 Simone Hettmer
Simone Hettmer Roderick T. Bronson5
Roderick T. Bronson5- 1Division of Pediatric Hematology and Oncology, Department of Pediatric and Adolescent Medicine, University Medical Center Freiburg, Freiburg, Germany
- 2Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, Boston, MA, USA
- 3Howard Hughes Medical Institute, Chevy Chase, MD, USA
- 4Joslin Diabetes Center, Boston, MA, USA
- 5Department of Biomedical Sciences, Tufts University Veterinary School, North Grafton, MA, USA
Rhabdomyosarcomas (RMS) are heterogeneous cancers with myogenic differentiation features. The cytogenetic and mutational aberrations in RMS are diverse. This study examined differences in the malignant behavior of two genetically distinct and disease-relevant mouse myogenic tumor models. Kras; p1619null myogenic tumors, initiated by expression of oncogenic Kras in p16p19null mouse satellite cells, were metastatic to the lungs of the majority of tumor-bearing animals and repopulated tumors in seven of nine secondary recipients. In contrast, SmoM2 tumors, initiated by ubiquitous expression of a mutant Smoothened allele, did not metastasize and repopulated tumors in 2 of 18 recipients only. In summary, genetically distinct myogenic tumors in mice exhibit marked differences in malignant behavior.
Introduction
Rhabdomyosarcomas (RMS) are heterogeneous cancers with myogenic differentiation (1). Fusion-positive RMS tumors carry exclusive chromosomal translocations at t(2;13)(q35;q14) or t(1;13)(p36;q14) and exhibit aggressive clinical behavior (2, 3). The remaining, fusion-negative spectrum of human RMS comprises a diverse group of tumors with frequent RAS pathway activation (4, 5) and variable mutations, including loss of heterozygosity at the PTCH1 locus (6, 7) in a subset of fusion-negative RMS. PTCH1 serves as a Hedgehog (Hh) receptor, and loss of PTCH1 function results in de-repression of downstream Hh pathway signaling. The contributions of RMS-relevant oncogenic pathways, including RAS and Hh signaling, to myogenic tumor formation were previously tested in mice (8, 9). This report highlights the distinct phenotypes of two mouse myogenic tumor models – those initiated by combined Cdkn2a (p16p19) disruption and Kras expression in transplanted mouse muscle satellite cells (10) and those arising in the skeletal muscle of mice with activated Hh signaling due to expression of a mutant, constitutively active smoothened (SmoM2) allele (11, 12). We demonstrate significant differences in tumor-repopulating activity and prevalence of lung metastases between Kras-driven and Hh-driven myogenic tumors in mice. These observations reveal marked differences in malignant behavior between genetically distinct mouse myogenic tumors, suggesting that an understanding of the distinct oncogenetic underpinnings of tumors on the fusion-negative RMS spectrum may be informative for clinical prognosis and treatment.
Materials and Methods
Mice
R26-SmoM2 (mixed genetic background including 129/Sv and Swiss Webster as main components) (11), CAGGS-CreER (11), and NOD.CB17-Prkdcscid/J (NOD.SCID) mice were purchased from The Jackson Laboratory. p16p19null mice (B6.129 background) were obtained from the NIH/Mouse Models of Human Cancer Consortium. Mice were bred and maintained at the Joslin Diabetes Center Animal Facility. All animal experiments were approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee.
Sarcoma Induction
Kras; p16p19null myogenic tumors were initiated by fluorescence-activated cell sorting of p16p19null satellite cells, followed by lentiviral transduction to introduce oncogenic Kras(G12v) and implantation in the gastrocnemius muscles of NOD.SCID mice as previously described (10). R26-SmoM2;CAGGS-CreER were injected with Tamoxifen (1 mg/40 g) on postnatal day 10 to activate expression of CRE recombinase and SMOM2. R26-SmoM2;CAGGS-CreER spontaneously developed multifocal skeletal muscle tumors (SmoM2 tumors) as previously described (11, 12).
Histopathology
Tumor tissue was dissected, fixed in 4% paraformaldehyde for 2 h, and embedded in paraffin. Standard H&E stained sections were prepared. Staining for Actin (Dako, M0635, 1:200), Desmin (Dako, M0760, 1:50), and Ki67 Ki67 (Vector Labs, VP-K451, 1:250) was performed as previously described (10).
Lung Metastases
Tumor-bearing mice were monitored at least twice weekly for health problems, and were sacrificed once tumors reached a volume of 1 cm3 or were ill. Lungs were dissected, fixed in 4% paraformaldehyde for 2 h, and embedded in paraffin. Standard H&E stained sections were prepared and evaluated for the presence of metastases by Roderick T. Bronson.
