 David A. Sallman
David A. Sallman Sheng Wei
Sheng Wei- Immunology Program and Malignant Hematology Program, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA
Myelodysplastic syndromes (MDS) represent a hematologically diverse group of myeloid neoplasms, however, one subtype characterized by an isolated deletion of chromosome 5q [del(5q)] is pathologically and clinically distinct. Patients with del(5q) MDS share biological features that account for the profound hypoplastic anemia and unique sensitivity to treatment with lenalidomide. Ineffective erythropoiesis in del(5q) MDS arises from allelic deletion of the ribosomal processing S-14 (RPS14) gene, which leads to MDM2 sequestration with consequent p53 activation and erythroid cell death. Since its approval in 2005, lenalidomide has changed the natural course of the disease. Patients who achieve transfusion independence and/or a cytogenetic response with lenalidomide have a decreased risk of progression to acute myeloid leukemia and an improved overall survival compared to non-responders. Elucidation of the mechanisms of action of lenalidomide in del(5q) MDS has advanced therapeutic strategies for this disease. The selective cytotoxicity of lenalidomide in del(5q) clones derives from inhibition of a haplodeficient phosphatase whose catalytic domain is encoded within the common deleted region on chromosome 5q, i.e., protein phosphatase 2A (PP2Acα). PP2A is a highly conserved, dual specificity phosphatase that plays an essential role in regulation of the G2/M checkpoint. Inhibition of PP2Acα results in cell-cycle arrest and apoptosis in del(5q) cells. Targeted knockdown of PP2Acα using siRNA is sufficient to sensitize non-del(5q) clones to lenalidomide. Through its inhibitory effect on PP2A, lenalidomide stabilizes MDM2 to restore p53 degradation in erythroid precursors, with subsequent arrest in G2/M. Unfortunately, the majority of patients with del(5q) MDS develop resistance to lenalidomide over time associated with PP2Acα over-expression. Targeted inhibition of PP2A with a more potent inhibitor has emerged as an attractive therapeutic approach for patients with del(5q) MDS.
Introduction
Myelodysplastic syndromes (MDS) represent both a clinical and genetically heterogeneous group of clonal hematopoietic stem cell disorders characterized by progressive cytopenias, dysplasia, and risk of transformation into acute myeloid leukemia (AML). Cytogenetic characterization of MDS is a cornerstone of prognostic assessment for overall survival and risk of AML evolution. Conventional cytogenetic data are incorporated into both the International Prognostic Scoring System (IPSS) and the revised-IPSS (R-IPSS) (1, 2). However, chromosomal abnormalities are only found in 52% of patients (3). Over recent years, there has been considerable progress in the molecular characterization of MDS with somatic mutations found in 80% or more of cases (4). The molecular characterization of MDS adds further prognostic information with mutations involving TP53, EZH2, ETV6, RUNX1, and ASXL1 associated with decreased overall survival and higher risk of AML transformation (5, 6).
Myelodysplastic syndromes with isolated chromosome 5q deletion [del(5q)] represents a distinct clinical and pathological entity recognized in the World Health Organization (WHO) classification. An interstitial deletion involving chromosome 5q is the most common cytogenetic abnormality in MDS, accounting for approximately 15% of MDS cases (3, 7). Of these, 50% have isolated del(5q) while the remaining have either an additional cytogenetic abnormality or a complex karyotype (3, 8). Del(5q) MDS is characterized by hypoproliferative anemia with dysplastic megakaryocytes and a rather indolent clinical course (9). Red blood cell (RBC) transfusion dependence develops early in the disease course and represents the principle driver of morbidity and mortality. With the advent of exome sequencing, molecular profiling has shown that 20% of patients with isolated del(5q) MDS and 72% of patients with del(5q) accompanying a complex karyotype harbor TP53 gene mutations (7). This genetic heterogeneity accounts for the prognostic heterogeneity in clinical course and treatment outcome (8, 10). The following review is structured chronologically as each major breakthrough in the field has further defined the phenotype of del(5q) MDS and the critical role of PP2A in the treatment of these patients. Together, these data highlight novel therapeutic strategies in order to target the underlying pathogenesis of this disease.
Lenalidomide and DEL(5q) MDS
Lenalidomide represents the first therapeutic agent in MDS, which targets a cytogenetically defined disease subset. The initial evidence of its clinical activity was based on a high clinical and cytogenetic response rate in del(5q) MDS patients in the initial safety and efficacy study (11). Lenalidomide was approved by the Food and Drug Administration (FDA) in 2005 for the treatment of transfusion-dependent IPSS low or intermediate (int)-1 risk, del(5q) MDS. The approval was based on results of the MDS-003 multicenter phase 2 trial in which 67% of patients achieved transfusion independence (TI) with lenalidomide therapy with a median TI duration of 2.2 years (12). In addition, 73% of patients had at least a partial cytogenetic response with 45% of patients achieving a complete response (CR). A recently published long-term follow up of this study found that the median overall survival was significantly increased in patients who reached TI, 4.3 vs. 2 years in non-responders, and in cytogenetic responders, 4.9 vs. 3.1 years, respectively (13). Achievement of TI or cytogenetic response also led to a decreased risk of progression to AML.
