 Andrew Burgess
Andrew Burgess Mina Rasouli
Mina Rasouli Samuel Rogers
Samuel Rogers- 1The Kinghorn Cancer Centre, Garvan Institute of Medical Research, Sydney, NSW, Australia
- 2St. Vincent’s Clinical School, Faculty of Medicine, UNSW Australia, Sydney, NSW, Australia
The final stage of cell division (mitosis), involves the compaction of the duplicated genome into chromatid pairs. Each pair is captured by microtubules emanating from opposite spindle poles, aligned at the metaphase plate, and then faithfully segregated to form two identical daughter cells. Chromatids that are not correctly attached to the spindle are detected by the constitutively active spindle assembly checkpoint (SAC). Any stress that prevents correct bipolar spindle attachment, blocks the satisfaction of the SAC, and induces a prolonged mitotic arrest, providing the cell time to obtain attachment and complete segregation correctly. Unfortunately, during mitosis repairing damage is not generally possible due to the compaction of DNA into chromosomes, and subsequent suppression of gene transcription and translation. Therefore, in the presence of significant damage cell death is instigated to ensure that genomic stability is maintained. While most stresses lead to an arrest in mitosis, some promote premature mitotic exit, allowing cells to bypass mitotic cell death. This mini-review will focus on the effects and outcomes that common stresses have on mitosis, and how this impacts on the efficacy of mitotic chemotherapies.
Introduction
The cell cycle is driven by the activity of the cyclin dependent kinases (Cdk), and their associated regulatory cyclin subunits. Each cell cycle phase is dependent on the sequential activation and deactivation of unique cyclin and Cdk complexes, with mitosis dependent on cyclin B bound with Cdk1 (1). To ensure the cell division process occurs with absolute fidelity, cells have developed numerous cell cycle checkpoints that delay progression in the presence of a wide variety of cellular and environmental stresses. During interphase (G1, S, and G2) stress activates checkpoints, which block cell cycle progression by increasing the translation of Cdk inhibitory proteins and activation of checkpoint kinases (Chk) that phosphorylate and inhibit Cdk (2). However, in mitosis the situation is reversed, the spindle assembly checkpoint (SAC) is on by default, which maintains high Cdk activity, thereby preventing cells from exiting mitosis. The primary role of the SAC is to block the activity of the anaphase promoting complex (APC), an E3 ubiquitin ligase responsible for targeting cyclin B1 (and many other key mitotic proteins) for degradation by the proteasome (3). This inhibition is achieved by the recruitment of several SAC proteins to the kinetochores, a protein structure located on the centromere of each chromosome (Figure 1). This localization allows the formation of the mitotic checkpoint complex (MCC) consisting of Cdc20, Mad2, Bub3, and BubR1, which then binds to and potently inhibits the APC, blocking degradation and preventing cells from entering anaphase (4). Once each kinetochore is attached to the mitotic spindle, the SAC proteins are displaced, and Cdc20 is released, allowing the APC to target proteins for degradation. However, the SAC arrest can be overcome by premature degradation of cyclin B1 (5), or direct inhibition of Cdk1 activity (6, 7) (Figure 1). This process is referred to as mitotic slippage and results in aberrant segregation of chromosomes and failure of abscission during cytokinesis, which can drive polyploidy, chromosome instability, and cancer formation (8). Therefore, during mitosis it is critical that interphase checkpoint pathways are turned off to prevent the deleterious effects of premature Cdk1 inactivation.
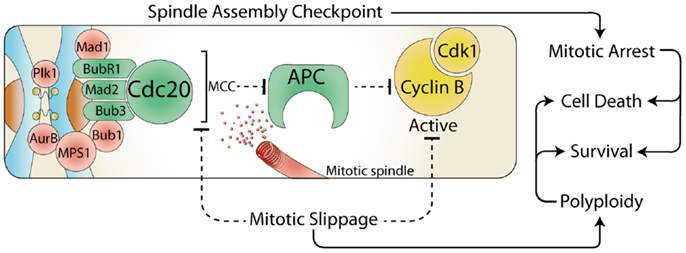
Figure 1. The spindle assembly checkpoint and cell fate. During mitosis, the constitutively active spindle assembly checkpoint (SAC) delays anaphase until all chromosomes are attached to the mitotic spindle. Any stress that prevents satisfaction of the SAC results in a prolonged mitotic arrest, which often leads to cell death. However, the SAC can be over-come by the release of Cdc20 from the mitotic checkpoint complex (MCC) or by direct inhibition of Cdk1. This mitotic slippage can result in polyploidy, increased cell survival, and provides a potential mechanism for escaping mitotic cell death.
Inhibition of Interphase Checkpoints
The inhibition of interphase checkpoints is achieved primarily by inhibition of transcription (9) and down regulation of the majority (60–80%) of protein translation (10). In addition, Cdk1 and other mitotic kinases phosphorylate and disable key effectors of interphase checkpoint pathways, providing a feedback loop that restricts this inhibition to mitosis (11).
Transcription
The inhibition of transcription is a critical mechanism for preventing the upregulation of Cdk inhibitor proteins, such as p21. The expression of p21 is strongly upregulated during interphase in response to a variety of cellular stresses. For example, during interphase, DNA single and double strand breaks induced by exposure to ultraviolet light (UV) or ionizing radiation (IR) respectively, results in the recruitment and activation of ataxia-telangiectasia mutated and related (ATM and ATR) kinases to the sites of damage. ATM/ATR then activate p53, which in concert with the transcription factor Sp1, increases p21 expression (12, 13). However, during mitosis the majority of proteins involved in transcription are removed from the DNA, inhibiting the production of new mRNA (14, 15). Surprisingly, transcription factors and other structural proteins can still gain access to the highly compacted chromosome structure (16), and are actively removed by mitotic kinases (17). For example, Cdk1 phosphorylates Sp1 and CUX1 resulting in their dissociation from chromatin during mitosis (18, 19), thereby preventing upregulation of p21 (20).
