 Lucia Centurione
Lucia Centurione Francesca B. Aiello
Francesca B. Aiello- Department of Medicine and Aging Sciences, G. d’Annunzio University of Chieti-Pescara, Chieti, Italy
Double strand breaks (DSBs) induced by radiotherapy are highly cytotoxic lesions, leading to chromosomal aberrations and cell death. Ataxia-telangiectasia-mutated (ATM)-dependent DNA-damage response, non-homologous end joining, and homologous recombination pathways coordinately contribute to repairing DSBs in higher eukaryotes. It is known that the expression of DSB repair genes is increased in tumors, which is one of the main reasons for radioresistance. The inhibition of DSB repair pathways may be useful to increase tumor cell radiosensitivity and may target stem cell-like cancer cells, known to be the most radioresistant tumor components. Commonly overexpressed in neoplastic cells, cytokines confer radioresistance by promoting proliferation, survival, invasion, and angiogenesis. Unfortunately, tumor irradiation increases the expression of various cytokines displaying these effects, including transforming growth factor-beta and interleukin-6. Recently, the capabilities of these cytokines to support DNA repair pathways and the ATM-dependent DNA response have been demonstrated. Thrombopoietin, essential for megakaryopoiesis and very important for hematopoietic stem cell (HSC) homeostasis, has also been found to promote DNA repair in a highly selective manner. These findings reveal a novel mechanism underlying cytokine-related radioresistance, which may be clinically relevant. Therapies targeting specific cytokines may be used to improve radiosensitivity. Specific inhibitors may be chosen in consideration of different tumor microenvironments. Thrombopoietin may be useful in fending off irradiation-induced loss of HSCs.
Introduction
Approximately half of cancer patients receive radiation as part of the treatment (1). Radiation sensitivity is influenced by neoplastic cell proliferation or quiescence status, resistance to apoptosis, levels of free-radical scavengers, and the ability to repair highly cytotoxic DNA double strand breaks (DSBs) caused by radiation therapy (RT) (2–4). DSBs are the main contributors to RT-induced cell killing through the formation of chromosomal aberrations that lead to cell death. Improperly repaired DSBs increase genomic instability, chromosomal translocation, and cancer risk (1). Thus, the ability to repair DSBs in cancer cells confers radioresistance, while RT reaching normal cells induces, among other side effects, the development of secondary malignancies. Biological approaches exploiting differences in cellular responses to RT between tumor and normal cells are desirable to specifically radiosensitize tumor cells and protect normal cells.
A role of cytokines in promoting tumorigenesis is recognized as an essential component in all tumors (5). Cytokines promote proliferation, survival, invasion, and angiogenesis, which confer tumor cell radioresistance (6). Although RT induces DNA damage, it also upregulates the expression of various interleukins (IL), including IL-1, IL-6, IL-8, transforming growth factor-beta (TGF-β), and tumor necrosis factor (TNF) (6), which, through the activity of the transcription factor nuclear factor κB (NF- κB), further increases the expression of IL-6 and IL-8 (7). Early studies in mice demonstrated the radioprotective effect of IL-6 (8). High levels of serum IL-6 produced by neoplastic plasma cells and bone marrow stromal cells in multiple myeloma are associated with poor prognosis (9, 10). Lung tumors express IL-6 and the transcription factor Stat3 that mediates IL-6-dependent proliferation and survival effects (11). In these tumors and in squamous esophageal carcinoma, Stat3 overexpression confers radioresistance (11–13). Overexpression of TGF-β is associated with aggressive tumor growth in head and neck cancer and breast and prostate cancer (14, 15). We observed TGF-β expression in all non-small-cell (NSC) lung tumors tested (16). TGF-β inhibitors increase radiosensitivity in breast cancer and glioblastoma in animal experimental models (17, 18). TPO regulates megakaryopoieis and supports hematopoietic stem cell (HSC) quiescence and expansion post-transplantation (19). Importantly, mutations resulting in constitutive activation of the TPO receptor are involved in myeloproliferative neoplasms (19). The effects of TGF-β, IL-6, and TPO on DSB repair (summarized in Table 1) will be the focus of this mini-review.
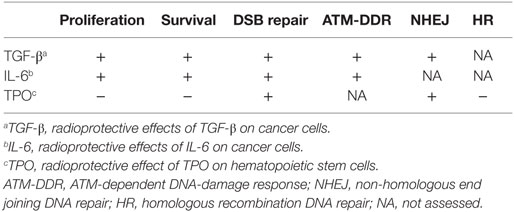
Table 1. Radioprotective effects induced by TGF-β, IL-6, and TPO.