DEL(5q) MDS Pathobiology
The initial molecular understanding stemmed from genetic mapping experiments that identified two common deleted regions (CDRs) in the 5q− syndrome (14, 15). The 5q− syndrome is characterized by isolated del(5q), female predominance, refractory macrocytic anemia accompanied by erythroid hypoplasia, normal to increased platelet counts, normal to mildly decreased neutrophil counts, and hypolobated micromegakaryocytes (16, 17). In patients with MDS or AML (excluding patients with 5q− syndrome), Horrigan et al. (15) identified a CDR at 5q31 (proximal CDR) with a minimal deletion of 1 MB leading to presumed loss of a tumor suppressor gene(s). In 5q− syndrome patients, deletion mapping of the CDR identified a 1.5 MB interval at 5q32–33 (distal CDR) (14). The distal CDR contains 40 genes, 33 of which were found to be expressed in CD34+ cells. Based on Knudson’s two-hit hypothesis, extensive sequencing of 5q− syndrome patients was performed to identify a possible second genetic event leading to the malignant phenotype. Boultwood et al. (18) compared the transcriptome of CD34+ cells from 5q− syndrome patients to the transcriptome of CD34+ cells in healthy controls but did not identify silenced genes in the remaining allele. A recent study utilizing whole-exome sequencing on del(5q) patients at baseline and at the time of leukemic transformation also found no mutations in the residual alleles of the CDR (19).
Despite the absence of identifiable mutations to account for the second-hit, expression of genes encoded within the CDR was consistent with monoallelic expression (18). Of the candidate genes, haploinsufficiency was evident in secreted protein acidic and rich in cysteine (SPARC), a tumor suppressor gene, and ribosomal processing S14 gene [RPS14 (component of 40S ribosomal subunit)]. RPS14 haploinsufficiency was of particular interest given that RPS19 mutations represent the most common genetic mutation in patients with Diamond–Blackfan anemia (DBA), a congenital bone marrow failure syndrome with profound erythroid hypoplasia (20). Furthermore, Gazda et al. (21) had shown that haploinsufficiency of RPS19 was the underlying defect in a proportion of patients with DBA (21). To determine the possible role of RPS14 haploinsufficiency in the pathogenesis of del(5q) MDS, Ebert and colleagues (22) designed lentiviral expressed short hairpin RNA (shRNA) as an RNA interference screen against each of the 40 genes identified in the CDR (22). Utilizing this novel methodology, only shRNA to RPS14 re-capitulated erythroid features of the 5q− phenotype, producing erythroid maturation arrest and impaired viability (22). RPS14 over-expression, in contrast, selectively rescued erythropoiesis in CD34+ cells of del(5q) patients but not in non-del(5q) patients. Of importance, shRNA to RPS14 down-regulated gene expression by 60%, comparable to the haploinsufficient state of del(5q) patients. Furthermore, RPS14 haploinsufficiency was confirmed as the underlying genetic abnormality underlying the hypoplastic anemia as RPS14 gene sequencing in MDS patients showed absence of point mutations, biallelic deletion, or epigenetic modification (i.e., methylation) (22).
The question of how RPS14 haploinsuffiency led to such a dramatic deleterious effect on erythroid maturation without negative consequences on myeloid or megakaryocyte lineages has recently been elucidated. Specifically, RPS14 haploinsufficiency (via lentiviral shRNA) induced p53 accumulation in erythroid precursors and subsequent cell-cycle arrest in a lineage specific fashion (23). Impaired ribosome biogenesis as a result of RPS14 deficiency liberates free ribosomal proteins (RP) in the nucleoplasm, particularly RPL11, that bind to and sequester the human homolog of the E3 ubiquitin ligase, mouse double minute 2 protein (MDM2), the major negative regulator of p53 (24). Degradation of MDM2 drives p53 stabilization and activation (25). Interference with the assembly of the 40S ribosomal subunit, as occurs in RPS14 haploinsufficiency, induces up-regulation of the RPL11 transcript (23, 24). Treatment of CD34+ cells with Nutlin-3, a direct inhibitor of MDM2–p53 interaction (26), leads to p53 accumulation and cell death primarily in the erythroid lineage, providing a mechanistic rationale for the profound anemia in del(5q) MDS (23). These data were reproduced in vivo and occurred independent of ribosomal dysfunction, further supporting heightened sensitivity to p53-mediated apoptosis in erythroid precursors. Inhibition of p53 activity, via pifithrin-α, abrogates this pathway, restoring normal erythropoiesis. Peller and colleagues (27) had previously described p53 activation as a critical molecular event involved in normal erythroid maturation. The additional hit of ribosomal haploinsufficiency leading to p53 up-regulation in combination with a heightened baseline activation of the p53 pathway in erythroid precursors provide strong biological rationale for the erythroid phenotype in del(5q) MDS.
In addition to significant macrocytic anemia, del(5q) patients commonly have moderate neutropenia and thrombocytosis. The mechanism underlying these hematological features has been ascribed to deficiency of two microRNAs (miRNAs) encoded in the distal CDR, miR-145 and miR-146a (28). Targeted knockdown of these miRNAs in CD34+ cells leads to a pro-inflammatory state with activation of tumor necrosis factor receptor-associated factor-6 (TRAF6), inducing IL-6 and subsequent neutropenia, thrombocytosis, and the dysplastic megakaryocytes characteristic of del(5q) MDS (28, 29). Furthermore, down-regulation of miR-145 induces Fli-1, a megakaryocyte and erythroid transcription factor, accompanied by thrombocytosis (30). Combined knockdown of RPS14 and miR-145 was sufficient to re-capitulate the hematologic and pathologic phenotype of del(5q) MDS (30).