Phosphorylation
During interphase, stress often triggers a kinase phosphorylation cascade, which culminates in the inhibitory phosphorylation of the interphase Cdk. To ensure that Cdk1 is not inhibited during mitosis, these checkpoint kinases (Chk) must be inhibited. Surprisingly, Cdk1 itself disables many of these, for example, it phosphorylates Chk1/2 preventing activation by ATM/ATR (21). Furthermore, Cdk1 phosphorylation of the DNA damage signaling and repair proteins 53BP1 and BRCA1, blocks their recruitment to sites of DNA damage (22). In addition, many of these interphase Chks are repurposed and required for normal progression through mitosis. For example, Chk2 localizes to kinetochores during mitosis, stabilizing MPS1 and phosphorylating Aurora B (23). Active Aurora B then phosphorylates ATM (24), which then phosphorylates γ-H2AX and Bub1 at kinetochores (24), promoting the accumulation of Mad2 and Cdc20 (25). Consequently, ATM activity is required to ensure correct centrosome and mitotic spindle formation (26, 27).
Translation
The translation of mRNA into proteins is actively inhibited during mitosis (10). During interphase, the majority of mRNA is guided to ribosomes by cap-dependent translation, however as cells enter mitosis this process is repressed (9) by phosphorylation of cap-binding proteins (28). As a result, translation switches from the cap-dependent system to mRNA that contains an internal ribosomal entry site (IRES) (29). The mRNA of several important mitotic proteins contain IRES sites (30, 31), which ensures their continued translation during mitosis. In addition, the mRNA of critical mitotic factors such as cyclin B, are restricted temporally to mitosis, and locally at the mitotic spindle, by polyadenylation (32, 33).
SAC and the Response to Stress in Mitosis
Any stress that directly or indirectly prevents the satisfaction of the SAC prevents cells from progressing past metaphase. However, some stresses are able to deactivate the SAC and induce mitotic slippage, therefore bypassing mitotic cell death. Interestingly, mitotic slippage has been suggested as a possible mechanism for resistance to mitotic chemotherapies, in particular the microtubule poison Taxol (34). Therefore, understanding exactly how common environmental and cellular stresses affect mitosis is critical for understanding how and why some cancer cells are sensitive and others are resistant to this important class of chemotherapies.
DNA damage
Attempting to repair DNA during mitosis is highly dangerous for cells and can result in the fusion of telomeres, failed separation of chromatids during anaphase, and the promotion of genomic instability and cancer (22). Therefore, some have suggested that the primary mitotic response to DNA damage is to mark sites of damage (with γ-H2AX), but not to arrest in mitosis (35). Instead, damaged cells are allowed to exit to the next G1 phase where repair or death can be triggered (36). However, many cells do arrest for varying amounts of time in response to an array of DNA damaging stresses. The length of arrest roughly correlates with the level of damage, with higher levels that disrupt kinetochore–microtubule function being more efficient at blocking mitotic exit in a SAC dependent manner (37). Furthermore, a prolonged arrest can itself damage telomeres (38), suggesting that mitotic cells damage their DNA on purpose. The point of this self inflicted damage is still unclear, but it may act as a backup pathway, ensuring even minor mitotic DNA damage is fully detected in the following G1 thereby preventing defects being passed on to subsequent generations.
DNA decatenation. During replication in interphase, sister chromatid pairs become interwound, and must be untangled prior to metaphase by decatenation, a process that requires topoisomerase II (Topo II). In addition, DNA decatenation is also required for correct chromatid and telomere separation during anaphase (39). Inhibition of Topo II during mitosis produces different mitotic responses, which are dependent on the inhibitor used, and specifically if DNA damage is produced. For example, doxorubicin creates significant levels of DNA damage (γ-H2AX foci), and consequently cells arrest in metaphase for up to 9 h (40). In contrast, ICRF-193 generally produces mild damage, and results in cells only delaying in mitosis for 1–2 h (37, 41) although ultrafine DNA bridges are formed during anaphase causing cells to fail abscission and form polyploid cells (41, 42). In all cases, the arrest during mitosis is dependent on the SAC, and is likely due to direct damage of kinetochore structure preventing stable microtubule attachments (Figure 2). For example, the delay induced by ICRF-193 requires inhibition of the APC by Mad2, but surprisingly Mad2 does not accumulate at kinetochores (35, 43). This may explain why this delay is short lived. Unfortunately, the inhibition of Topo II prior to mitosis blocks cells in G2 phase (44), consequently its use in combination with mitotic chemotherapies such as Taxol is often counter-intuitive as cells never enter mitosis and are resistant to Taxol induced death (45, 46).
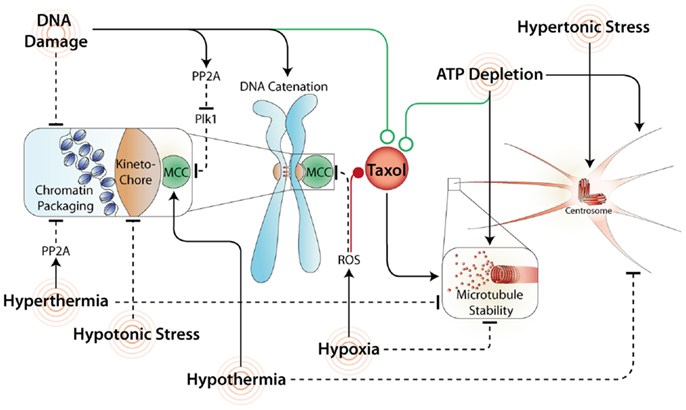
Figure 2. Common stresses and their effects on mitosis and Taxol response. A variety of stresses affect mitosis by acting directly or indirectly on the SAC. Stresses that maintain the SAC and/or increase microtubule stability often synergize (green line) with Taxol. In contrast, stresses that inhibit the MCC and/or disrupt microtubule formation commonly antagonize (red line) Taxol induced mitotic arrest and promote mitotic slippage.
