 Geir Åge Løset1,2,3*
Geir Åge Løset1,2,3* Gøril Berntzen1
Gøril Berntzen1 Terje Frigstad1Sylvie Pollmann1
Terje Frigstad1Sylvie Pollmann1 Kristin S. Gunnarsen2
Kristin S. Gunnarsen2 Inger Sandlie2,3
Inger Sandlie2,3
- 1Nextera AS, Oslo, Norway
- 2Centre for Immune Regulation, Oslo University Hospital, University of Oslo, Oslo, Norway
- 3Department of Biosciences, University of Oslo, Oslo, Norway
Cancer immunotherapy has finally come of age, demonstrated by recent progress in strategies that engage the endogenous adaptive immune response in tumor killing. Occasionally, significant and durable tumor regression has been achieved. A giant leap forward was the demonstration that the pre-existing polyclonal T cell repertoire could be re-directed by use of cloned T cell receptors (TCRs), to obtain a defined tumor-specific pool of T cells. However, the procedure must be performed with caution to avoid deleterious cross-reactivity. Here, the use of engineered soluble TCRs may represent a safer, yet powerful, alternative. There is also a need for deeper understanding of the processes that underlie antigen presentation in disease and homeostasis, how tumor-specific peptides are generated, and how epitope spreading evolves during tumor development. Due to its plasticity, the pivotal interaction where a TCR engages a peptide/MHC (pMHC) also requires closer attention. For this purpose, phage display as a tool to evolve cloned TCRs represents an attractive avenue to generate suitable reagents allowing the study of defined pMHC presentation, TCR engagement, as well as for the discovery of novel therapeutic leads. Here, we highlight important aspects of the current status in this field.
Introduction
T cells initiate and regulate adaptive immune responses to infections, are major components of allergic and autoimmune responses as well as transplant rejection, and play a pivotal role in cancer immune surveillance (1). The cancer-prone phenotypes of mice that lack components of the adaptive immune system strongly points to lymphocytes as critical factors in the anti-tumor activity (2). That the T cells represent the critical lymphocyte population is further underscored by a correlation between the presence of tumor infiltrating lymphocytes (TILs) and ability to control tumor growth. The CD45RO+ memory sub-group of the CD3 T cell compartment appears responsible of this activity (3, 4), and the CD8+ and CD4+ T cells probably act in concert (5). Furthermore, the observation that selective CD4+ TH cell silencing may abrogate the anti-tumor response points to the CD4+ TH cells as crucial (6, 7). It is also clear that adoptive cell therapy (ACT) through the use of CD8+ cytotoxic T lymphocyte (CTL) or CD4+ TH cells may both result in durable anti-tumor activity (8–10). This is not merely a consequence of specific T cell target recognition, nor the affinity by which the T cell receptor (TCR) recognizes the target (11–13). Thus, to further delineate the mechanisms that lead to successful anti-tumor responses and how these can be exploited, it becomes imperative to further characterize the TCR–peptides bound to MHC molecules (pMHC) interaction, both at the cellular and the molecular level. The latter has posed a challenge to the field, since recombinant soluble TCRs have proven difficult to manufacture and work with. Consequently, our ability to study this pivotal interaction still depends on technology development (14). As such, protein engineering using combinatorial technologies is a powerful tool (15). Here, we focus on examples derived from the most prevalent combinatorial platform technology, namely phage display (16).
T Cell Specificity at the Molecular Level
T cell function relies on productive binding between TCRs and antigens, which are proteolytically derived pMHC displayed on the surface of a variety of antigen presenting cells (APCs). Most TCRs bind pMHC ligands in a semi-conserved diagonal orientation with the somatically derived CDR3 loops located centrally atop the bound peptide, and the germ-line encoded variable CDR1 and CDR2 loops positioned over the MHC α helices (17). Upon activation, T cells may proliferate, differentiate, release cytokines, kill target cells, or carry out other effector functions. Thus, the ability of T cells to orchestrate the adaptive anti-tumor response depends on the TCR–pMHC interaction and downstream signaling events (18, 19). Productive interactions between TCRs and pMHCs are among the weakest known to initiate a biological response (20–22). Thus, a T cell needs to discriminate between foreign and self-peptides bound to MHC molecules even though the differences in affinity and binding kinetics may be minute (21, 22). Nonetheless, the earliest events in TCR signaling are characterized by high sensitivity and selectivity toward agonist pMHC (19). This is remarkable considering the apparent promiscuity of TCR binding, which in extreme cases have been suggested to be in the range of 106 different peptides, yet still in a HLA restricted context (23). Such scaffold-dependent ligand binding promiscuity may partly be attributed to germ-line encoded HLA interaction signatures that ensure preservation of HLA restriction (24–27). It could also be an important feature explaining how a limited number of TCR germ line segments in combination with somatically generated CDR3 loops serve as versatile building blocks that generate a supply of TCRs able to promptly respond to a universe of pathogens (28–30). Clearly, multi-epitope specificity can also be a characteristic of tumor-specific TCRs, as shown in the study of Chinnasamy et al. focusing on HLA-A2/MAGE-A3 targeting (31). However, during ACT, such lack of mono-specificity may translate into fatal toxicity, underscoring the need for improved procedures for pre-clinical testing (32). Also, there is a need for a very precise delineation of how a TCR actually sees its cognate pMHC target, since minute structural changes may translate into very different cellular responses (33). Here, elucidating the underlying thermodynamic parameters governing the interaction may give clues to the rules that dictate TCR specificity (34, 35). Such biophysical insight may be further complemented by precise delineation of docking modes and binding studies that mimic the cellular topology (36, 37). In either case, one will need access to sufficient amounts of pure and stable soluble TCR and pMHC proteins.