Lenalidomide Selective Cytotoxicity in DEL(5q) MDS Results from Inhibition of Haplodeficient Phosphatases
Just as allelic haplodeficiency of specific genes accounts for the del(5q) MDS phenotype, gene dosage of two dual specificity phosphatases encoded within or adjacent to the proximal CDR at 5q31, CDC25C, and PP2Acα, underlies the selective suppression of del(5q) clones by lenalidomide (31). Both of these phosphatases play a pivotal co-regulatory role at the G2M checkpoint (32, 33). Phosphorylation of tyrosine (Tyr15) and threonine (Thr14) residues within the ATP binding domain of cyclin-dependent kinase-1 (CDK-1 or cdc2) inhibit CDK-1/Cyclin B complexes, thereby blocking cell-cycle progression from G2 to M. Entry into mitosis is mediated through PP2A dephosphorylation of Cdc25C at the regulatory serine substrate (Ser216), leading to dissociation from the 14-3-3 binding protein and nuclear translocation of the Cyclin B/cdc2 complex (34, 35). The above regulatory pathway is depicted in Figure 1. Del(5q) cells in MDS or AML patients were demonstrated to be haplodeficient for PP2Acα and CDC25C. Lenalidomide-induced apoptosis in a concentration-dependent fashion in del(5q) AML cells while demonstrating no apoptotic effects in non-del(5q) patient cells or non-del(5q) cell lines (31). Lenalidomide was shown to inhibit the activity of Cdc25C and PP2Acα (as evidenced by retention of critical phosphate residues) by direct and indirect mechanisms, respectively. To determine if inhibition of these phosphatases is directly responsible for the cytoselective toxicity of lenalidomide in del(5q) MDS, knockdown of CDC25C and PP2cα using lentiviral shRNA was performed in non-del(5q) cells to levels commensurate with haplodeficiency. Down-regulation of CDC25C or PP2cα markedly increased sensitivity to lenalidomide, and the pro-apoptotic effects were additive with the dual gene knockdown. Of greater importance, these findings were replicated in non-del(5q) MDS bone marrow mononuclear cells (MNC) with a threefold increase in apoptosis and G2/M cell-cycle arrest.
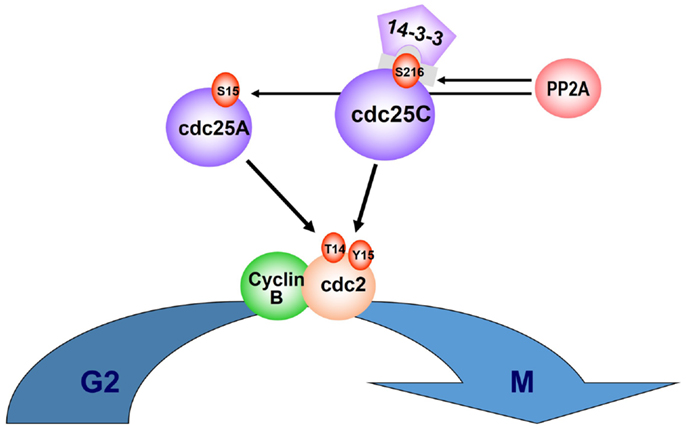
Figure 1. CDC25C and PP2Acα regulatory role on cell cycle. Entry into mitosis is mediated via PP2A dephosphorylation of Cdc25C, at the regulatory serine substrate (Ser216), leading to 14-3-3 displacement and nuclear translocation of the CyclinB/cdc2 complex. PP2A also regulates phosphorylation of Cdc25A at Ser15. Phosphorylation of tyrosine (Tyr15) and threonine (Thr14) residues within the ATP binding domain of cyclin-dependent kinase-1 (CDK-1, or cdc2) inhibit CDK-1/Cyclin B complexes and cell-cycle progression from G2 to M.
PP2Acα: Critical Target of Lenalidomide
Even though inhibition of the haplodeficient phosphatases Cdc25C and PP2Acα can alone account for the selective cytotoxicity of lenalidomide in del(5q) cells, how this effect could be reconciled in clones arrested in G1 by p53 activation warranted further investigation (22, 31). Although cellular p53 expression is up-regulated in bone marrow erythroid precursors of del(5q) patients, lenalidomide stabilizes MDM2 to release cells from G1 arrest by modifying its phosphorylation as a result of PP2Acα inhibition (36). Specifically, in Namalwa cells harboring a del(5q) chromosomal abnormality and primary del(5q) MDS bone marrow MNC, lenalidomide induced MDM2 expression and consequent p53 degradation. Over-expression of PP2Acα suppressed MDM2 induction and restored p53 stabilization upon lenalidomide treatment, indicating that expression level of PP2Acα is a key determinant of drug-induced p53 degradation.
The RPS14–MDM2 interaction arising from nucleolar stress in del(5q) is disrupted by lenalidomide to foster transition to subsequent G2/M arrest. PP2A is also recognized to modulate p53 through dephosphorylation of Thr55 and Ser46 residues, thereby preventing proteasome targeted degradation (37, 38). Lenalidomide treatment promotes retention of the Thr55 and Ser46 phosphorylated residues with consequent down-regulation of cellular p53 (36). In addition, lenalidomide specifically increased phosphorylation at critical regulatory sites at Ser166 and Ser186, inhibiting auto-ubiquitination of MDM2 thereby leading to MDM2 nuclear translocation and p53 degradation. Phosphorylation of Ser166 and Ser186 occurs through the kinase activity of Akt, which is in turn regulated by PP2A (39, 40). Treatment with lenalidomide activated Akt as evidenced by increased phosphorylation at Thr308 (36). PP2Acα association with MDM2 increased in a dose-dependent manner after lenalidomide treatment while having no direct effects on PP2Acα mRNA or protein expression. Together, these data provide a biological rationale of lenalidomide mediated p53 down-regulation in PP2Acα-haplodeficient del(5q) erythroid precursors through PP2Acα inhibition and subsequent modulation of MDM2 phosphorylation. To further validate PP2Acα as the key regulator of MDM2 modulation by lenalidomide, lentiviral PP2Acα shRNA were infected into non-del(5q) U937 cells (36). Whereas knockdown of PP2Acα stabilized MDM2, CDC25c down-regulation had no effect on MDM2 expression. Similarly, decreased MDM2 expression using siRNA prevents lenalidomide-mediated p53 degradation. The mechanism of lenalidomide’s dual actions to suppress del(5q) clones while restoring and promoting normal erythropoiesis is illustrated in Figure 2.