Double strand breaks. Extensive double strand breaks during mitosis produce a strong SAC dependent arrest with cells delaying for more than 5 h over the normal 30–60 min transit time (37). Furthermore, extensive DNA damage has also been shown to inhibit the activity of Polo like kinase 1 (Plk1) (47), a mitotic kinase that plays a key role in mediating attachments between the kinetochore and mitotic spindle (48). This inhibition occurs independently of ATM (49), primarily through PP2A mediated dephosphorylation of Plk1 (50), and likely strengthens the mitotic arrest induced by extensive double strand breaks by preventing satisfaction of the SAC (Figure 2). Consequently, combining DNA damaging agents with Taxol, especially in p53 mutant cancer cells with dysfunctional interphase DNA damage checkpoints (51), may greatly enhance the amount of damage produced and promote a prolonged mitotic arrest resulting in increased levels of cell death. Accordingly, Taxol is commonly used in combination with platinum-based chemotherapeutics as a first line treatment for ovarian cancer (52), and is being trialed in combination with DNA damaging agents in several other cancer types including small cell lung cancer, melanoma, and pancreatic cancer (53–55).
Chromatin structure. Disruption of the mitotic chromosome architecture also produces a temporary mitotic delay. Treatment with histone deacetylase inhibitors (HDACi) prevents correct chromosome condensation and increases the access of transcription factors to the DNA, disrupting correct kinetochore formation (56). This delays the correct capture and alignment of chromosomes by the mitotic spindle, leading to SAC-dependent mitotic arrest (44, 57). However, if this damage is too severe, SAC proteins fail to remain at kinetochores, leading to silencing of the SAC and premature exit (slippage) from mitosis (58, 59). Interestingly, although HDACi have been highly successful in treatment of lymphoma, they have not been as successful with solid tumors, which could be due to SAC dysfunction (e.g., BubR1 mutation) in these cancers and an increased rate of mitotic slippage.
Hypoxia and oxidative stress
Reduced oxygen supply especially within the core of solid tumors results in a hypoxic environment within the tumor mass. Hypoxia is a poor prognostic factor, and correlates with resistance to radiation and many chemotherapeutic agents (60). Exposure to hypoxia during mitosis results in the rapid disruption and destabilization of microtubules (61), which delays mitotic progression. However, this arrest is unstable and cyclin B levels decrease rapidly (62), in turn inactivating Cdk1, and promoting mitotic slippage, providing an explanation for why hypoxia induces tetraploidization in melanoma (63). However, hypoxia induces a wide variety of intracellular responses including formation of reactive oxygen species (ROS), and a switch to anaerobic glycolysis resulting in decreased levels of ATP. The effects of hypoxia on mitosis are most likely due to the increased formation of ROS. In support, exposing mitotic cells to hydrogen peroxide (H2O2) to mimic ROS, induces mitotic slippage and the formation of hypertetraploid cells (64). The mechanism for this slippage is yet to be full elucidated, however, in yeast, H2O2 exposure depletes the SAC protein BubR1 from kinetochores, silencing the SAC allowing cells to exit mitosis prematurely (65). In addition, H2O2 also depolymerizes microtubules (66), which results in need for higher doses of Taxol to stabilize microtubules and induce cell death (67). These effects may explain why hypoxia reduces toxicity to Taxol in cancer cells (Figure 2). Consequently, reducing ROS with antioxidants has long been proposed as a co-treatment to enhance the effects of Taxol, with some limited success (68). The inconsistent results are likely due to the specific antioxidant used. For example, the popular dietary antioxidants Resveratrol and Fisetin (found in red wine), inhibit Cdks, induce a G2 arrest and prevent entry into mitosis (69, 70), providing an explanation for why they antagonize Taxol (71, 72). Therefore, finding methods that specifically reduce ROS without off target effects will be critical for the future success of co-treatment regimes.
ATP depletion
Hypoxia can also cause depletion of ATP pools, however the mitotic effects of ATP depletion are opposite to that of hypoxia and oxidative stress. Specific depletion of ATP pools with DNP, Azide, or AMP-PNP, results a rapid prolonged mitotic arrest in mammalian cells (73). ATP is needed for microtubule disassembly (74), and therefore depletion of ATP stabilizes microtubules (75). In addition, depletion of ATP activates AMP-activated protein kinase, which phosphorylates myosin regulatory light chain, and promotes astral microtubule growth (76) (Figure 2). Surprisingly, depletion of ATP also depletes Mad2 and BubR1 from kinetochores, with both proteins accumulating at spindle poles, however this does not appear to affect their ability to bind Cdc20 and inhibit the APC (77–79). Taken together, this area has significant potential for future novel therapeutic approaches, with some metabolic inhibitors already showing synergy with Taxol (80).
Thermal shock
Heat-shock (hyperthermia) has been commonly used as adjunctive cancer therapy to augment radiotherapy and chemotherapy, with varying levels of success (81). The initial mitotic response to acute (42°C) heat-shock is to arrest in mitosis (82). This delay is most likely SAC dependent due to effects on microtubules and centrosomes, which become permanently disorganized and destabilized upon exposure to heat (83). In addition, heat can increase the binding of the heat-shock transcription factor 2 (HSF2) to DNA during mitosis (84). HSF2 binding attracts PP2A, which dephosphorylates condensin, thereby reducing the compaction of the chromosomes (85), and acting similar to HDACi treatment (Figure 2). Consequently, the mitotic delay is only temporary, and cells rapidly reform a nuclear envelope around chromosomes, and undergo mitotic slippage (86, 87), even in the presence of Taxol (88). Hyperthermia has been shown to both antagonize (89) and synergize (88) with Taxol, with the outcome dependent on the functionality of the apoptotic pathway (90, 91).