Reductionist Approach to Understanding the pMHC–TCR Interaction – The TCR Expression Problem
T cell receptors are membrane anchored proteins, and it is challenging to obtain sufficient quantities of recombinant soluble TCRs for molecular studies. A variety of approaches have therefore been adopted, including formation of single chain (sc) TCR, an analog to scFv antibody (Ab) fragments, and fusion of the extracellular TCR domains to other proteins; i.e., maltose binding protein, thioredoxin, human constant kappa domain, or leucine zippers (38–42). However, all of these strategies have had limited success due to low production yield and poor functionality. The most widely applied format as of today is the disulfide-bond linked TCRs (dsTCRs), which have a non-native disulfide bridge between the TCR constant domains (43). The method has significantly increased the stability and improved the folding characteristics of several human TCRs (44) when refolded from inclusion bodies, whereas direct soluble expression appears of limited utility (44, 45). An alternative approach is periplasmic expression with simultaneous over-expression of the chaperone FkpA, which has a huge impact on both the yield and functionality of the TCRs expressed (46). However, despite the optimized and improved methods, all are laborious and the expression levels vary extensively between individual clones. Thus, in many cases engineering of the TCR scaffold for higher stability, solubility and clone independent expression levels appears needed to obtain high quality protein.
TCR Stability Can be Engineered by Use of Phage Display
Evolution of recombinant proteins by random mutagenesis and subsequent in vitro selection has been successfully applied to a wide range of protein classes (47), and in particular antibodies (48). One such strategy has utilized selection of mutated heavy chain variable domains in combination with thermal challenge to obtain aggregation-resistant domains (49). Recently, guided by the study of Jespers et al., molecular evolution of a TCR for increased stability and expression was carried out by use of phage display (50). Libraries of randomly mutated scTCRs were produced as fusion to protein III on the surface of M13 phage. High valence display allowed stress-induced aggregation after thermal challenge (Figure 1). Variants characterized by markedly increased soluble expression levels and reduced aggregation propensity were obtained after rapid heating and cooling, followed by capture of aggregation-resistant scTCRs (Figures 1A,D). Importantly, over-expression of the periplasmic chaperone FkpA resulted in even display levels among the TCR library members, which proved imperative for successful selection. Thus, the previously identified folding assistance to soluble and phage displayed scTCRs offered by FkpA now allows for extended engineering opportunities to TCRs in conjunction with high-throughput soluble screening (Figures 1A,B,D). The list of strategies used for engineering of increased protein biophysical stability employing destabilization challenges in combination with multivalent phage display selection has been further extended. Famm et al. reported selection of Ig domains resistant to e.g., acidic pH induced aggregation with increased thermodynamic stability (51, 52). Furthermore, Christ et al. have reported a method for generation of Ab sub repertoires, based on combinatorial assembly of CDRs from an aggregation-resistant repertoire (53). Repeated cycles of selection and thermal denaturation generated domains with remarkable aggregation-resistant properties. Similar strategies may well be employed to obtain soluble TCR scaffolds with even higher expression levels and increased stability than reported to date (50, 54).
