 Dorien Haesen
Dorien Haesen Ward Sents1†
Ward Sents1† Yana Hoorne
Yana Hoorne Veerle Janssens
Veerle Janssens- 1Laboratory of Protein Phosphorylation and Proteomics, Department Cellular and Molecular Medicine, University of Leuven, Leuven, Belgium
- 2Gene Expression Unit, Department Cellular and Molecular Medicine, University of Leuven, Leuven, Belgium
Reversible protein phosphorylation plays a crucial role in regulating cell signaling. In normal cells, phosphoregulation is tightly controlled by a network of protein kinases counterbalanced by several protein phosphatases. Deregulation of this delicate balance is widely recognized as a central mechanism by which cells escape external and internal self-limiting signals, eventually resulting in malignant transformation. A large fraction of hematologic malignancies is characterized by constitutive or unrestrained activation of oncogenic kinases. This is in part achieved by activating mutations, chromosomal rearrangements, or constitutive activation of upstream kinase regulators, in part by inactivation of their anti-oncogenic phosphatase counterparts. Protein phosphatase 2A (PP2A) represents a large family of cellular serine/threonine phosphatases with suspected tumor suppressive functions. In this review, we highlight our current knowledge about the complex structure and biology of these phosphatases in hematologic cells, thereby providing the rationale behind their diverse signaling functions. Eventually, this basic knowledge is a key to truly understand the tumor suppressive role of PP2A in leukemogenesis and to allow further rational development of therapeutic strategies targeting PP2A.
Introduction
With 518 kinases encoded by the human genome and up to 70% of all eukaryotic proteins undergoing phosphorylation on a Ser, Thr, or Tyr residue, nearly every cellular process is controlled by this key modification (1). The covalent attachment of the bulky, negatively charged phosphoryl moiety to a protein markedly affects protein function through conformational changes that alter catalytic activity (for enzymes), affinity for ligands, subcellular localization, or stability (2). Several decades of biochemical and genetic studies have revealed crucial roles for protein kinases in the processes leading to tumor cell proliferation, survival, and migration in hematologic and other malignancies. In particular, genetic alterations that lead to constitutive activation of kinases, uncoupled from extracellular regulatory inputs, are well-characterized drivers of cancer development, a knowledge, which has emerged in the development of small-molecule kinase inhibitors for anti-cancer therapy (3). Kinase inhibitors have been extremely successful in the treatment of cancers driven by a single oncogenic kinase, such as chronic myeloid leukemia (CML) (4), but several challenges remain, including the development of drug resistance, lack of inhibitor selectivity or efficacy, and difficulty in drug target validation, particularly in cancers that do not exhibit such oncogenic kinase addiction (5).
Obviously, because protein phosphatases antagonize the action of protein kinases, they should be considered as equally important players in maintaining the correct phosphorylation balance of a given protein. Nonetheless, persistent misconceptions regarding the supposed lack of specificity and regulation of protein phosphatases as opposed to protein kinases, have contributed to a general underestimation of their critical role in the regulation of signal transduction (6, 7). Hence, much less is known about their role in cancer development and progression. Research over the past decade has begun to highlight the importance of the tumor suppressive activities of protein phosphatases, which, upon functional inactivation, contribute to persistent kinase or oncogene activation, and perhaps even more importantly, to drug resistance development (8, 9). Therefore, protein phosphatases may represent valuable novel drug targets for alternative cancer therapies, either in their own right or as part of combination therapies with kinase inhibitors (10–13).
Protein phosphatase 2A (PP2A) represents the prototype of a highly regulated phosphatase family with suspected critical tumor suppressive properties in several human tissues (14–16). Recent reports have demonstrated that modulation of PP2A activity can be beneficial for the treatment of cancer, particularly of hematologic malignancies (17, 18). Increasing evidence from cellular and clinical studies has indeed underscored the tumor suppressive role of PP2A in leukemogenesis, although the complex biology of these enzymes in hematologic cells remains incompletely understood. Here, we will provide insights into the basics of PP2A structure and regulation in hematologic cells and tissues, and highlight how proper PP2A function or activity is affected in hematologic malignancies. This knowledge is not only imperative to understand the protective role of PP2A in leukemogenesis but also equally important to allow for rational design of PP2A-directed drugs, and thus, to fully exploit PP2A as anti-cancer target in these devastating diseases.
PP2A Family
PP2A Enzymes: Structural and Functional Centipedes
“PP2A” refers to a large, highly conserved family of ubiquitously expressed Ser/Thr phosphatases that, together with PP1, constitutes the bulk of Ser/Thr phosphatase activity in a given cell or tissue (19). The prototypic PP2A holoenzyme is a heterotrimeric complex of a catalytic C subunit, a scaffolding A subunit, and a regulatory B-type subunit (Figure 1). In human cells, B-type subunits are encoded by 15 different genes, which give rise to 23 different isoforms through use of alternative gene promoters, alternative splicing events, or alternative translation (20). Based on sequence homology, they are divided into four distinct families, called B (or B55, or PR55, or by gene name: PPP2R2), B′ (or B56, or PR61, or by gene name: PPP2R5), B″ (or PR72, or by gene name: PPP2R3), and B‴ (or the striatins, STRN) (Figure 1). The B-type subunits are true “regulatory” subunits, in the sense that they dictate substrate specificity of the associated PP2A C subunit and can directly modulate PP2A catalytic activity. They are often expressed in a cell- or tissue-specific way, and can be found at distinct subcellular locations (cytoplasm, nucleus, plasma membrane, mitochondria, Golgi apparatus, endoplasmic reticulum, and cytoskeleton), thus, restricting PP2A activity to cell- or tissue-specific substrates present at specific subcellular sites (20, 21). Also, the C and A subunits are encoded by two different genes each, giving rise to two nearly identical Cα and Cβ isoforms (encoded by PPP2CA and PPP2CB), and two highly related Aα and Aβ isoforms (encoded by PPP2R1A and PPP2R1B). Despite an extremely high degree of sequence identity, there is evidence that these isoforms, remarkably, do not serve redundant functions (22–25). Besides their assembly into trimeric PP2A complexes, A and C subunits can form active A–C heterodimers (Figure 1), which are estimated to represent about one-third of cellular PP2A in a given cell (26).
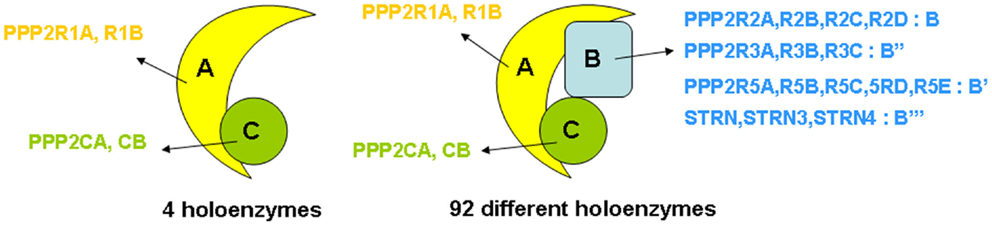
Figure 1. Structure of PP2A holoenzymes. The majority of PP2A enzymes have a heterotrimeric structure and consist of one catalytic C subunit, one scaffolding A subunit, and one regulatory B-type subunit. Owing to the existence of various isoforms of each of these subunits – in human tissues, two C (encoded by PPP2CA and PPP2CB), two A (encoded by PPP2R1A and PPP2R1B), and 23 B-type isoforms (encoded by 15 different genes) – 92 different PP2A trimeric complexes can be assembled, each characterized by its own catalytic properties, substrate specificities, tissue or cell-specific expression, and subcellular localization. In addition, about one-third of PP2A occurs as a dimer of one A and one C subunit (four holoenzymes).
The combinatorial assembly of one C and one A, or one C, one A, and one B-type subunit can theoretically give rise to 4 different heterodimers and 92 different heterotrimers (Figure 1), all exhibiting potentially different physiological functions. Thus, the broad diversity in PP2A composition creates specificity and constitutes the basis for the highly diverse and multiple cellular and physiological functions of these phosphatases. PP2A has indeed been implicated in a wide range of signaling pathways, many of which are involved in the control of cell proliferation and death (16, 27), cell division (28, 29), differentiation (28), adhesion and migration (30), and metabolism (31, 32). Besides function, PP2A composition also largely defines regulation by upstream factors, including specific second messengers (cAMP, Ca2+-ions, lipids) (20), cellular PP2A inhibitors (33) (see further), and phosphorylation by specific kinases (20). Most of these regulatory inputs are again largely determined by the nature of the specific B-type subunit present in the complex.
