 Simone Patergnani
Simone Patergnani Sonia Missiroli
Sonia Missiroli Saverio Marchi
Saverio Marchi Carlotta Giorgi
Carlotta Giorgi- Laboratory for Technologies of Advanced Therapies (LTTA), Section of Pathology, Oncology and Experimental Biology, Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, Ferrara, Italy
Autophagy is a tightly regulated catabolic pathway that terminates in the lysosomal compartment after the formation of a cytoplasmic vacuole that engulfs macromolecules and organelles. Notably, autophagy is associated with several human pathophysiological conditions, playing either a cytoprotective or cytopathic role. Many studies have investigated the role of autophagy in cancer. However, whether autophagy suppresses tumorigenesis or provides cancer cells with a rescue mechanism under unfavorable conditions remains unclear. Mitochondria-associated membranes (MAMs) are juxtaposed between the endoplasmic reticulum and mitochondria and have been identified as critical hubs in the regulation of apoptosis and tumor growth. One key function of MAMs is to provide asylum to a number of proteins with tumor suppressor and oncogenic properties. Accordingly, mechanistic studies during tumor progression suggest a strong involvement of these proteins at various steps of the autophagic process. This paper discusses the present state of our knowledge about the intimate molecular networks between MAMs and autophagy in cancer cells and addresses how these networks might be manipulated to improve anticancer therapeutics.
Cell Death and Autophagy
Cell death is a fundamental biological process that is highly evolutionarily conserved and occurs in all multicellular organisms during the course of normal development and during adulthood. Programmed cell death (PCD) is required to maintain tissue homeostasis, to remove abnormal or damaged cells, and to remodel and sculpt tissues during morphogenesis (1, 2).
Perturbations to the normal patterns and regulation of cell death can contribute to several pathologies, such as neurodegenerative diseases, autoimmune diseases, and cancer. Not surprisingly, more than one form of cell death exists. Caspase-dependent apoptosis is considered the principal pathway in mammals. However, various additional forms of cell death have been described more recently and include necroptosis, pyroptosis, and autophagic cell death (3, 4).
Apoptosis and macroautophagy (hereafter, referred to as autophagy) play major roles in determining cellular fate and are discrete cellular processes that are mediated by distinct groups of regulatory and executioner molecules (4–6).
Apoptosis is the best-described PCD mechanism and is characterized by a series of morphological changes and plasma membrane blebbing, leading to the formation of apoptotic bodies.
The molecular biology of apoptosis has been studied over the last 40 years. The two most prominent molecular mechanisms that regulate the cell death process are the extrinsic and intrinsic pathways (7).
In the extrinsic pathway, the ligation of ligands (e.g., FasL, also known as CD95L), tumor necrosis factor-α (TNF-α), TNF-α-related apoptosis-inducing ligand (TRAIL, also known as APO2L), or TNF-α ligand superfamily member 10 (TNFSF10), with their respective death receptors, stimulate the recruitment of adaptor proteins, such as Fas-associated via death domain (FADD), which then recruits the initiator procaspase-8 or -10, causing their dimerization and activation (8).
Unlike the extrinsic pathway that regulates apoptosis through specialized death signals, the intrinsic pathway (also known as “mitochondrial apoptosis”) is mediated by several intracellular and extracellular signals, including endoplasmic reticulum (ER) stress, loss of survival/trophic factors, toxins, radiation, hypoxia, oxidative stress, ischemia–reperfusion injury, and DNA damage. As a result, these signals converge on mitochondria to induce mitochondrial outer membrane permeabilization (MOMP), which contributes to multiple redundant lethal events, including caspase cascades, the activation of caspase-independent pathways for cellular dismantling and bioenergetic failure (9, 10) via mitochondrial permeability transition pore opening (11). In particular, MOMP proapoptotic factors (apoptogens) that are normally found in the intermembrane space (IMS) are irreversibly released into the cytoplasm. A critical apoptogen that is released from the intermembrane mitochondria space is cytochrome c (Cyt c). In the cytosol, Cyt c induces the apoptosis protease-activating factor 1 (APAF-1) and ATP/dATP to assemble the apoptosome, a molecular platform that promotes the proteolytic maturation of caspase-9. Finally, caspase-9 recruits and activates effector caspases.
Autophagy is a lysosome-mediated, multistep self-degradation process and is distinct from other degradative pathways such as proteasomal degradation, in which components are degraded to supply energy (12, 13). During autophagy, an isolation membrane sequesters a small portion of the cytoplasm, including cytosolic materials and organelles, to form the autophagosome. Autophagosomes fuse with lysosomes to yield autolysosomes, which degrade internalized materials.
This process is executed and regulated by a large group of distinct autophagy-related (Atg) proteins, which direct the four major steps of the autophagic machinery: initiation, nucleation, cycling, and expansion/closure. More than 30 Atg genes are known. Readers interested in the specialized role of Atg proteins should consult recent reviews (14–16). The cofactors involved in autophagosome processing are described as follows. Upon induction of autophagy, the main negative regulator of the catabolic process, MTOR (mechanistic target of rapamycin), is suppressed and inactivated. This event activates the ULK1 (unc-51 like kinase 1)-complex (including ULK1, Atg13, FIP200, and Atg101), resulting in the ULK1-dependent phosphorylation of Atg13, FIP200, and ULK1 itself and translocation of the complex to the ER, the major site of autophagosome formation (17). At these activation levels, ULK1 also regulates the activity of the class III phosphatidylinositol (PtdIns) 3-kinase complex (including Beclin1, Atg14(L)/barkor, Vps15, Vps34, and Ambra1). This complex generates PI3P, which interacts with DFCP1, Atg2, and WIPI1, and recruits other Atg proteins involved in membrane elongation. Additionally, other complexes are fundamental for the autophagosome elongation, such as two ubiquitin-like proteins. Atg7 and Atg10 mediate the association of the Atg5–Atg12 complex with Atg16L1. The Atg12–Atg5–Atg16L1 complex and the LC3 (Atg8 homolog)–phosphatidylethanolamine (PE) conjugate play important roles in the elongation and closure of the isolation membrane. The Atg12–Atg5–Atg16L1 complex is also required for the formation of a covalent bond between LC3 and PE. When autophagosome formation is concluded, the outer autophagosomal membrane fuses with lysosomes, releasing autophagic cargo into the lysosomal lumen (18).
