 Amelie S. Schober
Amelie S. Schober Edurne Berra
Edurne Berra- 1Centro de Investigación Cooperativa en Biociencias, CIC bioGUNE, Derio, Spain
- 2Faculty of Health and Life Sciences, Center for Cell Imaging, Institute of Integrative Biology, University of Liverpool, Liverpool, UK
Cellular protein homeostasis is tightly regulated by ubiquitination. Responsible for target protein ubiquitination is a class of enzymes, the so-called ubiquitin E3 ligases. They are opposed to a second class of enzymes, called deubiquitinating enzymes (DUBs), which can remove polyubiquitin chains from their specific target proteins. The coaction of the two sets of enzymes allows the cell to adapt its overall protein content and the abundance of particular proteins to a variety of cellular and environmental stresses, including hypoxia. In recent years, DUBs have been highlighted to play major roles in many diseases, including cancer, both as tumor suppressors and oncogenes. Therefore, DUBs are emerging as promising targets for cancer-cell specific treatment. Here, we will review the current understanding of DUBs implicated in the control of hypoxia-inducible factor, the regulation of DUBs by hypoxia, and the use of DUB-specific drugs to target tumor hypoxia-signaling.
Introduction
Like most other posttranslational modifications (PTMs), ubiquitin (Ub) conjugation is a reversible modification (1). Ub E3 ligases covalently attach monomers of Ub to lysine (and also cysteine) residues of their target proteins. Furthermore, ligases also convert monoubiquitination into polyubiquitin chains by attaching one by one further Ub monomers to one of the seven internal lysine residues (K6/K11/K27/K29/K33/K48/K63) of the preceding Ub molecule. In contrast, the family of DeUBiquitinating enzymes (DUBs) breaks down those mono- and polyubiquitin chains from the target protein. Besides counteracting the action of the Ub E3 ligases, DUBs are proteases that process Ub precursors.
Of the nearly 100 DUBs encoded by the human genome, 79 are predicted to be active and mostly cleave particular types of Ub chain linkages from their respective target proteins. DUBs can be grouped into six families based on sequence and structure similarity: ubiquitin-specific proteases (USPs) that comprise the largest and most diverse subfamily, ubiquitin carboxyl-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Josephins, JAB1/MPN/MOV34 (JAMMs), and the more recently discovered monocyte chemotactic protein-induced proteins (MCPIP). With the exception of JAMMs, which belong to the Zn2+-dependent metalloproteases, all the rest use the classical cysteine protease triad in the catalytical side (2).
Classically, the reversal of the polyubiquitination protects the target protein from being degraded by the proteasome, but ubiquitination has also been shown to have a broad range of non-catabolic functions (3). Thus, it is not surprising that DUB activity or inappropriate expression impacts on the regulation of multiple biological processes and several signaling pathways that are frequently altered in many disorders from cancer over neurodegenerative pathologies to inflammatory diseases [for more details, please refer to Ref. (4)]. Because of their direct or indirect implications in those diseases and because of their potential druggability, DUBs have become of increasing interest in recent years.
Hypoxia is a feature of most human cancers (5). The cancer cells and their environment adapt to and survive under low oxygen availability. The activation of the hypoxia-inducible factor (HIF) that orchestrates the hypoxia-signaling pathway is instrumental to this adaptation. HIF is a heterodimeric transcription factor that consists of a constitutively expressed β-subunit (HIF-β) and HIF-α, whose expression is tightly regulated through the ubiquitin-proteasome system (UPS) (6–8). HIF triggers the expression of hundreds of direct target genes, indirect transcription factors, and non-coding RNAs that enable cancer cell survival and tumor progression by promoting, among others, angiogenesis, metabolic rewiring, genomic instability, drug resistance, and the self-renewal capacity of cancer stem cells. HIFs are also activated by genetic alterations in human cancers, such as the von Hippel–Lindau protein (pVHL) loss of function in clear-cell renal carcinoma (9). Accordingly, sustained expression of HIF-α in tumors has been associated with higher aggressiveness, migratory, and metastasis-initiating potential and therefore worse prognosis (10, 11).
In this review, we will summarize the current knowledge about the action of DUBs on HIF-α and the regulation of those enzymes by hypoxia. We will also discuss the potential of exploiting DUBs to target tumor hypoxia signaling.