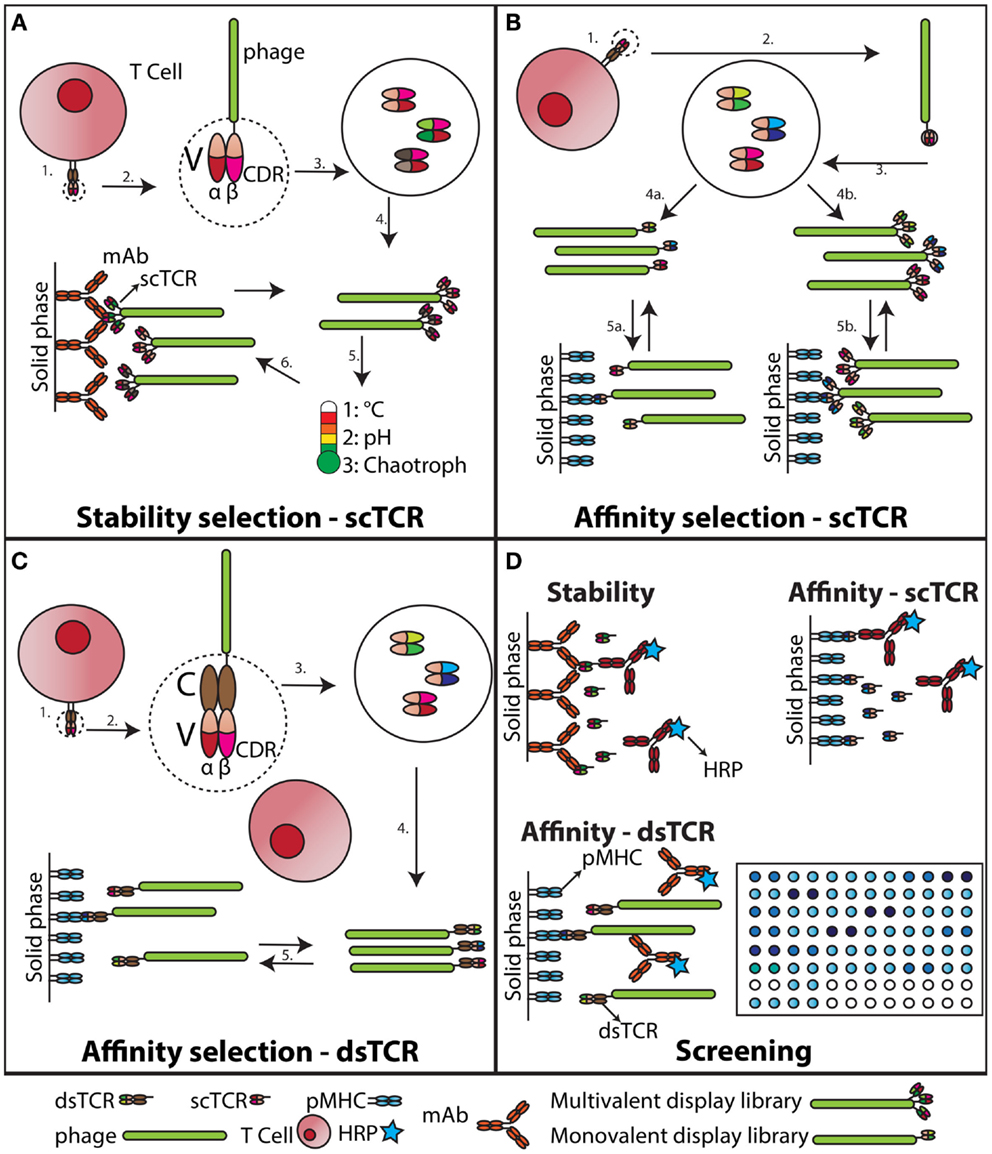
Figure 1. Stability engineering and affinity maturation of soluble TCRs. (A) Stability engineering of scTCR. The variable (V) regions of an individual TCR (1) are cloned and connected via a synthetic linker (55). This scTCR is expressed as fusion to the M13 pIII capsid protein (2). The scTCR is then diversified by in vitro mutagenesis (3). This collection of mutagenized scTCRs are expressed as a high valence display phage library (4), which is challenged with increased temperature, unfavorable acid/base, or chaotropic conditions (5). Stabilized scTCR resisting aggregation despite the challenge is retrieved by capture on a conformation-specific ligand, such as an Ab (6). (B) Affinity maturation of scTCR. The V regions of an individual TCR (1) are cloned and expressed as a scTCR fusion to either the M13 pIII (55), or pIX capsid protein (56) (2). Individual TCR α- and β-chain CDR loops of the scTCR are randomized to create diversity (3). This collection of mutagenized scTCRs is then expressed as a low (4a) or high (4b) valence display phage library, which is selected against pMHC (5a and b). (C) Affinity maturation of dsTCR. The V regions of an individual TCR (1) are cloned and expressed as fusions to prototypic constant (C) domains stabilized by an artificial disulfide bridge, hence reconstituting the complete TCR ectodomain architecture (43). This recombinant dsTCR is then expressed as fusion to the M13 pIII capsid protein (2). Individual TCR α- and β-chain CDR loops of the dsTCR are randomized to create diversity (3). Usually this process is confined to the in vivo pMHC specificity-determining CDR3 loops (57), but has also been successfully applied to the germ-line encoded CDR2 only (58, 59). This collection of mutagenized dsTCRs is then expressed as a low valence display phage library (57), which is selected against pMHC (5). (D) Screening of engineered dsTCR and scTCR. The stability engineered (A), or affinity matured (B) scTCR is reformatted to soluble, periplasmic expression (46), and individual mutated scTCRs screened for functionality against target immobilized on solid phase. The screening for desired binders following dsTCR selection is done on phage due to incompatibility with high-throughput soluble dsTCR screening (45).