Basal levels of autophagy are important for maintaining cellular homeostasis. The physiological importance of autophagy in maintaining tissue homeostasis has been demonstrated in different organs, including brain, liver, heart, muscle, kidney, and adipose tissue (12, 19, 20). Nutrient deprivation is the main activator for autophagy induction, but is not the only mechanism. For example, decreased levels of specific amino acids and increased levels of glucocorticosteroids and thyroid hormones also stimulate the catabolic process (21). Furthermore, it is clear that autophagy is an important energy generator. Indeed, amino acids obtained by autophagy can be converted into intermediates of the tricarboxylic acid (TCA) cycle and contribute to ATP production. The breakdown of lipid droplets by autophagy (i.e., lipophagy) may also account for this energy-producing role, especially in the liver (22). At molecular levels, nutrient failure modulates the activity of important energy-sensing proteins. One of these proteins is AMPK (5′ AMP-activated protein kinase), a conserved sensor of intracellular adenosine nucleotide levels. AMPK is activated when levels of AMP or ADP increase in response to slight decreases in ATP production. This kinase is significantly involved in autophagic regulation via mechanisms wherein MTOR activity is suppressed. Furthermore, MTOR regulates mitochondrial ATP production and determines the health of the mitochondrial population by a process termed mitophagy. Previous studies have shown that these events are also regulated by AMPK-dependent MTOR inactivation (23).
In addition, a function for autophagy in cell death has long been proposed. Cell death is often attributed with high levels of autophagosomes and active autophagy, and hence, the term “autophagic cell death” is often used.
Many publications have reported that death stimuli in mammalian cells and non-mammalian systems are caused by autophagy (24). However, it is unclear whether autophagy occurs prior to apoptosis or during the dismantling of cellular mechanisms (25).
Furthermore, autophagy has been implicated in cell death for various pathological conditions, such as neurodegeneration, immunity, and aging and especially in cancer (26).
Tumor-Suppressing and Tumor-Promoting Roles of Autophagy in Cancer
A number of recent studies have revealed that autophagy can function as either a pro-death or pro-survival mechanism in cancer cells. Depending on the different stages of tumor development and the cell type, autophagy acts in two opposing capacities. During early stages of cancer, autophagy has preventive effects; however, after sufficient tumor development, cancer cells utilize autophagy mechanisms to provide energy for sustained growth (Table 1).
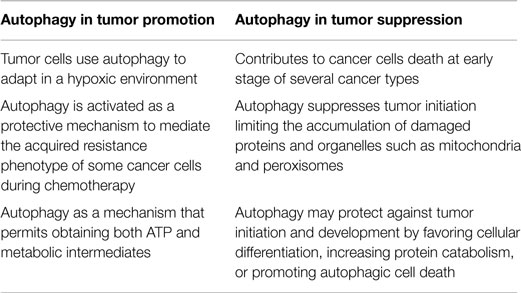
Table 1. Functions of autophagy in cancer.
The role of autophagy as a potent tumor suppressor mechanism was inferred from the observation that its molecular pathways were frequently ablated in tumor cells.
During tumor initiation, autophagy is a protective process that limits the accumulation of harmful proteins and organelles through its intrinsic quality control activities. These tumor suppressor properties are primarily attributed to protection against genotoxic stress, such as the elimination of reactive oxygen species (ROS) (27). Autophagy-mediated ROS removal inhibits the deleterious effect of ROS on DNA mutations that have been extensively shown to induce tumorigenesis (28). Autophagy primarily prevents ROS accumulation through the elimination of damaged mitochondria via a selective form of autophagy (i.e., mitophagy) (29). The tumor-suppressing roles of autophagy are also related to the regulation of the inflammatory process, which has been compared with the initiation of cancer (30–32).
The role of autophagy in tumor suppression can also be observed during the upregulation of members of the phosphatidylinositol 3-kinase (PI3K) family to similar levels as the MTOR and AKT kinases (33, 34). These positive regulators of cell growth inhibit PTEN (phosphatase and tensin homolog deleted on chromosome 10) activity and other tumor suppressors, such as LKB1 (liver kinase B1). These proteins are deregulated in cancer cells and are potent activators of the autophagic machinery (35).
Furthermore, AKT inhibits autophagy in an MTOR-independent manner via the phosphorylation of the essential autophagy and tumor suppressor protein BECLIN-1 (36). A BECLIN-1 mutation has been shown to confer resistance to AKT-dependent phosphorylation, increased autophagy, and inhibited AKT-driven tumorigenesis (36). As for AKT, the implications in cancer of the oncoprotein BCL (B-cell lymphoma)-2 and its antiapoptotic homologs (e.g., BCL-XL and MCL-1) are attributed to their regulatory roles in cell death and the autophagic process. In particular, antiapoptotic BCL-2 family members have been shown to suppress autophagy through their interaction with or inhibition by BECLIN-1 (37, 38). Thus, the inhibition of autophagy contributes to the transformation of a normal cell into a cancerous cell and favors a state of genomic instability, i.e., the ideal trigger for tumor initiation.
Another protagonist of this signaling pathway involving BCL-2 family members during autophagy is the putative tumor suppressor AMBRA1 (autophagy/beclin-1 regulator 1), which is a crucial factor in regulating autophagy in vertebrates. AMBRA-1 enhances the activity of BECLIN-1, and thus mediates autophagosome nucleation (39, 40).
BECLIN-1-mediated autophagy is negatively regulated through a direct interaction between BECLIN-1 and ER-BCL-2. Past studies have shown that the AMBRA-1-BECLIN-1 complex re-localizes to the ER upon autophagy induction and that AMBRA-1 is able to bind to the antiapoptotic factor BCL-2 (41).
A further potential molecular link between defective autophagy and tumorigenesis involves the autophagic-dependent protein p62/SQSTM1, responsible for removing damaged organelles and misfolded proteins that may cause DNA damage and genomic instability. p62/SQSTM1−/− mice have been shown to be protected from RAS-induced lung carcinomas.
The predominant role of autophagy in tumor cells is to confer stress resistance and thereby maintain cancer cell survival. Indeed, tumor cells may use autophagy to increase the supply of limited nutrients. In addition, autophagy has been induced in hypoxic tumor cells distal from blood vessels, and cancer cells have been shown to upregulate the autophagic process to prevent metabolic stress induced by chemical and radiological therapies. Based on these studies, there are a number of potential anticancer approaches targeting the autophagic process. As such, autophagy-modulating agents have been studied in preclinical models (Table 2) (42).