The Canonical HIF Signaling Pathway
The adaptive cellular program in response to low oxygen availability is mainly triggered by two HIF-α subunits (HIF-1α and HIF-2α), which share several common targets but also exhibit non-redundant functions (12). Anyhow, the levels of both HIF-α subunits result from the dynamic interplay between their ubiquitination and deubiquitination. In well-oxygenated cells, HIF-α is very unstable, as it is degraded by the proteasome within approximately <5 min after translation, whereas HIF-α’s half-life is greatly increased in hypoxia (7, 8, 13). Proteasomal degradation is triggered by the continuous polyubiquitination of HIF-α by pVHL (9). pVHL is part of an E3 ligase complex and binds to HIF-α after the hydroxylation of two designated proline residues in HIF-α’s oxygen-dependent degradation domain (ODDD), the central regulatory domain that confers its oxygen sensibility (8). This binding can be stabilized by SSAT2, therefore enhancing HIF-α ubiquitination (14). The family of prolyl hydroxylase domain-containing proteins (PHDs), the oxygen sensors also referred to as EGLNs or HPHs, catalyze the hydroxylation of HIF-α (Pro402 and Pro564, in the case of HIF-1α) (15–19). HIF-α also harbors an N-terminal basic helix-loop-helix (bHLH) domain that mediates HIF-binding to the target DNA after heterodimerization with HIF-β/ARNT via the adjacent PAS domain. Of the two transactivation domains (TAD), the N-terminal TAD (N-TAD) lies within the ODDD, while the C-terminal TAD (C-TAD) is responsible for the recruitment of CBP/p300 needed to successfully induce the transcription of the HIF target genes that are characterized by having one or more HREs (hypoxia response elements) (20, 21). This C-TAD contains an asparagine residue (Asn803, in the case of HIF-1α) that upon oxygen-dependent hydroxylation by FIH (factor inhibiting HIF) hinders the successful interaction of HIF with CBP/p300 and therefore, HIF’s transactivation activity is reduced (22). Interestingly, HIF induces the expression of two of its negative regulators, PHD2 and PHD3, in order to ensure its own rapid degradation upon reoxygenation (19, 23). However, in conditions of chronic hypoxia, once the transcriptional adaptive program has been triggered, HIF-α levels drop again to avoid sustained HIF signaling and assure cell survival (24).
In the context of the canonical HIF signaling pathway, so far there are relatively few DUBs reported in the literature, and reports are mostly focused on the impact on HIF-1α (Figure 1 upper part). USP20 (also called pVHL interacting DUB2, VDU2) was the first DUB to be described to reverse pVHL-mediated HIF-1α ubiquitination (25). In turn, USP20 is a pVHL target (26). MCPIP1 also deubiquitinates HIF-1α to promote angiogenesis (27). In the context of ciliogenesis, USP8 has been found to bind to HIF-1α’s PAS domain and to partially protect HIF-1α from degradation (28). More recently, UCHL1 has been shown to be a positive regulator of HIF-1α protein stability acting on HIF-1α’s ODDD (29).
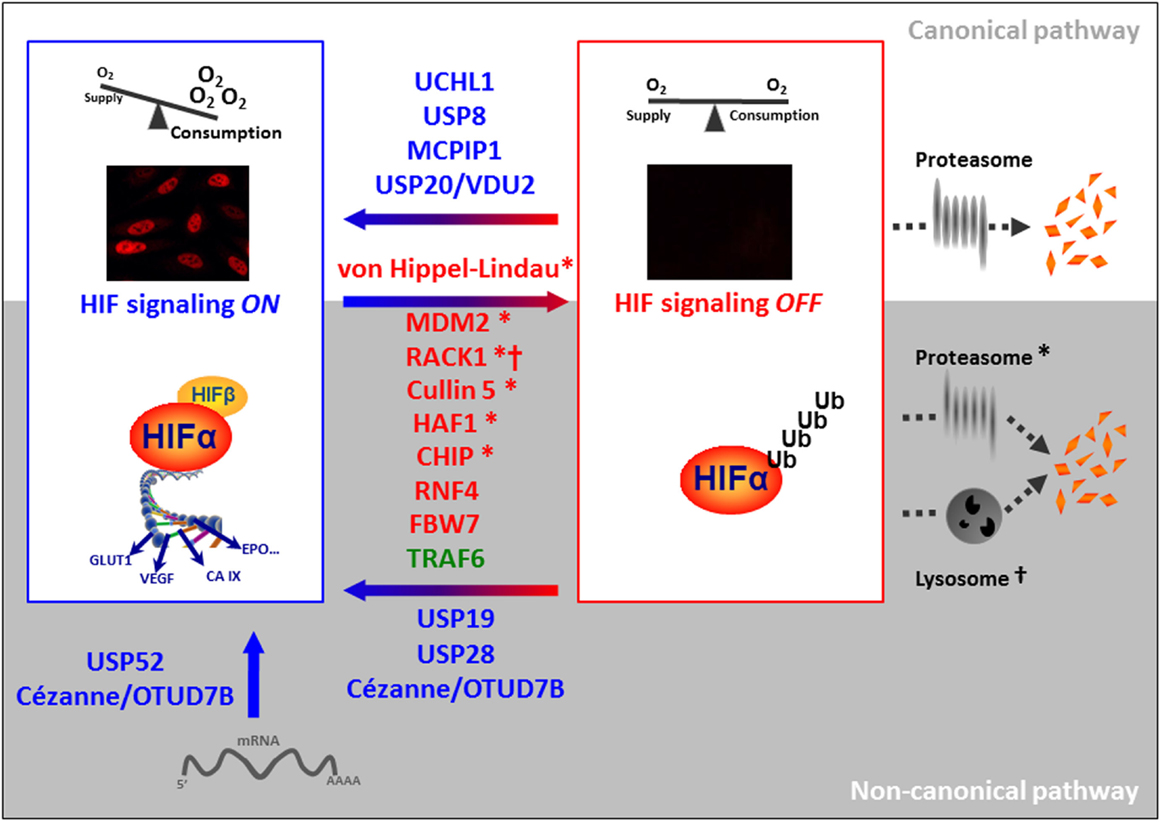