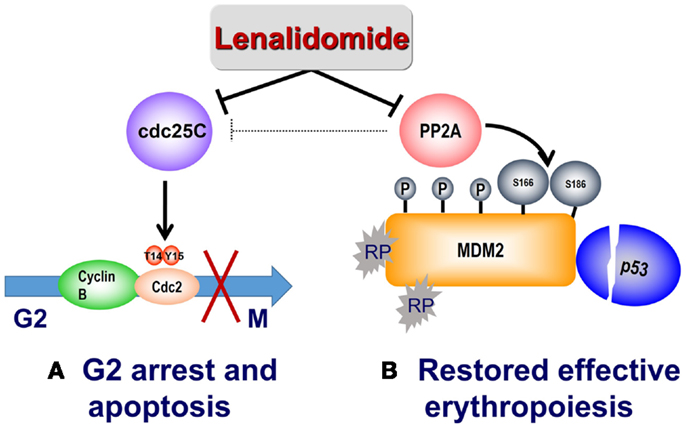
Figure 2. Mechanism of action of lenalidomide in del(5q) MDS. (A) Lenalidomide directly inhibits Cdc25C, resulting in G2/M arrest and apoptosis in del(5q) cells. Lenalidomide also indirectly inhibits PP2A with consequent Cdc25C inactivation. (B) Reduced expression of RPS14 results in binding of free ribosomal proteins (RP) to MDM2, thereby triggering its auto-ubiquitination and p53 accumulation. Lenalidomide inhibition of the phosphatase PP2A results in hyperphosphorylation of MDM2 at Ser166 and Ser186 and stabilization of MDM2, with consequent p53 degradation and restored cell-cycle re-entry.
Lenalidomide Resistance in DEL(5q) MDS is Associated with PP2Acα Over-Expression
PP2Acα and p53 expression in bone marrow trephine biopsies were analyzed prior to therapy, at the time of TI/cytogenetic response and at the time of treatment failure in a series of 22 del(5q) MDS patients treated with lenalidomide (36). Mean cellular expression of p53 and PP2Acα significantly decreased with treatment response. More importantly, at the time of treatment failure, PP2Acα was markedly up-regulated with restored expression of p53 in erythroid precursors, supporting our above in vitro data that PP2Acα over expression promotes resistance to lenalidomide. PP2Acα up-regulation in lenalidomide-resistant patients arose from a threefold increase in expression of the residual allele with no evidence for gene amplification and/or mutations in PP2Acα. Change in cellular PP2Acα expression emerged as a biomarker predictive for quality of response to lenalidomide, with the magnitude of suppression at treatment response correlating with duration of TI.
Primary resistance to lenalidomide has been linked to the presence of TP53 mutations in lower risk del(5q) MDS (41, 42). In an analysis of the French and Spanish compassionate treatment programs involving a total of 107 patients, cytogenetic response to lenalidomide in del(5q) MDS ranged from 0 to 12% in patients with mutated TP53 compared to 73% in patients with wild-type (WT) TP53 (41–43). Similarly, all patients who experienced a cytogenetic response also achieved TI. Whereas 73% of patients with WT-TP53 achieved TI, only 43% of patients with mutated TP53 achieved TI. These findings have important clinical implications as recent studies indicate that TP53 gene mutations are demonstrable by deep sequencing in 18–20% of patients with isolated del(5q), and in 70–100% in del(5q) in combination with aberrations of chromosome 7 and/or 17 (7, 43). Moreover, ability to reach TI and cytogenetic response are directly related to OS and risk of AML transformation in patients treated with lenalidomide (13). Molecular stratification of MDS patients by TP53 mutation status has shown decreased survival in patients harboring a TP53 mutant clone (5), indicating that this is a critical prognostic marker that is also predictive for potential benefit from lenalidomide treatment. To this end, Saft and colleagues (44) recently reported a retrospective study of the prognostic significance of cellular p53 expression detected by immunohistochemical staining and TP53 gene mutation in a series of IPSS low or int-1 risk del(5q) MDS patients treated with lenalidomide in the MDS-004 trial (44, 45). Strong cellular p53 expression by IHC in ≥1% of bone marrow precursors strongly correlated with decreased cytogenetic response (P = 0.009), higher risk of leukemic transformation (P = 0.00006) and shorter overall survival (P = 0.0175) (44). Strong p53 positivity directly correlated with TP53 mutation whereas moderate positivity correlated with WT-TP53. Presence of strong p53 staining was the most important prognostic co-variate for risk of leukemic transformation, which accounts in part for the apparent heterogeneity of risk in isolated del(5q) patients. This was exemplified by the MDS-004 trial where 3-year OS and AML transformation rates were 56 and 25%, respectively, which correlates with high percentage of strongly p53+ patients in this trial (35%) (44, 45). Furthermore, sequential whole-exome sequencing of two del(5q) patients at baseline and at AML transformation identified TP53 mutations in both patients at the time of leukemic transformation [but not in two non-del(5q) patients], suggesting that TP53 mutation is the critical event in progression of del(5q) MDS (19). The precise reason for the disproportionate prevalence of TP53 gene mutations in del(5q) MDS is unknown, but one could speculate that mutant clones may be selected for under the pressure of constitutive activation of this pathway in this particular cytogenetic MDS subset, analogous to other cancers in which such mutations emerge in the presence of ongoing cellular stress or DNA-damage response activation (46). Targeting the RPS14–MDM2–p53 pathway by PP2A inhibition or alternative therapeutic strategies could prove to alter the natural course of the disease by preventing clonal evolution and transformation to leukemia. As sustained suppression of p53 would lead to genomic instability (46), careful clinical investigation needs to be performed to balance clinical benefit with potential risk.