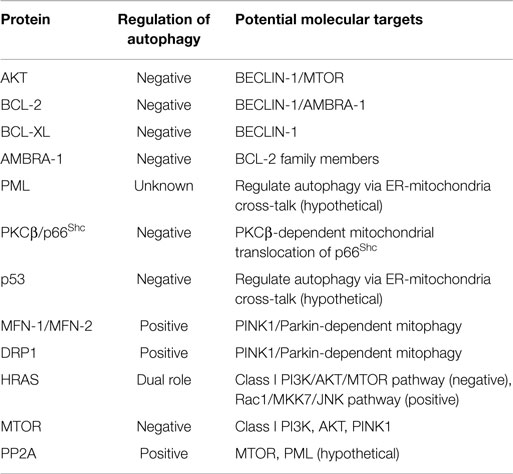
Table 2. MAM cofactors regulating autophagic machinery.
In particular, the most promising anticancer agents are autophagy inhibitors (such as chloroquine, CQ, and its derivative hydroxychloroquine, HCQ), which are able to enhance cell cytotoxicity when combined with different anticancer drugs (Table 3) (43). Interestingly, several studies have shown how rapamycin and its analogs, i.e., allosteric MTOR inhibitors, in combination with autophagy inhibitors (e.g., HCQ, bafilomycin A1, and methyladenine) increased in vitro and in vivo cytotoxicity of human cancers (44, 45). The efficacy of this combination may be explained as follows. The aberrant expressions of PI3K/AKT/MTOR are found in several tumor types and are important to the initiation and progression of cancers. These cofactors are critical negative regulators of the autophagic machinery. When their activity or expression is blocked, autophagy may be induced and may trigger consequent autophagic-related resistance that promotes cancer cell survival. The resistance of cancer cells to AKT/MTOR inhibitors is a significant issue in a number of different tumor cells, such as relapsed mantle cell lymphomas. Pretreatments with autophagic inhibitors can effectively overcome this resistance by inhibiting AKT/MTOR activity, blocking the autophagic process, and activating the apoptotic pathway. The targeting of the PI3K/AKT/MTOR autophagy pathways can overcome cancer cell resistance to chemotherapy and radiotherapy. However, the CQ and HCQ studies should be interpreted carefully because the reported effects may be mediated by mechanisms unrelated to autophagy inhibition. For example, a recent study showed that a CQ treatment reduced hypoxia and cancer cell metastasis and improved chemotherapy efficacy rates and responses in an autophagic-independent manner (46).
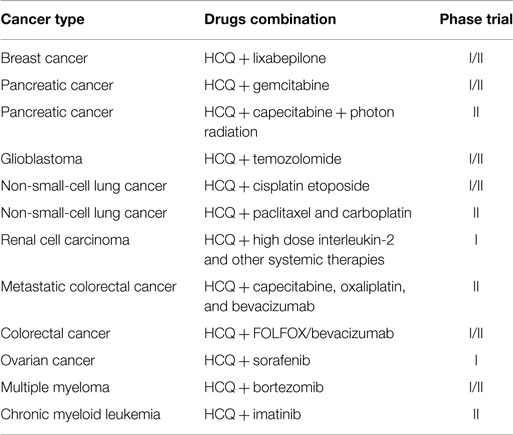
Table 3. Some of the clinical trials combining the autophagy inhibitor HCQ.
Recently, various cancer and autophagy-related factors, including the aforementioned proteins, have been shown to localize at mitochondria-associated ER membranes (MAMs), which are the primary intracellular platforms that detect extracellular inputs and stressful conditions (47, 48). We next discuss the roles of oncogenes and oncosuppressors at MAMs in autophagy and consider the structural functions of the ER-mitochondria interface in the regulation of autophagy (49).
MAM Structure
MAMs are ER membranes that are similar to mitochondria. However, MAMs are not simply static bridges between the ER and mitochondria. On the contrary, interorganelle communication between the ER and mitochondria is crucial for processes such as lipid synthesis and transport, mitochondrial functions, the regulation of calcium homeostasis, and apoptosis (48). A large number of ER and mitochondria-associated proteins have been identified in MAMs, demonstrating the crucial involvement of MAMs in all physiological processes (Figure 1A). These proteins include various chaperones [e.g., glucose-regulated protein 75 (Grp75)], enzymes involved in ER redox regulation [e.g., ER oxidoreductase 1 alpha (Ero1α)] and protein kinases [e.g., ER stress sensor double-stranded RNA-activated protein kinase (PKR)-like ER kinase (PERK)]. Other proteins are also responsible for regulating mitochondrial dynamics and morphology, such as the mitofusins MFN-1 and MFN-2 and the dynamin-like protein DRP1 (50).
