 Nicole Renee Parker1†
Nicole Renee Parker1† Peter Khong1†
Peter Khong1† Jonathon Fergus Parkinson1,2
Jonathon Fergus Parkinson1,2 Viive Maarika Howell1*‡
Viive Maarika Howell1*‡ Helen Ruth Wheeler1,3‡
Helen Ruth Wheeler1,3‡
- 1Bill Walsh Translational Cancer Research Laboratory, Kolling Institute of Medical Research, Royal North Shore Hospital, University of Sydney, St Leonards, NSW, Australia
- 2Department of Neurosurgery, Royal North Shore Hospital, St Leonards, NSW, Australia
- 3Department of Medical Oncology, Royal North Shore Hospital, St Leonards, NSW, Australia
Glioblastomas, (grade 4 astrocytomas), are aggressive primary brain tumors characterized by histopathological heterogeneity. High-resolution sequencing technologies have shown that these tumors also feature significant inter-tumoral molecular heterogeneity. Molecular subtyping of these tumors has revealed several predictive and prognostic biomarkers. However, intra-tumoral heterogeneity may undermine the use of single biopsy analysis for determining tumor genotype and has implications for potential targeted therapies. The clinical relevance and theories of tumoral molecular heterogeneity in glioblastoma are discussed.
Introduction
Malignant gliomas are the most common intrinsic primary brain tumors in adults. Despite advances in neurosurgery, chemotherapy, and radiation, the median survival for the most aggressive tumors, glioblastoma remains less than 2 years. Although glioblastomas share common histological features, at a molecular level these tumors are highly variable from patient to patient and within the same tumor can display significant regional heterogeneity. Molecular analysis of a single biopsy specimen for diagnosis and determination of therapeutic options has profound clinical implications for targeted therapeutic treatment strategies until more is known about molecular pathways and driver mutations in glioblastoma.
Histological Diagnosis and Classification of Malignant Glioma Subtypes
Histopathology remains the gold standard for the diagnosis and classification of gliomas and currently determines adjuvant therapy. Gliomas include astrocytomas (with cells that resemble astrocytes), oligodendrogliomas (predominantly of cells that resemble oligodendrocytes), and mixed oligoastrocytomas. The current 2007 World Health Organization (WHO) classification of astrocytoma (including glioblastoma) depends on cellular morphology to determine tumor grade, focusing on the presence or absence of nuclear atypia, mitotic activity, microvascular proliferation, and necrosis (1). This histopathological classification system is fraught with error, and inter-observer subjectivity is common (2). Regional heterogeneity is common, as reflected by the original name for WHO Grade IV astrocytoma, glioblastoma multiforme (GBM) (3). Pathological diagnosis is based on the area of the highest grade visible in multiple sections, even though less aggressive areas may also be present in the specimen. Tumors with the same histopathological classification also exhibit widely variable clinical presentation, magnetic resonance imaging (MRI) features, response to therapy and outcome. Primary glioblastoma most commonly presents de novo in elderly patients, typically with a short clinical history. In contrast, secondary glioblastoma progresses from lower grade astrocytic lesions, occur in younger patients, are more likely to be located in the frontal lobes, and have a significantly improved outcome (4). Molecular analyses show they arise from different genomic alterations, which may influence response to therapy (5).
Molecular Classification of Glioblastoma Subtypes
Recent large-scale genomic analyses have identified extensive inter-patient heterogeneity, further refining histopathological classification of this disease. The Cancer Genome Atlas (TCGA) project and others (5–7) have shown that glioblastoma can be subclassified into at least four molecular subtypes, featuring distinct genetic, epigenetic, and transcriptional alterations (5, 8). Tumor variants can be classified on the basis of somatic mutations in isocitrate dehydrogenase (IDH) 1/2 and TP53; transcriptional signature (classical, mesenchymal, neural, or proneural); copy number variation, including co-deletion of chromosomes 1p and 19q; and amplification or mutation of the epidermal growth factor receptor (EGFR) and increased DNA hypermethylation of promoter-associated CpG islands (Figure 1) (5–10).
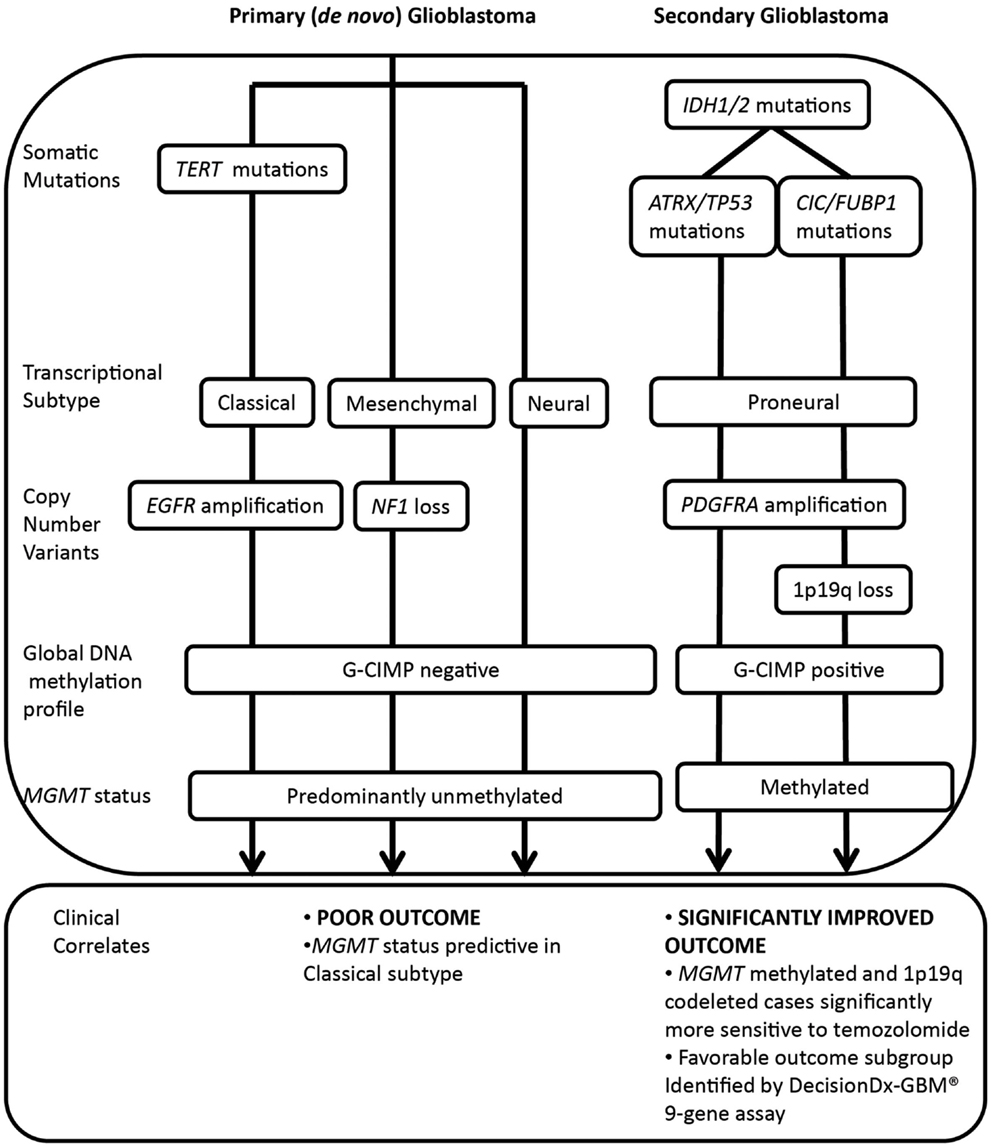