This being said, it should come of little surprise that “PP2A” (i.e., the large family of distinct PP2A complexes) may exert collaborating as well as opposing functions within a given signaling pathway by acting at different levels in the cascade. This is, for instance, the case in growth factor-induced ERK signaling, TGFβ signaling, or in canonical and non-canonical Wnt signaling (16, 27, 34). In addition, different PP2A complexes may dephosphorylate the same substrate, even on the same site, depending on the regulatory stimulus involved, the cell type or the broader physiological context (35–37). In contrast, functional redundancies, particularly between PP2A complexes harboring a B-type subunit from the same subfamily, have also been reported (38), further illustrating the complexity of PP2A holoenzyme function and substrate selection. It is clear though that in order to fully understand the role and regulation of “PP2A” in any (patho)physiological context, it is of utmost importance to identify which particular holoenzymes are involved in a non-redundant way. Nonetheless, and despite their general importance in PP2A biology, the specific PP2A regulatory subunits controlling dephosphorylation of a given substrate in a given mammalian cell or tissue remain poorly defined, particularly in the physiological context of a whole organism (39–41). Additional “PP2A” knockout mice are eagerly being awaited to overcome this lack of in vivo knowledge, and eventually, to advance the rational development of PP2A as a druggable target in the relevant cancer types.
Expression of PP2A Subunits in Spleen, Thymus, and Bone Marrow
To truly understand the biology of “PP2A” in hematologic cells and tissues, one should ideally know which PP2A complexes occur in these tissues. Because of general lack of sufficient isoform-specific antibodies and because only fragmented relevant information can be found in the currently available PP2A literature, we have analyzed, for the purpose of this review, mRNA expression of all PP2A subunit genes via microarray in mouse spleen (n = 3), thymus (n = 3), and bone marrow (n = 4) (Figure 2). Brain cortex (n = 3) and heart (n = 3) were included as “controls” (Figure 2) as in these tissues, expression of most PP2A subunits has been investigated and reported before. If we presume that hybridization of the PP2A mRNAs to their respective gene probes on the chip occurs with comparable efficiency, we find overall significantly higher expression of the α isoforms of both C and A subunits as opposed to their respective β isoforms (Figure 2A), fully in accordance with published data (19, 24). When considering expression of B-type subunits, most of them are expressed in all three hematologic tissues, except Ppp2r2b and Ppp2r2c (encoding Bβ and Bγ), which were reported to be exclusively expressed in brain (42), Ppp2r3a (encoding B″α), which was reported to be predominantly expressed in heart (43), and Ppp2r5b (encoding B′β) whose hematologic expression is extremely low (Figure 2B). Highest expression is seen for Ppp2r5a and Ppp2r5c (encoding B′α and B′γ), followed by Ppp2r5e (encoding B′ε), Strn3 (encoding B‴/SG2NA), Ppp2r3c (encoding B″γ/G5PR), Ppp2r2a, and Ppp2r2d (encoding Bα and Bδ). Lowest expression is seen for Ppp2r5d (encoding B′δ), Strn, and Strn4 (encoding B‴/striatin and B‴/zinedin) (Figure 2B). Expression of Ppp2r3d could not be analyzed because it was not represented on the microarray chip. For most PP2A subunits present in these tissues, expression is comparable between spleen, thymus, and bone marrow, except for Ppp2r2a (Bα), which is approximately two times more abundant in spleen and thymus as opposed to bone marrow, and for Ppp2r5a (B′α), Ppp2r5d (B′δ), and Strn4 (B‴/zinedin), which are least abundant in spleen as opposed to thymus and bone marrow (Figure 2B). Thus, these data illustrate in a qualitative and semi-quantitative way the repertoire of PP2A B-type subunits expressed in the three main hematologic tissues in mice.
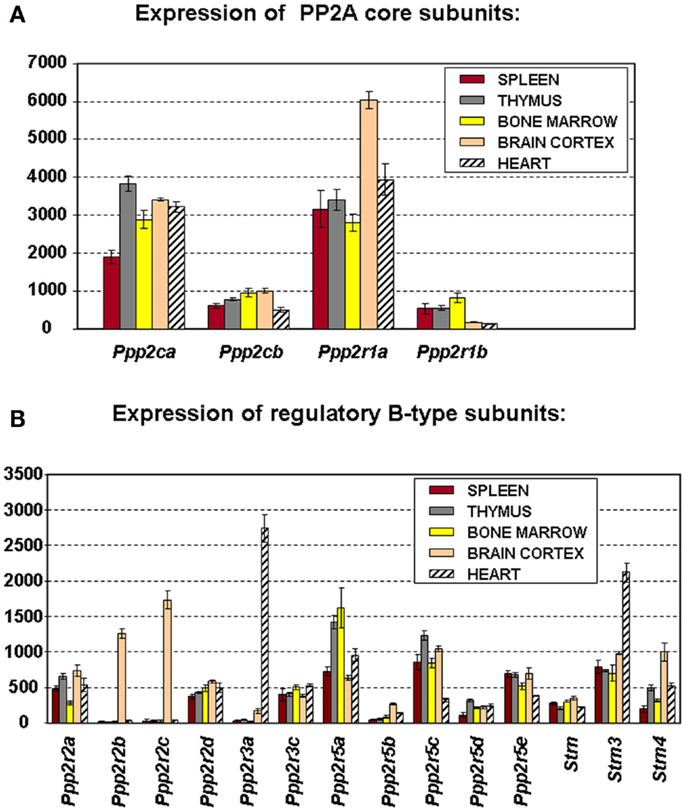
Figure 2. Microarray expression profiles of PP2A subunit encoding genes in mouse tissues. Spleen, thymus, bone marrow, brain cortex, and heart were hand-dissected from 10- to 12-week-old C57Bl6 mice. Total RNA was extracted, labeled, and hybridized to the Affymetrix mouse MOE 430 2.0 array (44). Scanning, quality control, data processing, and statistical analysis of the data were as described (44). Shown is the mean mRNA expression signal ±SD of three (spleen, thymus, brain, and heart) or four (bone marrow) biological replicate experiments. (A) Expression the PP2A core subunit encoding genes. (B) Expression of the genes encoding PP2A regulatory B-type subunits. Expression of Ppp2r3d could not be analyzed because it was not present on the array used.
Inactive PP2A Complexes and PP2A Holoenzyme Assembly
Besides the prototypical PP2A holoenzymes described above, several “atypical” PP2A complexes have been identified that can occur within cells as catalytically inactive PP2A complexes. For example, the interaction between the C subunit and the α4 protein (encoded by IGBP1) stabilizes the C subunit as a latent, inactive form (45, 46), although there is also some evidence that this complex might be active toward very specific cellular substrates [reviewed in Ref. (47)]. Another example is the catalytically inactive complex between the C subunit, the A subunit, and PME-1 (PP2A Methyl Esterase 1, encoded by PPME1) (Figure 3A) that has been estimated to represent up to 25% of the cellular PP2A C pool (48, 49). It is thought that these atypical, inactive PP2A complexes constitute intermediate, but stable complexes during the process of PP2A holoenzyme biogenesis (47) or holoenzyme disassembly (45). Interestingly, increased expression of α4 or PME-1 has been found in several human cancers [hepatocellular carcinoma (50), lung carcinoma (50, 51), breast cancer (50), glioma (52), and endometrial cancer (53)], indicative for a relative increase in inactive PP2A complexes as opposed to active holoenzymes in these transformed cells.
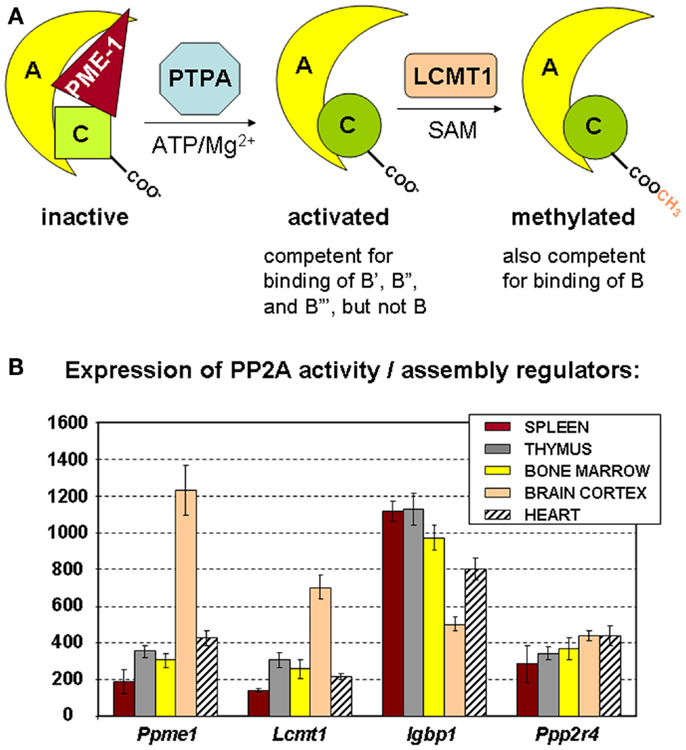
Figure 3. Regulators of PP2A holoenzyme biogenesis and assembly. (A) Simplified schematic of the roles of PME-1, PTPA, and LCMT1 in the biogenesis of active PP2A trimers. The PP2A methylesterase PME-1 serves to stabilize the inactive PP2A C subunit in a complex with the A subunit, at the same time preventing PP2A C methylation. With ATP/Mg2+ as necessary cofactors, PTPA promotes folding of PP2A C in an active conformation, and thereby, indirectly, PP2A C carboxymethylation by LCMT1. The latter modification is absolutely required for binding of B subunits, facilitates interaction of all B′ subunits but the δ isoform, is of no apparent importance for binding of B′δ and the B″ subunits, and is disliked by the striatin subunits. α4 (not depicted here) is another regulator that stabilizes PP2A C in a latent form. It is currently unclear if and how this inactive α4–C complex might become activated by similar mechanisms (47). (B) Expression of PP2A biogenesis regulators in hematologic tissues, brain, and heart. The mean mRNA expression signal ±SD for Ppme1 (PME-1), Lcmt1 (LCMT1), Igbp1 (α4), and Ppp2r4 (PTPA) is shown of three (spleen, thymus, brain, and heart) or four (bone marrow) biological replicate experiments.
