi:10.1186/2049-3002-1-5
98. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al., Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol (2013) 14(11):1173–82. doi:10.1038/ni.2714
99. Ascierto PA. Ipilimumab in the treatment of metastatic melanoma: a summary of recent studies. Tumori (2013) 99(6):302e–5e. doi:10.1700/1390.15474
100. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al., The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi:10.1016/S1074-7613(02)00323-0
101. Robert C, Thomas L, Bondarenko I, O’Day S, M DJW, Garbe C, et al., Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi:10.1056/NEJMoa1104621
102. Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al., Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol (2007) 25(9):1035–44. doi:10.1038/nbt1328
103. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al., Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med (2011) 17(9):1094–100. doi:10.1038/nm.2438
104. Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood (2004) 104(4):1094–9. doi:10.1182/blood-2003-12-4266
105. Nika K, Soldani C, Salek M, Paster W, Gray A, Etzensperger R, et al., Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity (2010) 32(6):766–77. doi:10.1016/j.immuni.2010.05.011
106. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al., Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi:10.1172/JCI69589
107. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al., Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res (2007) 67(14):6745–52. doi:10.1158/0008-5472.CAN-06-4447
108. Ben-Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al., Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res (2011) 71(13):4366–72. doi:10.1158/0008-5472.CAN-10-1769
109. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al., Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun (2012) 3:865. doi:10.1038/ncomms1859
110. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32(1):67–78. doi:10.1016/j.immuni.2009.10.010
111. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al., mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi:10.1038/nature08155
112. Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, et al., Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol (2006) 177(8):5163–8. doi:10.4049/jimmunol.177.5.3406
113. Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, et al., High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle (2011) 10(22):3948–56. doi:10.4161/cc.10.22.18124
114. Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, et al., Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med (2002) 8(2):128–35. doi:10.1038/nm0202-128
115. Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem (1998) 31(5):335–40. doi:10.1016/S0009-9120(98)00045-9
116. Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol (2005) 56(1):47–60. doi:10.1016/j.critrevonc.2004.09.009
117. Nam JH. Rapamycin: could it enhance vaccine efficacy? Expert Rev Vaccines (2009) 8(11):1535–9. doi:10.1586/erv.09.115
118. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112(12):1785–8. doi:10.1172/JCI200320514
119. Ramos-Nino ME. The role of chronic inflammation in obesity-associated cancers. ISRN Oncol (2013) 2013:697521. doi:10.1155/2013/697521
120. Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm (2006) 74:443–77. doi:10.1016/S0083-6729(06)74018-3
121. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol (2009) 1(5):a000141. doi:10.1101/cshperspect.a000141
122. Lutz CT, Quinn LS. Sarcopenia, obesity, and natural killer cell immune senescence in aging: altered cytokine levels as a common mechanism. Aging (Albany NY) (2012) 4(8):535–46.
123. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
124. Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med (2013) 64:45–57. doi:10.1146/annurev-med-121211-091527
125. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
Keywords: immunometabolism, cancer, T cells, metabolic targeting agents, obesity
Citation: Mockler MB, Conroy MJ and Lysaght J (2014) Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front. Oncol. 4:107. doi: 10.3389/fonc.2014.00107
Received: 03 March 2014; Accepted: 28 April 2014;
Published online: 16 May 2014.
Edited by:
Juan José Lasarte, Centro de Investigación Médica Aplicada, SpainReviewed by:
Elizabeth Ann Repasky, Roswell Park Cancer Institute, USAHeather Lynne Wilson, Vaccine and Infectious Disease Organization – International Vaccine Centre, Canada
Copyright: © 2014 Mockler, Conroy and Lysaght. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne Lysaght, Department of Surgery, Trinity Centre for Health Sciences, St. James’s Hospital, Dublin 8, Ireland e-mail: jlysaght@tcd.ie