TCR Affinity Can be Engineered by Use of Phage Display
To overcome the intrinsically low binding affinity of the TCR–pMHC interaction, two approaches have been utilized, namely multimerization and affinity maturation. Tetrameric forms of soluble TCRs have been produced by capturing biotinylated TCRs onto avidin, which have four binding sites for biotin (60, 61). The overall increased avidity greatly increases the half-life of the TCR–pMHC interaction. Such reagents are used in cellular binding assays, as they stably adhere to the cell surface. Crucial information may be collected that allows for deduction of biologically relevant information (61). However, for example direct assessment of peptide presentation at stoichiometric levels requires stronger binding between the TCR and pMHC than what is possible to reach with native TCRs (60). Therefore, affinity maturation of TCRs for increased binding has been performed. Again, phage display technology has been efficient (15, 57), and selection from mutant TCR display libraries can yield TCRs with dramatically increased affinities toward the cognate pMHCs without concomitant increase in cross-reactivity (Figures 1B–D). Crystallographic data show that this can be explained by a loss of flexibility in the otherwise entropically unfavorable TCR–pMHC interaction interphase, as well as an overall increase in shape complementarity (62). The degenerate pMHC interaction mode of TCRs could suggest that engineering must be restricted to the somatically derived CDR3 loops to preserve MHC restriction (26). However, this appears not to be the case as also the germ-line encoded CDR2 loop has been targeted by mutagenesis resulting in increased affinity (58, 63). Such engineered high-affinity TCRs have been used to study low level tumor associated pMHC presentation at physiological levels to obtain information that has previously been unattainable (64–66).
Lessons Learnt – Translation to the Tumor pMHC Complexes and Cancer Therapy
Conformational plasticity in the CDR loops upon pMHC binding appears to be a driving mechanism upon TCR–pMHC complex formation, whereas, rigid “lock and key” interaction modes also have been reported (67). This energetic diversity reflects the multiple binding strategies implemented by the TCR during pMHC engagement. However, in spite of the described diversity (68), step by step, we are unveiling the mechanism by which a TCR deciphers a pMHC complex.
The low level of molecular shape complementarity in the TCR–pMHC complex gives rich opportunities for in vitro affinity maturation (57, 58, 62). This feature is likely to be generic to most TCRs (26). In the case of the affinity maturation of a HLA-A2/MART-1 specific TCR, the increase in shape complementarity was focused primarily onto the MHC portion of the complex essentially without affecting the peptide interaction (69). Thus, loss of peptide specificity could potentially be expected. However, structural and thermodynamic investigations suggested that this was not the case. In stark contrast, the opposite pMHC interaction strategy was employed by a different TCR recently reported, which was evolved toward high affinity against the HLA-A2/Tax complex. Here, a peptide-focused mechanism was found to underlie the enhanced affinity (59). Thus, the authors suggest an alternative interaction mode to the generally accepted two-step TCR–pMHC binding model (19). Here, instead of first docking the CDR1 and CDR2 onto the MHC, followed by kinetic proofreading of the peptide by the CDR3s, the opposite order of interaction is suggested. This scan-clamp model fits well with how weak, but specific protein–protein interactions have been stabilized by affinity clamping in other trimeric complexes analogous to TCR–pMHC (70, 71). It also explains how exquisite peptide specificity can be preserved both in natural and engineered systems. In either case, the picture is not consistent, and the observation that complementary structural fluctuations of both the antigenic peptide and the CDR3s of the TCR prevail even after final complex formation, underscores the remarkable flexibility of the interaction (35). Thus, at present it is challenging to validate both naturally and artificially evolved TCRs e.g., for safe use in therapy. Despite rigorous classical pre-clinical validation, a human clinical pilot study resulted in fatal cardiac toxicity due to unforeseen cross-reactivity when an affinity matured TCR against HLA-A1/MAGE-A3 was employed in specificity redirected ACT (32).