Figure 1. Involvement of ubiquitinating and deubiquitinating enzymes in the regulation of HIF-α. Destabilizing and stabilizing E3 ligases targeting HIF-α are pictured in red and green, respectively and DUBs are depicted in blue. E3 ligases that have been described to target HIF-α for proteasomal degradation are marked with a *; † refers to lysosomal HIF-α degradation.
The Non-Canonical HIF Signaling
Not surprisingly because of HIF’s crucial role in cell fate, many more proteins have been described to be involved in the control of its stability (Figure 1 lower part). The heat-shock protein 90 (HSP90) that interacts with the PAS domain of HIF-α regulates its degradation in an O2/PHD/pVHL-independent manner (30). HSP90 competes with RACK1 for binding to HIF-α and prevents the recruitment of the elongin C/B Ub E3 ligase complex (31). A similar mechanism has been proposed for HIF-α activation by ErbB4 (32). As for other HSP90 client proteins, Cullin5 also regulates HIF-α degradation independently of elongin C/B function (33). The tumor suppressors p53, TAp73, and pTEN promote the Ub-mediated degradation of HIF-1α via recruitment of the Ub E3 ligase Mdm2 (34–36). Furthermore, Fbw7 ubiquitinates and induces HIF-1α degradation following phosphorylation by GSK3β (37, 38). Interestingly, this degradation can be antagonized by the Ub-specific protease (USP28) (38). Until now, this is the only non-canonical Ub E3 ligase–DUB pair identified for proteasomal degradation of HIF-α. HAF, the hypoxia-associated factor, seems to play a dual role in the control of HIF-α stability and/or activity. While HAF acts as an Ub E3 ligase targeting HIF-1α for degradation independently of oxygen availability, hypoxia-induced SUMOylated HAF promotes HIF-2α transactivation without affecting its stability (39, 40). Furthermore, RNF4 controls the levels of SUMOylated HIF-2α (41). USP19 seems to be required for the hypoxic accumulation of HIF-1α, though the effect is not dependent on its deubiquitinase activity (42). USP19 is further substrate of Siah-1 and Siah-2 Ub E3 ligases, which also control the stability of PHD1, PHD3, and FIH (43–45). Thus, further studies are necessary to clarify the direct impact of USP19 in HIF-1α ubiquitination.
The chaperone-dependent Ub ligase CHIP targets HIF-1α but not HIF-2α for degradation either by the proteasome or by the autophagic machinery, the second big protein degradation and recycling pathway that has been implicated in the elimination of ubiquitinated HIF-α (46–49). In this regard, Cezanne (OTUD7B), a deubiquitinase targeting K11 Ub chains (50), has been reported to protect HIF-1α from lysosomal degradation. While this process is independent of HIF-1α prolyl hydroxylation, it depends on the presence of pVHL (51).
Calpain and the activation of the forkhead transcription factor FOXO4 destabilize HIF-α, although the underlying molecular mechanisms are unknown (52, 53). Further studies are also needed to characterize the role of Parkin in the regulation of HIF-α, based on its identification within the Parkin-dependent ubiquitinome by a proteomic approach (54). In contrast with all the previous reports, it is worth mentioning the role played by the Ub E3 ligase TRAF6. TRAF6 increases HIF-1α but not HIF-2α polyK-63 ubiquitination and protects the protein from proteasomal degradation (55).