Novel Therapeutic Strategies for DEL(5q) MDS
As described above, PP2Acα is a key target of lenalidomide leading to modulation of the RP–MDM2–P53 pathway, clonal suppression, and restoration of normal erythropoiesis. Unfortunately, approximately 50% of patients develop resistance to lenalidomide after 2–3 years of treatment and there are currently no alternative karyotype selective therapeutic agents (13, 47). Given that PP2Acα over-expression underlies lenalidomide resistance, novel strategies targeting this pathway are of pivotal importance (Figure 3). This is reinforced by our findings that duration of TI to lenalidomide was directly related to the magnitude of PP2Acα suppression (36). Specifically, median duration of TI was not reached in patients with PP2Acα suppression from baseline (1507+ days) versus 679 days in patients without (P = 0.006, log rank).
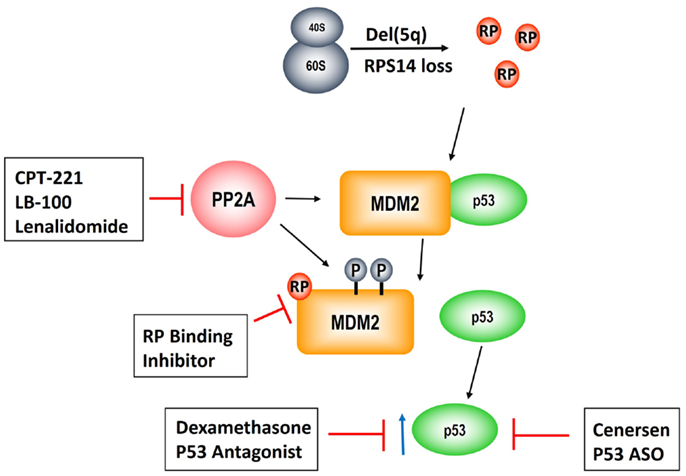
Figure 3. Pathogenesis of the del(5q) MDS hematologic phenotype and novel therapeutic strategies to rescue erythropoiesis. Allelic deletion of the genes encoded in the del(5q) CDR disrupts ribosomal integrity via haploinsufficiency of RPS14 leading to RP, e.g., RPL11, sequestration of MDM2 and consequent p53 activation. PP2A is a negative regulatory phosphatase of MDM2 whose inhibition results in hyperphosphorylation at phosphate residues that protect it from ubiquitination. Lenalidomide abrogates the anemia in del(5q) MDS through PP2A inhibition. CP2-221 is a deuterium-modified analog of S-lenalidomide enantiomer with greater potency than the parent compound. LB-100 is a novel direct PP2A inhibitor that has entered clinical testing. Lastly, p53 modulation via dexamethasone and/or cenersen (p53 antisense) are exciting potential clinical strategies that are currently under clinical investigation.
One potential strategy is development of more potent and specific inhibitors of PP2A. Okadaic acid, a derivative of shellfish toxin, inhibits both PP2A and PP1 (48). LB-100 and LB-102 are synthetic derivatives of cantharadin, a demethylated homolog of cantharadin (extract of beetle juice), which is an inhibitor of PP2A with relative specificity in vitro and in vivo and acceptable toxicity (49–52). LB-102 was shown to increase Akt phosphorylation and decrease p53 expression in malignant glioma cells and xenografts (50). LB-100/LB-102 blocked cell-cycle arrest and led to chemotherapy sensitization to temozolomide and doxorubicin (50, 52). LB-100 was also shown to induce tumor differentiation and/or cell death in glioblastoma multiforme (53). LB-100 has now entered a phase 1 clinical trial as a chemotherapy sensitizer in solid tumors (NCT01837667) (54). A phase 1 clinical trial of LB-100 is planned in lower risk MDS patients with symptomatic anemia, including del(5q) MDS patients who are resistant and/or intolerant to lenalidomide. This would be a first in class novel inhibitor as there are currently no FDA approved phosphatase inhibitors. Another potential strategy to enhance PP2Acα inhibition would be using the lenalidomide analog, CPT-221. CP2-221 is a deuterium-modified analog of S-lenalidomide (lenalidomide is a racemic mixture of S- and R-enantiomers). In vitro studies have shown that CPT-221 is threefold to fivefold more potent than the lenalidomide racemic mixture (55). Investigation of CPT-221 in the treatment of MDS patients has not yet been initiated.