Also, there are still many questions to be answered regarding the difference between MHC class I and class II restricted TCRs. In particular, it is important to understand the significance of co-receptors in creating a fully functional immunological synapse (72). For instance, it has been shown that CD8 plays a stabilizing role in the TCR–pMHC class I interaction (73), whereas, CD4 does not appear to play a role in the corresponding TCR–pMHC class II interaction (36). Notably, these two co-receptor interactions differ significantly in MHC binding strength, which may possibly elude to their differential importance (72). Thus, an affinity threshold has been observed for the CD8 T cell compartment that limits the benefits of very high intrinsic affinity between TCR–pMHC class I (11, 74–77). So far, this has not been observed for TCR–pMHC class II. Even though fewer examples have been reported with respect to TCRs reactive toward pMHC class II, it appears that different functional rules govern this interaction (74, 77, 78), and pilot trials have shown promising results in pMHC class II restricted ACT (10, 79).
Concluding Remarks
The ability to engineer stable and high-affinity TCRs offers a unique ability to harness the immune system with an improved ability to respond to a given pMHC. However, our current understanding is still incomplete with respect to how this can safely be translated into durable cancer immunotherapy (9). One would expect improved affinity to translate into improved killing ability, but the empirical data suggest otherwise. Rather, an affinity threshold limiting any additional benefit in cellular responses above a certain TCR–pMHC binding strength has been reported, as outlined above. Moreover, the affinity threshold appears to be largely confined to the CD8 T cell compartment, as nearly all high-affinity engineered CD4 T cells have responded with both improved peptide sensitivity and preserved specificity. This gives clues as to how one might differentially exploit TCRs derived from the two distinct T cell compartments. On one hand, engineered high-affinity MHC class I and II restricted TCRs may both serve as very potent cytotoxic drugs in a soluble format (80). On the other hand, the most potent avenue for redirected cell therapy might in some cases be limited to the MHC class II restricted compartment due to the CD8 T cell affinity threshold (10, 74).
A final question is whether or not one actually needs to apply ACT to achieve optimal clinical benefit. ACT is demanding as it relies on massive ex vivo autologous cell expansion, which will be difficult in major patient groups for example due to cellular senescence (81–83). Epitope spreading appears to be the signature of successful anti-tumor immune responses (10, 13, 84, 85). Now it appears that this can also be achieved by the use of soluble TCRs harnessed with the ability to recruit the endogenous adaptive effector apparatus (80, 86). Such soluble TCRs appear attractive compared to the cellular approaches in light of patient convenience and safety issues (32). The use of a soluble TCR obviates the need for ex vivo cell expansion and a single drug may be used by a genetically heterogeneous patient population sharing the target MHC allele only. Putative off target toxicity may also be better controlled, and quenched if needed, due to tunable dosing and limited drug half-life. A soluble drug would also be less prone to efficacy variation due to in vivo regulatory mechanisms than ACT. How well the soluble TCR approach is reduced to clinical practice is currently under investigation through a first in man phase I/II clinical trial in late-stage malignant melanoma targeting a HLA-A2/gp100 complex (http://www.clinicaltrials.gov/ and IMCgp100).
Undoubtedly, improved phage display technology will continue to be a driver in providing engineered TCRs, which will be powerful tools to monitor and elucidate specific pMHC complexes, as well as creating novel specificities suitable for safe use in the clinic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme (project number 179573). The authors apologize for omission of publications that could not be included due to limited space.