Interestingly, like hyperthermia, cold-shock (hypothermia) has also shown some success in synergizing with radiotherapy and a variety of chemotherapeutics (92, 93). Exposure to cold induces a transient mitotic delay in cells, however cells eventually complete mitosis and segregate their chromosomes normally (94). Hypothermia reversibly destabilizes non-kinetochore microtubules (95, 96), but this still allows chromosomes to be captured by kinetochore microtubules and positioned at the metaphase plate (94). However, a reduction in microtubule dynamics and loss of astral microtubules results in reduced tension at kinetochores leading to the retention of Bub1 and BubR1 at kinetochores and a SAC-dependent mitotic delay (97). The ability of cells to recover from hypothermia and complete mitosis may explain why cold-shock can reduce the number of mitotic defects induced by chemotherapies (98), and minimize side effects (e.g., hair loss) of Taxol in cancer patients (99). However, given that the delay is transient and reversible, it also explains why co-treatment regimes have not shown any significant synergy and are unlikely to be useful for enhancing the killing of cancer cells.
Mechanical stress
As cells enter mitosis they transform their architecture to create a spherical shape, which is driven by changes in the actin cytoskeleton (100), and by regulation of osmotic pressure (101). The small GTPase RhoA is critical for cortical retraction during mitotic cell rounding (102). During early prophase RhoA promotes remodeling of the actin cytoskeleton, increasing the mechanical stiffness of the cell (103). Cell rounding is achieved by combining RhoA-mediated cellular rigidity with increased hydrostatic pressure inside the cell. This occurs by increasing intracellular sodium levels resulting in an influx of water (101). Failure to round up, and/or disruption of the RhoA pathway prevents mitotic exit in a SAC-dependent manner by inducing spindle pole fragmentation (104), disruption of astral microtubule organization and spindle function (105, 106) (Figure 2). Interestingly, placing cells in hypertonic solution (preventing water influx) stably arrests cells in mitosis and was originally used in the 1970s as a method for enriching mammalian cells in mitosis (107). After several hours most arrested cells die, although some escape via mitotic slippage to form polyploid cells (108). Interestingly, in yeast, hypertonic stress can promote activation of Cdc14 phosphatase (109), which then dephosphorylates Cdk substrates driving cells out of mitosis, suggesting that phosphatases can drive slippage. However, in humans the role of Cdc14 is not conserved (110), and PP2A appears to be the primary phosphatase responsible for removing mitotic Cdk1 phosphorylations (111, 112). If PP2A is directly activated in response to hypertonic stress it could promote mitotic slippage in human cells, providing a rational for future research focusing on the effectiveness of PP2A inhibitors in combination with mitotic chemotherapies.
Exposure of mitotic cells to hypotonic conditions increases water influx, rising internal pressure and a swelling of mitotic cell size, with weak hypotonic solutions arresting cells in pro-metaphase (113). However, unlike hypertonic stress, this arrest is far less stable and cells rapidly undergo mitotic slippage, characterized by chromosome decondensation, disrupted kinetochore and spindle structure, and reformation of the nuclear envelope around un-segregated chromosomes (114, 115), which all promote chromosome aberrations and polyploidy (116). The effects of hypotonic stress in combination with Taxol have not been studied in detail, however, hypotonic solutions can increase the uptake of chemotherapies in cells (117), and have shown some promise in enhancing response to platinum-based treatments (118). Consequently, it is likely that similar to hyperthermia, local hypotonic conditions could be used to enhance Taxol response in tumors with a functional apoptotic pathway.
Conclusion/Perspectives
In summary, the ability of cells to arrest during mitosis in response cellular and environmental stresses is dependent on the presence of a functional SAC, the correct suppression of transcription and translation, and critically the maintenance of Cdk1 activity. Stress that prevents the satisfaction of the SAC results in a mitotic arrest, while those stresses that disrupt Cdk1 activity or directly disable the SAC force cells to prematurely exit mitosis. Future research on the role mitotic phosphatases, such as PP2A, play in stress response and slippage will be critical for fully elucidating the mechanisms of how a specific cancer will response or can be sensitized to mitotic chemotherapies such as Taxol.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Liz Caldon, Ms. Rachael McCloy, and Dr. Andrew Stone for their helpful comments and advice. Andrew Burgess is a Cancer Institute of NSW FRL fellow.