Tumor Transplantation
Tumors were harvested, digested in DMEM + 0.2% collagenase type II (Invitrogen) + 0.05% dispase (Invitrogen) for 90 min at 37°C in a shaking waterbath, triturated to disrupt the remaining tumor pieces, and filtered through a 70 mm cell strainer. Red blood cells were lysed from tumor cell preparations by 3 min incubation in 0.15 M ammonium chloride, 0.01 M potassium bicarbonate solution on ice. Defined numbers of tumor cells were resuspended in 10–15 ml of HBSS with 2% FBS and injected into the gastrocnemius muscles of 1- to 3-month-old, anesthetized NOD.SCID mice using a transdermally inserted dental needle attached to a Hamilton syringe via polyethylene tubing. Recipient muscles were preinjured 24 h before cell implantation by injection of 25 ml of a 0.03 mg/ml solution of cardiotoxin (from Naja mossambica, Sigma) in order to enhance cell engraftment. Mice were screened once weekly for the development of tumors at the injection sites.
Statistics
Differences between Kras; p16p19null and SmoM2 mouse myogenic tumors were evaluated by T-test (Ki67 indices), Fisher’s Exact test (prevalence of lung metastases), and Kaplan–Meier analysis (tumor-repopulating activity).
Results
Kras; p16p19null and SmoM2 Mouse Tumors Exhibit a Myogenic Tumor Phenotype
Kras; p16p19null mouse myogenic tumors were induced by intramuscular implantation of Kras(G12v)-expressing p16p19null muscle satellite cells (10). In contrast, SmoM2 mouse myogenic tumors were initiated by ubiquitous activation of a mutant, constitutively active smoothened (SmoM2) allele in R26-SmoM2;CAGGS-CreER mice (11, 12). The phenotypes of Kras; p16p19null and SmoM2 myogenic tumors were previously described (10–12). In brief, Kras; p16p19null tumors contained bundles of cells with large, atypical nuclei, frequent mitotic figures, and occasional multinucleated giant cells. Subsets of cells (<50% of all tumor cells) expressed terminal muscle differentiation markers such as desmin and actin (Figure 1A), and the proliferative index as evidenced by the percentage of Ki67-expressing nuclei was 41.6 ± 12.5% (range 30.5–59.3%; four tumors evaluated) (Table 1). SmoM2 tumors contained many multinucleated, elongated cells with abundant cytoplasm interspersed with small round cells. SmoM2 tumors lacked cellular atypia and diffusely expressed desmin and actin in many tumor cells (more than 75% of all tumor cells; Figure 1B). As previously reported (12), the Ki67 index of SmoM2 tumors was 19.1 ± 15.9% (range 3.4–41.8%; six tumors evaluated) and lower than that observed in Kras; p16p19null tumors (p = 0.05; Table 1).
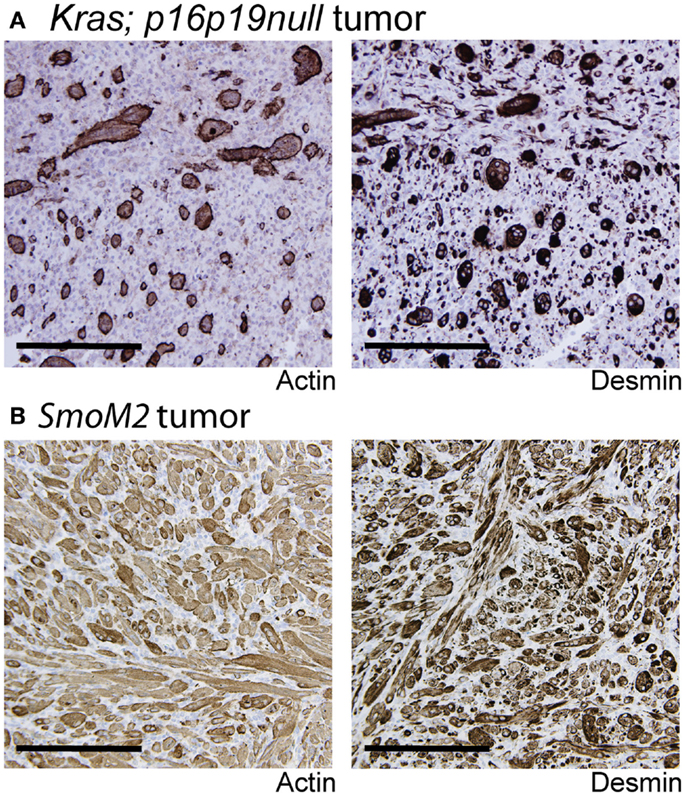
Figure 1. Terminal myogenic differentiation in Kras; p16p19null and SmoM2 mouse tumors. (A) Subsets of Kras; p16p19null tumors cells express terminal muscle differentiation markers, actin and desmin. (B) The majority of SmoM2 tumor cells express actin and desmin. Images were taken at 20× (scale bars indicate 100 μm).