In mammalian cells, non-homologous end joining (NHEJ) and homologous recombination (HR) pathways repair RT-induced DSBs. They differ in the requirement of a homologous DNA template and in the fidelity of the repair. The HR pathway utilizes DNA sequences of the undamaged chromatid as a template (20, 21). Thus, it is an accurate form of repair that functions in the late S and G2 phase of the cell cycle when an identical sister chromatid is available. The NHEJ pathway promotes direct ligation of the DSBs in all phases of the cell cycle. This is an error-prone mechanism that may result in insertions, deletions, or substitutions at the break sites and translocations, when DSBs from different chromosomes are joined.
Different subtypes of these pathways and the ataxia-telangiectasia-mutated (ATM)-dependent DNA-damage response to DSBs have been thoroughly reviewed elsewhere (20–25). Here, pathways utilized by eukaryotic cells in response to RT will be only briefly summarized.
NHEJ Pathway
The NHEJ pathway is considered the major repair pathway for DSBs in human cells (26) and is divided into three stages: (1) end detection and tethering, (2) processing, and (3) ligation (22). In the first step, the heterodimeric protein Ku70/Ku80 binds to DSB ends, encircles the DNA duplex, and recruits the DNA protein kinase catalytic subunit (PKcs), a member of the phosphoinositide 3 (PI3)-kinase-like family, which contacts the DNA termini. The binding of PKcs promotes the tethering of the two ends allowing two DNA PKcs molecules to interact across DSBs in a “synaptic complex.” This promotes the phosphorylation in “trans” across the DSB and the autophosphorylation of PKcs (20) which induce conformational changes and improve the access of processing proteins and their functions (22). In the second phase, non-ligatable ends are processed to remove blocking end groups and damaged DNA, or to fill in gaps, thus, this is an error-prone process. Different enzymes are required; the most important is the NHEJ-specific nuclease Artemis, whose activity is regulated by PKcs and ATM kinase-mediated phosphorylation (22, 26). Other enzymes, also involved in base excision and nucleotide excision DNA repair pathways, participate in processing, indicating that cells have evolved to a coordinated cascade of events to be protected by DNA damage. These include the phosphatase polynucleotide kinase and exonucleases Exo1 and WRN (21, 22). WRN is mutated in Werner syndrome, characterized by premature aging and genomic instability (27). The nucleotide gaps are repaired by two members of the X family polymerases, pol λ and pol μ.
Finally, ligation of DNA ends is carried out by ligase IV, bound to the scaffolding X-ray cross-complementing protein (XRCC)4, which stabilizes the ligase and stimulates its activity (20–22).
The XRCC4-like factor (XLF) is required to promote ligation of blunt ends and mismatched non-cohesive ends, and to re-adenylate the ligase, a necessary step for ligation (22). Mutations inactivating XLF or ligase IV are associated with growth retardation, radiation sensitivity, and immunodeficiency (28, 29). Noteworthy, the NHEJ pathway is critical in the repair of physiological DSBs created during immunoglobulin V(D)J recombination and class switch recombination. Thus, patients lacking normal NHEJ are sensitive to radiation and immunodeficiency (30).
HR Pathways
Homologous recombination pathways require undamaged homologous DNA to repair DSBs. In general, HR can also be divided into three stages: presynapsis, synapsis, and postsynapsis (21). During presynapsis, an extensive and complex 5′ to 3′ resection of broken DNA ends occurs to generate 3′ ended single strand DNA.
The heterotrimeric complex MRN formed by Mre11, NBS 1, and Rad 50 cooperates with the C-terminal binding protein interacting protein (CtIp) to remove about 100 nt (23). MRN is also an initial DSB sensor and unwinds DNA ends (25, 31). Breast cancer-associated protein (BRCA)1 is recruited at this site and regulates this step activating end resection and promoting the activity of CtIp (1).