Figure 1. Molecular classification of major glioblastoma subtypes and correlation with treatment response and outcome. IDH1/2 mutations are major prognostic biomarkers, stratifying primary and secondary pathways of gliomagenesis. Primary glioblastoma features a high frequency of TERT mutations, whereas IDH1/2 mutated glioma (including secondary glioblastoma and low grade glioma) may be further subdivided on the basis of co-mutations in either ATRX and TP53 or CIC and FUBP1, occurring at high frequency in astrocytic or oligodendroglial tumor subtypes. Co-deletion of 1p and 19q, a marker of enhanced chemosensitivity, also clusters with mutations in CIC and FUBP1. Primary glioblastomas display classical, mesenchymal, and neural phenotypes, whereas secondary glioblastomas tend to display a proneural phenotype that shifts toward a mesenchymal phenotype with recurrence. The significantly improved outcome of the proneural subset is due to the G-CIMP phenotype, established by IDH1/2-mediated metabolic reprogramming of the epigenome. Primary glioblastomas and a subset of proneural tumors are glioma-CpG island hypermethylator phenotype (G-CIMP) negative, and a large proportion are MGMT unmethylated. Both classical and mesenchymal transcriptional subtypes benefit from concurrent chemoradiotherapy, however, MGMT status is only predictive of treatment response in the classical subset.
In particular, the TCGA subtypes have been increasingly applied for their prognostic ability and ease of testing for a single typical molecular alteration associated with that subtype, namely IDH1/2 mutation (proneural), EGFR amplification (classical), or Neurofibromin 1(NF1) loss (mesenchymal) (5). Also associated with the proneural subtype are 1p and 19q co-deletion, TP53 mutations, and platelet-derived growth factor receptor, alpha polypeptide (PDGFRA) amplification. These and other molecular aberrations associated with the transcriptional subtypes are shown in Figure 1.
The identified molecular heterogeneity may underlie differences in patient sensitivity to therapy and prognosis. In current clinical practice, standard histopathology is complemented by molecular testing generally performed on single tumor biopsies. Key biomarkers commonly assessed include IDH1/2 mutations, O6-methylguanine DNA methyltransferase (MGMT) promoter methylation, co-deletion of 1p and 19q, and EGFR amplification/truncation.
Molecular Biomarkers in Glioblastoma
IDH1/2 mutation status
Somatic mutations in IDH1/2 are routinely screened by Sanger sequencing and/or immunohistochemistry. These heterozygous point mutations occur commonly at arginine residues, at codon 132 of IDH1 and at codon 172 of IDH2, and are definitive markers of secondary glioblastoma and a significantly improved prognosis (9). IDH1 mutations occur at high frequency in WHO grade II and III astrocytoma (>80% of cases), precursor lesions of secondary glioblastoma, whereas IDH2 mutations occur largely in oligodendroglial tumors, with much lower frequency. Both alterations are rare in de novo glioblastoma (occurring in <5% of cases). IDH-mutant glioblastoma occurs predominantly in the frontal lobe, whereas the anatomical distribution of IDH-wildtype glioblastoma is more heterogeneous (11). More recently, loss of alpha-thalassemia/mental retardation syndrome X-linked (ATRX) has been shown to further refine IDH-mutant astrocytic tumors with IDH/ATRX mutant-tumors carrying a more favorable prognosis (10).
In addition to its role as a biomarker, IDH1 mutation may provide the basis for a targeted immunotherapy. A peptide containing mutant (R132H) IDH1 has been shown to be immunogenic suggesting the potential for a mutation-specific vaccine for IDH1-mutant gliomas (12).
In both low- and high-grade gliomas with IDH mutations, combined loss of the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q; 1p/19q co-deletion) is prognostic of a more favorable outcome than equivalent tumors without this co-deletion (13–15). It is most commonly found in oligodendrogliomas or tumors with oligodendroglial features. The 1p/19q co-deletion and ATRX mutations are generally mutually exclusive, ATRX mutations being markers for astrocytic lineage tumors (16).
Mutations in IDH1/2 are also strongly associated with a distinct epigenetic signature. Mutant IDH1 and IDH2 alter glioma metabolism, favoring the reduction of α-ketoglutarate to 2-hydroxyglutarate (2-HG) (17), which in turn inhibits DNA and histone demethylases and establishes a glioma-CpG island hypermethylator phenotype (G-CIMP) (5), featuring hypermethylation at a large number of loci. The combination of IDH mutation, 1p/19q co-deletion and G-CIMP is prognostic of a favorable outcome for anaplastic oligodendroglial tumors and predictive of chemotherapy response (14, 15). IDH-mutant human gliomas also have reduced levels of hypoxia-inducible factor (HIF) 1α and the glycolytic enzyme lactate dehydrogenase A (LDHA) (18). This deficit in glycolytic ability may also contribute to the slower growth of IDH-mutant tumors and their improved prognosis.