Table 1. Differences in the malignant behavior of Kras; p16p19null and SmoM2 mouse tumors.
Kras; p16p19null and SmoM2 Mouse Myogenic Tumors have Different Metastatic Potential
The lung is the primary organ affected by distant sarcoma metastases in humans. To assess the metastatic potential of Kras; p16p19null and SmoM2 tumors, random lung sections obtained from tumor-bearing animals were screened for the presence of metastases. Six of seven mice with Kras; p16p19null myogenic tumors were found to have lung metastases at the time of death (mice were sacrificed 17–28 days after detection of palpable tumors) (Figure 2). In contrast, 0 of 8 mice with SmoM2 myogenic tumors had lung metastases at the time of death (mice were sacrificed at 38–55 days of age and 5–21 days after detection of palpable tumors). The prevalence of lung metastases in Kras; p16p19null and SmoM2 myogenic tumor-bearing mice was significantly different (p = 0.001).
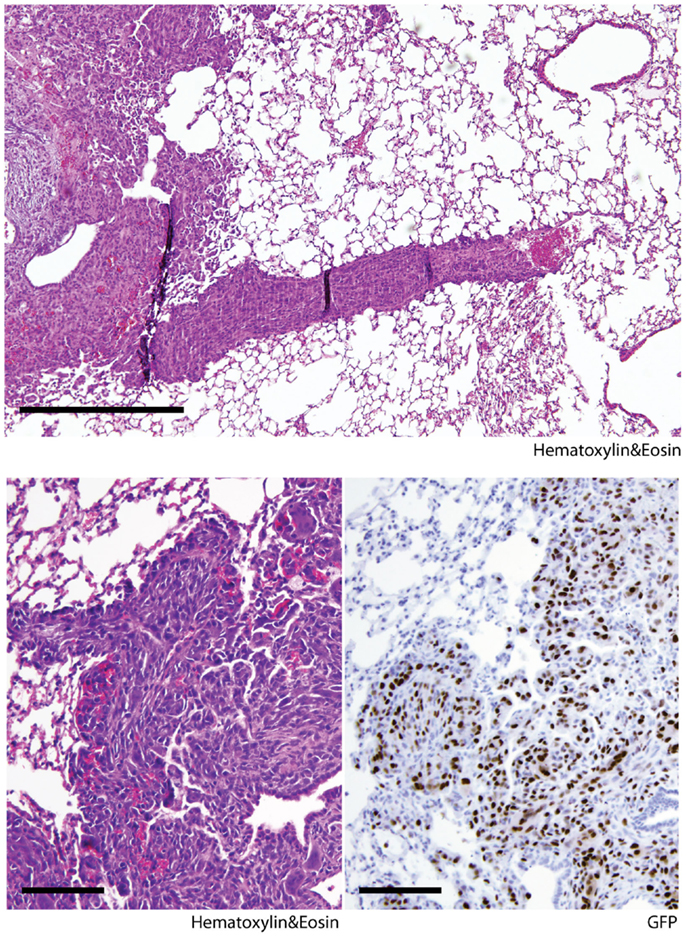
Figure 2. Kras; p16p19null mouse myogenic tumors metastasize to the lungs of tumor-bearing animals. Random lung sections from Kras; p16p19null tumor-bearing mice show metastases. Tumor cells invade lung capillaries (top panel). Similar to primary tumors arising from GFP+ Kras-expressing; p16p19null satellite cells, lung metastases are GFP+ (bottom right panel). Images were taken at 10× and 20× (scale bars indicate 100 μm)
Kras; p16p19null and SmoM2 Mouse Myogenic Tumors Differ in Tumor-Repopulating Activity
Most malignant tumors contain cells that have the capacity to repopulate secondary tumors when transplanted into a susceptible secondary environment, and this assay has been used as a test of the malignancy of distinct tumors and tumor cell subsets (13). To evaluate the tumor-repopulating activity of Kras; p16p19null and SmoM2 mouse myogenic tumors, viable tumor cells were transplanted into the cardiotoxin-pre-injured gastrocnemius muscles of NOD.SCID mice. The Kras; p16p19null tumor cell pool contains approximately 70% GFP+ cells and 30% GFP− cells (10). Because tumor-repopulating activity in Kras; p16p19null tumors resides within the Kras-expressing, GFP+ subset of tumor cells descended from virally infected satellite cells (Figure S1 in Supplementary Material), Kras; p16p19null tumor cells were sorted for transplantation from two Kras; p16p19null primary tumors as GFP+, Pi−, Calcein+ cells. Seven of nine mice injected with only 50 GFP+, Pi−, Calcein+ Kras; p16p19null tumor cells developed secondary tumors at the injection site 26–39 days after tumor cell injection. For SmoM2 tumors, viable tumor cells were sorted as PI− Calcein+ cells from primary tumors obtained from four mice. Surprisingly, despite significantly higher numbers of cells transplanted (100,000 to 150,000 PI−, Calcein+ SmoM2 tumor cells per recipient), only 2 of 18 recipient mice developed secondary tumors, which were detected 71 and 127 days after cell injection. These experiments indicate marked differences in tumor-repopulating activity of Kras; p16p19null and SmoM2 tumors (p < 0.001, Figure 3), in terms of both the frequency of tumor-repopulating cells and the latency of secondary tumor formation.