In addition to HIF-α stability, mRNA expression and activity of the transcriptional complex fine-tune HIF regulation. In this regard, USP52 is required for the protection of HIF-1α (but not HIF-2α) mRNA from premature degradation and therefore, allows the normal hypoxic induction of HIF-1α (56). The case of USP52 is somewhat special as this protein, although structurally related to the family of USPs, lacks the catalytic cysteine (57). Besides protecting HIF-1α protein from its degradation, Cezanne’s catalytical activity is also required for maintaining basal levels of the E2F1 transcription factor. Moniz et al. demonstrated that E2F1 controls the expression of HIF-2α mRNA and therefore, established an indirect role of the DUB Cezanne in HIF-2α expression (58).
Finally, a number of DUBs have been shown to regulate transcription factors and signaling pathways that cross talk with HIFs, likely contributing to the complexity and specificity of the cellular hypoxic response, even though they go beyond the scope of this review (59–61).
Regulation of DUBs by Hypoxia
As for other enzymes, there are several possible layers of regulation of DUB activity. Next to the transcriptional regulation, the stability and translation of the mRNA can be regulated by mRNA-processing enzymes. The turnover and therefore, the availability of the mature protein can be set by a variety of PTMs. PTMs can also interfere with the binding of the DUBs to their target proteins or other interactors, as well as modulate reversibly and irreversibly the (auto) catalytical activity of the DUB. Hypoxia, being an extreme cellular stress condition, should be able to regulate deubiquitinating activity on all the possible different layers in order to adapt DUB functions to the cell’s needs. However, the literature about the regulation of specific DUBs by hypoxia (1% O2, if not specified differently) is still scarce and almost exclusively restricted to transcriptional regulation. For instance, the expression of USP13 is reduced upon treatment with as little as 6 h of 2% O2 in melanoma cell lines (59). The reduction of the mRNA also translates to the protein level and causes the loss of Siah-2 stabilization. Similarly, in colon cancer cells hypoxia reduces USP46 mRNA and protein levels and, therefore, diminishes USP46’s stabilizing effect on the tumor suppressors PHLPP1 and PHLPP2, conferring to the colon cancer cells an increased paclitaxel resistance (62, 63). Guo et al. provide more detailed information about the hypoxia-mediated transcriptional regulation of the UCH CYLD. They suggest that the decrease of CYLD mRNA and protein seen in glioblastoma cells is due to the hypoxia-induced increase of the transcriptional repressors Snail and Hes1 (64). In contrast, hypoxia has been shown to increase Cezanne via p38 MAPK (65).
An et al. claimed that CYLD is targeted for proteasomal degradation after interaction with the HPV E6 protein in hypoxia (66). This is to date the only report of a posttranslational regulation of DUB activity by hypoxia. However, Lee et al. present evidence that the activity of many, if not most, DUBs depends on the redox state of the cell. They show that the catalytically active cysteine residue can be oxidized, for instance, by intracellular hydrogen peroxide, leading to the abolishment of the deubiquitinating activity. The inactivating oxidation can be reversed in the presence of reducing agents, such as DTT, or prevented by antioxidants (67). As hypoxia and mitochondrial ROS production are intrinsically linked it might not be too far-fetched to propose that hypoxia directly modulates DUB activity via ROS.
Dysregulation of Hypoxia-Related DUBs in Cancer
Given the importance of Ub-mediated changes in protein function and homeostasis, it is not by chance that the entire process is highly regulated. Disruption of the ubiquitination cycle by mutations or altered expression of specific components within the cascade has been associated with several disorders. In particular, more than 30 DUBs have been associated with cancer directly or indirectly. Both, the loss of a specific DUB activity or its hyperactivity are non-desired events if the targets are tumor suppressors or oncogenes, respectively. Recurrent mutations of DUBs are rare in cancer with only few exceptions. Gene fusions with RUNX are reported for USP42 and USP16 in hematologic diseases, such as chronic myelomonocytic leukemia and acute myeloid leukemia. However, dysregulated mRNA levels of DUBs are implicated in many malignancies. Here, we will focus only on a few examples of hypoxia-related DUBs, for a more extensive overview please refer to the very comprehensive review by D’Arcy et al. (68).
Germline mutations of the tumor-suppressor gene CYLD are prevalent in familial cylindromatosis, a genetic condition that leads to predisposition for developing multiple skin tumors (69, 70). In addition, CYLD deubiquitinating activity has been seen to be abolished in different cancers on the protein level by inactivating phosphorylations or destabilizing polyubiquitination (71). More recently, it has been reported that USP8 is frequently mutated in adenomas causing Cushing’s disease (72).