In the second stage, the heterotrimeric replication protein A (RPA), also involved in the nucleotide excision repair pathway (21), binds to single strand tails preventing internal base pairing (23). Then, RPA is replaced by Rad 51 recombinase in conjunction with BRCA2 and a group of proteins known as Rad 51 paralogs required for RPA replacement (1) (Rad 51B, C, and D, XRCC2, and XRCC3), which yield the Rad51 nucleoprotein filament (1, 21, 23). This filament invades the double strand DNA molecule to search sequence homology and forms a structure termed displacement loop (D-loop). Immunofluorescence staining detects Rad51 nucleoproteins as distinct subnuclear foci (23). Following Rad51 removal from the 3′ end to reveal the 3′-OH group, necessary for priming, DNA synthesis starts elongating the invading strand and forming a cross-shaped structure: the Holliday junction (1, 21, 23). HR repair can proceed differently from this point (1). In the synthesis-dependent strand-annealing (SDSA) repair, after DNA synthesis, performed by polymerase δ, the new DNA strand is displaced and re-ligated with the original DNA, thus, SDSA repair is not associated with crossover. Alternatively, in the double strand break repair (DSBR), two independent strand invasions from both DSB ends are followed by simultaneous DNA synthesis (performed by polymerase η), and generate a double Holliday junction (1, 23). Specific enzymes cleave this junction, and depending on which pair of strands is cut the DSBR pathway can lead to a crossover or non-crossover outcome (1, 21, 23).
ATM-Dependent DNA-Damage Response
In response to DNA damage, including RT, eukaryotic cells activate cell cycle checkpoints: they arrest the cell cycle allowing DNA repair or triggering apoptosis if repair is impossible (25). Kinase-dependent signaling networks regulate checkpoint activation. In parallel to promoting cell cycle arrest, checkpoint signaling mediates the recruitment of DNA repair pathways (25). ATM kinase, containing a PI3 kinase-like sequence, is encoded by the ATM gene, mutated in patients affected by ataxia-telangiectasia (24, 32), whose cells exhibit decreased survival and increased radiation sensitivity (24). ATM signaling is induced by DSBs and also by chromatin perturbations that do not directly cause DSBs (33). ATM exists as an inactive multimer that dissociates into active monomers upon a conformational change associated with autophosphorylation and acetylation by the acetyl transferase Tip 60, which binds the above-mentioned MRN complex (25). MRN recruits ATM at the DSB sites crucially enhancing its activity (31, 34). The minor histone H2A variant contributes to the suppression of genomic instability preventing the separation of cleaved DNA strands (35). ATM phosphorylates H2AX histones surrounding DSBs and a multitude of substrates, including the MRN complex (25). Substrates of ATM also include the checkpoint kinase (CHK)2, CHK1, and p53 (25, 36). Activation of CHK1 and CHK2 contributes to cell cycle arrest at the G1/S and G2/M phases of the cell cycle (25). CHK2 also promotes p53-dependent and -independent apoptosis pathways (37). Thus, ATM activity regulates cell cycle arrest (CHK1, CHK2, p53), apoptosis (p53 and CHK2), and DNA repair (MRN complex and H2AX). H2AX phosphorylation on serine 139 (γH2AX) can be visualized by immunofluorescence as discrete spots or foci or by Western blot analysis. Detection of γH2AX is widely used as an indicator of the incidence of DSBs (38).
TGF-β, IL-6, and TPO Differently Influence Radioresistance and DNA Repair
Transforming growth factor-beta is a pleiotropic cytokine that regulates proliferation, angiogenesis, and immune responses (39). In normal epithelial cells, it negatively regulates cell cycle progression by activating cyclin-dependent kinase inhibitors, such as p15 and p21 (40). By contrast, it promotes cancer progression and metastasis by a variety of mechanisms, including induction of angiogenesis, cell motility and invasion, and repression of the immune system (39). In vitro studies, performed in 2006 showed that the ATM-dependent DNA-damage response to irradiation was impaired in TGFβ-deficient murine mammary cells (41). Subsequently, in human breast cancer cell lines, it was shown that inhibition of TGF-β signaling using the TGF-β type I receptor kinase inhibitor Ly 364947 decreased the clonogenic cell growth prior to irradiation, and blocked irradiation-induced γH2AX foci formation and p53 phosphorylation (18). Moreover, an anti-TGF-β antibody decreased the number of irradiation-induced γH2AX foci and the growth of neoplastic cells injected in immunocompromised mice (18). Similar results were obtained by different laboratories using TGF-β type I receptor kinase inhibitors and glioblastoma murine and human cell lines, and in murine experimental models (17, 42). In addition, an inhibitor was shown to decrease growth and apoptosis of glioblastoma cancer stem cell-like cells (CSCLC), as well as tumor invasion and angiogenesis (42). In murine prostate cancer, it was confirmed that irradiation increased TGF-β expression while its inhibition by a silencing vector increased the level of nuclear phospho-ATM (Figure 1) and the number of nuclear γH2AX foci (43). Recently, in human epidermoid carcinoma cells and in embryonic kidney cells, it was demonstrated that TGF-β pre-treatment not only protected the cell lines from irradiation-induced apoptosis and decreased the amount of nuclear γ-H2AX foci but also increased the expression of ligase IV and promoted the nuclear retention of Ku70/Ku80, ligase IV, and XRCC4 (44). SMAD proteins are intracellular mediators of TGF-β signaling. TGF-β stimulation leads to phosphorylation of SMAD2 and SMAD3, which form complexes with SMAD4 and regulate the transcription of target genes in the nucleus (39). RNA silencing of SMAD2/3 proteins confirmed that ligase IV levels depended on canonical SMAD-dependent signaling. Importantly, ligase IV RNA silencing decreased TGF-β-induced protection against irradiation, underlining the important role of NHEJ repair (44). These data indicate that TGF-β increases radioresistance by multiple mechanisms, including effects on DNA repair, and suggest that specific inhibitors administered before RT might improve radiosensitivity.