The precise mechanism of assembly of active PP2A holoenzyme is still incompletely understood (47). A major insight came from the finding that the PP2A C subunit is synthesized/translated as an inactive enzyme (54) that is subsequently activated in a way that is strictly coupled to its incorporation into the complete holoenzyme (55). Like that, promiscuous and unregulated phosphatase activity of the free C subunit can be avoided (54, 55). There is evidence that proteins such as α4 and PME-1 can stabilize such inactive PP2A C subunits within cells, either in the absence (for α4) (46) or the presence (for PME-1) of the A subunit (47). To generate active PP2A holoenzymes from these inactive complexes, at least two additional PP2A regulating enzymes are needed. First, PTPA (or “PP2A Activator,” encoded by PPP2R4) may activate the PME-1-bound PP2A complex in the presence of ATP/Mg2+ as necessary cofactors (Figure 3A) (49). In accordance, in vivo data in yeast have shown that PTPA-dependent generation of active C subunit requires a functional interaction with the A subunit and is regulated by PME-1 (55). Crystallographic data suggested that PTPA may act as an ATP/Mg2+-dependent prolyl-peptidyl cis/trans isomerase of a single prolyl-peptidyl bond in PP2A C (56), as well as an ATP/Mg2+-dependent chaperone promoting the incorporation of catalytic metal ions into the PP2A C active site (57). Regardless of its precise mechanism-of-action, several in vivo studies have underscored the importance of PTPA as a physiological activator of PP2A [reviewed in Ref. (47)]. The second enzyme important in PP2A biogenesis is LCMT1 (leucine carboxyl methyl transferase 1, encoded by LCMT1), an S-adenosylmethionine-dependent methyltransferase catalyzing the carboxymethylation of the PP2A C subunit (58). This unusual post-translational modification of PP2A C is reversible through the presence of PME-1, the PP2A methylesterase (59), which may thus serve a dual function. Interestingly, PP2A C carboxymethylation requires an active PP2A C conformation (60), is facilitated by the presence of the A subunit (61), and enhances the affinity of the core dimer for PP2A regulatory subunits (Figure 3A). Specifically, PP2A C methylation is an absolute prerequisite to bind subunits of the B family, it facilitates interaction of all members but the δ isoform of the B′ family, is of no apparent importance for binding of B′δ and the B″ subunits, and is disliked by the B‴ subunits [reviewed in Ref. (62)]. Intriguingly, all regulators involved in PP2A holoenzyme biogenesis (α4, PME-1, PTPA, and LCMT1) are indispensable for mammalian survival (63–65, unpublished work), indicative for their physiological importance. In accordance, they are all expressed in spleen, thymus, and bone marrow, the three hematologic tissues analyzed here by microarray (Figure 3B). Our data also show a relatively higher expression of PME-1 and LCMT1 in brain, as opposed to other tissues analyzed (Figure 3B).
Cellular PP2A Inhibitory Proteins
Although the first cellular PP2A inhibitors were discovered almost two decades ago (66), their role in direct regulation of PP2A activity has only during the recent years come into considerable focus, not the least because some of them commonly suppress PP2A activity in hematologic and other cancers, and thus, may constitute novel therapeutic targets (33). These inhibitors either directly bind to the PP2A catalytic subunit or target very specific PP2A holoenzymes, thereby preventing dephosphorylation of a large variety of PP2A substrates (Figure 4A).
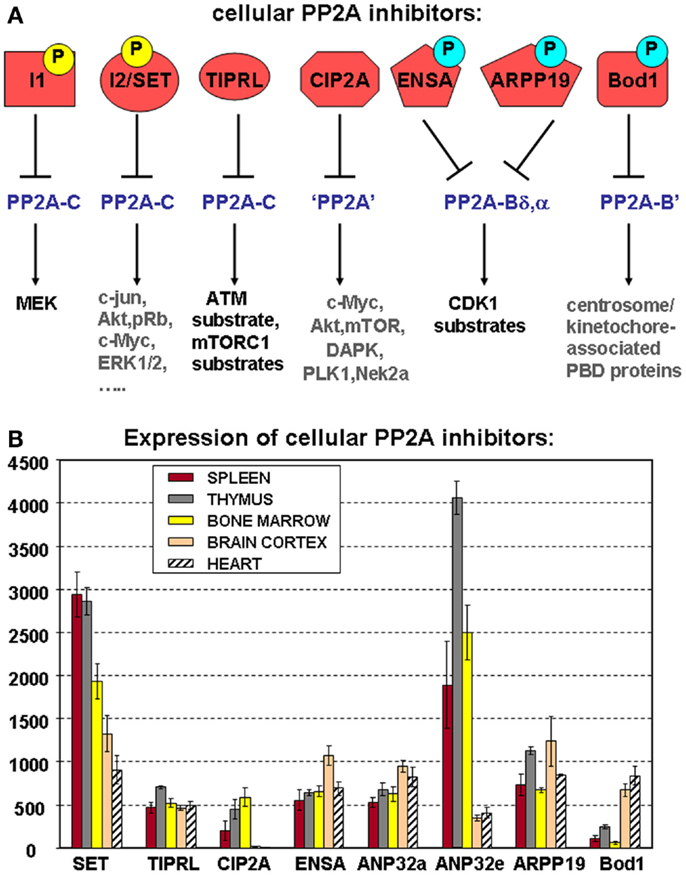
Figure 4. Cellular PP2A inhibitors. (A) Schematic representation of known cellular PP2A inhibitors, highlighting their potential regulation by (yellow) or dependence on (blue) phosphorylation, their holoenzyme specificity (if known), and the PP2A substrates they affect. Best characterized so far, in terms of phosphorylation dependence and holoenzyme specificity, are the mitotic inhibitors ENSA, ARPP-19, and Bod1. Both I1 (ANP32a, e) and I2 (SET) are established phosphoproteins, but depending on the specific site of modification, these phosphorylations may increase as well as decrease their PP2A inhibitory abilities. Phosphoregulation of CIP2A or TIPRL has not yet been described. I1, I2, and TIPRL are thought to interact with PP2A complexes through the C subunit, while holoenzyme specificity of CIP2A-mediated inhibition remains undefined. PBD: polo-box domain. (B) Expression of cellular PP2A inhibitors in hematologic tissues, brain, and heart. The mean mRNA expression signal ±SD for the eight indicated cellular PP2A inhibitory proteins is shown of three (spleen, thymus, brain, and heart) or four (bone marrow) biological replicate experiments.
ANP32a and SET
PP2A inhibitor 1 (also called ANP32a, or by gene name: ANP32a) and inhibitor 2 (also called TAF-Iβ, or PHAP1, or by gene name: SET) were originally de novo purified from bovine kidney as two potent, heat-stable PP2A-specific inhibitors (66) and subsequently cloned from cDNA libraries (67, 68). ANP32a belongs to a large family of at least nine members (ANP32a–h), of which only ANP32a and ANP32e show PP2A inhibitory ability in an in vitro phosphatase assay (69, 70). Free PP2A C subunit, the core A–C dimer and a trimeric PP2A complex with B subunit were inhibited in this assay, suggestive for direct binding of ANP32a to the C subunit, and thus, for no specific holoenzyme selectivity. Essentially, the same observations were made for SET in in vitro PP2A phosphatase assays (68). SET exists as two splice variants (SETα and β) that are both capable of inhibiting PP2A (71). ANP32a/e and SET are all phosphoproteins and can be found in the nucleus as well as the cytoplasm. Tyrosine phosphorylation of ANP32a releases its binding to PP2A and relieves PP2A inhibition toward MEK (72). In SETα, Ser9, Ser24, Ser93, and Ser171 have been identified as phosphorylation sites of functional importance (73–75). Phosphorylation of Ser9 functionally disrupts a nuclear localization signal and promotes SET retention in the cytoplasm (76, 77), while Ser171 phosphorylation decreases its PP2A inhibitory potential (74) and Ser9/Ser93 phosphorylation increases its ability to inhibit PP2A (75). Proteolytic cleavage of SET, either by Granzyme A at K176 (78) or by asparaginyl endopeptidase at N175 (79) is another mechanism that promotes its translocation into the cytoplasm (80), while the SET-binding protein SETBP1 stabilizes full-length SET inside the nucleus (81). Several PP2A substrates are known to be affected by SET, either directly or indirectly, including ERK1/2 (82), Akt (82–84), PTEN (83), Mcl-1 (85, 86), c-Myc (82, 84, 87), c-jun (88), and pRb (82), just to name a few (Figure 4A).