USP28 is a DUB whose overexpression has been reported in breast and colon cancer and glioblastoma (73, 74). A recent publication has proposed USP28 to be a potential predictive marker in bladder cancer, as they found correlation of USP28 with tumor histological grade, clinical stage, recurrence, and survival (75). Similar to USP28, UCHL1 has also been proposed to be a useful biomarker, being overexpressed in gastric cancer (76) and in myeloma (77), and epigenetically down-regulated in colorectal cancer (78). As mentioned above, downregulation of USP46 may serve as a biomarker of resistance to chemotherapy in colon cancer (63). Finally, despite being inconsistent to its role in the regulation of HIF-1α and HIF-2α, decreased Cezanne expression is associated with the progression and poor prognosis in hepatocellular carcinoma (79).
DUBs as Druggable Targets for Therapy
Modulators of individual UPS components are emerging as a novel class of anticancer drugs. The initial research focus had been directed toward targeting the proteasome, with activity described for many compounds with proteasome inhibitory activity, including bortezomib. Because Ub E3 ligases provide substrate specificity, their direct targeting may avoid the deleterious side effects associated with the global inhibition of the proteasome, making them interesting candidates as drug targets. Nutlin-3 and JNJ-26854165 are classic examples directed against the Ub E3 ligase MDM2 and are currently undergoing clinical evaluation as anticancer therapy.
Newly arising, DUBs may serve as equally or more useful targets. Indeed, DUBs are highly specialized and evolutionary linked to proteases, a typified pharmaceutical target class for drug discovery, thanks to their well-characterized catalytical domain. Several partial and specific inhibitors against a small number of DUBs have been developed, have proved active in preclinical studies as reviewed recently by D’Arcy and Linder (80), and have provided feasibility for targeting these enzymes for anticancer purposes. Among them, HBX 41,108 is a partially selective USP inhibitor because it inhibits USP5, USP7, USP8, and UCHL3 in addition to caspase 3 (81). This is to our knowledge the only DUB inhibitor so far described as targeting one of the DUBs linked to the HIF signaling pathway. Interestingly, the inhibition of USP8 suppresses growth of gefitinib-resistant non-small cell lung cancer cells, though no link to the potential impact on HIF-1α is reported (82). It is tempting to speculate about new drugs directed against hypoxia-related DUBs that succeed to fight intratumoral hypoxia-signaling in the coming years.
Conclusion
HIF-α protein homeostasis is tightly controlled in healthy cells in order to avoid inappropriate activation of HIF signaling. A variety of E3 ligases and DUBs are involved in this task by triggering and protecting HIF-α from its degradation, respectively. Permanent activation of the HIF signaling pathway has been found in many tumors and seems to be beneficial for tumor growth and cancer progression. In most cases, the reason for sustained HIF-α protein levels in the tumor cells are still not revealed, but a possible mechanism is the pathological increase of HIF-α specific DUB activity. In recent years, the dysregulation of deubiquinating enzymes in cancer (and other diseases) has become of increasing interest, and alterations of their expression and activities have been shown to have diagnostic value. Whether cancer-related events that lead to the upregulation of DUB activity are the primary cause of uncontrolled HIF signaling, or whether initial hypoxia upregulates DUB expression as a positive feed-back-loop is not determined. But in the light of DUBs being druggable enzymes, it is important to understand their implications in HIF and tumor hypoxia-signaling.
Author Contributions
ASS and EB contributed with review writing, editing, and final approval of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all the laboratory members for discussions and comments. We apologize to the many research groups whose work was not cited due to space constraints.
Funding
Our research is supported by the Plan Nacional of I+D BFU-2013-46647. ASS is a Liverpool-bioGUNE partnership fellow.
References
1. Clague MJ, Heride C, Urbe S. The demographics of the ubiquitin system. Trends Cell Biol (2015) 25:417–26. doi: 10.1016/j.tcb.2015.03.002
2. Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene (2012) 31:2373–88. doi:10.1038/onc.2011.443
3. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem (2012) 81:203–29. doi:10.1146/annurev-biochem-060310-170328
4. Heideker J, Wertz IE. DUBs, the regulation of cell identity and disease. Biochem J (2015) 467:191. doi:10.1042/bj4670191
5. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33:207–14. doi:10.1016/j.tips.2012.01.005
6. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem (1995) 270:1230–7. doi:10.1074/jbc.270.3.1230
7. Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem (1997) 272:22642–7. doi:10.1074/jbc.272.36.22642
8. Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A (1998) 95:7987–92. doi:10.1073/pnas.95.14.7987
9. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399:271–5. doi:10.1038/20459
10. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res (1999) 59:5830–5.
11. Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouyssegur J, et al. HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer (2007) 120:1451–8. doi:10.1002/ijc.22436
12. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2012) 12:9–22. doi:10.1038/nrc3183
13. Berra E, Roux D, Richard DE, Pouyssegur J. Hypoxia-inducible factor-1 alpha (HIF-1 alpha) escapes O(2)-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep (2001) 2:615–20. doi:10.1093/embo-reports/kve130
14. Baek JH, Liu YV, McDonald KR, Wesley JB, Hubbi ME, Byun H, et al. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1alpha. J Biol Chem (2007) 282:23572–80. doi:10.1074/jbc.M703504200
15. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science (2001) 294:1337–40. doi:10.1126/science.1066373
16. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi:10.1016/S0092-8674(01)00507-4
17. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (2001) 292:464–8. doi:10.1126/science.1059817
18. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
19. Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J (2003) 22:4082–90. doi:10.1093/emboj/cdg392
20. Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A (1993) 90:4304–8. doi:10.1073/pnas.90.9.4304
21. Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A (1996) 93:12969–73. doi:10.1073/pnas.93.23.12969
22. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science (2002) 295:858–61. doi:10.1126/science.1068592
23. Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landázuri MO, et al. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J (2005) 390:189–97. doi:10.1042/bj20042121
24. Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci U S A (2008) 105:4745–50. doi:10.1073/pnas.0705680105
25. Li Z, Wang D, Messing EM, Wu G. VHL protein-interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha. EMBO Rep (2005) 6:373–8. doi:10.1038/sj.embor.7400377
26. Li Z, Wang D, Na X, Schoen SR, Messing EM, Wu G. Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem Biophys Res Commun (2002) 294:700–9. doi:10.1016/S0006-291X(02)00534-X
27. Roy A, Zhang M, Saad Y, Kolattukudy PE. Antidicer RNAse activity of monocyte chemotactic protein-induced protein-1 is critical for inducing angiogenesis. Am J Physiol Cell Physiol (2013) 305:C1021–32. doi:10.1152/ajpcell.00203.2013
28. Troilo A, Alexander I, Muehl S, Jaramillo D, Knobeloch K-P, Krek W. HIF1α deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep (2014) 15:77–85. doi:10.1002/embr.201337688
29. Goto Y, Zeng L, Yeom CJ, Zhu Y, Morinibu A, Shinomiya K, et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat Commun (2015) 6:6153. doi:10.1038/ncomms7153
30. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem (2002) 277:29936–44. doi:10.1074/jbc.M204733200
31. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell (2007) 25:207–17. doi:10.1016/j.molcel.2007.01.001
32. Paatero I, Jokilammi A, Heikkinen PT, Iljin K, Kallioniemi OP, Jones FE, et al. Interaction with ErbB4 promotes hypoxia-inducible factor-1alpha signaling. J Biol Chem (2012) 287:9659–71. doi:10.1074/jbc.M111.299537
33. Ehrlich ES, Wang T, Luo K, Xiao Z, Niewiadomska AM, Martinez T, et al. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci U S A (2009) 106:20330–5. doi:10.1073/pnas.0810571106
34. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev (2000) 14:34–44. doi:10.1101/gad.14.1.34
35. Joshi S, Singh AR, Durden DL. MDM2 regulates hypoxic hypoxia-inducible factor 1alpha stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem (2014) 289:22785–97. doi:10.1074/jbc.M114.587493
36. Amelio I, Inoue S, Markert EK, Levine AJ, Knight RA, Mak TW, et al. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1alpha degradation. Proc Natl Acad Sci U S A (2015) 112:226–31. doi:10.1073/pnas.1410609111
37. Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem (2011) 112:3882–90. doi:10.1002/jcb.23321
38. Flugel D, Gorlach A, Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood (2012) 119:1292–301. doi:10.1182/blood-2011-08-375014
39. Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol (2008) 28:7081–95. doi:10.1128/MCB.00773-08