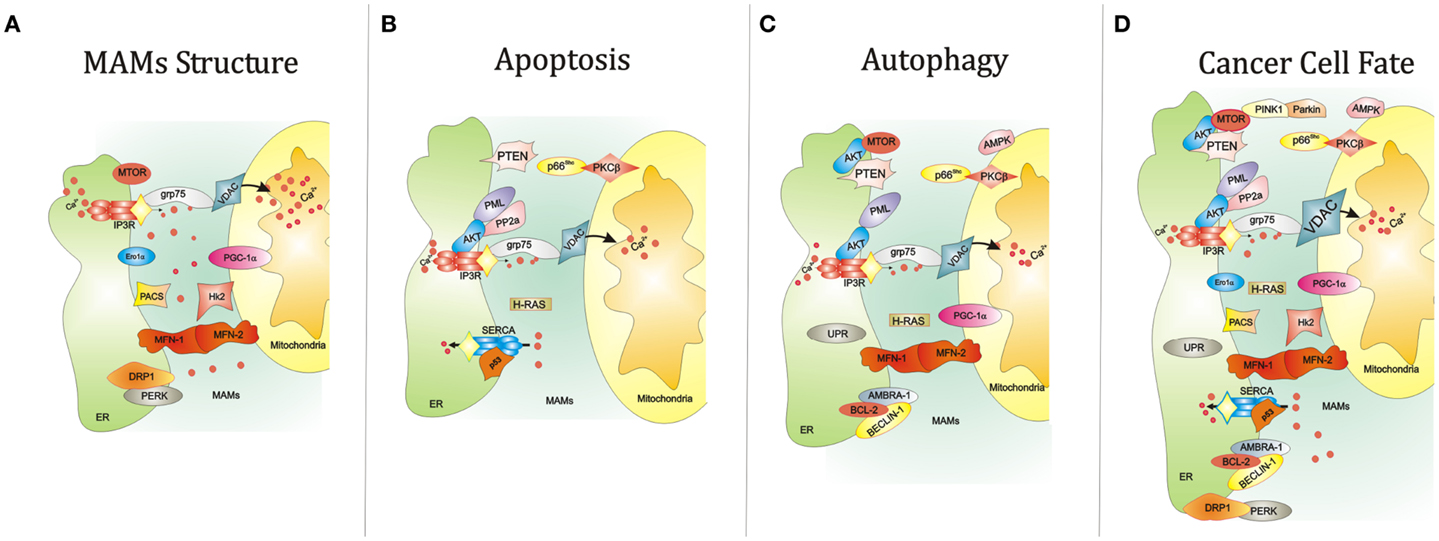
Figure 1. Summary of the key functions of the ER-MAMs resident proteins. (A) Several proteins reside in MAMs compartment and regulate the juxtaposition between ER and mitochondria, including grp75, Ero1α, PERK, and MFN-1/2. At the same time, other MAM resident proteins control the cell survival by governing apoptosis (B) and autophagy (C). For example, it has been reported that PML, IP3R3, and AKT mutually interact to allow the correct Ca2+-movement between ER and mitochondria, an essential proapoptotic signal. Interestingly, the maintenance of this interorganelle Ca2+-communication is also important for the autophagic process. Of relevance, reductions of mitochondrial Ca2+ accumulation may trigger autophagy. In parallel, most of MAM proteins also govern the autophagic machinery. Of relevance, AKT and MTOR regulate negatively autophagic process. The same work is executed by the molecular axis composed by p66Shc and PKCβ. It is widely accepted that autophagy is strictly linked to several human diseases, in particular cancer. Think about a possible link between autophagy, cancer, and MAMs is not so difficult. Several findings may suggest this. For example, the corrected maintenance of MAMs integrity by MFN-1/-2 and DRP1 is also crucial for autophagosome formation and tumor cell growth. (D) The autophagic regulator AMBRA-1 has been found to interact with both the oncogene BCL-2 and the tumor suppressor BECLIN-1. Again, ER stress mediated by UPR a potent autophagic activator: at the same time ER stress is a critical signal capable to drive cell death. Also, the activity of the main proteins involved in Ca2+ release and reuptake at MAM levels, SERCA and IP3R3, is reported to be involved in apoptosis and tumorigenesis. Interestingly, the functioning of these channels is intimately regulated by several oncogenes (like AKT) and tumor suppressor (such as p53), which are also involved in regulation of autophagy. Abbreviations: grp75, glucose-regulated protein 75; IP3R3, inositol 1,4,5-trisphosphate receptor type 3; MFN-1/-2, mitofusin-1/-2; Hk2, Hexokinase 2; PACS, phosphofurin acidic cluster sorting protein; VDAC, voltage-dependent anion-selective channel; Ca2+, calcium; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; PTEN, phosphatase and tensin homolog; p66Shc, 66 kDa proto-oncogene Src homologous-collagen homolog; PKCβ, protein kinase C beta; PP2a, protein phosphatase 2; AKT, protein kinase B; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase; MTOR, mechanistic target of rapamycin; PML, promyelocytic leukemia; UPR, unfolded protein response; PINK1, PTEN-induced putative kinase 1; PERK, protein kinase-like ER kinase; DRP1, dynamin-related protein; Ero1a, ER oxidoreductase 1 alpha; IP3R3, inositol 1,4,5-trisphosphate receptor type 3; AMBRA-1, Beclin1-regulated autophagy; BCL-2, B-cell lymphoma 2; BECLIN-1, BCL-2-interacting protein;, AMPK, 5′ adenosine monophosphate-activated protein kinase).
The ER is also the major site of Ca2+ storage in mammalian cells. The movement of Ca2+ between the ER and mitochondria is an essential component of the cell survival processes.
Several studies have examined the distribution of Ca2+ between the ER and mitochondria (51–53). A study by Csordas and colleagues provided direct evidence for the existence of high-Ca2+ microdomains at ER-mitochondria contact sites that are regulated by the area and gap width of ER-mitochondria interconnections. Furthermore, they described a novel approach for determining the [Ca2+]ER-mt in microdomains (54).
Based on these studies, MAMs regulate several Ca2+-dependent cellular processes. The movement of Ca2+ between the ER and mitochondria is essential for the correct execution of both apoptotic (55) and autophagy pathways (56). Several Ca2+ channels regulating the apoptotic program are located in the MAM compartment. For example, IP3R3 (located at the ER) and VDAC (situated at the outer mitochondrial membrane) are highly concentrated in MAMs (57).
Furthermore, growing bodies of evidence shows that the ER-mitochondria contact sites contribute to autophagosome formation and that proteins in the MAM compartments are indispensable for proper autophagic vesicle formation (49, 58–60).
MAMs, Novel Regulators of Cancer Cell Fate?
Although the ER–mitochondria interface is known to assist the autophagic pathway, the presumed link between the regulation of the autophagy/MAMs axis and cancer progression has not been addressed.
The role of MAMs in the regulation of apoptotic cell death in cancer is partially understood. In particular, several proteins are known to exert their pro- and antiapoptotic functions between the ER and mitochondria (57) (Figure 1B).
Furthermore, most of these proteins possess tumor suppressor and oncogenic properties and are mutated or deleted in various types of human cancers.
One classical example is the tumor suppressor PML (promyelocytic leukemia protein). This protein performs its proapoptotic functions through the assemblage of nuclear macromolecular structures called PML nuclear bodies (PML-NBs) (61). A recent study found that this protein localizes to the ER and MAMs, where it regulates Ca2+-dependent apoptosis by blocking the activity of IP3R3 (62). PML is one component of a complex composed of PP2A (protein phosphatase 2), IP3R3 (inositol 1,4,5-trisphosphate receptor, type 3), and AKT. Several studies have demonstrated that AKT mediates the phosphorylation of IP3R3 with a consequent decrease in Ca2+ transfer from the ER to the mitochondria (63–65).
The significant accumulation of mitochondrial Ca2+ amplifies the apoptotic signal. Key proteins (such as IP3R3, VDAC, and the permeability transition pore-complex) have also been demonstrated to be critical components for the ER-triggered, proapoptotic mitochondrial membrane permeabilization process (66).
Other tumor suppressors have also been observed to localize in the MAM compartment and regulate Ca2+ flux and apoptosis by regulating AKT activity.
In fact, PTEN was recently shown to interact with the AKT/IP3R complex, leading to a reduction in its phosphorylation and an increase in Ca2+ release (67).