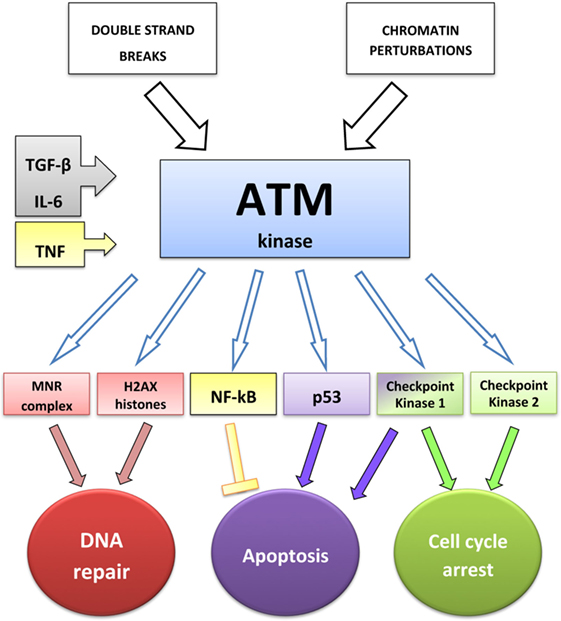
Figure 1. ATM-dependent DNA-damage response: interactions between ATM and cytokines. TGF-β increases the level of phospho-ATM (43), IL-6 increases the expression of both ATM and phospho-ATM (45), and TNF induces ATM activation, required to stimulate NF-κB transcriptional activity (46).
Interleukin-6 is a multifunctional cytokine involved in inflammatory processes; it stimulates acute phase protein synthesis, hematopoiesis, survival, and cell growth (12). It can cross the blood–brain barrier and the resulting synthesis of prostaglandin E2 in the hypothalamus changes the body temperature set point (12). Stat3 is the critical regulator of IL-6-dependent cell growth, differentiation, and survival signals. It promotes the transcription of pro-survival regulatory genes: c-myc, B-cell lymphoma (Bcl)-extra large (Bcl-XL), and myeloid cell leukemia 1 (Mcl-1) anti-apoptotic genes, and binds p53 inhibiting its function (12, 13). Transfection of dominant-negative Stat3 abolishes the pro-survival effect of IL-6 (47). IL-6 is involved in proliferation, survival, and differentiation of almost all tumors studied, and is overexpressed in multiple myeloma, oral squamous carcinoma, and in breast, ovarian, prostate, endometrial, colorectal, renal, and lung cancers (12).
Previous studies showed that the administration of anti-IL6 antibody in mice enhanced radiation-induced mortality (8). Other authors confirmed that irradiation enhanced IL-6 expression and showed that the increased growth and angiogenesis of murine hormone resistant versus hormone sensitive prostate cancer cells was attributable to higher IL-6 production (48). It has recently been demonstrated using human NSC lung cancer cell lines that following irradiation CD133+ CSCL-like cells proliferated and survived better than CD133− cells. Silencing of IL-6 reduced proliferation and survival in both groups of cells. IL-6 silencing in CD133+ cells resulted in a higher number of DSBs compared with CD133+ non-silenced cells 3 h after irradiation, indicating a difference in DNA repair. By contrast, no difference was observed between IL-6 silenced and non-silenced CD133− cells. The expression level of ATM, phosphorylated ATM, CHK2, and phosphorylated p53 was lower in IL-6 silenced CD133+ cells than in non-silenced CD133+ cells, whereas no difference was found between CD133− silenced and non-silenced cells. IL-6 upregulated the transcription of ATM (Figure 1) and, as expected, the expression of anti-apoptotic genes, such as Bcl-2 and Mcl-1 (45). These data indicate that IL-6 specifically affects the DNA-damage response in CSCL cells and suggest the hypothesis of other effects on DNA repair pathways. The evidence that IL-6 induces c-myc (49) lends support to this hypothesis. Radioresistance of nasopharyngeal carcinoma cells is dependent on c-myc-mediated overexpression of CHK1 and CHK2 genes, which display c-myc binding sites on their promoters (48). Moreover, c-myc transcriptional activity promotes NHEJ repair (50, 51), and its silencing in irradiated embryonal rhabdomyosarcoma cells increases the number of γH2AX foci and decreases the expression of DNA PKcs, Ku/70, and RAD51 (51). IL-6-targeted biological therapies are available. The human–mouse chimeric monoclonal antibody CNTO 328 has shown promising results in phase II clinical trials concerning patients with ovarian and renal cancers (12). Patients with multicentric Castleman disease treated with this antibody as a single agent showed high rates of clinical response (52). In addition, clinical trials are ongoing in ovarian cancer patients utilizing the humanized anti-IL-6 receptor monoclonal antibody Tocilizumab in combination with chemotherapy (53).