References
1. Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, Mcmahon R, et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity (2009) 30:348–57. doi: 10.1016/j.immuni.2009.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353:2654–66. doi:10.1056/NEJMoa051424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early- stage colorectal cancer. J Clin Oncol (2009) 27:5944–51. doi:10.1200/jco.2008.19.6147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+T cells maintain effector and memory tumor-specific CD8+T cells. Eur J Immunol (2014) 44:69–79. doi:10.1002/eji.201343718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Snook AE, Magee MS, Schulz S, Waldman SA. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur J Immunol (2014) 44:1956–66. doi:10.1002/eji.201444539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:126–9. doi:10.1126/science.1129003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12:269–81. doi:10.1038/nri3191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi:10.1126/science.1251102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol (2010) 184(9):4936–46. doi:10.4049/jimmunol.1000173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood (2012) 119:3420–30. doi:10.1182/blood-2011-09-377051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJC, Bolotin DA, Chen X, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med (2013) 19:1534–41. doi:10.1038/nm.3359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Su LF, Han A, Mcguire HM, Furman D, Newell EW, Davis MM. The promised land of human immunology. Cold Spring Harb Symp Quant Biol (2013) 78:203–13. doi:10.1101/sqb.2013.78.022905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Birnbaum ME, Dong S, Garcia KC. Diversity-oriented approaches for interrogating T-cell receptor repertoire, ligand recognition, and function. Immunol Rev (2012) 250:82–101. doi:10.1111/imr.12006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Løset GÅ, Sandlie I. Next generation phage display by use of pVII and pIX as display scaffolds. Methods (2012) 58:40–6. doi:10.1016/j.ymeth.2012.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and co-receptors. Annu Rev Immunol (2006) 24:419–66. doi:10.1146/annurev.immunol.23.021704.115658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity (1998) 8:403–11. doi:10.1016/S1074-7613(00)80546-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lever M, Maini PK, Van Der Merwe PA, Dushek O. Phenotypic models of T cell activation. Nat Rev Immunol (2014) 14:619–29. doi:10.1038/nri3728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol (2007) 178:5727–34. doi:10.4049/jimmunol.178.9.5727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Deng L, Langley RJ, Brown PH, Xu G, Teng L, Wang Q, et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol (2007) 8:398–408. doi:10.1038/ni1447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang K-M, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol (2012) 42:3174–9. doi:10.1002/eji.201242606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Wooldridge L, Ekeruche-Makinde J, Van Den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287:1168–77. doi:10.1074/jbc.M111.289488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, et al. Crossreactive T cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity (2008) 28(3):324–34. doi:10.1016/j.immuni.2008.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu Rev Immunol (2008) 26:171–203. doi:10.1146/annurev.immunol.26.021607.090421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol (2009) 10:143–7. doi:10.1038/ni.f.219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Scott-Browne JP, White J, Kappler JW, Gapin L, Marrack P. Germline-encoded amino acids in the alphabeta T-cell receptor control thymic selection. Nature (2009) 458:1043–6. doi:10.1038/nature07812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, et al. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol (2003) 4:241–7. doi:10.1038/ni891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol (2012) 12:669–77. doi:10.1038/nri3279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Stadinski BD, Trenh P, Duke B, Huseby PG, Li G, Stern LJ, et al. Effect of CDR3 sequences and distal V gene residues in regulating TCR-MHC contacts and ligand specificity. J Immunol (2014) 192:6071–82. doi:10.4049/jimmunol.1303209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol (2010) 186(2):685–96. doi:10.4049/jimmunol.1001775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5:197ra103. doi:10.1126/scitranslmed.3006034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Kersh GJ, Miley MJ, Nelson CA, Grakoui A, Horvath S, Donermeyer DL, et al. Structural and functional consequences of altering a peptide MHC anchor residue. J Immunol (2001) 166:3345–54. doi:10.4049/jimmunol.166.5.3345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Krogsgaard M, Prado N, Adams EJ, He X-L, Chow D-C, Wilson DB, et al. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell (2003) 12:1367–78. doi:10.1016/S1097-2765(03)00474-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Hawse WF, De S, Greenwood AI, Nicholson LK, Zajicek J, Kovrigin EL, et al. TCR scanning of peptide/MHC through complementary matching of receptor and ligand molecular flexibility. J Immunol (2014) 192:2885–91. doi:10.4049/jimmunol.1302953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature (2010) 463:963–7. doi:10.1038/nature08746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity (2011) 35:681–93. doi:10.1016/j.immuni.2011.