In addition, the most studied tumor suppressor (p53) regulates tumorigenesis via a Ca2+-dependent pathway. A recent study found that p53 localizes to the ER and MAMs, where it modulates the ER-mitochondria cross-talk and the Ca2+ transfer from the ER to the mitochondria. As a result, mitochondria accumulate significant amounts of Ca2+, leading to an alteration in the morphology of this organelle and the induction of apoptosis (68, 69).
Other indications of the close relationship between MAMs and cell death can be found in the link between the SHC1 gene (encoding for the p66Shc protein) and the putative oncogene PRKCB (encoding for the protein kinase C, beta, PKCβ). p66Shc is a MAM protein (70) that is regulated by ROS and involved in the regulation of Ca2+ homeostasis (71, 72). Once activated, p66Shc translocates into mitochondria where it is reported to influence the cell life span and apoptosis through the perturbation of mitochondrial functions. PKCβ is required for the phosphorylation and activation of p66Shc at Ser36 and for its translocation into the mitochondrial compartment (72).
One of the most mutated oncogenes in human cancer, H-RAS, is localized in MAMs, where it exerts its antiapoptotic function by modulating Ca2+-dependent apoptosis. Notably, the activation of oncogenic H-RAS leads to a perturbation in Ca2+-handling and dysfunction in mitochondrial physiology (73).
Intriguingly, these proteins are also known to regulate the autophagic pathway. Thus, the localization of these players in the MAM compartment could be important for the regulation of autophagy (Figure 1C).
Okadaic acid (OA) is a PP2A-blocking drug. Several works have identified a direct role for this compound in the regulation of autophagy (74–76).
Interestingly, PML maintains an appropriate Ca2+ flux in the MAM compartment by mediating the recruitment of PP2A (62). Given that treatment with OA interferes with this molecular pathway, PML may regulate autophagy via ER-mitochondria cross-talk. The same issue is relevant to AKT. Several class I PI3K inhibitors (such as LY294002 and wortmannin) negatively regulate autophagy through AKT inhibition (77). Preclinical studies have also suggested the use of these compounds in cancer treatment (78). The main inhibitor of the MTOR kinase, rapamycin, was found to regulate AKT, and its use has been approved in several clinical trials. Considering the important role of regulating AKT activity to modulate cell death during cancer, the specific and direct roles of MAM compartment-localized AKT needs to be addressed (78). In addition, AKT may be directly phosphorylated and activated by MTOR.
MTOR also modulates the cellular distribution of mitochondria (79, 80) and the fate of this organelle when associated with the MTOR complex 2 (MTORC2) (81). This complex is activated by PINK1 (PTEN-induced kinase 1) (82), the main regulator of selective mitophagy (83). Interestingly, MTORC2 is also physically associated with MAMs, where it interacts with and phosphorylates AKT. Once activated, MTORC2-AKT signaling controls MAM integrity, mitochondrial metabolism, and cell survival by regulating the activity of MAM resident proteins PACS2, IP3R, and HK2 (84).
Thus, these findings suggest a mutual regulation between the MTORC2-AKT complex and the integrity and functionality of MAMs. Moreover, the pharmacological modulation of this MAM-localized complex could be used as a novel therapeutic intervention to modulate cell fate in cancers with AKT/MTOR mutations.
As reported earlier, the protein AMBRA-1 seems to be a fundamental regulator during autophagy induction, as evidenced by its interactions with BCL-2 family members at the ER and mitochondrial compartments.
Interestingly, several molecular and pharmacological agents capable of interfering with BCL-2 activity (such as BH3-mimetic compounds) are already used in cancer therapies (85). Thus, these drugs may also modulate AMBRA-1 activities at the ER–mitochondrial interfaces.
Several works report that a few of these chemotherapeutic drugs modulate autophagy by regulating BCL-2 family member activities (86), which shows a possible and crucial involvement of AMBRA-1 protein activity in cancer therapies.
A growing body of evidence suggests the antidiabetic drug metformin as a novel candidate for cancer therapy (87, 88). However, the mechanism of action remains unknown. This compound drives mitochondrial bioenergetics, models the ER-mitochondria contact site, and acts as a selective inhibitor of PKCβ (89, 90). Because the role of p66Shc-PKCβ in the regulation of autophagy has been recently shown (91) and metformin can be used to manipulate the autophagic process (35, 92), metformin may also directly affect the p66Shc-PKCβ-mediated regulation of the cell life span at MAM sites.
A broad range of chemotherapeutic agents has been used to improve or restore p53 activity (93). Given the autophagic-modulating properties of this tumor suppressor and considering its novel apoptotic function in the ER-MAM compartment, a novel therapy based on the simultaneous modulation of the autophagic mechanism and the p53-dependent transfer of Ca2+ from the ER to mitochondria appears to be possible.
However, proteins with tumor suppressor and oncogenic properties are not the only proteins that have been linked to MAMs and cancer progression. As reported earlier, a large number of MAM proteins are required to maintain the proper communication channels between the ER and mitochondria. Most of these proteins play pivotal roles during the autophagic process.
Notably, the correct assembly of MAMs is also guaranteed by fusion and fission events in the mitochondrial compartment.
Mitochondrial fusion is primarily orchestrated by mitofusins-1 and -2 (MFN-1/-2) and DRP1 (94). Remarkably, these proteins are crucial for tethering the ER to the mitochondria and stabilizing MAMs. Fusion proteins are critical elements for the transfer of outer mitochondrial membrane proteins to autophagosomes (49). The proteins are also indispensable elements for the execution of PINK1/Parkin-dependent mitophagy (95, 96). Importantly, the modulation of MFNs and DRP1 activity has been proposed to prevent tumor cell growth in several human cancers (97). Mitochondrial biogenesis is a critical aspect of the integrity of MAMs. Over the past few years, transcriptional coactivators of the peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family have been shown to directly mediate the transcription of mitochondrial genes responsible for the biogenesis of the organelle. This family includes PGC-1α and PGC-1β, which are present in many cell types, including cells found in the heart, skeletal muscles, and brain (98). Interestingly, both isoforms were significantly upregulated in the chemo-resistant cells. In particular, the silencing of PGC-1β isoform has been shown to reestablish the sensitivity of cancer cells carrying mtDNA mutations to chemotherapy agents (99). However, activity of the alpha isoform has been linked to autophagic modulation (100). Thus, the action of PGC-1α may be modulated by several pharmacological compounds (101), many of which also regulate the autophagic machinery (such as AMPK activators). In addition, several works have reported that the disruption of PGC-1α expression could be a critical aspect of cancer progression (102).