Most IDH mutated glioblastomas feature a proneural transcriptional profile, including a high frequency of TP53 mutations and amplification of PDGFRA (5). In contrast IDH-wildtype glioblastoma tends to be subclassified into mesenchymal, classical, and neural transcriptional subtypes. Upon recurrence, proneural tumors shift toward a more aggressive mesenchymal phenotype, enriched for expression of mesenchymal markers including chitinase 3-like 1 (CHI3L1 or YKL40), CD44 and signal transducer and activator of transcription 3 (STAT3) (6).
Colman et al. developed a robust 9-gene expression assay (now a proprietary test called DecisionDx-GBM®) with the ability to discriminate between glioblastoma patients with a more favorable outcome, associated with high-level expression of proneural genes, and those with a poor outcome, with enriched expression of mesenchymal and angiogenesis genes (19). Prognostic subgroups are identified by profiling tumor RNA extracted from formalin-fixed paraffin embedded (FFPE) tissue blocks and gene expression levels converted to a metagene score, using a proprietary algorithm. Multivariate analyses show that this 9-gene prognostic score is a stronger predictor of overall outcome and progression-free survival than other clinically significant variables, including age, Karnofsky performance score (KPS), and MGMT. This 9-gene expression assay independently predicts response to standard first line therapy and has been used to prognostically stratify patients and as a secondary endpoint in two clinical trials (20). However, its lack of validation in larger independent patient cohorts means that it has not been integrated into routine clinical testing.
MGMT promoter methylation
The current standard treatment for newly diagnosed glioblastoma involves surgery and radiation, in conjunction with the alkylating agent temozolomide (Stupp regimen), achieving a median survival of 14.6 months (21). Response to temozolomide is mediated by MGMT, encoding a DNA repair enzyme involved in the repair of cytotoxic adducts produced by this alkylating agent. Hypermethylation or epigenetic silencing of the MGMT disables DNA repair capacity, rendering cells more sensitive to treatment (7). MGMT promoter methylation is a common feature of IDH1/2 mutant/G-CIMP positive glioma, however, is less prevalent in G-CIMP negative tumors, such as primary glioblastoma, where MGMT methylation occurs in approximately 40% of cases (22–24).
Several clinical trials, including the landmark studies by Stupp and Hegi have shown that a methylated MGMT promoter is an independent predictor of response to therapy and outcome (21, 25–28). Patients with a methylated MGMT promoter exhibited a 6.4-month survival benefit following concurrent treatment with temozolomide and radiotherapy, compared to cases receiving radiotherapy alone (25), whereas the benefit for unmethylated cases was less than 1 month (25). The NOA-08 and Nordic clinical trials in elderly patients presenting with primary glioblastoma have also shown that a methylated MGMT promoter is predictive and prognostic in response to therapy (29, 30). The Nordic study found that patients more than 60 years of age with methylated MGMT gained an overall survival benefit following temozolomide therapy, whereas methylation status did not correlate with overall survival following radiotherapy (30). In the NOA-08 trial, patients more than 65 years of age with methylated MGMT and a favorable Karnofsky performance status similarly showed a greater overall survival benefit following dose dense temozolomide therapy than patients with unmethylated MGMT (29).
However, the use of MGMT methylation as a predictive and prognostic biomarker in the routine clinical environment is not straightforward. Some patients with methylated MGMT do poorly, and occasionally patients with unmethylated MGMT have prolonged survival. This is likely due to additional molecular changes. For example, it has recently been reported that MGMT methylation status was only predictive in glioblastoma cases with a classical transcriptional gene signature (7). There is also evidence that defects in the mismatch repair (MMR) pathway can confer increased resistance to temozolomide independent of MGMT methylation status (31) with mutations in mutS homolog 6 (MSH6) (32) and aberrant expression of MMR proteins MSH6, mutS homolog 2 (MSH2), PMS2 postmeiotic segregation increased 2 (PMS2), and mutL homolog 1 (MLH1) reported in some glioblastoma specimens (33, 34). The lack of correlation between MGMT status and treatment response in some cases may be related to technical aspects of current analytical assays. Differences in the efficiency of the initial bisulfite conversion step and also the region of the MGMT promoter selected for methylation analysis may contribute to variability in MGMT results. Everhard et al. performed a comprehensive study of CpG islands with the MGMT promoter, correlating methylation status with MGMT mRNA levels in 54 glioma specimens (35). This analysis identified 6 out of the 52 CpG sites as having the strongest correlation with MGMT expression (p < 0.0001), indicating that methylation at some sites may be more informative than others. MGMT expression may be induced by glucocorticoids (36), cAMP and protein kinase C activators, and radiation to a moderate extent (37, 38). Detection of MGMT expression by immunohistochemistry has proved unreliable in predicting response (25, 39, 40).
EGFR amplification/truncation
EGFR is an attractive therapeutic target in glioblastoma, with gene amplification noted in 40–60% of patients (1, 41, 42). A constitutively active mutation of EGFR (EGFRvIII) is found in 20–30% of glioblastomas and typically occurs in the presence of over-expression (amplification) of the wild type transcript. EGFRv111 is an in-frame genomic deletion of exons 2–7 resulting in a truncated protein with constitutive tyrosine kinase activity, pro-oncogenic effects, and increased chemotherapeutic and radiotherapy resistance. It is an independent marker of poor prognosis in glioblastoma (42–44).
EGFR alterations increase cell signaling through multiple pathways, including the phosphatidylinositol-3-kinase (PI3K)/Akt/mTOR pathway, and ultimately accelerate tumor growth and progression. However, attempts to block EGFR signaling in the clinic using small molecule inhibitors, such as erlotinib and gefitinib, or monoclonal antibodies, such as cetuximab have been largely unsuccessful, even after molecular preselection of patients (45). An alternate strategy targeting EGFRv111 with a vaccine (Rindopepimut) (46) is currently in Phase 3 clinical trials for patients with newly diagnosed (ACT IV) and with relapsed (ReACT) EGFRv111-positive glioblastoma following promising phase II trials (47, 48).