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Chang H-C, Bao Z-Z, Yao Y, Tse AGD, Goyarts EC, Madsen M, et al. A general method for facilitating heterodimeric pairing between two proteins: application to expression of [alpha] and [beta] T-cell receptor extracellular segments. Proc Natl Acad Sci U S A (1994) 91:11408–12. doi:10.1073/pnas.91.24.11408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Chung S, Wucherpfennig KW, Friedman SM, Hafler DA, Strominger JL. Functional three-domain single-chain T-cell receptors. Proc Natl Acad Sci U S A (1994) 91:12654–8. doi:10.1073/pnas.91.26.12654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Schodin BA, Schlueter CJ, Kranz DM. Binding properties and solubility of single-chain T cell receptors expressed in E. coli. Mol Immunol (1996) 33:819–29. doi:10.1016/0161-5890(96)00038-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Willcox BE, Gao GF, Wyer JR, O’Callaghan CA, Boulter JM, Jones EY, et al. Production of soluble alphabeta T-cell receptor heterodimers suitable for biophysical analysis of ligand binding. Protein Sci (1999) 8:2418–23. doi:10.1110/ps.8.11.2418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Maynard J, Adams EJ, Krogsgaard M, Petersson K, Liu CW, Garcia KC. High-level bacterial secretion of single-chain a/b T-cell receptors. J Immunol Methods (2005) 306:51–67. doi:10.1016/j.jim.2005.07.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng (2003) 16:707–11. doi:10.1093/protein/gzg087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Molloy PE, Sewell AK, Jakobsen BK. Soluble T cell receptors: novel immunotherapies. Curr Opin Pharmacol (2005) 5:438–43. doi:10.1016/j.coph.2005.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Liddy N, Molloy PE, Bennett AD, Boulter JM, Jakobsen BK, Li Y. Production of a soluble disulfide bond-linked TCR in the cytoplasm of Escherichia coli trxB gor mutants. Mol Biotechnol (2010) 45(2):140–9. doi:10.1007/s12033-010-9250-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Gunnarsen KS, Lunde E, Kristiansen PE, Bogen B, Sandlie I, Løset GÅ. Periplasmic expression of soluble single chain T cell receptors is rescued by the chaperone FkpA. BMC Biotechnol (2010) 10:8. doi:10.1186/1472-6750-10-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci (2006) 15:14–27. doi:10.1110/ps.051817606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Ewert S, Honegger A, Pluckthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods (2004) 34:184–99. doi:10.1016/j.ymeth.2004.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol (2004) 22:1161–5. doi:10.1038/nbt1000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Gunnarsen KS, Kristinsson SG, Justesen S, Frigstad T, Buus S, Bogen B, et al. Chaperone-assisted thermostability engineering of a soluble T cell receptor using phage display. Sci Rep (2013) 3:1162. doi:10.1038/srep01162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Famm K, Winter G. Engineering aggregation-resistant proteins by directed evolution. Protein Eng Des Sel (2006) 19:479–81. doi:10.1093/protein/gzl032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol (2008) 376:926–31. doi:10.1016/j.jmb.2007.10.075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Christ D, Famm K, Winter G. Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel (2007) 20:413–6. doi:10.1093/protein/gzm037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol (2000) 18:754–9. doi:10.1038/77325
55. Løset GÅ, Lunde E, Bogen B, Brekke OH, Sandlie I. Functional phage display of two murine a/b T-cell receptors is strongly dependent on fusion format, mode and periplasmic folding assistance. Protein Eng Des Sel (2007) 20:461–72. doi:10.1093/protein/gzm044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Løset GÅ, Roos N, Bogen B, Sandlie I. Expanding the versatility of phage display II: improved affinity selection of folded domains on protein VII and IX of the filamentous phage. PLoS One (2011) 6:e17433. doi:10.1371/journal.pone.0017433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol (2005) 23:349–54. doi:10.1038/nbt1070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci (2006) 15:710–21. doi:10.1110/ps.051936406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Cole DK, Miles KM, Madura F, Holland CJ, Schauenburg AJA, Godkin AJ, et al. T-cell receptor (TCR)-peptide specificity overrides affinity-enhancing TCR-major histocompatibility complex interactions. J Biol Chem (2014) 289:628–38. doi:10.1074/jbc.M113.522110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Laugel B, Boulter JM, Lissin N, Vuidepot A, Li Y, Gostick E, et al. Design of soluble recombinant T cell receptors for antigen targeting and T cell inhibition. J Biol Chem (2005) 280:1882–92. doi:10.1074/jbc.M409427200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol (2008) 9:282–91. doi:10.1038/ni1559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Sami M, Rizkallah PJ, Dunn S, Molloy P, Moysey R, Vuidepot A, et al. Crystal structures of high affinity human T-cell receptors bound to peptide major histocompatibility complex reveal native diagonal binding geometry. Protein Eng Des Sel (2007) 20:397–403. doi:10.1093/protein/gzm033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Cole DK, Miles KM, Madura F, Holland CJ, Schauenburg AJA, Godkin AJ, et al. TCR-peptide specificity overrides affinity enhancing TCR-MHC interactions. J Biol Chem (2013) 289(2):628–38. doi:10.1074/jbc.M113.522110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol (2006) 176:7308–16. doi:10.4049/jimmunol.176.12.7308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Purbhoo MA, Li Y, Sutton DH, Brewer JE, Gostick E, Bossi G, et al. The HLA A*0201-restricted hTERT540-548 peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol Cancer Ther (2007) 6:2081–91. doi:10.1158/1535-7163.mct-07-0092