A functioning and healthy ER is a critical component of the MAM integrity. When this aspect is lacking, ER stress increases and results in unfolded protein response (UPR), ER-associated degradation (ERAD), MAM structure alteration, and Ca2+ release. For prolonged or extreme stresses, these signaling pathways can lead to cell death (103). Accumulating data indicate that ER stress is also a potent trigger of autophagy. Depending on the context, autophagy counterbalances ER stress-induced ER expansion enhances cell survival or commits the cell to non-apoptotic death. In a cancerous environment, this subject is a matter of debate. Previous reports have indicated that the inhibition of autophagy in colon epithelial cells prevents cell death induced by an ER-MAM stressor (e.g., Ca2+, thapsigargin, and tunicamycin) (104). However, in other human cancers, certain ER-MAM-disrupting treatments (e.g., photodynamic therapy and vitamin D analog EB1089) may kill cancer cells by a mechanism that depends on autophagy (105–107).
Overall, these findings suggest that the correct maintenance of the ER-mitochondria interface is a critical part of the autophagic process. Because catabolic process and several proteins linked to MAMs are deeply involved in cancer progression, a number of novel therapeutic approaches based on the manipulation of MAMs can be proposed (Figures 1D and Figure 2).
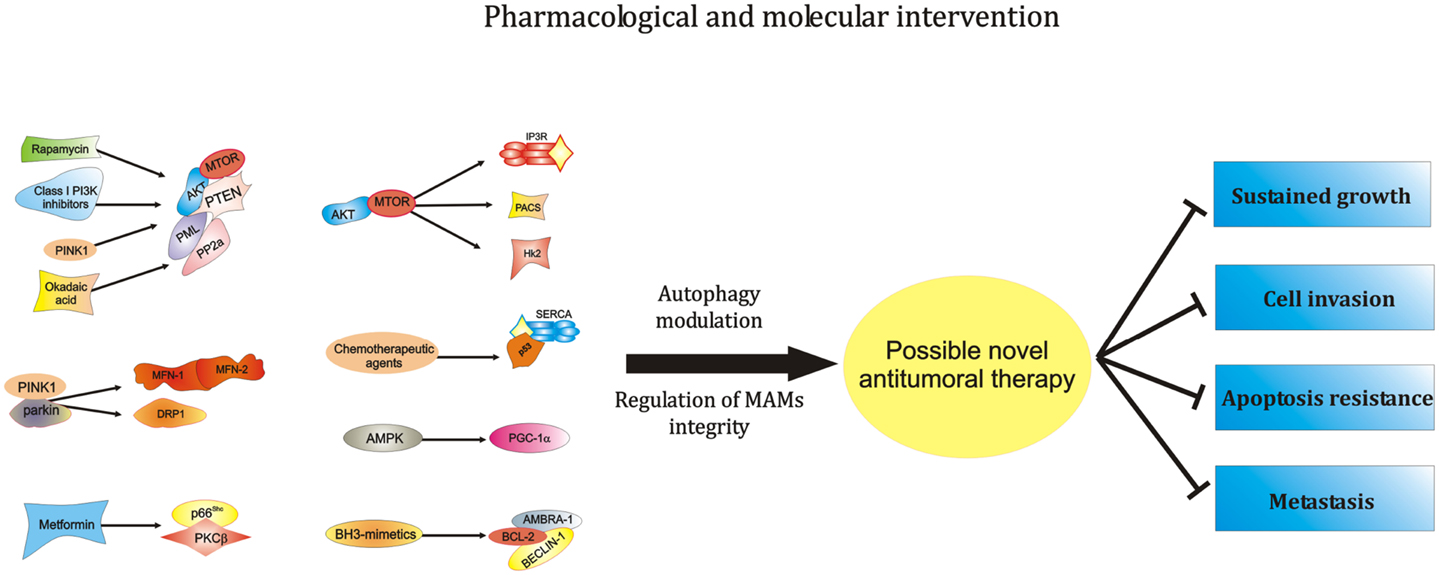
Figure 2. Modulation of the activity of ER-MAMs protein may represent a novel therapeutical strategy against carcinogenesis. MTOR, the main regulator of autophagic process, was found to modulate MAMs structure and activities. In addition, other PI3K family members, like AKT and PTEN, are located at ER-MAMs sites and represent critical regulator of autophagy and MTOR activity. Of relevance, these PI3K family members may be modulated by rapamycin and class I PI3K inhibitors. Furthermore, since the interaction of AKT-PTEN axis with the tumor suppressor PML and the protein PP2A at MAMs level is well described, it is possible to hypothesize a pharmacological intervention at this level to modulate the autophagic process during tumorigenesis. Also, the most studied tumor suppressor, p53, recently found to be located at ER-MAMs sites, was found to regulate autophagy. It is so clear that chemotherapic agents activating p53 may exert their antitumor activities at ER-mitochondria contact sites in an autophagy-dependent manner. Another critical regulator of autophagy, AMPK, was recently found to destabilize the correct juxtaposition between ER and mitochondria. Interestingly, the pharmacological AMPK-activator Metformin is found to regulate the activity of PKCβ. It is easy to think that metformin may interfere with the recent discovered negative modulator of autophagy, the p66Shc–PKCβ axis. Likewise, the potent autophagic regulator AMBRA-1 is found to be a potent controller of BCL-2 family member activities. Interestingly, the employment of BH3-mimetics is often used in chemotherapy, suggesting a possible role of AMBRA-1 during therapeutical approaches against cancer. Finally, the most important proteins involved in the mitophagic process, PINK1 and Parkin interact and regulate the activity of different ER-MAMs resident protein, thus suggesting the possibility of intercede to the activity of PINK-1-Parkin to develop novel antitumor approaches. Abbreviations: MFN-1/-2, mitofusin-1/-2; Hk2, hexokinase 2; PACS, phosphofurin acidic cluster sorting protein; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; PTEN, phosphatase and tensin homolog; p66Shc, 66 kDa proto-oncogene Src homologous-collagen homolog; PKCβ, protein kinase C beta; PP2a, protein phosphatase 2; AKT, protein kinase B; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase; MTOR, mechanistic target of rapamycin; PML, promyelocytic leukemia; PINK1, PTEN-induced putative kinase 1; DRP1, dynamin-related protein; AMPK, 5′ adenosine monophosphate-activated protein kinase; PI3K, phosphoinositide-3-kinase; AMBRA-1, Beclin1-regulated autophagy; BCL-2, B-cell lymphoma 2.