CIP2A
CIP2A (or cancerous inhibitor of PP2A, or by gene name KIAA1524) is an oncoprotein, originally identified as a novel co-precipitating partner of the PP2A A subunit (89). CIP2A is barely detectable in normal cells, but becomes specifically upregulated in a large variety of human cancers, hence its name [reviewed in Ref. (90)]. CIP2A knockout mice show no overt phenotypes, except for a defect in spermatogenesis (91). In cancer cells, CIP2A upregulation is mediated by several oncogenic transcription factors, including Ets (92), Myc (93), and E2F (94), and often correlates with cancer aggressiveness and poor prognosis (90). At the signaling level, increased CIP2A expression has been associated with increased c-Myc stability and Ser62 phosphorylation (89), increased Akt signaling (95), inhibition of dependence receptor-dependent apoptosis (96), and more recently, with changes in regulation of cell cycle kinases such as Plk1 (97) and NEK2 (98), and activation of the TOR pathway (99, 100) (Figure 4A). The biochemistry of CIP2A remains, however, largely undefined; in particular, it remains to be determined which PP2A complexes it may specifically inhibit and how this is achieved.
TIPRL1
TIPRL1 (also called TIP, or two A inhibitory protein, gene name TIPRL) is a ubiquitously expressed PP2A inhibitory protein that has been shown to inhibit free PP2A C and the PP2A A–C dimer by an allosteric mechanism (101, 102). TIPRL1 directly interacts with PP2A C, as well as with the C subunits of the PP2A-like phosphatases PP4 and PP6 (103). Notably, TIPRL1 may play an important role in DNA damage and repair signaling as it regulates PP2A enzymes that oppose ATM/ATR-dependent phosphorylation events (101). In addition, it may facilitate mTORC1 signaling and increase protein translation by sustaining phosphorylation of the mTORC1 substrates S6K1 and 4E-BP1 (104). In cancer cells, highly elevated TIPRL1 expression was reported in hepatocellular carcinoma, correlating with decreased pro-apoptotic MKK7/JNK signaling and contributing to resistance to TRAIL-induced apoptosis (105). The physiological role of TIPRL1 remains, however, undefined.
Mitotic PP2A Inhibitors: ENSA, ARPP-19, and Bod1
cAMP-regulated phosphoproteins ARPP-16 and ARPP-19 are splice variants and members of an evolutionary conserved protein family, to which ENSA (α-endosulfine) is closely related. ENSA and ARPP-19 are mitotic PP2A inhibitors that promote the G2/M transition and the mitotic state (106, 107). Intriguingly, they strongly bind to Bα and Bδ, but no other B-type subunits, dimeric PP2A or monomeric PP2A C, suggestive for a strong PP2A holoenzyme specificity (106–108) (Figure 4A). Moreover, these proteins require prior phosphorylation by the mitotic Greatwall/MASTL kinase, the Cdk1/cyclinB kinase, or potentially other mitotic kinases to exert their PP2A inhibitory effects (106, 107, 109, 110). Phosphorylation of ARPP-19 by cAMP-dependent kinase (PKA) serves to keep oocytes arrested in prophase (111), but how this may affect PP2A inhibition is unknown. More recently, yet another mitotic PP2A inhibitor was identified: Bod1, a protein required for proper chromosome alignment at mitosis. Bod1 shares sequence similarity with ENSA and ARPP-19, but intriguingly, specifically inhibits kinetochore- and centrosome-associated PP2A-B′ holoenzymes. Again, a phosphorylation of Bod1 by Cdk1/cyclinB is required to promote interaction with and inhibition of PP2A-B′ (112). Although many more needs to be discovered about the biochemistry and physiological roles of these mitotic PP2A inhibitors, they currently represent an exemplary mechanism of holoenzyme (family)-specific PP2A inhibition.
Expression of PP2A Inhibitors in Hematologic Tissues
As for the PP2A subunits (Figure 2) and the regulators of PP2A holoenzyme assembly (Figure 3B), we have analyzed mRNA expression of the above PP2A inhibitors in mouse spleen (n = 3), thymus (n = 3), and bone marrow (n = 4) (Figure 4B). Expression of SET, CIP2A, and ANP32e appears significantly higher in all three hematologic tissues analyzed, as opposed to terminally differentiated brain and heart tissues. In fact, CIP2A expression is completely undetectable in brain and heart, consistent with the idea that its expression is tightly coupled to cell proliferation, and potentially, stemness (90). Bod1 expression shows the opposite behavior and is significantly less expressed in spleen, thymus, and bone marrow as opposed to brain and heart, while TIPRL1, ANP32a, ENSA, and ARPP-19 expression is comparable in all tissues analyzed (Figure 4B).
PP2A Aberrations in Hematologic Malignancies
Several mechanisms of PP2A dysfunction in hematologic malignancies have been reported, including changes in expression of PP2A subunits and inhibitors (by epigenetic or other mechanisms), genomic alterations in PP2A subunit and regulator encoding genes (including mutations, deletions, splicing errors, chromosomal translocations), and alterations in subunit modifications affecting PP2A activity.
Alterations in PP2A Subunits
Although both PPP2R1A (Aα) and PPP2R1B (Aβ) have been identified as genuine tumor suppressor genes in solid cancers (17, 25), few reports have currently documented their inactivation in hematologic malignancies. Decreased A subunit expression is observed in myeloid cells expressing activated c-KIT mutants (113), while loss of Aβ function occurs with low frequency in ALL (G90D mutation, 3/150) (114), B-CLL (exon skipping and reduced mRNA expression) (115, 116), and AML (117). Decreased expression of Cα is one of the hallmarks of del(5q) myelodysplastic syndromes (MDS) and AML, and interestingly, predicts a favorable therapy response to lenalidomide (118), suggestive for its use as a stratification marker. Increased Y307 phosphorylation of PP2A C occurred in 29/37 AML cases, correlating with significantly decreased PP2A activity toward Akt and ERK (117).
PP2A B-type subunit alterations occur more frequently, particularly in AML. Reduced expression of Bα in AML blasts, correlating with increased Akt, p70S6K, and PKCα phosphorylation and deregulated expression of specific microRNAs (miRs), is associated with significantly reduced complete remission duration (119, 120). In c-KIT mutant AML, reduced expression of Bα is observed, along with decreased expression of several B′ subunits (α,γ,δ), correlating with overall decreased PP2A activity (113). Genomic deletion of PPP2R5B/C (B′β,γ) (117) and downregulation of B′ε by an elusive non-genomic mechanism (121) do also frequently occur in AML, correlating with increased oncogenicity of the leukemic cells. In lymphocytic leukemia, PPP2R5C (B′γ) downregulation is a hallmark of progressive as opposed to stable B-CLL (122), while in Notch-induced T-ALL, PPP2R5E (B′ε) was identified as one of the targets for miR-19, an oncomiR that promotes leukemogenesis in vivo (123). In childhood T-ALL and B-ALL, PPP2R3A (B″α) is epigenetically inactivated by increased methylation with high frequency (69 and 82%, respectively) (124). Genomic deletion of PPP2R2A/B (Bα, β) is sporadically observed in primary plasma cell leukemia and multiple myeloma (125, 126).
Alterations in PP2A Regulators
The large majority of PP2A aberrations in hematologic malignancies involve abnormalities (overexpression, genetic modifications) in the proto-oncogenic PP2A inhibitors CIP2A and SET.