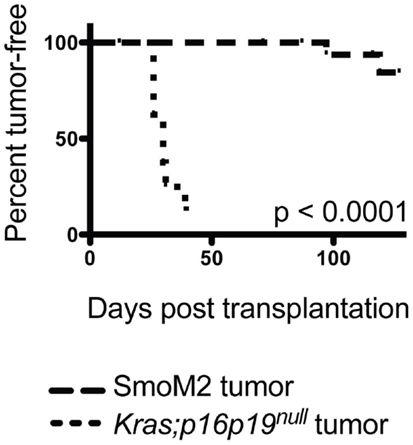
Figure 3. Kras; p16p19null tumor cells repopulate tumors in secondary recipients more effectively than SmoM2 mouse tumor cells. Pi−Ca+GFP+ Kras; p16p19null tumor cells were sorted independently from two primary tumors and injected into the cardiotoxin-pre-injured gastrocnemius muscles of NOD.SCID mice (50 cells per injection). Pi-Ca+ SmoM2 tumor cells were sorted independently from four primary tumors and injected into the cardiotoxin-pre-injured gastrocnemius muscles of NOD.SCID mice (100,000–150,000 cells per injection). Recipient mice were monitored for the occurrence of secondary tumors at the injection site for up to 4 months.
Discussion
Our findings highlight differences in the malignant phenotype and behavior of mouse myogenic tumors driven by activation of distinct RMS-relevant oncogenic pathways. Kras; p1619null myogenic tumors were metastatic to the lungs of the majority of tumor-bearing animals and contained high tumor-repopulating activity. In contrast, SmoM2 tumors did not metastasize and were substantially less effective in repopulating tumors in secondary recipients. These observations indicate that genetically distinct myogenic tumors in mice display marked differences in their malignant behavior.
The two model systems described in this study were induced by different experimental methods. SmoM2 tumors originated from Cre-mediated activation of a conditionally expressed transgene. Kras; p16p19null mouse tumors, on the other hand, were initiated by viral transduction and intramuscular implantation of target satellite cells. We note that Kras; Tp53−/−mouse myogenic tumors (14, 15), induced by Cre-mediated activation of oncogenic hits instead of viral transduction, exhibit a phenotype that closely resembles the Kras; p16p19null mouse tumors described here. For example, Kras; p16p19null share their propensity to metastasize to the lungs of tumor-bearing animals with Kras; Tp53−/−mouse tumors (14). Nevertheless, it is possible that differences in the tumor induction strategy (such as off-target effects of viral transduction) could contribute to the observed differences in malignant behavior between SmoM2 and Kras; p16p19null mouse myogenic tumors.
Similar to mouse myogenic tumors, human fusion-negative RMS comprises a group of tumors with clear differences in histology, myogenic differentiation state, oncogenic pathway activation, and genetic background. In recent years, subsets of human RMS tumors that exhibit a combination of specific genetic and phenotypic characteristics were distinguished. For example, a subset of human fusion-negative RMS with spindle cell/sclerosing histology was recently found to exhibit diffuse MyoD expression, carry frequent somatic MyoD mutations, and portend a poor prognosis (16, 17). Also, children with TP53 germline mutations are predisposed to develop anaplastic RMS at a young age (18), and germline mutations in DICER1 were linked to a genetic susceptibility to develop RMS of the genitourinary tract (19). Future extended (epi-)genotype/phenotype correlations might pinpoint clinically/biologically distinct subgroups of human fusion-negative RMS and identify biomarkers to facilitate prognostication and/or stratification of therapy.