References
2. Pearce AK, Humphrey TC. Integrating stress-response and cell-cycle checkpoint pathways. Trends Cell Biol (2001) 11:426–33. doi:10.1016/S0962-8924(01)02119-5
3. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol (2012) 22:R966–80. doi:10.1016/j.cub.2012.10.006
4. Overlack K, Krenn V, Musacchio A. When Mad met Bub. EMBO Rep (2014) 15:326–8. doi:10.1002/embr.201438574
5. Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol (2006) 16:1194–200. doi:10.1016/j.cub.2006.04.043
6. Potapova TA, Daum JR, Pittman BD, Hudson JR, Jones TN, Satinover DL, et al. The reversibility of mitotic exit in vertebrate cells. Nature (2006) 440:954–8. doi:10.1038/nature04652
7. McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle (2014) 13:1400–12. doi:10.4161/cc.28401
8. Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. The mitotic origin of chromosomal instability. Curr Biol (2014) 24:R148–9. doi:10.1016/j.cub.2014.01.019
9. Konrad CG. Protein synthesis and RNA synthesis during mitosis in animal cells. J Cell Biol (1963) 19:267–77. doi:10.1083/jcb.19.2.267
10. Fan H, Penman S. Regulation of protein synthesis in mammalian cells. II. Inhibition of protein synthesis at the level of initiation during mitosis. J Mol Biol (1970) 50:655–70. doi:10.1016/0022-2836(70)90091-4
11. van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong S-E, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to Inactivate the G2/M DNA damage checkpoint. PLoS Biol (2010) 8:e1000287. doi:10.1371/journal.pbio.1000287
12. Kim H-S, Heo J-I, Park S-H, Shin J-Y, Kang H-J, Kim M-J, et al. Transcriptional activation of p21(WAF1/CIP1) is mediated by increased DNA binding activity and increased interaction between p53 and Sp1 via phosphorylation during replicative senescence of human embryonic fibroblasts. Mol Biol Rep (2014) 41:2397–408. doi:10.1007/s11033-014-3094-9
13. Koutsodontis G, Tentes I, Papakosta P, Moustakas A, Kardassis D. Sp1 plays a critical role in the transcriptional activation of the human cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by the p53 tumor suppressor protein. J Biol Chem (2001) 276:29116–25. doi:10.1074/jbc.M104130200
14. Martínez-Balbás MA, Dey A, Rabindran SK, Ozato K, Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell (1995) 83:29–38. doi:10.1016/0092-8674(95)90231-7
15. Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin (2013) 6:6. doi:10.1186/1756-8935-6-6
16. Chen D, Dundr M, Wang C, Leung A, Lamond A, Misteli T, et al. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J Cell Biol (2005) 168:41–54. doi:10.1083/jcb.200407182
17. Long JJ, Leresche A, Kriwacki RW, Gottesfeld JM. Repression of TFIIH transcriptional activity and TFIIH-associated cdk7 kinase activity at mitosis. Mol Cell Biol (1998) 18:1467–76.
18. Chuang J-Y, Wang S-A, Yang W-B, Yang H-C, Hung C-Y, Su T-P, et al. Sp1 phosphorylation by cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding activity during mitosis in cancer cells. Oncogene (2012) 31:4946–59. doi:10.1038/onc.2011.649
19. Sansregret L, Gallo D, Santaguida M, Leduy L, Harada R, Nepveu A. Hyperphosphorylation by cyclin B/CDK1 in mitosis resets CUX1 DNA binding clock at each cell cycle. J Biol Chem (32843) 285(2010):32834–43. doi:10.1074/jbc.M110.156406
20. Vadnais C, Davoudi S, Afshin M, Harada R, Dudley R, Clermont P-L, et al. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res (2012) 40:4483–95. doi:10.1093/nar/gks041
21. Shiromizu T, Goto H, Tomono Y, Bartek J, Totsukawa G, Inoko A, et al. Regulation of mitotic function of Chk1 through phosphorylation at novel sites by cyclin-dependent kinase 1 (Cdk1). Genes Cells (2006) 11:477–85. doi:10.1111/j.1365-2443.2006.00955.x
22. Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science (2014) 344:189–93. doi:10.1126/science.1248024
23. Petsalaki E, Zachos G. Chk2 prevents mitotic exit when the majority of kinetochores are unattached. J Cell Biol (2014) 205:339–56. doi:10.1083/jcb.201310071
24. Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, et al. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol Cell (2011) 44:597–608. doi:10.1016/j.molcel.2011.09.016
25. Eliezer Y, Argaman L, Kornowski M, Roniger M, Goldberg M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J Biol Chem (2014) 289:8182–93. doi:10.1074/jbc.M113.532739
26. Oricchio E, Saladino C, Iacovelli S, Soddu S, Cundari E. ATM is activated by default in mitosis, localizes at centrosomes and monitors mitotic spindle integrity. Cell Cycle (2006) 5:88–92. doi:10.4161/cc.5.1.2269
27. Palazzo L, Della Monica R, Visconti R, Costanzo V, Grieco D. ATM controls proper mitotic spindle structure. Cell Cycle (2014) 13:1091–100. doi:10.4161/cc.27945
28. Pyronnet S, Pradayrol L, Sonenberg N. A cell cycle-dependent internal ribosome entry site. Mol Cell (2000) 5:607–16. doi:10.1016/S1097-2765(00)80240-3
29. Pyronnet S, Dostie J, Sonenberg N. Suppression of cap-dependent translation in mitosis. Genes Dev (2001) 15:2083–93. doi:10.1101/gad.889201
30. Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol Cell (2000) 5:597–605. doi:10.1016/S1097-2765(00)80239-7
31. Qin X, Sarnow P. Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J Biol Chem (2004) 279:13721–8. doi:10.1074/jbc.M312854200