Myelosuppression and loss of HSCs are important side effects of RT. TPO, essential for megakaryopoiesis, increases proliferation, survival, quiescence, and expansion post-transplantation of HSCs (19). TPO receptor-deficient mice exhibit 10–20% of the normal HSC number. TPO has been shown to exhibit important and selective DNA repair promoting activity (54). HSCs from TPO receptor-deficient mice were more radiosensitive and exhibited decreased DSB repair than cells from wild-type mice. Removal of TPO but not of stem cell factor or Fms-related tyrosine kinase 3 ligand from the medium impaired DSB repair in normal HSC, underlining TPO specificity. Importantly, TPO increased PKcs phosphorylation and promoted NHEJ repair. The number of irradiation-induced Rad51 foci was not modified, suggesting that TPO had no effect on HR repair. HSCs from TPO-receptor-deficient mice showed genomic instability after irradiation, whereas TPO treatment of normal HSC before irradiation protected from genomic instability and improved HSC reconstitution capacity in secondary transplants. Surprisingly, TPO did not modify the cell cycle or survival of irradiated HSCs (54).
Other cytokines support tumor radioresistance, however, whether they promote DNA repair is unknown. These cytokines produced by neoplastic cells and in response to RT include IL-1, IL-8, and TNF (6). The role of IL-1 in enhancing radioresistance was known more than 20 years ago (55). IL-1 activates NF-κB, which mediates the expression of more than 200 genes promoting survival proliferation, invasion, and radioresistance (6). Interestingly, IL-1 may promote DNA repair, since it induces TGF-β expression (56, 57).
Interleukin-8, a pro-inflammatory chemokine enhances proliferation and survival of endothelial cells, leading to neoangiogenesis (58). High levels of IL-8 correlate with a poor prognosis in hepatocarcinoma, colon, and nasopharyngeal cancer (58, 59). In prostate cancer cells and in nasopharyngeal carcinoma, high IL-8 expression confers radioresistance (59, 60), mediated, in nasopharyngeal cancer, by PI3 kinase and Stat3-dependent signaling pathways. Importantly, a Stat3 inhibitor inhibited radiosensitivity (59, 61).
Tumor necrosis factor, a pro-inflammatory cytokine with potent antitumor effects binds to two structurally related but functionally distinct receptors, TNF receptors 1 and 2. Binding to these receptors initiates a complex array of signaling pathways (62, 63), including one that induce activation of NF-κB, leading to cell survival, while the other, through regulation of Fas-associated protein with death domain, can lead to apoptosis (62). Therefore, TNF-α affects radioresistance in a complex manner, cell type dependent, mainly influencing cell survival (62, 63). For instance, in human neuroblastoma cells TNF-α expression induced by RT resulted in sustained NF-κB activation, survival advantage, and radioprotection (64). The ATM kinase activity can promote NF-κB activation following various genotoxic stimuli, including irradiation (46, 65). In the human A549 lung epithelial cell line, TNF induced simultaneously DSBs due to free-radical formation and NF-κB activation (66). Interestingly, NF-κB activation depended on the activation of ATM, which was an unexpected nuclear damage response signal, activated by TNF (46, 65) (Figure 1). NF-κB inhibitors may represent novel radiosensitizing strategies targeting both TNF pro-survival signals and NF-κB-mediated IL-6 and IL-8 production induced by TNF (7, 62).
Conclusion
The recently demonstrated activity of cytokines on DNA repair pathways may be clinically relevant, suggesting that therapies targeting cytokines could be employed to improve the results of RT in very different contexts. TGF-β and IL-6 confer radioresistance using multiple mechanisms that may be desirable to counteract with specific inhibitors before RT administration. It should be taken into account that TGF-β effects on normal versus neoplastic cells are different, and sometimes opposite, whereas IL-6 has similar effects on normal and neoplastic cells.