Focal homozygous deletion of cyclin-dependent kinase inhibitor 2A (CDKN2A) is a frequent finding in EGFR amplified tumors and these, together with the EGFRvIII, are features of the classical transcriptional subtype of glioblastoma (5). More recently, the presence of activating telomerase reverse transcriptase (TERT) promoter mutations have been reported in over 80% of primary glioblastomas. In this cohort, EGFR amplification occurred only in TERT-mutated tumors (49).
Limitations of Single-Biopsy Based Diagnosis in Glioblastoma
Despite the major advances in the molecular profiling of glioblastoma, improvements to patient outcome overall have been modest. Patient stratification and treatment have generally been performed on the basis molecular biomarkers present in a single tumor specimen. However, this approach may be too simplistic, as recent studies have unraveled yet further layers of complexity, with striking molecular heterogeneity also present within individual tumor specimens. Sottoriva et al. (50) employed a novel fluorescence-guided multiple sampling approach to collect spatially distinct tumor fragments from 11 glioblastomas, demonstrating the presence of multiple transcriptional tumor subtypes within the same tumor mass (50). Spatial heterogeneity in glioblastoma not only confounds histopathological classification but can also make treatment decisions based on samples obtained from limited areas difficult, especially as we enter the realm of personalized medicine and targeted therapies (2, 51, 52). There is also evidence of temporal heterogeneity, with selective expansion or regression of particular tumor cell subpopulations resulting from various treatments (53).
The sampling of glioma presents a challenge unique to brain tumors (54–56). Extracranial tumors can usually be resected en-bloc to preserve the geographical map from which spatially distinct tumor areas can be characterized and tumor margins and draining lymph nodes examined to assess extent of disease. Some gliomas are located in eloquent areas limiting sampling to a single biopsy. Larger gliomas are generally resected in a piecemeal fashion in order to minimize damage to surrounding brain and preserve function and quality of life of the individual. Most surgery is guided by gadolinium enhanced MRI scans. High-grade tumors are usually resected up to and including the macroscopically abnormal margin or to the area of contrast enhancement on T1 weighted MRI. It is well known, however, that aggressive infiltrating malignant cells lie well beyond this definable margin (54–56). Newer techniques using fluorescent dyes such as 5-aminolevulinic acid (ALA) potentially allow surgeons to identify areas of residual high-grade tumor at surgical margins making it possible to remove more tumor, prolonging patient progression-free survival (57).Using newer guidance technology may be able to separate these tumors spatially for analysis and potentially provide better guides to therapy. However, it is not clear from which location and how many biopsies constitute a bona fide representative sample. The co-existence of genetically divergent tumor cell clones may lead to a misinterpretation of the overall genetic tumor landscape if only a single sample is utilized.
Tumor Heterogeneity
Theories of Tumor Heterogeneity
There are a number of theories regarding tumor evolution and the generation of heterogeneous tumor cell subpopulations (Figure 2). In the Darwinian theory of clonal evolution and natural selection, successive waves of tumor cells acquire genetic changes, some of which convey a proliferative advantage (58). Selection and proliferation of therapy resistant clones lead to tumor progression and resistance to therapy (59). Such branched tumor evolution underscores the importance of targeting ubiquitous alterations in the trunk of the phylogenetic tree (60). In this theory, “driver mutations” allow the progression of a cancer, whereas “passenger mutations” are neutral or only slightly deleterious (61). More recently, with the use of high-resolution “next-generation” sequencing platforms, the definition of driver events has been further refined to encompass “Mut-driver genes” and “Epi-driver genes.” (62). Mut-driver genes tend to display a non-random mutation pattern and are recurrently mutated at the same amino acid position (for example, IDH). Epi-driver genes may be aberrantly expressed as a result of gene amplification/loss or epigenetic alterations. These early somatic events drive tumor growth and can be considered as “trunk events,” with branched separation of tumor subclones giving rise to tumor heterogeneity (63). This theory has been studied in colon cancer where the evolution of genomic changes from benign polyps to invasive carcinoma can be tracked from colonic biopsies. Study of secondary glioblastoma, which usually develops from IDH mutated low-grade lesions to invasive glioblastoma, may be possible, however primary glioblastoma appears to develop rapidly, with some patients having had a normal MRI just months before presentation. This group poses a particular challenge.