Given that p53 accumulation in erythroid precursors is responsible for the hypoplastic anemia in del(5q) MDS, targeting p53 directly could restore terminal differentiation and effective erythropoiesis in the del(5q) clone. Cenersen, a clinically active 20-mer antisense phosphorothioate oligonucleotide complementary to exon 10, down-regulates both WT and mutant TP53 expression in vitro and in vivo when administered in combination with chemotherapy in patients with hematologic malignancies (56–58). Cenersen treatment of RPS14 deficient erythroblasts, to simulate the haplodeficient state in del(5q) cells, decreased cellular expression of TP53, leading to improved cell survival (59). Treatment of bone marrow CD34+ cells from lenalidomide resistant, del(5q) MDS with cenersen dramatically increased erythroid colony formation with less improvement in non-del(5q) or normal controls. Of interest, cenersen had no suppressive effect on the del(5q) clone, thereby supporting the notion that p53 suppression restored erythropoietic potential within the malignant clone (Figure 2). A phase 1 clinical trial testing cenersen in del(5q) MDS patients resistant and/or intolerant to lenalidomide therapy is now underway. As proof of this biological principle, Caceres et al. (59) tested the addition of dexamethasone, a glucocorticoid-receptor-dependent p53 antagonist, to lenalidomide treatment in del(5q) MDS patients who acquired resistance to lenalidomide. In this pilot study, 20 mg of weekly dexamethasone down-regulated p53 expression in erythroid progenitors and consequently restored TI in five of eight patients. The potential to achieve targeted suppression of WT and mutant TP53 with cenersen is exciting, given the decreased efficacy of lenalidomide in patients with a TP53 mutation (41–43). Of particular interest, Woll and colleagues (60) recently discovered that acquired driver mutations in the malignant stem cells of del(5q) MDS patients, especially TP53, emerge prior to leukemic transformation. This builds on prior work, which also showed that chromosome 5q deletion is the initial inciting event and is present in both the progenitor and malignant stem cells of these patients (60, 61). These stem cells that harbor del(5q) are intrinsically resistant to lenalidomide secondary to their quiescent state (61). Acquisition of TP53 mutations could foster propagation of myeloid progenitor cells leading to hematologic relapse and disease progression in these patients. Targeting of WT and mutated TP53 could prove to be a promising therapeutic strategy in possibly achieving and/or prolonging remissions in del(5q) MDS patients treated with lenalidomide.
Conclusion
Myelodysplastic syndromes with isolated del(5q) represents a distinct disease subtype distinguished by clinical presentation and unique sensitivity to lenalidomide. These pathognomonic features are directly linked to haplodeficiency of critical targets in the CDR on chromosome 5q. Severe anemia constitutes the major morbidity/mortality for del(5q) MDS patients and arises from RPS14 haplodeficiency leading to MDM2 sequestration, p53 stabilization, and erythroid cell death. Lenalidomide inhibits this pathway through its inhibitory effect on PP2A, rescuing erythropoiesis. Lenalidomide’s selective cytotoxity to del(5q) cells arises from inhibition of two haplodeficient phosphatases, PP2Acα and Cdc25C. Clonal suppression of del(5q) is of pivotal importance in preventing leukemic transformation and death. However, resistance to treatment emerges over time and future studies targeting PP2A and/or the RP–MDM2–p53 axis should be a priority to improve outcomes in this patient population.
Conflict of Interest Statement
Dr. David A. Sallman and Dr. Alan List have patent pending for PP2A inhibitor in MDS. Dr. Alan List is a consultant for Celgene. Dr. Sheng Wei has no conflict of interest
References
1. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood (1997) 89(6):2079–88.
2. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood (2012) 120(12):2454–65. doi: 10.1182/blood-2012-03-420489
3. Haase D, Germing U, Schanz J, Pfeilstocker M, Nosslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood (2007) 110(13):4385–95. doi:10.1182/blood-2007-03-082404
4. Kulasekararaj AG, Mohamedali AM, Mufti GJ. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. Br J Haematol (2013) 162(5):587–605. doi:10.1111/bjh.12435
5. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med (2011) 364(26):2496–506. doi:10.1056/NEJMoa1013343
6. Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol (2012) 30(27):3376–82. doi:10.1200/JCO.2011.40.7379
7. Kulasekararaj AG, Smith AE, Mian SA, Mohamedali AM, Krishnamurthy P, Lea NC, et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol (2013) 160(5):660–72. doi:10.1111/bjh.12203
8. Mallo M, Cervera J, Schanz J, Such E, Garcia-Manero G, Luno E, et al. Impact of adjunct cytogenetic abnormalities for prognostic stratification in patients with myelodysplastic syndrome and deletion 5q. Leukemia (2011) 25(1):110–20. doi:10.1038/leu.2010.231
9. Giagounidis AA, Germing U, Haase S, Hildebrandt B, Schlegelberger B, Schoch C, et al. Clinical, morphological, cytogenetic, and prognostic features of patients with myelodysplastic syndromes and del(5q) including band q31. Leukemia (2004) 18(1):113–9. doi:10.1038/sj.leu.2403189
10. Patnaik MM, Lasho TL, Finke CM, Knudson RA, Ketterling RP, Chen D, et al. Isolated del(5q) in myeloid malignancies: clinicopathologic and molecular features in 143 consecutive patients. Am J Hematol (2011) 86(5):393–8. doi:10.1002/ajh.21984
11. List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med (2005) 352(6):549–57. doi:10.1056/NEJMoa041668
12. List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med (2006) 355(14):1456–65. doi:10.1056/NEJMoa061292
13. List AF, Bennett JM, Sekeres MA, Skikne B, Fu T, Shammo JM, et al. Extended survival and reduced risk of AML progression in erythroid-responsive lenalidomide-treated patients with lower-risk del(5q) MDS. Leukemia (2014) 28(5):1033–40. doi:10.1038/leu.2013.305
14. Boultwood J, Fidler C, Strickson AJ, Watkins F, Gama S, Kearney L, et al. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood (2002) 99(12):4638–41. doi:10.1182/blood.V99.12.4638
15. Horrigan SK, Westbrook CA, Kim AH, Banerjee M, Stock W, Larson RA. Polymerase chain reaction-based diagnosis of del (5q) in acute myeloid leukemia and myelodysplastic syndrome identifies a minimal deletion interval. Blood (1996) 88(7):2665–70.