Concluding Remarks
Half a century ago, Christian de Duve coined the term “autophagy” to describe a process in which the cell digests its cytoplasmic materials via lysosomal degradation.
More than 20 years ago, Jean Vance biochemically isolated an intracellular structure representing the physical contact between the ER and mitochondria and termed this membrane fraction MAMs.
In the last decade, dysregulated autophagy has been associated with various types of disease-like phenotypes, including cancer.
In recent years, it has been revealed that the function and behavior of MAMs are central to the main molecular pathways of human cells, and consequently, MAM dysfunction has been associated with several types of cancer. In addition, a growing body of evidence suggests a key role for MAMs in the regulation of autophagy.
Together, these findings suggest that the MAM environment could be a fundamental background for the regulation of autophagic-dependent cancers. However, this intricate network has not been well addressed.
Future work will be required to better understand how this finely tuned compartment participates in various stages of autophagy during cancer development. These studies will help facilitate the rational design of combinatorial strategies aimed at modulating autophagy or MAM structures and resident proteins to elicit maximum therapeutic benefits against various types of human cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol (2002) 3:779–87. doi:10.1038/nrm931
2. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell (2011) 147:742–58. doi:10.1016/j.cell.2011.11.045
3. Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev (2010) 24:2592–602. doi:10.1101/gad.1984410
4. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ (2012) 19:107–20. doi:10.1038/cdd.2011.96
5. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ (2009) 16:966–75. doi:10.1038/cdd.2009.33
6. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34:856–80. doi:10.15252/embj.201490784
7. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev (2007) 87:99–163. doi:10.1152/physrev.00013.2006
8. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene (2006) 25:4798–811. doi:10.1038/sj.onc.1209608
9. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med (2000) 6:513–9. doi:10.1038/74994
10. Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol (2005) 6:268–75. doi:10.1038/nrm1573
11. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle (2013) 12:674–83. doi:10.4161/cc.23599
12. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell (2011) 147:728–41. doi:10.1016/j.cell.2011.10.026
13. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24:24–41. doi:10.1038/cr.2013.168
14. Mizushima N. Autophagy: process and function. Genes Dev (2007) 21:2861–73. doi:10.1101/gad.1599207
15. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi:10.1146/annurev-cellbio-092910-154005
16. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol (2013) 14:759–74. doi:10.1038/nrm3696
17. Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol (2012) 32:2–11. doi:10.1128/MCB.06159-11
18. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell (2010) 40:310–22. doi:10.1016/j.molcel.2010.09.026
19. Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov (2012) 11:709–30. doi:10.1038/nrd3802
20. Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal (2014) 20:519–37. doi:10.1089/ars.2013.5363
21. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med (2013) 368:651–62. doi:10.1056/NEJMra1205406
22. Weidberg H, Shvets E, Elazar Z. Lipophagy: selective catabolism designed for lipids. Dev Cell (2009) 16:628–30. doi:10.1016/j.devcel.2009.05.001
24. Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ (2012) 19:87–95. doi:10.1038/cdd.2011.146
25. Yonekawa T, Thorburn A. Autophagy and cell death. Essays Biochem (2013) 55:105–17. doi:10.1042/bse0550105
26. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science (2004) 306:990–5. doi:10.1126/science.1099993
27. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol (2015) 4:184–92. doi:10.1016/j.redox.2014.12.003
28. Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta (2009) 1793:1524–32. doi:10.1016/j.bbamcr.2009.01.006
29. Patergnani S, Pinton P. Mitophagy and mitochondrial balance. Methods Mol Biol (2015) 1241:181–94. doi:10.1007/978-1-4939-1875-1_15
30. Gomes M, Teixeira AL, Coelho A, Araujo A, Medeiros R. The role of inflammation in lung cancer. Adv Exp Med Biol (2014) 816:1–23. doi:10.1007/978-3-0348-0837-8_1
31. Radons J. The role of inflammation in sarcoma. Adv Exp Med Biol (2014) 816:259–313. doi:10.1007/978-3-0348-0837-8_11
32. Senol K, Ozkan MB, Vural S, Tez M. The role of inflammation in gastric cancer. Adv Exp Med Biol (2014) 816:235–57. doi:10.1007/978-3-0348-0837-8_10
33. Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell (2004) 7:179–92. doi:10.1016/j.devcel.2004.07.005
34. Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med (2005) 11:353–61. doi:10.1016/j.molmed.2005.06.007
35. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol (2011) 13:1016–23. doi:10.1038/ncb2329
36. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through beclin 1 phosphorylation. Science (2012) 338:956–9. doi:10.1126/science.1225967
37. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell (2005) 122:927–39. doi:10.1016/j.cell.2005.07.002
38. Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy (2008) 4:600–6. doi:10.4161/auto.6260
39. Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature (2007) 447:1121–5. doi:10.1038/nature05925
40. Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J (2011) 30:1195–208. doi:10.1038/emboj.2011.49
41. Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol (2010) 191:155–68. doi:10.1083/jcb.201002100
42. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis (2013) 4:e838. doi:10.1038/cddis.2013.350
43. Poklepovic A, Gewirtz DA. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy (2014) 10:1478–80. doi:10.4161/auto.29428
44. Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy (2014) 10:1391–402. doi:10.4161/auto.29119
45. Chiarini F, Evangelisti C, Mccubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci (2015) 36:124–35. doi:10.1016/j.tips.2014.11.004
46. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell (2014) 26:190–206. doi:10.1016/j.ccr.2014.06.025
47. Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun Signal (2011) 9:19. doi:10.1186/1478-811X-9-19
48. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta (2014) 1837:461–9. doi:10.1016/j.bbabio.2013.10.015
49. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495:389–93. doi:10.1038/nature11910
50. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta (2014) 1843:2253–62. doi:10.1016/j.bbamcr.2014.03.009
51. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280:1763–6. doi:10.1126/science.280.5370.1763
52. Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol (2006) 174:915–21. doi:10.1083/jcb.200604016
53. Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta (2014) 1841:595–609. doi:10.1016/j.bbalip.2013.11.014
54. Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell (2010) 39:121–32. doi:10.1016/j.molcel.2010.06.029
55. Giorgi C, Wieckowski MR, Pandolfi PP, Pinton P. Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Commun Integr Biol (2011) 4:334–5. doi:10.4161/cib.4.3.15021
56. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
57. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal (2015) 22:995–1019. doi:10.1089/ars.2014.6223
58. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol (2008) 182:685–701. doi:10.1083/jcb.200803137
59. Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy (2009) 5:1180–5. doi:10.4161/auto.5.8.10274
60. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell (2010) 141:656–67. doi:10.1016/j.cell.2010.04.009
61. Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell (2006) 24:331–9. doi:10.1016/j.molcel.2006.09.013
62. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330:1247–51. doi:10.1126/science.1189157
63. Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem (2006) 281:3731–7. doi:10.1074/jbc.M509262200
64. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun (2008) 375:501–5. doi:10.1016/j.bbrc.2008.07.153
65. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis (2012) 3:e304. doi:10.1038/cddis.2012.45
66. Deniaud A, Sharaf El Dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene (2008) 27:285–99. doi:10.1038/sj.onc.1210638
67. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ (2013) 20(12):1631–43. doi:10.1038/cdd.2013.77
68. Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget (2014) 6(3):1435–45.
69. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112:1779–84. doi:10.1073/pnas.1410723112
70. Lebiedzinska M, Duszynski J, Rizzuto R, Pinton P, Wieckowski MR. Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch Biochem Biophys (2009) 486:73–80. doi:10.1016/j.abb.2009.03.007
71. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell (2005) 122:221–33. doi:10.1016/j.cell.2005.05.011
72. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science (2007) 315:659–63. doi:10.1126/science.1135380
73. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene (2014) 33:2329–40. doi:10.1038/onc.2013.192
74. Banreti A, Lukacsovich T, Csikos G, Erdelyi M, Sass M. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy (2012) 8:623–36. doi:10.4161/auto.19081
75. Magnaudeix A, Wilson CM, Page G, Bauvy C, Codogno P, Leveque P, et al. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol Aging (2013) 34:770–90. doi:10.1016/j.neurobiolaging.2012.06.026
76. Yu HC, Lin CS, Tai WT, Liu CY, Shiau CW, Chen KF. Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J Biol Chem (2013) 288:18249–59. doi:10.1074/jbc.M112.446385
77. Buontempo F, Ersahin T, Missiroli S, Senturk S, Etro D, Ozturk M, et al. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Invest New Drugs (2011) 29:1303–13. doi:10.1007/s10637-010-9486-3
78. Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev (2013) 65:1162–97. doi:10.1124/pr.112.007120
79. Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci U S A (2002) 99:4319–24. doi:10.1073/pnas.261702698
80. Ramanathan A, Schreiber SL. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci U S A (2009) 106:22229–32. doi:10.1073/pnas.0912074106
81. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy (2012) 8:445–544. doi:10.4161/auto.19496
82. Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami J, Yamada H, et al. A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem (2011) 286:7182–9. doi:10.1074/jbc.M110.179390
83. Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy (2013) 9:1677–86. doi:10.4161/auto.24795
84. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A (2013) 110:12526–34. doi:10.1073/pnas.1302455110
85. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther (2013) 12:1691–700. doi:10.1158/1535-7163.MCT-13-0058
86. Fu LL, Cheng Y, Liu B. Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol (2013) 45:921–4. doi:10.1016/j.biocel.2013.02.007
87. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther (2010) 9:1092–9. doi:10.1158/1535-7163.MCT-09-1186
88. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res (2014) 159:355–76. doi:10.1007/978-3-642-38007-5_21
89. Gallo A, Ceolotto G, Pinton P, Iori E, Murphy E, Rutter GA, et al. Metformin prevents glucose-induced protein kinase C-beta2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes (2005) 54:1123–31. doi:10.2337/diabetes.54.4.1123
90. Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes (2014) 63:3279–94. doi:10.2337/db13-1751
91. Patergnani S, Marchi S, Rimessi A, Bonora M, Giorgi C, Mehta KD, et al. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy (2013) 9:1367–85. doi:10.4161/auto.25239
92. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther (2011) 10:1533–41. doi:10.1158/1535-7163.MCT-11-0047
93. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov (2014) 13:217–36. doi:10.1038/nrd4236
94. Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol (2014) 24:761–70. doi:10.1016/j.tcb.2014.08.005
95. Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet (2010) 19:R28–37. doi:10.1093/hmg/ddq143
96. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet (2010) 19:4861–70. doi:10.1093/hmg/ddq419
97. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J (2012) 26:2175–86. doi:10.1096/fj.11-196543
98. Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev (2008) 22:1948–61. doi:10.1101/gad.1661708
99. Yao Z, Jones AW, Fassone E, Sweeney MG, Lebiedzinska M, Suski JM, et al. PGC-1beta mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene (2013) 32:2592–600. doi:10.1038/onc.2012.259
100. Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda) (2011) 26:214–24. doi:10.1152/physiol.00010.2011
101. Wenz T. PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life (2009) 61:1051–62. doi:10.1002/iub.261
102. Huang B, Cheng X, Wang D, Peng M, Xue Z, Da Y, et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1alpha signaling. Oncotarget (2014) 5:4732–45.
103. Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ (2007) 14:1576–82. doi:10.1038/sj.cdd.4402200
104. Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem (2007) 282:4702–10. doi:10.1074/jbc.M609267200
105. Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and beclin 1-mediated autophagic cell death. Cell Death Differ (2005) 12:1297–309. doi:10.1038/sj.cdd.4401651
106. Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J (2006) 20:756–8. doi:10.1096/fj.05-4305fje
Keywords: MAM, autophagy, apoptosis, calcium, cancer, cell death
Citation: Patergnani S, Missiroli S, Marchi S and Giorgi C (2015) Mitochondria-associated endoplasmic reticulum membranes microenvironment: targeting autophagic and apoptotic pathways in cancer therapy. Front. Oncol. 5:173. doi: 10.3389/fonc.2015.00173
Received: 06 May 2015; Accepted: 10 July 2015;
Published: 27 July 2015
Edited by:
Jerry Edward Chipuk, Icahn School of Medicine at Mount Sinai, USAReviewed by:
Catherine Brenner, University of Paris Sud, FranceYoshihiro Suzuki-Karasaki, Nihon University School of Medicine, Japan
Copyright: © 2015 Patergnani, Missiroli, Marchi and Giorgi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlotta Giorgi, Laboratory for Technologies of Advanced Therapies (LTTA), Section of Pathology, Oncology and Experimental Biology, Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, Via Fossato di Mortara, Ferrara 70 – 44121, Italy, carlotta.giorgi@unife.it