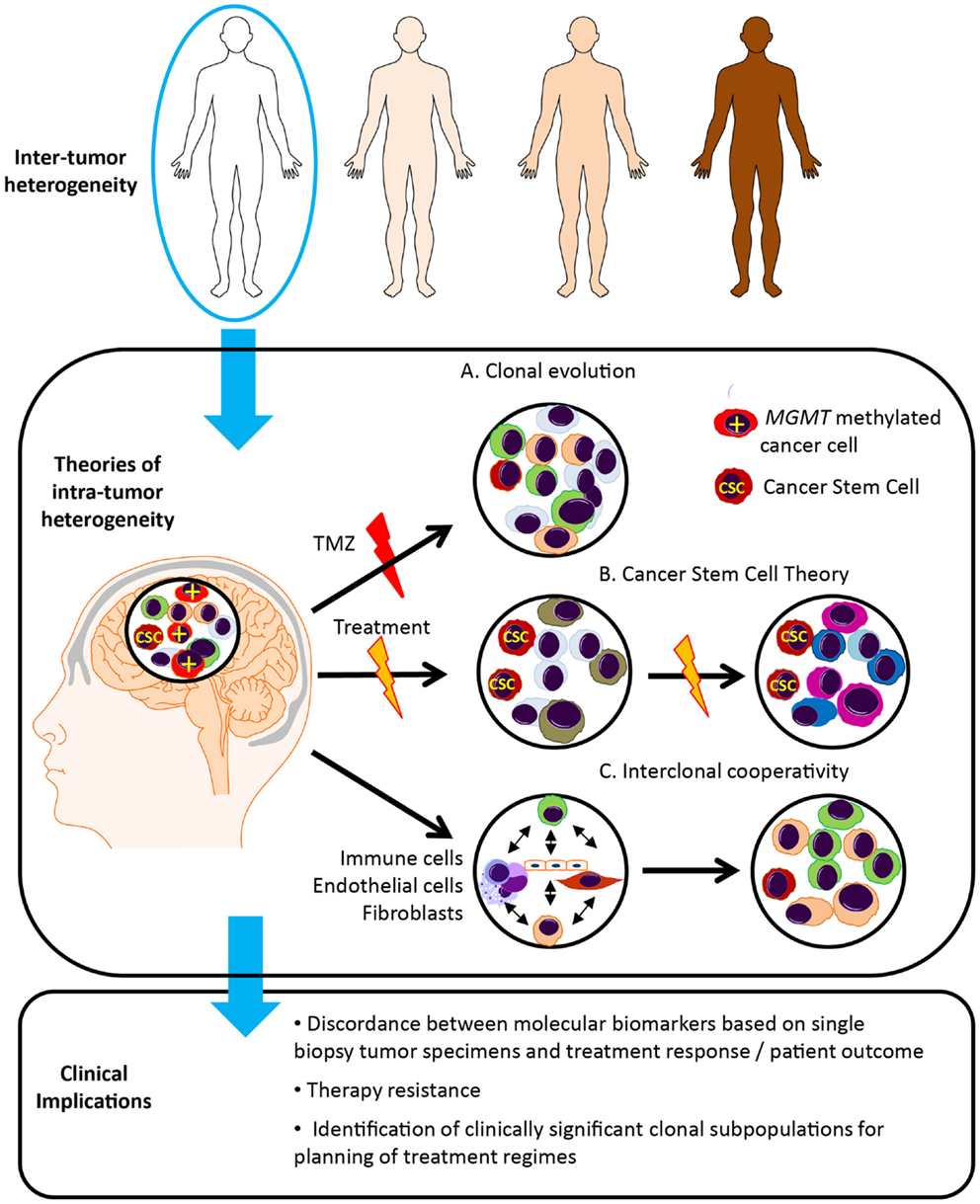
Figure 2. Glioblastoma tumor heterogeneity and implications for patient management. Tumor evolution and tumor heterogeneity may be promoted by clonal evolution, cancer stem cells and interclonal cooperativity. (A) According to the theory of clonal evolution, somatic alterations affecting the initial cell of origin give rise to multiple cancer clones, with different sensitivity to therapy and ability to survive and proliferate. These tumor cell clones are genetically unstable, undergoing successive waves of genetic alterations, and clones with the most aggressive phenotype are favored. For example, only the MGMT methylated cells sensitive to temozolomide (TMZ) disappear following TMZ treatment. (B) In contrast, according to the cancer stem cell theory, only a single subset of cells, known as cancer stem cells (CSC), possess the ability to self-renew, continuously proliferate and give rise to clones of variable genetic profiles, and are inherently resistant to therapy. (C) The theory of interclonal cooperativity suggests that tumor evolution and heterogeneity is promoted by interactions between tumor cell clones and their microenvironment, with immune/stromal factors influencing malignant progression. Significant clonal diversity within tumor specimens may explain the failure of molecularly targeted therapies in glioblastoma patients.
An alternative theory is the cancer stem cell theory, which postulates that only a subset of tumor cells possess the ability to self-renew, continuously proliferate and give rise to clones containing variable genetic profiles, and the heterogeneity within a tumor arises from proliferation of these genetically variable cancer stem cells (64, 65). Genetically distinct cancer clones may originate in specific anatomical locations, differing in neurochemistry, energy metabolism, and surrounding tissue architecture (66). Gliomas are thought to originate in the subventricular zone, which contains the highest density of astrocyte-like stem cells, and may be more susceptible to malignant transformation (56). Distinct tumor clones may harbor genetic alterations that promote invasion along blood vessels and white matter tracts and tumor expansion in particular cerebral lobes. The anatomical distribution of glioblastoma in the brain is heterogeneous, occurring with the highest frequency in the frontal lobe, followed by the temporal, parietal and occipital lobes (66).
More recently a third theory, “interclonal cooperativity” has emerged in which genetic subpopulations of tumor cell clones and immune/stromal factors co-operate to create a favorable microenvironment (67–69). Feedback from the microenvironment drives disease progression and a malignant phenotype. The complex tumor stroma, consisting of reactive astrocytes, microglial cells and immune infiltrate, aberrant microvascular proliferation and hypoxia, as well as cell populations of varying developmental stages, may also enhance the proliferation of specific cancer subclones. Specifically, the hypoxic perivascular niche has been shown to be critical for the self-renewal of glioblastoma cancer stem-like cells (70, 71), and may be important in driver of divergent tumor evolution and treatment resistance.
Unlike other tumors, gliomas rarely metastasize outside the brain and despite intense local radiotherapy, most commonly recur at the site of origin. Whether these recurrent tumors arise from incompletely resected primary tumor or distinct cancer clones that have evolved in response to treatment or other factors, such microenvironmental cues, is unknown. However, a recent whole exome sequencing study of low grade glioma supports the concept that recurrent disease originates from tumor cells present at tumor initiation that subsequently drive evolution of the tumor (72), with just under half of recurrent specimens sharing 50% of mutations with the primary tumor sample.
Evidence of Molecular Intra-Tumoral Heterogeneity in Glioblastoma
As described above, microscopic heterogeneity is a hallmark of glioblastoma. The evolution of more detailed molecular investigation and their potential implication in treatment decision making has resulted in the further verification of tumor heterogeneity at the molecular level. Tumor development and evolution has also been extensively investigated and Sottoriva et al. have demonstrated tumor heterogeneity in tumor evolution (50). Further to this, Meyer et al. by establishing in vitro functionally distinct clonal populations from glioblastoma specimens, have demonstrated the potential differential response to treatment of these various clones (73).