16. Van den Berghe H, Cassiman JJ, David G, Fryns JP, Michaux JL, Sokal G. Distinct haematological disorder with deletion of long arm of no. 5 chromosome. Nature (1974) 251(5474):437–8. doi:10.1038/251437a0
17. Heaney ML, Golde DW. Myelodysplasia. N Engl J Med (1999) 340(21):1649–60. doi:10.1056/NEJM199905273402107
18. Boultwood J, Pellagatti A, Cattan H, Lawrie CH, Giagounidis A, Malcovati L, et al. Gene expression profiling of CD34+ cells in patients with the 5q- syndrome. Br J Haematol (2007) 139(4):578–89. doi:10.1111/j.1365-2141.2007.06833.x
19. Pellagatti A, Fernandez-Mercado M, Di Genua C, Larrayoz MJ, Killick S, Dolatshad H, et al. Whole-exome sequencing in del(5q) myelodysplastic syndromes in transformation to acute myeloid leukemia. Leukemia (2014) 28(5):1148–51. doi:10.1038/leu.2013.381
20. Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet (1999) 21(2):169–75. doi:10.1038/5951
21. Gazda HT, Zhong R, Long L, Niewiadomska E, Lipton JM, Ploszynska A, et al. RNA and protein evidence for haplo-insufficiency in Diamond-Blackfan anaemia patients with RPS19 mutations. Br J Haematol (2004) 127(1):105–13. doi:10.1111/j.1365-2141.2004.05152.x
22. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature (2008) 451(7176):335–9. doi:10.1038/nature06494
23. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood (2011) 117(9):2567–76. doi:10.1182/blood-2010-07-295238
24. Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol (2009) 11(4):501–8. doi:10.1038/ncb1858
25. Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell (2003) 3(6):577–87. doi:10.1016/S1535-6108(03)00134-X
26. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol (2009) 49:223–41. doi:10.1146/annurev.pharmtox.48.113006.094723
27. Peller S, Frenkel J, Lapidot T, Kahn J, Rahimi-Levene N, Yona R, et al. The onset of p53-dependent apoptosis plays a role in terminal differentiation of human normoblasts. Oncogene (2003) 22(30):4648–55. doi:10.1038/sj.onc.1206541
28. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med (2010) 16(1):49–58. doi:10.1038/nm.2054
29. Oliva EN, Cuzzola M, Aloe Spiriti MA, Poloni A, Lagana C, Rigolino C, et al. Biological activity of lenalidomide in myelodysplastic syndromes with del5q: results of gene expression profiling from a multicenter phase II study. Ann Hematol (2013) 92(1):25–32. doi:10.1007/s00277-012-1569-0
30. Kumar MS, Narla A, Nonami A, Mullally A, Dimitrova N, Ball B, et al. Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q- syndrome. Blood (2011) 118(17):4666–73. doi:10.1182/blood-2010-12-324715
31. Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci USA (2009) 106(31):12974–9. doi:10.1073/pnas.0811267106
32. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J (2001) 353(Pt 3):417–39. doi:10.1042/0264-6021:3530417
33. Sartor H, Ehlert F, Grzeschik KH, Muller R, Adolph S. Assignment of two human cell cycle genes, CDC25C and CCNB1, to 5q31 and 5q12, respectively. Genomics (1992) 13(3):911–2.
34. Margolis SS, Perry JA, Forester CM, Nutt LK, Guo Y, Jardim MJ, et al. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell (2006) 127(4):759–73. doi:10.1016/j.cell.2006.10.035
35. Wilker EW, Grant RA, Artim SC, Yaffe MB. A structural basis for 14-3-3sigma functional specificity. J Biol Chem (2005) 280(19):18891–8. doi:10.1074/jbc.M500982200
36. Wei S, Chen X, McGraw K, Zhang L, Komrokji R, Clark J, et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene (2013) 32(9):1110–20. doi:10.1038/onc.2012.139
37. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med (2008) 14(4):152–60. doi:10.1016/j.molmed.2008.02.001
38. Mi J, Bolesta E, Brautigan DL, Larner JM. PP2A regulates ionizing radiation-induced apoptosis through Ser46 phosphorylation of p53. Mol Cancer Ther (2009) 8(1):135–40. doi:10.1158/1535-7163.MCT-08-0457
39. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol (2001) 3(3):245–52. doi:10.1038/35060032
40. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol (2001) 3(11):973–82. doi:10.1038/35060032
41. Bally C, Ades L, Renneville A, Mozziconacci MJ, Preudhomme C, DeThé H, et al. Prognostic value of TP53 gene mutations in higher-risk MDS treated with azacitidine (AZA). Leuk Res (2013) 37:S25. doi:10.1016/j.leukres.2014.03.012
42. Mallo M, del Rey M, Ibáñez M, Calasanz MJ, Arenillas L, Larráyoz MJ, et al. Response to lenalidomide in myelodysplastic syndromes with del(5q): influence of cytogenetics and mutations. Br J Haematol (2013) 162(1):74–86. doi:10.1111/bjh.12354
43. Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol (2011) 29(15):1971–9. doi:10.1200/JCO.2010.31.8576
44. Saft L, Karimi M, Ghaderi M, Matolcsy A, Mufti GJ, Kulasekararaj A, et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica (2014) 99:1041–9. doi:10.3324/haematol.2013.098103
45. Fenaux P, Giagounidis A, Selleslag D, Beyne-Rauzy O, Mufti G, Mittelman M, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood (2011) 118(14):3765–76. doi:10.1182/blood-2011-01-330126
46. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol (2010) 11(3):220–8. doi:10.1038/nrm2858
47. List AF, Estes M, Williams A, Sekharam M, Ozawa U, Gao G, et al. Lenalidomide (CC-5013; Revlimid(R)) promotes erythropoiesis in myelodysplastic syndromes (MDS) by CD45 protein tyrosine phosphatase (PTP) inhibition. Blood (2006) 108(11):1360.