The first time deregulated CIP2A expression was linked to blood cancer development was through the discovery of a chromosomal translocation, resulting in an MLL-KIAA1524 fusion protein in an isolated case of infant AML (127). In this fusion, exons 1–10 of MLL are coupled in frame to exons 17–21 of CIP2A, encompassing the CIP2A coiled coil domain. In addition, CIP2A overexpression occurs frequently in newly diagnosed AML (54/70) and relapsed AML (11/14) (128). In CML, a positive feedback loop between CIP2A and BCR/ABL has been described, implying that CIP2A overexpression may promote CML pathogenesis (129, 130). Importantly, and in contrast to expression of SET, CIP2A expression is a clear determinant of disease progression to blast crisis (129) and thus confers a poor prognosis in these patients. Mechanistically, high-CIP2A levels in primary CML correlate with high levels of S62-phosphorylated c-Myc (129) and increased resistance to bortezomib-induced apoptosis (131). Analysis of CIP2A expression levels in a panel of 105 B-cell lymphomas further demonstrated a link with clinical aggressiveness of the subtypes, with weak or absent CIP2A expression in indolent B-cell lymphomas and strongly positive signals in the more aggressive diffuse large B-cell and Burkitt lymphoma subtypes (132).
Increased SET expression is found in CML, where it correlates with blast crisis and resistance to therapeutic BCR/ABL tyrosine kinase inhibitors (TKI) (82), in Philadelphia chromosome-positive (Ph)-ALL (133), (c-KIT positive) AML (113, 134), and B-CLL (85). In AML and B-CLL, its expression is associated with disease severity and poor outcome. In leukemic progenitors, PP2A activity is substantially impaired as a result of SET overexpression (82). In CML and Ph-ALL, induction of SET expression is controlled by BCR/ABL (82), while in AML, overexpression of EVl1 or downregulation of miR199b may contribute (134). Restoration of PP2A activity in leukemic cells results in decreased phosphorylation of pRb, c-Myc, Stat5, ERK1/2, Akt, Bad, and Jak2, and induction of SHP1-mediated BCR/ABL inactivation and degradation (82, 135). In atypical CML, lacking the BCR/ABL fusion, recurrent SETBP1 mutations are found in 17/70 cases, some of which abrogate a site for ubiquitination, resulting in increased amounts of SETBP1 and SET protein, lower PP2A activity, and higher proliferation rates (136). In AML, overexpression of SETBP1 predicts poor outcome in elderly AML patients (81). Finally, SET is recurrently involved in chromosomal rearrangements and translocations, in particular, with the nucleoporin-encoding Nup214 gene (also called CAN) in AML, T-ALL, and acute undifferentiated leukemia (137–140).
PP2A (Re)Activation as a Novel Therapeutic Strategy in Hematologic Malignancies
The above findings, highlighting several mechanisms of PP2A inactivation in patients with hematologic malignancies, substantially underscore the tumor suppressor activities of (specific) PP2A holoenzymes and the proto-oncogenic properties of PP2A inhibitors CIP2A and SET. Importantly, some of these mechanisms may serve as biomarkers to improve current therapies (118), or may be directly amenable for therapeutic intervention. Several recent preclinical studies have shown that pharmacological restoration of PP2A tumor suppressor activity by PP2A-activating drugs (PADs) indeed effectively antagonizes cancer development and progression [reviewed in Ref. (17)]. Because PP2A complexes have so many cellular targets, these therapies may have the additional advantage, not to target just a single oncogene, but rather many different oncogenic pathways, contributing to their therapeutic efficacy. On the other hand, it is obvious that not all mechanisms of PP2A inhibition are suitable for restoration, particularly when subunit mutations are involved. Likewise, the development of small-molecule protein–protein interaction inhibitors targeting PP2A–SET or PP2A–CIP2A complexes remains, although attractive, extremely challenging.
Neviani et al. were the first to highlight the therapeutic relevance of using PP2A activators, such as FTY720 and forskolin, to target leukemia cells (82, 133). These observations prompted many others to test these compounds successfully in their own leukemic models (85, 113, 117, 141, 142). Treatment of AML patients with forskolin, in combination with standard induction therapy, gave an additive effect, highlighting therapeutic potential of PP2A activators in combination with standard chemotherapy (117). The mechanism of PP2A activation by these compounds remains somewhat obscure, but may involve direct binding of FTY720 to a ceramide-binding domain of SET (143, 144), resulting in SET dissociation from PP2A (143, 145). FTY720 also reduces SET Ser phosphorylation (144) and promotes SET nuclear localization (145), suggesting that its therapeutic effect may be largely attributable to restoration of cytoplasmic PP2A activity. The cell penetrating SET antagonistic peptides COG112 and OP449 (formerly COG449) directly bind SET to prevent SET–PP2A interaction and enhance PP2A activity (84). Like FTY720, they show significant therapeutic potential as PADs, as they induce apoptosis of human B-cell non-Hodgkin lymphoma and B-CLL in vitro and in vivo, without any discernable effects on normal B cells (85, 146). In models of human CML and AML (147) and canine T-cell lymphomas (148), OP449 also shows anti-tumoral effects, especially in combination with ABL TKI (147). The latter demonstrates the added benefit of combining TKIs and PADs for anti-leukemic therapy (17). Very recently, yet another class of FDA-approved drugs, the phenothiazines, were shown to act as PADs in models of T-ALL (149). These compounds induce rapid dephosphorylation of multiple PP2A targets, resulting in suppressed growth and increased apoptosis of T-ALL cells in vitro and in vivo. Mechanistically, a direct interaction with the Aα subunit is involved, but how this results in increased PP2A activity should still be further explored.
Together, these findings strongly encourage the inclusion of pharmacological PP2A activators with major anti-cancer activities and good safety profiles into current anti-cancer protocols in hematologic malignancies. The partially overlapping effects of existing drugs and PP2A stimulation predict that the inclusion of PADs in combination therapies with TKIs or other conventional therapeutics would represent particularly attractive therapeutic strategies to improve therapeutic outcome in these devastating malignancies. In the meantime, additional efforts to improve the potency and selectivity of existing PADs, and to identify alternative PP2A-activating strategies should be undertaken, in order to achieve their eventual use in the clinic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding was provided by the KU Leuven Research Fund (OT/13/094), the Research Foundation – Flanders (G.0582.11), and the IAP program of the Belgian federal government (P7/13). Dorien Haesen received a fellowship of the Flemish Agency for Innovation by Science and Technology. Ward Sents received a fellowship Emmanuel van der Schueren of the Flemish Cancer League.
References
1. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science (2002) 298:1912–34. doi:10.1126/science.1075762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Hunter T. Why nature chose phosphate to modify proteins. Philos Trans R Soc Lond B Biol Sci (2012) 367:2513–6. doi:10.1098/rstb.2012.0013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer (2009) 9:28–39. doi:10.1038/nrc2559
4. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol (2014) 89:547–56. doi:10.1002/ajh.23691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer (2010) 10:130–7. doi:10.1038/nrc2787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Brautigan DL. Protein Ser/Thr phosphatases: the ugly ducklings of cell signalling. FEBS J (2013) 280:324–45. doi:10.1111/j.1742-4658.2012.08609.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Tonks NK. Protein tyrosine phosphatases: from housekeeping enzymes to master regulators of signal transduction. FEBS J (2013) 280:346–78. doi:10.1111/febs.12077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Julien SG, Dubé N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer (2011) 11:35–49. doi:10.1038/nrc2980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev (2008) 27:159–68. doi:10.1007/s10555-008-9119-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. The therapeutic potential of phosphatase inhibitors. Curr Opin Chem Biol (2009) 13:272–83. doi:10.1016/j.cbpa.2009.03.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. McConnell JL, Wadzinski BE. Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol (2009) 75:1249–61. doi:10.1124/mol.108.053140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. De Munter S, Köhn M, Bollen M. Challenges and opportunities in the development of protein phosphatase-directed therapeutics. ACS Chem Biol (2013) 8:36–45. doi:10.1021/cb300597g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Zhang M, Yogesha SD, Mayfield JE, Gill GN, Zhang Y. Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J (2013) 280:4739–60. doi:10.1111/febs.12481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr Opin Genet Dev (2005) 15:34–41. doi:10.1016/j.gde.2004.12.004
15. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med (2008) 14:152–60. doi:10.1016/j.molmed.2008.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta (2009) 1795:1–15. doi:10.1016/j.bbcan.2008.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol (2013) 14:e229–38. doi:10.1016/S1470-2045(12)70558-2
18. Kalev P, Sablina AA. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med Chem (2011) 11:38–46. doi:10.2174/187152011794941172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J (2001) 353:417–39. doi:10.1042/0264-6021:3530417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lambrecht C, Haesen D, Sents W, Ivanova E, Janssens V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol (2013) 1053:283–305. doi:10.1007/978-1-62703-562-0_17
21. Slupe AM, Merrill RA, Strack S. Determinants for substrate specificity of protein phosphatase 2A. Enzyme Res (2011) 2011:398751. doi:10.4061/2011/398751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Götz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc Natl Acad Sci U S A (1998) 95:12370–5. doi:10.1073/pnas.95.21.12370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Gu P, Qi X, Zhou Y, Wang Y, Gao X. Generation of Ppp2Ca and Ppp2Cb conditional null alleles in mouse. Genesis (2012) 50:429–36. doi:10.1002/dvg.20815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Zhou J, Pham HT, Ruediger R, Walter G. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem J (2003) 369:387–98. doi:10.1042/BJ20021244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Sablina AA, Hahn WC. The role of PP2A A subunits in tumor suppression. Cell Adh Migr (2007) 1:140–1. doi:10.4161/cam.1.3.4986
26. Kremmer E, Ohst K, Kiefer J, Brewis N, Walter G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol Cell Biol (1997) 17:1692–701.