Both intra-tumoral and temporal heterogeneity in MGMT status have been reported in some (74) but not all (75) such investigations in glioma. Several authors have also demonstrated intra-tumoral heterogeneity of receptor tyrosine kinases (RTKs) in gliomas (76–78). Snuderl et al. (76) identified intermingled populations of tumor cells containing varying amplification of up to three different RTK genes (EGFR, MET, and PDGFRA), all of which were derived from a common precursor. Likewise, Szerlip et al. (77) found that multiple RTKs were likely to be maintaining distinct cell subpopulations. Little et al. (78) also observed a high degree of variability in gene copy number of EGFR and PDGFRA in individual cells across entire glioblastoma specimens.
More recently, Patel et al. performed single cell RNA sequencing on 430 cells from five primary glioblastomas, documenting intra-tumoral variation in the expression of a range of transcriptional programs, including oncogenic signaling, proliferation, immune response, and hypoxia (79).
Clinical Implications of Molecular Heterogeneity in Glioblastoma
Intra-tumoral heterogeneity has important implications for the development of prognostic and predictive biomarkers and their ability to guide personalized treatment regimens. Transcriptional profiling of glioblastoma into molecular subtypes is based on the average expression of genes across a sample irrespective of intra-tumoral heterogeneity (80) and may limit the usefulness of prognostic panels, such as DecisionDx-GBM®. Similarly, intra-tumoral molecular heterogeneity may underlie discordance between predictive markers, such as MGMT and treatment response.
Intra-tumoral heterogeneity may also contribute to the failure of targeted therapies in these patients. Glioblastomas possess genetic alterations that target three core signaling pathways, including the RTK/RAS/PI3K axis, p53/MDM2/MDM4 axis, and RB/CDK4/INK4A axis (7). However, targeting of these pathways with monotherapies (such as the first generation EGFR kinase inhibitors gefitinib and erlotinib) has failed to achieve any significant clinical benefit in the majority of patients (81, 82). Diversity of tumor subclones within the same individual, together with poor drug penetration and activation of other compensatory pathways in response to RTK inhibition (83, 84) is likely to account for this negative result. Molecular preselection of patients for clinical trials is challenging and tumor-sampling methods need to consider the regional diversity of tumor subclones. Technology is available to sequence genetic material from FFPE tumor blocks, however, information on the spatial orientation of specimens may not be available (80). Treatment of patients with more than one agent, targeting major clones may be possible but limited by drug toxicity and the ability of agents to penetrate the blood brain barrier (80).
The development of new treatment strategies for recurrent disease must consider temporal diversity of tumor clones, with molecular biomarkers assessed in recurrent tumor biopsies where possible, since they may differ substantially from initial diagnosis. Tumors harbor a complex hierarchy of clones, the proportions or dominance of which may change following therapy. Molecular biomarkers that are prognostic/predictive at initial diagnosis may not be informative at recurrence. For example, the TCGA transcriptional subtypes were determined using a cohort of primary biopsies and may not be robust classifiers of recurrent disease.
Monitoring of clonal dynamics during treatment and targeting of drug resistance clones may significantly improve patient outcomes. However, this may be difficult to implement in the clinic, with extensive/serial debulking not an option for glioblastoma patients. Serial measurements of circulating tumor DNA for tumor specific markers such as EGFRv111 (85), and the use of imaging modalities to non-invasively monitor molecular biomarkers may allow clinicians to monitor clonal evolution and treatment response.
Although major advances in sequencing approaches are enabling tumor heterogeneity to be studied at unprecedented resolution, the interpretation of results is still evolving, with the detection and the clinical significance of minor clones still in question. For example, heterogeneity may in some cases lead to treatment resistance. Clinically actionable mutations must be verified by independent analysis using an alternative technology, such as Sanger sequencing (86); however, this technology is limited by its sensitivity and may not detect important low frequency variants. Clinicians need to decide which mutations to prioritize for the purpose of treatment design. Alternatively, a measure of subclonal diversity, such as the Shannon index, which estimates the total number of tumor subclones and the relatively frequency of each clone, may itself may be a useful prognostic biomarker, as has been shown in the case of esophageal adenocarcinoma (87).
Conclusion
Our increased knowledge of molecular alterations in glioblastoma has improved patient diagnosis and identified many new potential therapeutic targets. Treatments directed at these specific alterations may improve patient outcomes and reduce toxicity. The introduction of molecular testing into routine diagnostic pathology will also enable clinicians to better stratify patients for clinical trials, selecting those that may benefit from targeted therapy.
While molecular profiling allows us to explore in depth, the biological characteristics of a tumor, performing this on a single biopsy specimen may be overly simplistic and inadequately reflect both the genomic landscape and overall behavior of a tumor. It is being increasingly recognized that glioblastoma contain multiple distinct populations of tumor cells with the potential to convey survival advantage and resistance to therapy, and these may be selected and enriched through successive cycles of treatment. However, many trials are conducted in relapsed patients whose tumors may have been altered by previous radiotherapy and alkylating agents which are known to cause additional mutations, with molecular preselection based on their original pathology specimen. Intra-tumoral heterogeneity, together with the concomitant activation of multiple oncogenic signaling pathways in glioblastoma, suggests that a single agent will be ineffective in treating this disease. Importantly, next generation sequencing technology may enable the identification of molecular alterations occurring early in gliomagenesis, the so-called “driver events,” which may represent better targets for the design of therapeutic regimes.
Glioblastoma poses some unique challenges to clinicians – tumor cells diffusely invade surrounding normal brain parenchyma, making complete surgical resection impossible, and the efficacy of therapeutic agents are often limited by the blood brain barrier. Resampling of tumors following treatment is also limited. The development of non-invasive imaging techniques, such as those associated with PET scanning, and identification of circulating tumor cells harboring molecular biomarkers may enable clinicians to non-invasively monitor clonal dynamics during treatment and the development of disease recurrence.
The link between glioblastoma molecular subtype and response to treatment is still incompletely understood. While the answer may be gleaned from the molecular analysis of multiple biopsies at each initial and subsequent resection, the use of such information is still yet to prove beneficial in conveying any meaningful survival advantage to those suffering from glioblastoma.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
NP was funded by the Sydney Neuro-Oncology Group; PK was funded by an Avant Scholarship.