66. Bossi G, Gerry AB, Paston SJ, Sutton DH, Hassan NJ, Jakobsen BK. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology (2013) 2:e26840. doi:10.4161/onci.26840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem J (2008) 415:183–96. doi:10.1042/BJ20080850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Armstrong KM, Insaidoo FK, Baker BM. Thermodynamics of T-cell receptor-peptide/MHC interactions: progress and opportunities. J Mol Recognit (2008) 21:275–87. doi:10.1002/jmr.896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Madura F, Rizkallah PJ, Miles KM, Holland CJ, Bulek AM, Fuller A, et al. T-cell receptor specificity maintained by altered thermodynamics. J Biol Chem (2013) 288(26):18766–75. doi:10.1074/jbc.M113.464560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci U S A (2008) 105:6578–83. doi:10.1073/pnas.0801097105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Huang J, Makabe K, Biancalana M, Koide A, Koide S. Structural basis for exquisite specificity of affinity clamps, synthetic binding proteins generated through directed domain-interface evolution. J Mol Biol (2009) 392:1221–31. doi:10.1016/j.jmb.2009.07.067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Davis SJ, Ikemizu S, Evans EJ, Fugger L, Bakker TR, Van Der Merwe PA. The nature of molecular recognition by T cells. Nat Immunol (2003) 4:217–24. doi:10.1038/ni0303-217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Wooldridge L, Van Den Berg HA, Glick M, Gostick E, Laugel B, Hutchinson SL, et al. Interaction between the CD8 coreceptor and MHC class I stabilizes TCR-antigen complexes at the cell surface. J Biol Chem (2005) 280:27491–501. doi:10.1074/jbc.M500555200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol (2007) 179:5845–54. doi:10.4049/jimmunol.179.9.5845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Nauerth M, Weißbrich B, Knall R, Franz T, Dössinger G, Bet J, et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci Transl Med (2013) 5:192ra187. doi:10.1126/scitranslmed.3005958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz De Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A (2013) 110:6973–8. doi:10.1073/pnas.1221609110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Oren R, Hod-Marco M, Haus-Cohen M, Thomas S, Blat D, Duvshani N, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol (2014) 193(11):5733–43. doi:10.4049/jimmunol.1301769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Frankel TL, Burns WR, Peng PD, Yu Z, Chinnasamy D, Wargo JA, et al. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol (2010) 184(11):5988–98. doi:10.4049/jimmunol.1000189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Friedman KM, Prieto PA, Devillier LE, Gross CA, Yang JC, Wunderlich JR, et al. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J Immunother (2012) 35:400–8. doi:10.1097/CJI.0b013e31825898c5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med (2012) 18(6):980–7. doi:10.1038/nm.2764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Naylor K, Li G, Vallejo AN, Lee W-W, Koetz K, Bryl E, et al. The influence of age on T cell generation and TCR diversity. J Immunol (2005) 174:7446–52. doi:10.4049/jimmunol.174.11.7446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Satram-Hoang S, Reyes C, Hoang K, Momin F, Skettino S. The unmet need in chronic lymphocytic leukemia: impact of comorbidity Burden on treatment patterns and outcomes in elderly patients. J Cancer Ther (2013) 4:1321–9. doi:10.4236/jct.2013.48156
83. van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509:439–46. doi:10.1038/nature13193
84. Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, et al. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol Immunother (2011) 60(6):809–18. doi:10.1007/s00262-011-0991-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol (2014) 32:798–808. doi:10.1200/jco.2013.51.5304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Bossi G, Buisson S, Oates J, Jakobsen BK, Hassan NJ. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol Immunother (2014) 63:437–48. doi:10.1007/s00262-014-1525-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: phage display, tumor immunity, antigen presentation, T cell receptor, immunotherapy
Citation: Løset GÅ, Berntzen G, Frigstad T, Pollmann S, Gunnarsen KS and Sandlie I (2015) Phage display engineered T cell receptors as tools for the study of tumor peptide–MHC interactions. Front. Oncol. 4:378. doi: 10.3389/fonc.2014.00378
Received: 10 November 2014; Paper pending published: 25 November 2014;
Accepted: 17 December 2014; Published online: 12 January 2015.
Edited by:
Paul Eggleton, Exeter University Medical School, UKReviewed by:
Arnaud Moris, Université Pierre et Marie Curie (UPMC), FranceBrion William Murray, Pfizer Worldwide Research and Development, USA
Armando Acosta, Universiti Sains Malaysia, Malaysia
Copyright: © 2015 Løset, Berntzen, Frigstad, Pollmann, Gunnarsen and Sandlie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geir Åge Løset, Department of Biosciences, University of Oslo, PO Box 1066, Oslo N-0316, Norway e-mail: g.a.loset@ibv.uio.no