Author Contributions
SH, RB, and AW conceived experiments, analyzed data, wrote, and approved of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank C. L. Unitt and T. Bowman at the DF/HCC Histopathology Core for help with immunohistochemistry, D. Tchessalova for excellent animal care, and Joyce LaVecchio, Girijesh Burizula, and Atsuya Wakayabashe in the Joslin Diabetes Center Flow Cytometry Core (supported by the Harvard Stem Cell Institute and NIH P30DK036836) for flow cytometry support. This work was funded in part by a Stand Up To Cancer-American Association for Cancer Research Innovative Research Grant (SU2C-AACR-IRG1111; to AW); by grants from the Burroughs-Wellcome Fund and the Harvard Stem Cell Institute (to AW), and by P.A.L.S. Bermuda/St. Baldrick’s, ALSF, and Bear Necessities (to SH). Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding agencies.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fonc.2015.00050/abstract
References
1. Parham DM. Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol (2001) 14:506–14. doi:10.1038/modpathol.3880339
2. Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M. Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol (2000) 13:988–93. doi:10.1038/modpathol.3880179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol (2002) 20:2672–9. doi:10.1200/JCO.2002.03.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell (2013) 24:710–24. doi:10.1016/j.ccr.2013.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov (2014) 4:216–31. doi:10.1158/2159-8290.CD-13-0639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Bridge JA, Liu J, Weibolt V, Baker KS, Perry D, Kruger R, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer (2000) 27:337–44. doi:10.1002/(SICI)1098-2264(200004)27:4<337::AID-GCC1>3.0.CO;2-1
7. Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol (2006) 208:17–25. doi:10.1002/path.1882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Rubin BP, Nishijo K, Chen HI, Yi X, Schuetze DP, Pal R, et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell (2011) 19:177–91. doi:10.1016/j.ccr.2010.12.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. O’Brien D, Jacob AG, Qualman SJ, Chandler DS. Advances in pediatric rhabdomyosarcoma characterization and disease model development. Histol Histopathol (2012) 27:13–22.
10. Hettmer S, Liu J, Miller CM, Lindsay MC, Sparks CA, Guertin DA, et al. Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc Natl Acad Sci U S A (2011) 108:20002–7. doi:10.1073/pnas.1111733108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res (2006) 66:10171–8. doi:10.1158/0008-5472.CAN-06-0657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Hettmer S, Teot LA, Van Hummelen P, Macconaill L, Bronson RT, Dall’osso C, et al. Mutations in Hedgehog pathway genes in fetal rhabdomyomas. J Pathol (2013) 231(1):44–52. doi:10.1002/path.4229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lapouge G, Beck B, Nassar D, Dubois C, Dekoninck S, Blanpain C. Skin squamous cell carcinoma propagating cells increase with tumour progression and invasiveness. EMBO J (2012) 31:4563–75. doi:10.1038/emboj.2012.312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med (2007) 13:992–7. doi:10.1038/nm1602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Blum JM, Ano L, Li Z, Van Mater D, Bennett BD, Sachdeva M, et al. Distinct and overlapping sarcoma subtypes initiated from muscle stem and progenitor cells. Cell Rep (2013) 5:933–40. doi:10.1016/j.celrep.2013.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet (2014) 46:595–600. doi:10.1038/ng.2969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Yasui N, Yoshida A, Kawamoto H, Yonemori K, Hosono A, Kawai A. Clinicopathologic analysis of spindle cell/sclerosing rhabdomyosarcoma. Pediatr Blood Cancer (2014). doi:10.1002/pbc.25367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hettmer S, Archer NM, Somers GR, Novokmet A, Wagers AJ, Diller L, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer (2014) 120:1068–75. doi:10.1002/cncr.28507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer (2012) 59:558–60. doi:10.1002/pbc.24020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: rhabdomyosarcoma, myogenic differentiation, metastasis, transplantation
Citation: Hettmer S, Bronson RT and Wagers AJ (2015) Distinct malignant behaviors of mouse myogenic tumors induced by different oncogenetic lesions. Front. Oncol. 5:50. doi: 10.3389/fonc.2015.00050
Received: 21 December 2014; Accepted: 11 February 2015;
Published online: 24 February 2015.
Edited by:
Thomas Grunewald, Ludwig Maximilian University of Munich, GermanyReviewed by:
Frederic Barr, National Cancer Institute, USARossella Rota, Ospedale Pediatrico Bambino Gesù, Italy
Copyright: © 2015 Hettmer, Bronson and Wagers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Hettmer, University Medical Center, Mathildenstrasse 1, 79106 Freiburg, Germany e-mail: simone.hettmer@uniklinik-freiburg.de