Moreover, the different biological behavior of different tumors in terms of different cytokine overexpression should also be considered to obtain specific therapies.
TPO-induced DNA repair activity in irradiated HSCs appears impressively selective, thus, a short treatment to avoid side effects (67) may be useful to protect HSCs before irradiation in solid tumors.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anand Merchant, Chiara di Lisio and Chiara Cerletti for helpful suggestions.
References
1. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol (2013) 3:113. doi:10.3389/fonc.2013.00113
2. Hittelman WN, Liao Y, Wang L, Milas L. Are cancer stem cells radioresistant? Future Oncol (2010) 6(10):1563–76. doi:10.2217/fon.10.121
3. Di Pietro R, Fang H, Fields K, Miller S, Flora M, Petricoin EC, et al. Peroxiredoxin genes are not induced in myeloid leukemia cells exposed to ionizing radiation. Int J Immunopathol Pharmacol (2006) 19(3):517–24.
4. Di Pietro R, Centurione L, Santavenere E, Centurione MA, Sanità Di Toppi G, Zamai L, et al. Ionizing radiation-induced apoptosis and DNA repair in murine erythroleukemia cells. Scanning Microsc (1996) 10(1):253–9.
5. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi:10.1016/j.cell.2010.01.025
6. Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol (2010) 80(12):1904–14. doi:10.1016/j.bcp.2010.06.039
7. Epanchintsev A, Shyamsunder P, Verma RS, Lyakhovich A. IL-6, IL-8, MMP-9 are overexpressed in Fanconi anemia cells through a NKkB/TNF-a dependent mechanism. Mol Carcinog (2014) 54:1686–99. doi:10.1002/mc.222.40
8. Neta R, Perlstein R, Vogel SN, Ledney GD, Abrams J. Role of interleukin 6 (IL-6) in protection from lethal irradiation and in endocrine responses to IL-1 and tumor necrosis factor. J Exp Med (1992) 175(3):689. doi:10.1084/jem.175.3.689
9. Bataille R, Harousseau JL. Multiple myeloma. N Engl J Med (1997) 336(23):1657–64. doi:10.1056/NEJM199706053362307
10. Klein B, Bataille R. Cytokine network in human multiple myeloma. Hematol Oncol Clin North Am (1992) 6(2):273–84.
11. Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small-cell lung cancer. Cancers (Basel) (2014) 6(2):708–22. doi:10.3390/cancers6020708
12. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev (2012) 38(7):904–10. doi:10.1016/j.ctrv.2012.04.007
13. Leu CM, Wong FH, Chang C, Huang SF, Hu CP. Interleukin-6 acts as an antiapoptotic factor in human esophageal carcinoma cells through the activation of both STAT3 and mitogen-activated protein kinase pathways. Oncogene (2003) 22(49):7809–18. doi:10.1038/sj.onc.1207084
14. Drabsch Y, ten Dijke P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev (2012) 31(3–4):553–68. doi:10.1007/s10555-012-9375-7
15. Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol (2013) 25(1):76–84. doi:10.1097/CCO.0b013e32835b6371
16. Colasante A, Mascetra N, Brunetti M, Lattanzio G, Diodoro M, Caltagirone S, et al. Transforming growth factor beta 1, interleukin-8 and interleukin-1, in non-small-cell lung tumors. Am J Respir Crit Care Med (1997) 156(3 Pt 1):968–73. doi:10.1164/ajrccm.156.3.9701122
17. Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res (2012) 72(16):4119–29. doi:10.1158/0008-5472
18. Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res (2011) 7(21):6754–65. doi:10.1158/1078-0432
19. Hitchcock IS, Kaushansky K. Thrombopoietin from beginning to end. Br J Haematol (2014) 165(2):259–68. doi:10.1111/bjh.12772
20. Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J (2009) 417(3):639–50. doi:10.1042/BJ20080413
21. Dexheimer TS. DNA repair pathways and mechanisms. In: Matthews LA, Cabarcas SM, Hurt EM, editors. DNA Repair of Cancer Stem Cells. Netherlands: Springer (2013). p. 19–32.