48. Park DM, Li J, Okamoto H, Akeju O, Kim SH, Lubensky I, et al. N-CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell Cycle (2007) 6(4):467–70. doi:10.4161/cc.6.4.3856
49. Hart ME, Chamberlin AR, Walkom C, Sakoff JA, McCluskey A. Modified norcantharidins; synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg Med Chem Lett (2004) 14(8):1969–73. doi:10.1016/j.bmcl.2004.01.093
50. Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, et al. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc Natl Acad Sci U S A (2009) 106(28):11697–702. doi:10.1073/pnas.0905930106
51. Zhuang Z, Lu J, Lonser R, Kovach JS. Enhancement of cancer chemotherapy by simultaneously altering cell cycle progression and DNA-damage defenses through global modification of the serine/threonine phospho-proteome. Cell Cycle (2009) 8(20):3303–6. doi:10.4161/cc.8.20.9689
52. Zhang C, Peng Y, Wang F, Tan X, Liu N, Fan S, et al. A synthetic cantharidin analog for the enhancement of doxorubicin suppression of stem cell-derived aggressive sarcoma. Biomaterials (2010) 31(36):9535–43. doi:10.1016/j.biomaterials.2010.08.059
53. Lu J, Zhuang Z, Song DK, Mehta GU, Ikejiri B, Mushlin H, et al. The effect of a PP2A inhibitor on the nuclear receptor corepressor pathway in glioma. J Neurosurg (2010) 113(2):225–33. doi:10.3171/2009.11.JNS091272
54. Chung VM, Mansfield AS, Kovach J. A phase 1 study of a novel inhibitor of protein phosphatase 2A alone and with docetaxel. J Clin Oncol (2014) 32(5s):TS2636.
55. Uttamsingh V, Gallegos R, Cheng C, Aslanian A, Liu JF, Tung R, et al. CTP-221, a deuterated S-enantiomer of lenalidomide, is greatly stabilized to epimerization and results in a more desirable pharmacokinetic profile than racemic lenalidomide. Cancer Res (2013) 73(8 Suppl):3357. doi:10.1158/1538-7445.AM2013-3357
56. Bishop MR, Iversen PL, Bayever E, Sharp JG, Greiner TC, Copple BL, et al. Phase I trial of an antisense oligonucleotide OL(1)p53 in hematologic malignancies. J Clin Oncol (1996) 14(4):1320–6.
57. Cortes J, Kantarjian H, Ball ED, Dipersio J, Kolitz JE, Fernandez HF, et al. Phase 2 randomized study of p53 antisense oligonucleotide (cenersen) plus idarubicin with or without cytarabine in refractory and relapsed acute myeloid leukemia. Cancer (2012) 118(2):418–27. doi:10.1002/cncr.26292
58. Lanasa MC, Davis PH, Datto M, Li Z, Gockerman JP, Moore JO, et al. Phase II study of cenersen, an antisense inhibitor of p53, in combination with fludarabine, cyclophosphamide and rituximab for high-risk chronic lymphocytic leukemia. Leuk Lymphoma (2012) 53(2):218–24. doi:10.3109/10428194.2011.610012
59. Caceres G, McGraw K, Yip BH, Pellagatti A, Johnson J, Zhang L, et al. TP53 suppression promotes erythropoiesis in del(5q) MDS, suggesting a targeted therapeutic strategy in lenalidomide-resistant patients. Proc Nat Acad Sci U S A (2013) 110(40):16127–32. doi:10.1073/pnas.1311055110
60. Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell (2014). doi:10.1016/j.ccr.2014.03.036
Keywords: PP2A, p53, MDM2, RPS14, lenalidomide, deletion 5q, myelodysplastic syndrome
Citation: Sallman DA, Wei S and List A (2014) PP2A: the Achilles heal in MDS with 5q deletion. Front. Oncol. 4:264. doi: 10.3389/fonc.2014.00264
Received: 05 August 2014; Accepted: 08 September 2014;
Published online: 23 September 2014.
Edited by:
Peter Ruvolo, University of Texas MD Anderson Cancer Center, USAReviewed by:
Gheath Alatrash, University of Texas MD Anderson Cancer Center, USAPeter Ruvolo, University of Texas MD Anderson Cancer Center, USA
Copyright: © 2014 Sallman, Wei and List. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan List, Malignant Hematology Program, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL 33612, USA e-mail: alan.list@moffitt.org