32. Groisman I, Huang YS, Mendez R, Cao Q, Theurkauf W, Richter JD. CPEB, maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local translational control of cell division. Cell (2000) 103:435–47. doi:10.1016/S0092-8674(00)00135-5
33. Blower MD, Feric E, Weis K, Heald R. Genome-wide analysis demonstrates conserved localization of messenger RNAs to mitotic microtubules. J Cell Biol (2007) 179:1365–73. doi:10.1083/jcb.200705163
34. Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ (2012) 19:369–77. doi:10.1038/cdd.2011.197
35. Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol Cell (2004) 15:977–90. doi:10.1016/j.molcel.2004.08.018
36. Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol (2010) 20:1666–71. doi:10.1016/j.cub.2010.08.018
37. Mikhailov A, Cole RW, Rieder CL. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr Biol (2002) 12:1797–806. doi:10.1016/S0960-9822(02)01226-5
38. Hayashi MT, Cesare AJ, Fitzpatrick JAJ, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol (2012) 19:387–94. doi:10.1038/nsmb.2245
39. Charbin A, Bouchoux C, Uhlmann F. Condensin aids sister chromatid decatenation by topoisomerase II. Nucleic Acids Res (2014) 42:340–8. doi:10.1093/nar/gkt882
40. Choi E, Lee H. Chromosome damage in mitosis induces BubR1 activation and prometaphase arrest. FEBS Lett (2008) 582:1700–6. doi:10.1016/j.febslet.2008.04.028
41. Wang LH-C, Mayer B, Stemmann O, Nigg EA. Centromere DNA decatenation depends on cohesin removal and is required for mammalian cell division. J Cell Sci (2010) 123:806–13. doi:10.1242/jcs.058255
42. d’Alcontres MS, Palacios JA, Mejias D, Blasco MA. TopoIIα prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle (2014) 13:1463–81. doi:10.4161/cc.28419
43. Toyoda Y, Yanagida M. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol Biol Cell (2006) 17:2287–302. doi:10.1091/mbc.E05-11-1089
44. Mikhailov A, Shinohara M, Rieder CL. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol (2004) 166:517–26. doi:10.1083/jcb.200405167
45. Tokalov SV, Abramyuk AM, Abolmaali ND. Protection of p53 wild type cells from taxol by genistein in the combined treatment of lung cancer. Nutr Cancer (2010) 62:795–801. doi:10.1080/01635581003605912
46. Smith PJ, Marquez N, Wiltshire M, Chappell S, Njoh K, Campbell L, et al. Mitotic bypass via an occult cell cycle phase following DNA topoisomerase II inhibition in p53 functional human tumor cells. Cell Cycle (2007) 6:2071–81. doi:10.4161/cc.6.16.4585
47. Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol (2000) 2:672–6. doi:10.1038/35023629
48. Liu D, Davydenko O, Lampson MA. Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J Cell Biol (2012) 198:491–9. doi:10.1083/jcb.201205090
49. Yuan J-H, Feng Y, Fisher RH, Maloid S, Longo DL, Ferris DK. Polo-like kinase 1 inactivation following mitotic DNA damaging treatments is independent of ataxia telangiectasia mutated kinase. Mol Cancer Res (2004) 2: 417–26.
50. Jang Y-J, Ji J-H, Choi Y-C, Ryu CJ, Ko S-Y. Regulation of polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J Biol Chem (2007) 282:2473–82. doi:10.1074/jbc.M605480200
51. Vasey PA, Jones NA, Jenkins S, Dive C, Brown R. Cisplatin, camptothecin, and taxol sensitivities of cells with p53-associated multidrug resistance. Mol Pharmacol (1996) 50:1536–40.
52. McGuire WP. Current status of taxane and platinum-based chemotherapy in ovarian cancer. J Clin Oncol (2003) 21:133s–5s. doi:10.1200/JCO.2003.01.066
53. Sculier JP, Lafitte JJ, Lecomte J, Alexopoulos CG, Van Cutsem O, Giner V, et al. A phase III randomised trial comparing sequential chemotherapy using cisplatin-based regimen and paclitaxel to cisplatin-based chemotherapy alone in advanced non-small-cell lung cancer. Ann Oncol (2007) 18:1037–42. doi:10.1093/annonc/mdm084
54. Papadopoulos NE, Bedikian A, Ring S, Kim KB, Hwu W-J, Gerber DL, et al. Phase I/II study of a cisplatin-taxol-dacarbazine regimen in metastatic melanoma. Am J Clin Oncol (2009) 32:509–14. doi:10.1097/COC.0b013e3181942a1f
55. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med (2013) 369:1691–703. doi:10.1056/NEJMoa1304369
56. Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE, et al. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle (2005) 4:717–26. doi:10.4161/cc.4.5.1690
57. Warrener R, Beamish H, Burgess A, Waterhouse NJ, Giles N, Fairlie D, et al. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J (2003) 17:1550–2. doi:10.1096/fj.02-1003fje
58. Magnaghi-Jaulin L, Eot-Houllier G, Fulcrand G, Jaulin C. Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer Res (2007) 67:6360–7. doi:10.1158/0008-5472.CAN-06-3012
59. Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene (2008) 27:1345–54. doi:10.1038/sj.onc.1210779
60. Semenza GL. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell (2004) 5:405–6. doi:10.1016/S1535-6108(04)00118-7
61. Pandey R, Heeger S, Lehner CF. Rapid effects of acute anoxia on spindle kinetochore interactions activate the mitotic spindle checkpoint. J Cell Sci (2007) 120:2807–18. doi:10.1242/jcs.007690
62. Dong XL, Xu PF, Miao C, Fu ZY, Li QP, Tang PY, et al. Hypoxia decreased chemosensitivity of breast cancer cell line MCF-7 to paclitaxel through cyclin B1. Biomed Pharmacother (2012) 66:70–5. doi:10.1016/j.biopha.2011.11.016
63. Rofstad EK, Johnsen NM, Lyng H. Hypoxia-induced tetraploidisation of a diploid human melanoma cell line in vitro. Br J Cancer Suppl (1996) 27:S136–9.