22. Wang C, Lees-Miller SP. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys (2013) 86(3):440–9. doi:10.1016/j.ijrobp.2013.01.011
23. Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol (2014) 6(9):a016428. doi:10.1101/cshperspect.a016428
24. Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol (2005) 70:99–109. doi:10.1101/sqb.2005.70.002
25. Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol (2009) 21(2):245–55. doi:10.1016/j.ceb.2009.01.018
26. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol (2008) 9(4):297–308. doi:10.1038/nrm2351
27. Brosh RM Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res (2005) 35(22):7527–44. doi:10.1093/nar/gkm1008
28. Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche MC, Sanal O, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell (2006) 124(2):287–99. doi:10.1016/j.cell.2005.12.030
29. O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell (2001) 8(6):1175–85. doi:10.1016/S1097-2765(01)00408-7
30. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi:10.1146/annurev.biochem.052308.093131
31. Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science (2005) 308(5721):551–4. doi:10.1126/science.1108297
32. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science (1995) J268(5218):1749–53. doi:10.1126/science.7792600
33. Bakkenist CJ, Kastan MB. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst) (2015) 36:8–12. doi:10.1016/j.dnarep.2015.09.002
34. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature (2005) 434(7033):605–11. doi:10.1038/nature03442
35. Yin B, Savic V, Juntilla MM, Bredemeyer AL, Yang-Iott KS, Helmink BA, et al. Histone H2AX stabilizes broken DNA strands to suppress chromosome breaks and translocations during V(D)J recombination. J Exp Med (2009) 206(12):2625–39. doi:10.1084/jem.20091320
36. Gatei M, Sloper K, Sorensen C, Syljuäsen R, Falck J, Hobson K, et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem (2003) 278(17):14806–11. doi:10.1074/jbc.M210862200
37. Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol (2014) 6(6):442–57. doi:10.1093/jmcb/mju045
38. Rao VA, Agama K, Holbeck S, Pommier Y. Batracylin (NSC 320846), a dual inhibitor of DNA topoisomerases I and II induces histone gamma-H2AX as a biomarker of DNA damage. Cancer Res (2007) 67(20):9971–9. doi:10.1158/0008-5472.CAN-07-0804
39. Tian M, Neil JR, Schiemann WP. Transforming growth factor-β and the hallmarks of cancer. Cell Signal (2011) 23(6):951–62. doi:10.1016/j.cellsig.2010.10.015
40. Iavarone A, Massagué J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature (1997) 387(6631):417–22. doi:10.1038/387417a0
41. Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick AB, Lavin MJ, et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res (2006) 66(22):10861–9. doi:10.1158/0008-5472.CAN-06-2565
42. Zhang M, Kleber S, Röhrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res (2011) 71(23):7155–67. doi:10.1158/0008-5472
43. Wu CT, Hsieh CC, Yen TC, Chen WC, Chen MF. TGF-β1 mediates the radiation response of prostate cancer. J Mol Med (Berl) (2015) 93(1):73–82. doi:10.1007/s00109-014-1206-6
44. Kim MR, Lee J, An YS, Jin YB, Park IC, Chung E, et al. TGFβ1 protects cells from γ-IR by enhancing the activity of the NHEJ repair pathway. Mol Cancer Res (2015) 13(2):319–29. doi:10.1158/1541-7786
45. Chen Y, Zhang F, Tsai Y, Yang X, Yang L, Duan S, et al. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat Oncol (2015) 10:227. doi:10.1186/s13014-015-0534-1
46. Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science (2006) 311(5764):1141–6. doi:10.1126/science.1121513
47. Bonner JA, Trummell HQ, Willey CD, Plants BA, Raisch KP. Inhibition of STAT-3 results in radiosensitization of human squamous cell carcinoma. Radiother Oncol (2009) 92(3):339–44. doi:10.1016/j.radonc.2009.06.022
48. Wu CT, Chen MF, Chen WC, Hsieh CC. The role of IL-6 in the radiation response of prostate cancer. Radiat Oncol (2013) 8:159. doi:10.1186/1748-717X-8-159
49. Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB, et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res (2013) 73(3):1219–31. doi:10.1158/0008-5472
50. Cui F, Fan R, Chen Q, He Y, Song M, Shang Z, et al. The involvement of c-Myc in the DNA double-strand break repair via regulating radiation-induced phosphorylation of ATM and DNA-PKcs activity. Mol Cell Biochem (2015) 406(1–2):43–51. doi:10.1007/s11010-015-2422-2
51. Gravina GL, Festuccia C, Popov VM, Di Rocco A, Colapietro A, Sanità P, et al. c-Myc sustains transformed phenotype and promotes radioresistance of embryonal rhabdomyosarcoma cell lines. Radiat Res (2016) 185(4):411–22. doi:10.1667/RR14237.1
52. Kurzrock R, Voorhees PM, Casper C, Furman RR, Fayad L, Lonial S, et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res (2013) 19(13):3659–70. doi:10.1158/1078-0432
53. Dijkgraaf EM, Santegoets SJ, Reyners AK, Goedemans R, Wouters MC, Kenter GG, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann Oncol (2015) 26(10):2141–9. doi:10.1093/annonc/mdv309
54. de Laval B, Pawlikowska P, Petit-Cocault L, Bilhou-Nabera C, Aubin-Houzelstein G, Souyri M, et al. Thrombopoietin-increased DNA-PK-dependent DNA repair limits hematopoietic stem and progenitor cell mutagenesis in response to DNA damage. Cell Stem Cell (2013) 12(1):37–48. doi:10.1016/j.stem.2012.10.012
55. Neta R, Oppenheim JJ, Schreiber RD, Chizzonite R, Ledney GD, MacVittie TJ. Role of cytokines (interleukin 1, tumor necrosis factor, transforming growth factor beta) in natural and lypopolisaccaride-enhanced radioresistance. J Exp Med (1991) 173(5):1177–82. doi:10.1084/jem.173.5.1177
56. Bristol LA, Ruscetti FW, Brody DT, Durum SK. IL-1 alpha induces expression of active transforming growth factor-beta in nonproliferating T cells via a post-transcriptional mechanism. J Immunol (1990) 145(12):4108–14.