27. Janssens V, Rebollo A. The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr Mol Med (2012) 12:268–87. doi:10.2174/156652412799218930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kolupaeva V, Janssens V. PP1 and PP2A phosphatases: cooperating partners in modulating retinoblastoma protein activation. FEBS J (2013) 280:627–43. doi:10.1111/j.1742-4658.2012.08511.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hunt T. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv Biol Regul (2013) 53:173–8. doi:10.1016/j.jbior.2013.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sontag JM, Sontag E. Regulation of cell adhesion by PP2A and SV40 small tumor antigen: an important link to cell transformation. Cell Mol Life Sci (2006) 63:2979–91. doi:10.1007/s00018-006-6300-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Reid MA, Kong M. Adaptation to metabolic stress: insights into a paradoxical Q. Cell Cycle (2013) 12:1807–8. doi:10.4161/cc.25113
32. Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol (2012) 84:591–7. doi:10.1016/j.bcp.2012.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Haesen D, Sents W, Ivanova E, Lambrecht C, Janssens V. Cellular inhibitors of protein phosphatase PP2A in cancer. Biomed Res (2012) 23:197–211.
34. Batut J, Schmierer B, Cao J, Raftery LA, Hill CS, Howell M. Two highly related regulatory subunits of PP2A exert opposite effects on TGF-beta/Activin/Nodal signalling. Development (2008) 135:2927–37. doi:10.1242/dev.020842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Cegielska A, Shaffer S, Derua R, Goris J, Virshup DM. Different oligomeric forms of protein phosphatase 2A activate and inhibit simian virus 40 DNA replication. Mol Cell Biol (1994) 14:4616–23.
36. Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci U S A (2007) 104:2979–84. doi:10.1073/pnas.0611532104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Ahn JH, Sung JY, McAvoy T, Nishi A, Janssens V, Goris J, et al. The B”/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc Natl Acad Sci U S A (2007) 104:9876–81. doi:10.1073/pnas.0703589104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Foley EA, Maldonado M, Kapoor TM. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol (2011) 13:1265–71. doi:10.1038/ncb2327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Louis JV, Martens E, Borghgraef P, Lambrecht C, Sents W, Longin S, et al. Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A (2011) 108:6957–62. doi:10.1073/pnas.1018777108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Varadkar P, Despres D, Kraman M, Lozier J, Phadke A, Nagaraju K, et al. The protein phosphatase 2A B56γ regulatory subunit is required for heart development. Dev Dyn (2014) 243:778–90. doi:10.1002/dvdy.24111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Ruediger R, Ruiz J, Walter G. Human cancer-associated mutations in the Aα subunit of protein phosphatase 2A increase lung cancer incidence in Aα knock-in and knockout mice. Mol Cell Biol (2011) 31:3832–44. doi:10.1128/MCB.05744-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Strack S, Zaucha JA, Ebner FF, Colbran RJ, Wadzinski BE. Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J Comp Neurol (1998) 392:515–27. doi:10.1002/(SICI)1096-9861(19980323)392:4<515::AID-CNE8>3.3.CO;2-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Zwaenepoel K, Louis JV, Goris J, Janssens V. Diversity in genomic organisation, developmental regulation and distribution of the murine PR72/B” subunits of protein phosphatase 2A. BMC Genomics (2008) 9:393. doi:10.1186/1471-2164-9-393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, et al. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res (2011) 21:95–105. doi:10.1101/gr.109173.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Kong M, Ditsworth D, Lindsten T, Thompson CB. Alpha4 is an essential regulator of PP2A phosphatase activity. Mol Cell (2009) 36:51–60. doi:10.1016/j.molcel.2009.09.025
46. Jiang L, Stanevich V, Satyshur KA, Kong M, Watkins GR, Wadzinski BE, et al. Structural basis of protein phosphatase 2A stable latency. Nat Commun (2013) 4:1699. doi:10.1038/ncomms2663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J (2013) 280:644–61. doi:10.1111/j.1742-4658.2012.08579.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Wepf A, Glatter T, Schmidt A, Aebersold R, Gstaiger M. Quantitative interaction proteomics using mass spectrometry. Nat Methods (2009) 6:203–5. doi:10.1038/nmeth.1302
49. Longin S, Jordens J, Martens E, Stevens I, Janssens V, Rondelez E, et al. An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem J (2004) 380:111–9. doi:10.1042/BJ20031643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Chen LP, Lai YD, Li DC, Zhu XN, Yang P, Li WX, et al. α4 is highly expressed in carcinogen-transformed human cells and primary human cancers. Oncogene (2011) 30:2943–53. doi:10.1038/onc.2011.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sakashita S, Li D, Nashima N, Minami Y, Furuya S, Morishita Y, et al. Overexpression of immunoglobulin (CD79a) binding protein1 (IGBP-1) in small lung adenocarcinomas and its clinicopathological significance. Pathol Int (2011) 61:130–7. doi:10.1111/j.1440-1827.2011.02644.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Puustinen P, Junttila MR, Vanhatupa S, Sablina AA, Hector ME, Teittinen K, et al. PME-1 protects extracellular signal-regulated kinase pathway activity from protein phosphatase 2A-mediated inactivation in human malignant glioma. Cancer Res (2009) 69:2870–7. doi:10.1158/0008-5472.CAN-08-2760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wandzioch E, Pusey M, Werda A, Bail S, Bhaskar A, Nestor M, et al. PME-1 modulates protein phosphatase 2A activity to promote the malignant phenotype of endometrial cancer cells. Cancer Res (2014) 74:4295–305. doi:10.1158/0008-5472.CAN-13-3130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Fellner T, Lackner DH, Hombauer H, Piribauer P, Mudrak I, Zaragoza K, et al. A novel and essential mechanism determining specificity and activity of protein phosphatase 2A (PP2A) in vivo. Genes Dev (2003) 17:2138–50. doi:10.1101/gad.259903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Hombauer H, Weismann D, Mudrak I, Stanzel C, Fellner T, Lackner DH, et al. Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol (2007) 5:e155. doi:10.1371/journal.pbio.0050155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Leulliot N, Vicentini G, Jordens J, Quevillon-Cheruel S, Schiltz M, Barford D, et al. Crystal structure of the PP2A phosphatase activator: implications for its PP2A-specific PPIase activity. Mol Cell (2006) 23:413–24. doi:10.1016/j.molcel.2006.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Guo F, Stanevich V, Wlodarchak N, Sengupta R, Jiang L, Satyshur KA, et al. Structural basis of PP2A activation by PTPA, an ATP-dependent activation chaperone. Cell Res (2014) 24:190–203. doi:10.1038/cr.2013.138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. De Baere I, Derua R, Janssens V, Van Hoof C, Waelkens E, Merlevede W, et al. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry (1999) 38:16539–47. doi:10.1021/bi991646a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, et al. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem (1999) 274:14382–91. doi:10.1074/jbc.274.20.14382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, et al. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol Cell (2011) 41:331–42. doi:10.1016/j.molcel.2010.12.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Stanevich V, Zheng A, Guo F, Jiang L, Wlodarchak N, Xing Y. Mechanisms of the scaffold subunit in facilitating protein phosphatase 2A methylation. PLoS One (2014) 9:e86955. doi:10.1371/journal.pone.0086955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci (2008) 33:113–21. doi:10.1016/j.tibs.2007.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kong M, Fox CJ, Mu J, Solt L, Xu A, Cinalli RM, et al. The PP2A-associated protein alpha4 is an essential inhibitor of apoptosis. Science (2004) 306:695–8. doi:10.1126/science.1100537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ortega-Gutiérrez S, Leung D, Ficarro S, Peters EC, Cravatt BF. Targeted disruption of the PME-1 gene causes loss of demethylated PP2A and perinatal lethality in mice. PLoS One (2008) 3:e2486. doi:10.1371/journal.pone.0002486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Lee JA, Pallas DC. Leucine carboxyl methyltransferase-1 is necessary for normal progression through mitosis in mammalian cells. J Biol Chem (2007) 282:30974–84. doi:10.1074/jbc.M704861200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry (1995) 34:1988–96. doi:10.1021/bi00006a020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Li M, Makkinje A, Damuni Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry (1996) 35:6998–7002. doi:10.1021/bi960581y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem (1996) 271:11059–62. doi:10.1074/jbc.271.19.11059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Matilla A, Radrizzani M. The Anp32 family of proteins containing leucine-rich repeats. Cerebellum (2005) 4:7–18. doi:10.1080/14734220410019020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Reilly PT, Yu Y, Hamiche A, Wang L. Cracking