64. Ohshima S. Abnormal mitosis in hypertetraploid cells causes aberrant nuclear morphology in association with H2O2-induced premature senescence. Cytometry (2008) 73A:808–15. doi:10.1002/cyto.a.20604
65. Ren Q, Liou L-C, Gao Q, Bao X, Zhang Z. Bir1 deletion causes malfunction of the spindle assembly checkpoint and apoptosis in yeast. Front Oncol (2012) 2:93. doi:10.3389/fonc.2012.00093
66. Lee C-F, Liu C-Y, Hsieh R-H, Wei Y-H. Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann N Y Acad Sci (2005) 1042:246–54. doi:10.1196/annals.1338.027
67. Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer (2010) 10:194–204. doi:10.1038/nrc2803
68. Lissoni P, Barni S, Mandalà M, Ardizzoia A, Paolorossi F, Vaghi M, et al. Decreased toxicity and increased efficacy of cancer chemotherapy using the pineal hormone melatonin in metastatic solid tumour patients with poor clinical status. Eur J Cancer (1999) 35:1688–92. doi:10.1016/S0959-8049(99)00159-8
69. Liang Y-C, Tsai S-H, Chen L, Lin-Shiau S-Y, Lin J-K. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem Pharmacol (2003) 65:1053–60. doi:10.1016/S0006-2952(03)00011-X
70. Lu X, Jung JI, Cho HJ, Lim DY, Lee HS, Chun HS, et al. Fisetin inhibits the activities of cyclin-dependent kinases leading to cell cycle arrest in HT-29 human colon cancer cells. J Nutr (2005) 135:2884–90.
71. Mao Q-Q, Bai Y, Lin Y-W, Zheng X-Y, Qin J, Yang K, et al. Resveratrol confers resistance against taxol via induction of cell cycle arrest in human cancer cell lines. Mol Nutr Food Res (2010) 54:1574–84. doi:10.1002/mnfr.200900392
72. Salmela A-L, Pouwels J, Varis A, Kukkonen AM, Toivonen P, Halonen PK, et al. Dietary flavonoid fisetin induces a forced exit from mitosis by targeting the mitotic spindle checkpoint. Carcinogenesis (2009) 30:1032–40. doi:10.1093/carcin/bgp101
73. Lee GM. Characterization of mitotic motors by their relative sensitivity to AMP-PNP. J Cell Sci (1989) 94(Pt 3):425–41.
74. Spurck TP, Pickett-Heaps JD. On the mechanism of anaphase A: evidence that ATP is needed for microtubule disassembly and not generation of polewards force. J Cell Biol (1987) 105:1691–705. doi:10.1083/jcb.105.4.1691
75. Wadsworth P, Salmon ED. Spindle microtubule dynamics: modulation by metabolic inhibitors. Cell Motil Cytoskeleton (1988) 11:97–105. doi:10.1002/cm.970110203
76. Thaiparambil JT, Eggers CM, Marcus AI. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol (2012) 32:3203–17. doi:10.1128/MCB.00418-12
77. Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, et al. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol (2001) 155:1159–72. doi:10.1083/jcb.200105093
78. Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol (2000) 150:1233–50. doi:10.1083/jcb.150.6.1233
79. Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Biol Cell (2001) 12:1995–2009. doi:10.1091/mbc.12.7.1995
80. Hanna RK, Zhou C, Malloy KM, Sun L, Zhong Y, Gehrig PA, et al. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol Oncol (2012) 125:458–69. doi:10.1016/j.ygyno.2012.01.009
81. Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol (2002) 3:487–97. doi:10.1016/S1470-2045(02)00818-5
82. Coss RA, Dewey WC, Bamburg JR. Effects of hyperthermia (41.5 degrees) on Chinese hamster ovary cells analyzed in motisis. Cancer Res (1979) 39:1911–8.
83. Pawlik A, Nowak JM, Grzanka D, Gackowska L, Michalkiewicz J, Grzanka A. Hyperthermia induces cytoskeletal alterations and mitotic catastrophe in p53-deficient H1299 lung cancer cells. Acta Histochem (2013) 115:8–15. doi:10.1016/j.acthis.2012.02.006
84. Vihervaara A, Sergelius C, Vasara J, Blom MAH, Elsing AN, Roos-Mattjus P, et al. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci U S A (2013) 110:E3388–97. doi:10.1073/pnas.1305275110
85. Xing H, Wilkerson DC, Mayhew CN, Lubert EJ, Skaggs HS, Goodson ML, et al. Mechanism of hsp70i gene bookmarking. Science (2005) 307:421–3. doi:10.1126/science.1106478
86. Coss RA, Dewey WC, Bamburg JR. Effects of hyperthermia on dividing Chinese hamster ovary cells and on microtubules in vitro. Cancer Res (1982) 42:1059–71.
87. Gupta RK, Srinivas UK. Heat shock induces chromosomal instability in near-tetraploid embryonal carcinoma cells. Cancer Biol Ther (2008) 7:1471–80. doi:10.4161/cbt.7.9.6428
88. Giovinazzi S, Bellapu D, Morozov VM, Ishov AM. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle (2013) 12:2598–607. doi:10.4161/cc.25591
89. Leal BZ, Meltz ML, Mohan N, Kuhn J, Prihoda TJ, Herman TS. Interaction of hyperthermia with taxol in human MCF-7 breast adenocarcinoma cells. Int J Hyperthermia (1999) 15:225–36. doi:10.1080/026567399285747
90. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature (2011) 471:110–4. doi:10.1038/nature09779
91. Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell (2008) 14:111–22. doi:10.1016/j.ccr.2008.07.002
92. Matijasevic Z. Selective protection of non-cancer cells by hypothermia. Anticancer Res (2002) 22:3267–72.
93. Zhang XM, Lv YG, Chen GB, Zou Y, Lin CW, Yang L, et al. Effect of mild hypothermia on breast cancer cells adhesion and migration. Biosci Trends (2012) 6:313–24. doi:10.5582/bst.2012.v6.6.313
94. Rieder CL. Effect of hypothermia (20–25°C) on mitosis in PtK1 cells. Cell Biol Int Rep (1981) 5:563–73. doi:10.1016/S0309-1651(81)80007-0
95. Wise D, Cassimeris L, Rieder CL, Wadsworth P, Salmon ED. Chromosome fiber dynamics and congression oscillations in metaphase PtK2 cells at 23°C. Cell Motil Cytoskeleton (1991) 18:131–42. doi:10.1002/cm.970180208
96. Cassimeris L, Rieder CL, Rupp G, Salmon ED. Stability of microtubule attachment to metaphase kinetochores in PtK1 cells. J Cell Sci (1990) 96(Pt 1):9–15.