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114(2):97–109. doi: 10.1007/s00401-007-0278-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer (2013) 108(3):479–85. doi:10.1038/bjc.2012.581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Kleihues P, Cavenee WK. Pathology and Genetics of Tumours of the Nervous System. Lyon: International Agency for Research on Cancer (2000).
4. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res (2004) 64(19):6892–9. doi:10.1158/0008-5472.CAN-04-1337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi:10.1016/j.ccr.2009.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell (2006) 9(3):157–73. doi:10.1016/j.ccr.2006.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi:10.1016/j.cell.2013.09.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell (2010) 17(5):510–22. doi:10.1016/j.ccr.2010.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi:10.1126/science.1164382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol (2013) 126(3):443–51. doi:10.1007/s00401-013-1156-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol (2011) 29(34):4482–90. doi:10.1200/JCO.2010.33.8715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature (2014) 512(7514):324–7. doi:10.1038/nature13387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol (2009) 27(35):5874–80. doi:10.1200/JCO.2009.23.6497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol (2013) 31(3):337–43. doi:10.1200/JCO.2012.43.2674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol (2013) 31(3):344–50.
16. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget (2012) 3(7):709–22.
17. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462(7274):739–44.
18. Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol (2013) 16(5):686–95. doi:10.1093/neuonc/not243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol (2010) 12(1):49–57.
20. Allingham-Hawkins D, Lea A, Levine S. DecisionDx-GBM gene expression assay for prognostic testing in glioblastoma multiform. PLoS Curr (2010) 2:RRN1186. doi:10.1371/currents.RRN1186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96.
22. Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer (2009) 101(1):124–31. doi:10.1038/sj.bjc.6605127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP, et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol (2010) 12(2):116–21.
24. Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, et al. MGMT testing-the challenges for biomarker-based glioma treatment. Nat Rev Neurol (2014) 10(7):372–85. doi:10.1038/nrneurol.2014.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med (2005) 352(10):997–1003.
26. Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res (2000) 60(9):2368–71.
27. Herrlinger U, Rieger J, Koch D, Loeser S, Blaschke B, Kortmann RD, et al. Phase II trial of lomustine plus temozolomide chemotherapy in addition to radiotherapy in newly diagnosed glioblastoma: UKT-03. J Clin Oncol (2006) 24(27):4412–7.
28. Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German glioma network. J Clin Oncol (2009) 27(34):5743–50. doi:10.1200/JCO.2009.23.0805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol (2012) 13(7):707–15.
30. Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol (2012) 13(9):916–26. doi:10.1016/S1470-2045(12)70265-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Shinsato Y, Furukawa T, Yunoue S, Yonezawa H, Minami K, Nishizawa Y, et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget (2013) 4(12):2261–70.
32. Nguyen SA, Stechishin OD, Luchman HA, Lun XQ, Senger DL, Robbins SM, et al. Novel MSH6 mutations in treatment-naive glioblastoma and anaplastic oligodendroglioma contribute to temozolomide resistance independently of MGMT promoter methylation. Clin Cancer Res (2014) 20(18):4894–903. doi:10.1158/1078-0432.CCR-13-1856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer (2011) 129(3):659–70.
34. Stark AM, Doukas A, Hugo HH, Mehdorn HM. The expression of mismatch repair proteins MLH1, MSH2 and MSH6 correlates with the Ki67 proliferation index and survival in patients with recurrent glioblastoma. Neurol Res (2010) 32(8):816–20. doi:10.1179/016164110X12645013515052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Everhard S, Tost J, El Abdalaoui H, Criniere E, Busato F, Marie Y, et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro Oncol (2009) 11(4):348–56. doi:10.1215/15228517-2009-001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Grombacher T, Mitra S, Kaina B. Induction of the alkyltransferase (MGMT) gene by DNA damaging agents and the glucocorticoid dexamethasone and comparison with the response of base excision repair genes. Carcinogenesis (1996) 17(11):2329–36. doi:10.1093/carcin/17.11.2329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Fritz G, Tano K, Mitra S, Kaina B. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol Cell Biol (1991) 11(9):4660–8.
38. Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer (2004) 4(4):296–307. doi:10.1038/nrc1319
39. Brell M, Ibanez J, Tortosa A. O6-Methylguanine-DNA methyltransferase protein expression by immunohistochemistry in brain and non-brain systemic tumours: systematic review and meta-analysis of correlation with methylation-specific polymerase chain reaction. BMC Cancer (2011) 11:35. doi:10.1186/1471-2407-11-35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol (2010) 6(1):39–51. doi:10.1038/nrneurol.2009.197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Patel M, Vogelbaum MA, Barnett GH, Jalali R, Ahluwalia MS. Molecular targeted therapy in recurrent glioblastoma: current challenges and future directions. Expert Opin Investig Drugs (2012) 21(9):1247–66. doi:10.1517/13543784.2012.703177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res (2005) 11(4):1462–6. doi:10.1158/1078-0432.CCR-04-1737
43. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol (2007) 25(16):2288–94. doi:10.1200/JCO.2006.08.0705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hegi ME, Rajakannu P, Weller M. Epidermal growth factor receptor: a re-emerging target in glioblastoma. Curr Opin Neurol (2012) 25(6):774–9. doi:10.1097/WCO.0b013e328359b0bc
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Gallego Perez-Larraya J, Ducray F, Chinot O, Catry-Thomas I, Taillandier L, Guillamo JS, et al. Temozolomide in elderly patients with newly diagnosed glioblastoma and poor performance status: an ANOCEF phase II trial. J Clin Oncol (2011) 29(22):3050–5.
46. Del Vecchio CA, Wong AJ. Rindopepimut, a 14-mer injectable peptide vaccine against EGFRvIII for the potential treatment of glioblastoma multiforme. Curr Opin Mol Ther (2010) 12(6):741–54.
47. Babu R, Adamson DC. Rindopepimut: an evidence-based review of its therapeutic potential in the treatment of EGFRvIII-positive glioblastoma. Core Evid (2012) 7:93–103.
48. Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, et al. Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer (2014) 134(10):2437–47. doi:10.1002/ijc.28576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proceedings of the National Academy of Sciences of the United States of America (2013) 110(15):6021–6.
50. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A (2013) 110(10):4009–14. doi:10.1073/pnas.1219747110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta (2010) 1805(1):105–17. doi:10.1016/j.bbcan.2009.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. De Sousa EMF, Vermeulen L, Fessler E, Medema JP. Cancer heterogeneity – a multifaceted view. EMBO Rep (2013) 14(8):686–95. doi:10.1038/embor.2013.92
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Nickel GC, Barnholtz-Sloan J, Gould MP, McMahon S, Cohen A, Adams MD, et al. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS One (2012) 7(4):e35262. doi:10.1371/journal.pone.0035262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Giese A, Westphal M. Treatment of malignant glioma: a problem beyond the margins of resection. J Cancer Res Clin Oncol (2001) 127(4):217–25. doi:10.1007/s004320000188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Mitchell P, Ellison DW, Mendelow AD. Surgery for malignant gliomas: mechanistic reasoning and slippery statistics. Lancet Neurol (2005) 4(7):413–22.
56. Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med (2005) 353(8):811–22. doi:10.1056/NEJMra043666
57. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol (2006) 7(5):392–401.
58. Nowell PC. The clonal evolution of tumor cell populations. Science (1976) 194(4260):23–8. doi:10.1126/science.959840
59. Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer (2010) 103(8):1139–43. doi:10.1038/sj.bjc.6605912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med (2012) 366(10):883–92. doi:10.1056/NEJMoa1113205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Sprouffske K, Merlo LM, Gerrish PJ, Maley CC, Sniegowski PD. Cancer in light of experimental evolution. Curr Biol (2012) 22(17):R762–71. doi:10.1016/j.cub.2012.06.065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339(6127):1546–58. doi:10.1126/science.1235122
63. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res (2012) 72(19):4875–82.
64. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi:10.1038/35102167
65. Cheshier SH, Kalani MY, Lim M, Ailles L, Huhn SL, Weissman IL. A neurosurgeon’s guide to stem cells, cancer stem cells, and brain tumor stem cells. Neurosurgery (2009) 65(2):237–49. doi:10.1227/01.NEU.0000349921.14519.2A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Larjavaara S, Mantyla R, Salminen T, Haapasalo H, Raitanen J, Jaaskelainen J, et al. Incidence of gliomas by anatomic location. Neuro Oncol (2007) 9(3):319–25. doi:10.1215/15228517-2007-016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Lyons JG, Lobo E, Martorana AM, Myerscough MR. Clonal diversity in carcinomas: its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin Exp Metastasis (2008) 25(6):665–77.
68. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res (2011) 71(12):4055–60. doi:10.1158/0008-5472.CAN-11-0153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer (2012) 12(5):323–34. doi:10.1038/nrc3261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell (2007) 11(1):69–82. doi:10.1016/j.ccr.2006.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Hu YY, Fu LA, Li SZ, Chen Y, Li JC, Han J, et al. HIF-1α and HIF-2α differentially regulate Notch signaling through competitive interaction with the intracellular domain of Notch receptors in glioma stem cells. Cancer Lett (2014) 349(1):67–76.
72. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science (2014) 343(6167):189–93. doi:10.1126/science.1239947
73. Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M, et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A (2015) 112(3):851–6. doi:10.1073/pnas.1320611111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Parkinson JF, Wheeler HR, Clarkson A, McKenzie CA, Biggs MT, Little NS, et al. Variation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation in serial samples in glioblastoma. J Neurooncol (2008) 87(1):71–8.
75. Grasbon-Frodl EM, Kreth FW, Ruiter M, Schnell O, Bise K, Felsberg J, et al. Intratumoral homogeneity of MGMT promoter hypermethylation as demonstrated in serial stereotactic specimens from anaplastic astrocytomas and glioblastomas. Int J Cancer (2007) 121(11):2458–64. doi:10.1002/ijc.23020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell (2011) 20(6):810–7. doi:10.1016/j.ccr.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A (2012) 109(8):3041–6. doi:10.1073/pnas.1114033109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Little SE, Popov S, Jury A, Bax DA, Doey L, Al-Sarraj S, et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res (2012) 72(7):1614–20. doi:10.1158/0008-5472.CAN-11-4069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344(6190):1396–401. doi:10.1126/science.1254257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature (2013) 501(7467):355–64.
81. Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE II, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol (2010) 96(2):219–30. doi:10.1007/s11060-009-9950-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European organisation for research and treatment of cancer brain tumor group study. J Clin Oncol (2008) 26(28):4659–65.
83. Mellinghoff IK, Schultz N, Mischel PS, Cloughesy TF. Will kinase inhibitors make it as glioblastoma drugs? Curr Top Microbiol Immunol (2012) 355:135–69. doi:10.1007/82_2011_178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American brain tumor consortium trial 04-02. Neuro Oncol (2014) 16(4):567–78.
85. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol (2008) 10(12):1470–6. doi:10.1038/ncb1800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol (2012) 30(11):1033–6.
87. Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet (2006) 38(4):468–73. doi:10.1038/ng1768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: glioma, glioblastoma, heterogeneity, molecular, intra-tumoral heterogeneity, biomarkers, transcriptional subtype
Citation: Parker NR, Khong P, Parkinson JF, Howell VM and Wheeler HR (2015) Molecular heterogeneity in glioblastoma: potential clinical implications. Front. Oncol. 5:55. doi: 10.3389/fonc.2015.00055
Received: 20 January 2015; Accepted: 18 February 2015;
Published online: 03 March 2015.
Edited by:
Kerrie Leanne McDonald, University of NSW, AustraliaReviewed by:
Justin Lathia, Lerner Research Institute, USAHan Shen, University of New South Wales, Australia
Copyright: © 2015 Parker, Khong, Parkinson, Howell and Wheeler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viive Maarika Howell, Bill Walsh Translational Cancer Research Laboratory, Kolling Institute of Medical Research, Royal North Shore Hospital, University of Sydney, E25, Level 8 Kolling Building, St Leonards, NSW 2065 Australia e-mail: viive.howell@sydney.edu.au
†Nicole Renee Parker and Peter Khong are joint first authors.
‡Viive Maarika Howell and Helen Ruth Wheeler have contributed equally to this work.