the ANP32 whips: important functions, unequal requirement, and hints at disease implications. Bioessays (2014) 36:1062–71. doi:10.1002/bies.201400058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Saito S, Miyaji-Yamaguchi M, Shimoyama T, Nagata K. Functional domains of template-activating factor-I as a protein phosphatase 2A inhibitor. Biochem Biophys Res Commun (1999) 259:471–5. doi:10.1006/bbrc.1999.0790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Yu LG, Packman LC, Weldon M, Hamlett J, Rhodes JM. Protein phosphatase 2A, a negative regulator of the ERK signaling pathway, is activated by tyrosine phosphorylation of putative HLA class II-associated protein I (PHAPI)/pp32 in response to the antiproliferative lectin, jacalin. J Biol Chem (2004) 279:41377–83. doi:10.1074/jbc.M400017200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Adachi Y, Pavlakis GN, Copeland TD. Identification of in vivo phosphorylation sites of SET, a nuclear phosphoprotein encoded by the translocation breakpoint in acute undifferentiated leukemia. FEBS Lett (1994) 340:231–5. doi:10.1016/0014-5793(94)80144-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Irie A, Harada K, Araki N, Nishimura Y. Phosphorylation of SET protein at Ser171 by protein kinase D2 diminishes its inhibitory effect on protein phosphatase 2A. PLoS One (2012) 7:e51242. doi:10.1371/journal.pone.0051242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Naga Prasad SV. Inhibition of protein phosphatase 2A activity by PI3Kγ regulates β-adrenergic receptor function. Mol Cell (2011) 41:636–48. doi:10.1016/j.molcel.2011.02.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. ten Klooster JP, Leeuwen Iv, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J (2007) 26:336–45. doi:10.1038/sj.emboj.7601518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Yu G, Yan T, Feng Y, Liu X, Xia Y, Luo H, et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol Aging (2013) 34:1748–58. doi:10.1016/j.neurobiolaging.2012.12.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Beresford PJ, Zhang D, Oh DY, Fan Z, Greer EL, Russo ML, et al. Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem (2001) 276:43285–93. doi:10.1074/jbc.M108137200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Liu Z, Jang SW, Liu X, Cheng D, Peng J, Yepes M, et al. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell (2008) 29:665–78. doi:10.1016/j.molcel.2008.02.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett (2011) 585:2653–9. doi:10.1016/j.febslet.2011.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Cristóbal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood (2010) 115:615–25. doi:10.1182/blood-2009-06-227363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell (2005) 8:355–68. doi:10.1016/j.ccr.2005.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Leopoldino AM, Squarize CH, Garcia CB, Almeida LO, Pestana CR, Polizello AC, et al. Accumulation of the SET protein in HEK293T cells and mild oxidative stress: cell survival or death signaling. Mol Cell Biochem (2012) 363:65–74. doi:10.1007/s11010-011-1158-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene (2011) 30:2504–13. doi:10.1038/onc.2010.622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Liu Q, Zhao X, Frissora F, Ma Y, Santhanam R, Jarjoura D, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood (2008) 111:275–84. doi:10.1182/blood-2006-10-053884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Christensen DJ, Chen Y, Oddo J, Matta KM, Neil J, Davis ED, et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood (2011) 118:4150–8. doi:10.1182/blood-2011-04-351072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J (2009) 23:751–63. doi:10.1096/fj.08-120550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Al-Murrani SW, Woodgett JR, Damuni Z. Expression of I2PP2A, an inhibitor of protein phosphatase 2A, induces c-Jun and AP-1 activity. Biochem J (1999) 341:293–8. doi:10.1042/0264-6021:3410293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell (2007) 130:51–62. doi:10.1016/j.cell.2007.04.044
90. Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res (2013) 73:6548–53. doi:10.1158/0008-5472.CAN-13-1994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Ventelä S, Côme C, Mäkelä JA, Hobbs RM, Mannermaa L, Kallajoki M, et al. CIP2A promotes proliferation of spermatogonial progenitor cells and spermatogenesis in mice. PLoS One (2012) 7:e33209. doi:10.1371/journal.pone.0033209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Khanna A, Okkeri J, Bilgen T, Tiirikka T, Vihinen M, Visakorpi T, et al. ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS One (2011) 6:e17979. doi:10.1371/journal.pone.0017979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Khanna A, Böckelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst (2009) 101:793–805. doi:10.1093/jnci/djp103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemelä M, et al. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov (2013) 3:182–97. doi:10.1158/2159-8290.CD-12-0292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Chen KF, Liu CY, Lin YC, Yu HC, Liu TH, Hou DR, et al. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene (2010) 29:6257–66. doi:10.1038/onc.2010.357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Guenebeaud C, Goldschneider D, Castets M, Guix C, Chazot G, Delloye-Bourgeois C, et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell (2010) 40:863–76. doi:10.1016/j.molcel.2010.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Kim JS, Kim EJ, Oh JS, Park IC, Hwang SG. CIP2A modulates cell-cycle progression in human cancer cells by regulating the stability and activity of Plk1. Cancer Res (2013) 73:6667–78. doi:10.1158/0008-5472.CAN-13-0888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Jeong AL, Lee S, Park JS, Han S, Jang CY, Lim JS, et al. Cancerous inhibitor of protein phosphatase 2A (CIP2A) protein is involved in centrosome separation through the regulation of NIMA (never in mitosis gene A)-related kinase 2 (NEK2) protein activity. J Biol Chem (2014) 289:28–40. doi:10.1074/jbc.M113.507954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jäättelä M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol (2014) 204:713–27. doi:10.1083/jcb.201304012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Puustinen P, Jäättelä M. KIAA1524/CIP2A promotes cancer growth by coordinating the activities of MTORC1 and MYC. Autophagy (2014) 10:1352–4. doi:10.4161/auto.29076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. McConnell JL, Gomez RJ, McCorvey LR, Law BK, Wadzinski BE. Identification of a PP2A-interacting protein that functions as a negative regulator of phosphatase activity in the ATM/ATR signaling pathway. Oncogene (2007) 26:6021–30. doi:10.1038/sj.onc.1210406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Smetana JH, Zanchin NI. Interaction analysis of the heterotrimer formed by the phosphatase 2A catalytic subunit, alpha4 and the mammalian ortholog of yeast Tip41 (TIPRL). FEBS J (2007) 274:5891–904. doi:10.1111/j.1742-4658.2007.06112.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, et al. A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol Cell Proteomics (2005) 4:1725–40. doi:10.1074/mcp.M500231-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Nakashima A, Tanimura-Ito K, Oshiro N, Eguchi S, Miyamoto T, Momonami A, et al. A positive role of mammalian Tip41-like protein, TIPRL, in the amino-acid dependent mTORC1-signaling pathway through interaction with PP2A. FEBS Lett (2013) 587:2924–9. doi:10.1016/j.febslet.2013.07.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Song IS, Jun SY, Na HJ, Kim HT, Jung SY, Ha GH, et al. Inhibition of MKK7-JNK by the TOR signaling pathway regulator-like protein contributes to resistance of HCC cells to TRAIL-induced apoptosis. Gastroenterology (2012) 143:1341–51. doi:10.1053/j.gastro.2012.07.103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Mochida S, Maslen SL, Skehel M, Hunt T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science (2010) 330:1670–3. doi:10.1126/science.1195689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Gharbi-Ayachi A, Labbé JC, Burgess A, Vigneron S, Strub JM, Brioudes E, et al. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science (2010) 330:1673–7. doi:10.1126/science.1197048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Manchado E, Guillamot M, de Cárcer G, Eguren M, Trickey M, García-Higuera I, et al. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55α,δ phosphatase. Cancer Cell (2010) 18:641–54. doi:10.1016/j.ccr.2010.10.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Mochida S. Regulation of α-endosulfine, an inhibitor of protein phosphatase 2A, by multisite phosphorylation. FEBS J (2014) 281:1159–69. doi:10.1111/febs.12685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Okumura E, Morita A, Wakai M, Mochida S, Hara M, Kishimoto T. Cyclin B-Cdk1 inhibits protein phosphatase PP2A-B55 via a Greatwall kinase-independent mechanism. J Cell Biol (2014) 204:881–9. doi:10.1083/jcb.201307160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Dupré A, Daldello EM, Nairn AC, Jessus C, Haccard O. Phosphorylation of ARPP19 by protein kinase A prevents meiosis resumption in Xenopus oocytes. Nat Commun (2014) 5:3318. doi:10.1038/ncomms4318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Porter IM, Schleicher K, Porter M, Swedlow JR. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat Commun (2013) 4:2677. doi:10.1038/ncomms3677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Roberts KG, Smith AM, McDougall F, Carpenter H, Horan M, Neviani P, et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res (2010) 70:5438–47. doi:10.1158/0008-5472.CAN-09-2544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Esplin ED, Ramos P, Martinez B, Tomlinson GE, Mumby MC, Evans GA. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer (2006) 45:182–90. doi:10.1002/gcc.20284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Zhu Y, Loukola A, Monni O, Kuokkanen K, Franssila K, Elonen E, et al. PPP2R1B gene in chronic lymphocytic leukemias and mantle cell lymphomas. Leuk Lymphoma (2001) 41:177–83. doi:10.3109/10428190109057968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Kalla C, Scheuermann MO, Kube I, Schlotter M, Mertens D, Döhner H, et al. Analysis of 11q22-q23 deletion target genes in B-cell chronic lymphocytic leukaemia: evidence for a pathogenic role of NPAT, CUL5, and PPP2R1B. Eur J Cancer (2007) 43:1328–35. doi:10.1016/j.ejca.2007.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Cristóbal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia (2011) 25:606–14. doi:10.1038/leu.2010.294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci U S A (2009) 106:12974–9. doi:10.1073/pnas.0811267106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Ruvolo PP, Qui YH, Coombes KR, Zhang N, Ruvolo VR, Borthakur G, et al. Low expression of PP2A regulatory subunit B55α is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia (2011) 25:1711–7. doi:10.1038/leu.2011.146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Ruvolo PP, Ruvolo VR, Jacamo R, Burks JK, Zeng Z, Duvvuri SR, et al. The protein phosphatase 2A regulatory subunit B55α is a modulator of signaling and microRNA expression in acute myeloid leukemia cells. Biochim Biophys Acta (2014) 1843:1969–77. doi:10.1016/j.bbamcr.2014.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Cristóbal I, Cirauqui C, Castello-Cros R, Garcia-Orti L, Calasanz MJ, Odero MD. Downregulation of PPP2R5E is a common event in acute myeloid leukemia that affects the oncogenic potential of leukemic cells. Haematologica (2013) 98:e103–4. doi:10.3324/haematol.2013.084731
122. Fält S, Merup M, Gahrton G, Lambert B, Wennborg A. Identification of progression markers in B-CLL by gene expression profiling. Exp Hematol (2005) 33:883–93. doi:10.1016/j.exphem.2005.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol (2010) 12:372–9. doi:10.1038/ncb2037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Dunwell TL, Hesson LB, Pavlova T, Zabarovska V, Kashuba V, Catchpoole D, et al. Epigenetic analysis of childhood acute lymphoblastic leukemia. Epigenetics (2009) 4:185–93. doi:10.4161/epi.4.3.8752
125. Mosca L, Musto P, Todoerti K, Barbieri M, Agnelli L, Fabris S, et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am J Hematol (2013) 88:16–23. doi:10.1002/ajh.23339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Kamada Y, Sakata-Yanagimoto M, Sanada M, Sato-Otsubo A, Enami T, Suzukawa K, et al. Identification of unbalanced genome copy number abnormalities in patients with multiple myeloma by single-nucleotide polymorphism genotyping microarray analysis. Int J Hematol (2012) 96:492–500. doi:10.1007/s12185-012-1171-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Coenen EA, Zwaan CM, Meyer C, Marschalek R, Pieters R, van der Veken LT, et al. KIAA1524: a novel MLL translocation partner in acute myeloid leukemia. Leuk Res (2011) 35:133–5. doi:10.1016/j.leukres.2010.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Wang J, Li W, Li L, Yu X, Jia J, Chen C. CIP2A is over-expressed in acute myeloid leukaemia and associated with HL60 cells proliferation and differentiation. Int J Lab Hematol (2011) 33:290–8. doi:10.1111/j.1751-553X.2010.01288.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood (2011) 117:6660–8. doi:10.1182/blood-2010-08-304477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Wang J, Huang T, Sun J, Yu Y, Liu Z, Li W, et al. CIP2A is overexpressed and involved in the pathogenesis of chronic myelocytic leukemia by interacting with breakpoint cluster region-Abelson leukemia virus. Med Oncol (2014) 31:112. doi:10.1007/s12032-014-0112-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Liu CY, Shiau CW, Kuo HY, Huang HP, Chen MH, Tzeng CH, et al. Cancerous inhibitor of protein phosphatase 2A determines bortezomib-induced apoptosis in leukemia cells. Haematologica (2013) 98:729–38. doi:10.3324/haematol.2011.050187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Lilja L, Haapasaari KM, Bloigu R, Salonen T, Ristimäki A, Turpeenniemi-Hujanen T, et al. Increased expression of CIP2A in aggressive subtypes of B-cell lymphoma. Histopathology (2013) 63:438–9. doi:10.1111/his.12162
133. Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest (2007) 117:2408–21. doi:10.1172/JCI31095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Cristóbal I, Garcia-Orti L, Cirauqui C, Cortes-Lavaud X, García-Sánchez MA, Calasanz MJ, et al. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica (2012) 97:543–50. doi:10.3324/haematol.2011.050542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene (2009) 28:1669–81. doi:10.1038/onc.2009.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet (2013) 45:18–24. doi:10.1038/ng.2495
137. von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3’ half to different genes: characterization of the set gene. Mol Cell Biol (1992) 12:3346–55.
138. Rosati R, La Starza R, Barba G, Gorello P, Pierini V, Matteucci C, et al. Cryptic chromosome 9q34 deletion generates TAF-Ialpha/CAN and TAF-Ibeta/CAN fusion transcripts in acute myeloid leukemia. Haematologica (2007) 92:232–5. doi:10.3324/haematol.10538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Quentmeier H, Schneider B, Röhrs S, Romani J, Zaborski M, Macleod RA, et al. SET-NUP214 fusion in acute myeloid leukemia- and T-cell acute lymphoblastic leukemia-derived cell lines. J Hematol Oncol (2009) 2:3. doi:10.1186/1756-8722-2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Van Vlierberghe P, van Grotel M, Tchinda J, Lee C, Beverloo HB, van der Spek PJ, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood (2008) 111:4668–80. doi:10.1182/blood-2007-09-111872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Wallington-Beddoe CT, Don AS, Hewson J, Qiao Q, Papa RA, Lock RB, et al. Disparate in vivo efficacy of FTY720 in xenograft models of Philadelphia positive and negative B-lineage acute lymphoblastic leukemia. PLoS One (2012) 7:e36429. doi:10.1371/journal.pone.0036429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Yang Y, Huang Q, Lu Y, Li X, Huang S. Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT tyrosine kinase domain mutation. J Cell Biochem (2012) 113:1314–22. doi:10.1002/jcb.24003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med (2013) 5:105–21. doi:10.1002/emmm.201201283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Oaks JJ, Santhanam R, Walker CJ, Roof S, Harb JG, Ferenchak G, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood (2013) 122:1923–34. doi:10.1182/blood-2013-03-492181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Pippa R, Dominguez A, Christensen DJ, Moreno-Miralles I, Blanco-Prieto MJ, Vitek MP, et al. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia (2014) 28:1915–8. doi:10.1038/leu.2014.141
146. Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, et al. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J Immunol (2011) 186:2535–42. doi:10.4049/jimmunol.1002847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res (2014) 20:2092–103. doi:10.1158/1078-0432.CCR-13-2575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Fujiwara N, Kawasaki H, Yabe R, Christensen DJ, Vitek MP, Mizuno T, et al. A potential therapeutic application of SET/I2PP2A inhibitor OP449 for canine T-cell lymphoma. J Vet Med Sci (2013) 75:349–54. doi:10.1292/jvms.12-0366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest (2014) 124:644–55. doi:10.1172/JCI65093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: PP2A, subunit, inhibitor, tumor suppressor reactivation therapy, PP2A-activating drugs
Citation: Haesen D, Sents W, Lemaire K, Hoorne Y and Janssens V (2014) The basic biology of PP2A in hematologic cells and malignancies. Front. Oncol. 4:347. doi: 10.3389/fonc.2014.00347
Received: 30 October 2014; Paper pending published: 07 November 2014;
Accepted: 20 November 2014; Published online: 11 December 2014.
Edited by:
Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Renae Barr, University of Western Australia, AustraliaPeter Ruvolo, The University of Texas MD Anderson Cancer Center, USA
Copyright: © 2014 Haesen, Sents, Lemaire, Hoorne and Janssens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veerle Janssens, Gasthuisberg O&N1, Herestraat 49, PO-Box 901, Leuven B-3000, Belgium e-mail: veerle.janssens@med.kuleuven.be
†Dorien Haesen and Ward Sents have contributed equally to this work.