97. Shannon KB, Canman JC, Salmon ED. Mad2 and BubR1 function in a single checkpoint pathway that responds to a loss of tension. Mol Biol Cell (2002) 13:3706–19. doi:10.1091/mbc.E02-03-0137
98. Vig BK. Effect of hypothermia and hyperthermia on the induction of chromosome aberrations by adriamycin in human leukocytes. Cancer Res (1978) 38:550–5.
99. Lemenager M, Lecomte S, Bonneterre ME, Bessa E, Dauba J, Bonneterre J. Effectiveness of cold cap in the prevention of docetaxel-induced alopecia. Eur J Cancer (1997) 33:297–300. doi:10.1016/S0959-8049(96)00374-7
100. Mitchison TJ. Actin based motility on retraction fibers in mitotic PtK2 cells. Cell Motil Cytoskeleton (1992) 22:135–51. doi:10.1002/cm.970220207
101. Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature (2011) 469:226–30. doi:10.1038/nature09642
102. Maddox AS, Burridge K. RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J Cell Biol (2003) 160:255–65. doi:10.1083/jcb.200207130
103. Matthews HK, Delabre U, Rohn JL, Guck J, Kunda P, Baum B. Changes in Ect2 localization couple actomyosin-dependent cell shape changes to mitotic progression. Dev Cell (2012) 23:371–83. doi:10.1016/j.devcel.2012.06.003
104. Lancaster OM, Le Berre M, Dimitracopoulos A, Bonazzi D, Zlotek-Zlotkiewicz E, Picone R, et al. Mitotic rounding alters cell geometry to ensure efficient bipolar spindle formation. Dev Cell (2013) 25:270–83. doi:10.1016/j.devcel.2013.03.014
105. Bakal CJ, Finan D, LaRose J, Wells CD, Gish G, Kulkarni S, et al. The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis. Proc Natl Acad Sci U S A (2005) 102:9529–34. doi:10.1073/pnas.0504190102
106. Heng Y-W, Lim HH, Mina T, Utomo P, Zhong S, Lim C-T, et al. TPPP acts downstream of RhoA-ROCK-LIMK2 to regulate astral microtubule organization and spindle orientation. J Cell Sci (2012) 125:1579–90. doi:10.1242/jcs.096818
107. Wheatley DN. Hypertonicity and the arrest of mammalian cells in metaphase: a synchrony technique for HeLa cells. J Cell Sci (1974) 15:221–37.
108. Wheatley DN. Cell-growth and division in hypertonic medium. Exp Cell Res (1974) 87:219–32. doi:10.1016/0014-4827(74)90474-1
109. Reiser V, D’Aquino KE, Ee L-S, Amon A. The stress-activated mitogen-activated protein kinase signaling cascade promotes exit from mitosis. Mol Biol Cell (2006) 17:3136–46. doi:10.1091/mbc.E05-12-1102
110. Mocciaro A, Schiebel E. Cdc14: a highly conserved family of phosphatases with non-conserved functions? J Cell Sci (2010) 123:2867–76. doi:10.1242/jcs.074815
111. Burgess A, Vigneron S, Brioudes E, Labbé J-C, Lorca T, Castro A. Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A (12569) 107(2010):12564–9. doi:10.1073/pnas.0914191107
112. Schmitz MH, Held M, Janssens V, Hutchins JR, Hudecz O, Ivanova E, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol (2010) 12:886–93. doi:10.1038/ncb2092
113. Huges A. Some effects of abnormal tonicity on dividing cells in chick tissue cultures. Q J Microsc Sci (1952) 93:207–19.
114. Ford JH, Congedi MM. Rapid induction of anaphase in competent cells by hypotonic treatment. Cytobios (1987) 51:183–92.
115. Brinkley BR, Cox SM, Pepper DA. Structure of the mitotic apparatus and chromosomes after hypotonic treatment of mammalian cells in vitro. Cytogenet Cell Genet (1980) 26:165–74. doi:10.1159/000131438
116. Nowak C. Studies on the ability of hypotonic solutions to induce chromosomal aberrations in V 79 cell. Teratog Carcinog Mutagen (1987) 7:515–25. doi:10.1002/tcm.1770070603
117. Stephen RL, Novak JM, Jensen EM, Kablitz C, Buys SS. Effect of osmotic pressure on uptake of chemotherapeutic agents by carcinoma cells. Cancer Res (1990) 50:4704–8.
Keywords: mitosis, SAC, spindle, kinetochore, checkpoint, metaphase, DNA damage, Cdk1
Citation: Burgess A, Rasouli M and Rogers S (2014) Stressing mitosis to death. Front. Oncol. 4:140. doi: 10.3389/fonc.2014.00140
Received: 28 March 2014; Accepted: 22 May 2014;
Published online: 04 June 2014.
Edited by:
Megan Chircop, Children’s Medical Research Institute, AustraliaReviewed by:
Rosella Visintin, European Institute of Oncology, ItalyMakoto T. Hayashi, Salk Institute for Biological Studies, USA
Copyright: © 2014 Burgess, Rasouli and Rogers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew Burgess, The Kinghorn Cancer Centre, Garvan Institute of Medical Research, 370 Victoria Street, Sydney, NSW 2010, Australia e-mail: a.burgess@garvan.org.au