40. Koh MY, Nguyen V, Lemos R Jr, Darnay BG, Kiriakova G, Abdelmelek M, et al. Hypoxia-induced SUMOylation of E3 ligase HAF determines specific activation of HIF2 in clear-cell renal cell carcinoma. Cancer Res (2015) 75:316–29. doi:10.1158/0008-5472.CAN-13-2190
41. van Hagen M, Overmeer RM, Abolvardi SS, Vertegaal AC. RNF4 and VHL regulate the proteasomal degradation of SUMO-conjugated hypoxia-inducible factor-2alpha. Nucleic Acids Res (2010) 38:1922–31. doi:10.1093/nar/gkp1157
42. Altun M, Zhao B, Velasco K, Liu H, Hassink G, Paschke J, et al. Ubiquitin-specific protease 19 (USP19) regulates hypoxia-inducible factor 1α (HIF-1α) during hypoxia. J Biol Chem (2012) 287:1962–9. doi:10.1074/jbc.M111.305615
43. Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell (2004) 117:941–52. doi:10.1016/j.cell.2004.06.001
44. Fukuba H, Yamashita H, Nagano Y, Jin HG, Hiji M, Ohtsuki T, et al. Siah-1 facilitates ubiquitination and degradation of factor inhibiting HIF-1alpha (FIH). Biochem Biophys Res Commun (2007) 353:324–9. doi:10.1016/j.bbrc.2006.12.051
45. Velasco K, Zhao B, Callegari S, Altun M, Liu H, Hassink G, et al. An N-terminal SIAH-interacting motif regulates the stability of the ubiquitin specific protease (USP)-19. Biochem Biophys Res Commun (2013) 433:390–5. doi:10.1016/j.bbrc.2013.02.094
46. Bento CF, Fernandes R, Ramalho J, Marques C, Shang F, Taylor A, et al. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1α for degradation in the presence of methylglyoxal. PLoS One (2010) 5:e15062. doi:10.1371/journal.pone.0015062
47. Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but not HIF-2alpha. J Biol Chem (2010) 285:3651–63. doi:10.1074/jbc.M109.068577
48. Ferreira JV, Fofo H, Bejarano E, Bento CF, Ramalho JS, Girao H, et al. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy (2013) 9:1349–66. doi:10.4161/auto.25190
49. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem (2013) 288:10703–14. doi:10.1074/jbc.M112.414771
50. Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol (2010) 17:939–47. doi:10.1038/nsmb.1873
51. Bremm A, Moniz S, Mader J, Rocha S, Komander D. Cezanne (OTUD7B) regulates HIF-1alpha homeostasis in a proteasome-independent manner. EMBO Rep (2014) 15:1268–77. doi:10.15252/embr.201438850
52. Tang TT-L, Lasky LA. The forkhead transcription factor FOXO4 induces the down-regulation of hypoxia-inducible factor 1 alpha by a von Hippel-Lindau protein-independent mechanism. J Biol Chem (2003) 278:30125–35. doi:10.1074/jbc.M302042200
53. Zhou J, Kohl R, Herr B, Frank R, Brune B. Calpain mediates a von Hippel-Lindau protein-independent destruction of hypoxia-inducible factor-1alpha. Mol Biol Cell (2006) 17:1549–58. doi:10.1091/mbc.E05-08-0770
54. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature (2013) 496:372–6. doi:10.1038/nature12043
55. Sun H, Li XB, Meng Y, Fan L, Li M, Fang J. TRAF6 upregulates expression of HIF-1alpha and promotes tumor angiogenesis. Cancer Res (2013) 73:4950–9. doi:10.1158/0008-5472.CAN-13-0370
56. Bett JS, Ibrahim AF, Garg AK, Kelly V, Pedrioli P, Rocha S, et al. The P-body component USP52/PAN2 is a novel regulator of HIF1A mRNA stability. Biochem J (2013) 451:185–94. doi:10.1042/BJ20130026
57. Quesada V, Diaz-Perales A, Gutierrez-Fernandez A, Garabaya C, Cal S, Lopez-Otin C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem Biophys Res Commun (2004) 314:54–62. doi:10.1016/j.bbrc.2003.12.050
58. Moniz S, Bandarra D, Biddlestone J, Campbell KJ, Komander D, Bremm A, et al. Cezanne regulates E2F1-dependent HIF2alpha expression. J Cell Sci (2015) 128:3082–93. doi:10.1242/jcs.168864
59. Scortegagna M, Subtil T, Qi J, Kim H, Zhao W, Gu W, et al. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains. J Biol Chem (2011) 286:27333–41. doi:10.1074/jbc.M111.218214
60. Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev (2014) 28:1429–44. doi:10.1101/gad.242131.114
61. Kim JO, Kim SR, Lim KH, Kim JH, Ajjappala B, Lee HJ, et al. Deubiquitinating enzyme USP37 regulating oncogenic function of 14-3-3gamma. Oncotarget (2015) 6:36551–76. doi:10.18632/oncotarget.5336
62. Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM, et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene (2013) 32:471–8. doi:10.1038/onc.2012.66
63. Wen YA, Stevens PD, Gasser ML, Andrei R, Gao T. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol Cell Biol (2013) 33:4594–605. doi:10.1128/MCB.00695-13
64. Guo J, Shinriki S, Su Y, Nakamura T, Hayashi M, Tsuda Y, et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma: possible link to acquired resistance to anti-VEGF therapy. Oncotarget (2014) 5:6353–64. doi:10.18632/oncotarget.2216
65. Luong le A, Fragiadaki M, Smith J, Boyle J, Lutz J, Dean JL, et al. Cezanne regulates inflammatory responses to hypoxia in endothelial cells by targeting TRAF6 for deubiquitination. Circ Res (2013) 112:1583–91. doi:10.1161/CIRCRESAHA.111.300119
66. An J, Mo D, Liu H, Veena MS, Srivatsan ES, Massoumi R, et al. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell (2008) 14:394–407. doi:10.1016/j.ccr.2008.10.007
67. Lee JG, Baek K, Soetandyo N, Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun (2013) 4:1568. doi:10.1038/ncomms2532
68. D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther (2015) 147:32–54. doi:10.1016/j.pharmthera.2014.11.002
69. Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet (2000) 25:160–5. doi:10.1038/76006
70. Takahashi M, Rapley E, Biggs PJ, Lakhani SR, Cooke D, Hansen J, et al. Linkage and LOH studies in 19 cylindromatosis families show no evidence of genetic heterogeneity and refine the CYLD locus on chromosome 16q12-q13. Hum Genet (2000) 106:58–65. doi:10.1007/s004399900227
71. Massoumi R. CYLD: a deubiquitination enzyme with multiple roles in cancer. Future Oncol (2011) 7:285–97. doi:10.2217/fon.10.187
72. Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet (2015) 47:31–8. doi:10.1038/ng.3166
73. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol (2007) 9:765–74. doi:10.1038/ncb1601
74. Wang Z, Song Q, Xue J, Zhao Y, Qin S. Ubiquitin-specific protease 28 is overexpressed in human glioblastomas and contributes to glioma tumorigenicity by regulating MYC expression. Exp Biol Med (Maywood) (2015) 241(3):255–64. doi:10.1177/1535370215595468
75. Guo G, Xu Y, Gong M, Cao Y, An R. USP28 is a potential prognostic marker for bladder cancer. Tumour Biol (2014) 35:4017–22. doi:10.1007/s13277-013-1525-1
76. Gu YY, Yang M, Zhao M, Luo Q, Yang L, Peng H, et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumour Biol (2015) 36(11):8379–87. doi:10.1007/s13277-015-3566-0
77. Hussain S, Bedekovics T, Chesi M, Bergsagel PL, Galardy PJ. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget (2015) 6(38):40704–18. doi:10.18632/oncotarget.5727
78. Abdelmaksoud-Dammak R, Saadallah-Kallel A, Miladi-Abdennadher I, Ayedi L, Khabir A, Sallemi-Boudawara T, et al. CpG methylation of ubiquitin carboxyl-terminal hydrolase 1 (UCHL1) and P53 mutation pattern in sporadic colorectal cancer. Tumour Biol (2015) 1–8. doi:10.1007/s13277-015-3902-4
79. Wang JH, Wei W, Guo ZX, Shi M, Guo RP. Decreased Cezanne expression is associated with the progression and poor prognosis in hepatocellular carcinoma. J Transl Med (2015) 13:41. doi:10.1186/s12967-015-0396-1
80. D’Arcy P, Linder S. Molecular pathways: translational potential of deubiquitinases as drug targets. Clin Cancer Res (2014) 20:3908–14. doi:10.1158/1078-0432.CCR-14-0568
81. Colombo M, Vallese S, Peretto I, Jacq X, Rain JC, Colland F, et al. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem (2010) 5:552–8. doi:10.1002/cmdc.200900409
Keywords: DUBs, ubiquitination, HIF, protein homeostasis, cancer
Citation: Schober AS and Berra E (2016) DUBs, New Members in the Hypoxia Signaling clUb. Front. Oncol. 6:53. doi: 10.3389/fonc.2016.00053
Received: 04 December 2015; Accepted: 22 February 2016;
Published: 09 March 2016
Edited by:
Christian Gomez, University of Mississippi Medical Center, USAReviewed by:
Valerio Donato, New York University Medical Center, USAKeith R. Laderoute, SRI International, USA
Copyright: © 2016 Schober and Berra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edurne Berra, eberra@cicbiogune.es