57. Luo DD, Fielding C, Phillips A, Fraser D. Interleukin-1 beta regulates proximal tubular cell transforming growth factor beta-1 signalling. Nephrol Dial Transplant (2009) 24(9):2655–65. doi:10.1093/ndt/gfp208
58. Gales D, Clark C, Manne U, Samuel T. The chemokine CXCL8 in carcinogenesis and drug response. ISRN Oncol (2013) 2013:859154. doi:10.1155/2013/859154
59. Qu JQ, Yi HM, Ye X, Li LN, Zhu JF, Xiao T, et al. MiR-23a sensitizes nasopharyngeal carcinoma to irradiation by targeting IL-8/Stat3 pathway. Oncotarget (2015) 6(29):28341–56. doi:10.18632/oncotarget.5117
60. Xu Y, Fang F, St Clair DK, St Clair WH. Inverse relationship between PSA and IL-8 in prostate cancer: an insight into a NF-κB-mediated mechanism. PLoS One (2012) 7(3):e32905. doi:10.1371/journal.pone.0032905
61. Qu JQ, Yi HM, Ye X, Zhu JF, Yi H, Li LN, et al. MiRNA-203 reduces nasopharyngeal carcinoma radioresistance by targeting IL8/AKT signaling. Mol Cancer Ther (2015) 14(11):2653–64. doi:10.1158/1535-7163.MCT-15-0461
62. Ding M, Zhang E, He R, Wang X. Newly developed strategies for improving sensitivity to radiation by targeting signal pathways in cancer therapy. Cancer Sci (2013) 104(11):1401–10. doi:10.1111/cas.12252
63. Du W, Erden O, Pang Q. TNF-α signaling in Fanconi anemia. Blood Cells Mol Dis (2014) 52(1):2–11. doi:10.1016/j.bcmd.2013.06.005
64. Veeraraghavan J, Natarajan M, Aravindan S, Herman TS, Aravindan N. Radiation-triggered tumor necrosis factor (TNF) alpha-NFkappaB cross-signaling favors survival advantage in human neuroblastoma cells. J Biol Chem (2011) 286(24):21588–600. doi:10.1074/jbc.M110.193755
65. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell (2009) 36(3):365–78. doi:10.1016/j.molcel.2009.09.032
66. Fang L, Choudhary S, Zhao Y, Edeh CB, Yang C, Boldogh I, et al. ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res (2014) 42(13):8416–32. doi:10.1093/nar/gku529
Keywords: radioresistance, cytokines, DNA repair, ATM-dependent DNA-damage response, cancer
Citation: Centurione L and Aiello FB (2016) DNA Repair and Cytokines: TGF-β, IL-6, and Thrombopoietin as Different Biomarkers of Radioresistance. Front. Oncol. 6:175. doi: 10.3389/fonc.2016.00175
Received: 20 May 2016; Accepted: 11 July 2016;
Published: 22 July 2016
Edited by:
Joel S. Greenberger, University of Pittsburgh, USAReviewed by:
Eddy S. Yang, University of Alabama at Birmingham, USAHyuk-Jin Cha, Sogang University, South Korea
Copyright: © 2016 Centurione and Aiello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca B. Aiello, francesca.aiello@unich.it