Hirschsprung’s disease and associated congenital heart defects: a prospective observational study from a single institution
 Giulia Tuo1*
Giulia Tuo1*  Alessio Pini Prato2
Alessio Pini Prato2
 Maria Derchi1
Maria Derchi1
 Manuela Mosconi2
Manuela Mosconi2
 Girolamo Mattioli2,3
Girolamo Mattioli2,3  Maurizio Marasini1
Maurizio Marasini1
- 1Department of Pediatric Cardiology, Istituto Giannina Gaslini, Genoa, Italy
- 2Department of Pediatric Surgery, Istituto Giannina Gaslini, Genoa, Italy
- 3Department of Neuroscience, Ophthalmology, Rehabilitation, Genetics and Maternal-Infant Science (DINOGMI), University of Genoa, Genoa, Italy
Objective: To define the prevalence and characteristics of associated congenital heart diseases (CHDs) in patients with Hirschsprung’s disease (HSCR).
Method: All patients with a histological diagnosis of HSCR admitted to our hospital between January 2010 and December 2013 were included in this prospective observational study and underwent cardiovascular screening. Cardiac anatomy was assessed by a segmental echocardiographic approach. Measurements of aortic root and left ventricular dimensions, wall thickness, and function were obtained. CHDs requiring a percutaneous or surgical intervention were described as major heart diseases.
Results: One hundred thirty-three consecutive patients were enrolled at median age of 2.3 years. Eleven patients (8.3%) presented an associated heart disease. Moreover, five patients had mild dilatation of aortic root. Six out of 11 (4.5%) patients had a major CHDs requiring surgical repair.
Conclusion: Prevalence of associated CHDs was slightly higher than in previous papers, and mostly represented by septal defects. Four out of six patients with major heart disease had also a chromosomal anomaly. If we do not consider the subpopulation of patients with a chromosomal anomaly, cardiac defects were present in 3.8% of the patients. Based on these results, we suggest to perform routine echocardiogram in all Hirschsprung patients, with or without associated chromosomal syndromes.
Introduction
Hirschsprung’s disease (HSCR) is the commonest congenital gut motility disorder and is characterized by the absence of ganglion cells in a variable length of the distal gut. This results in absent peristalsis in the affected bowel and it represents a frequent cause of functional intestinal obstruction especially in the newborn period (1–17). It occurs as a consequence of premature arrest of cranio-caudal migration of neural crest-derived neuroblasts (NCN) in the hindgut and is therefore described as a neurocristopathy (1, 17–19). Alterations of heterogenous genetic pathways involved in the enteric nervous system development may interfere with the colonization process of NCN and represents a primary etiology both for HSCR and for the anomalies of other organs and systems that often come together (13, 20–23).
According to literature, HSCR may be associated with a chromosomal abnormality or additional congenital anomalies in approximately 20% of cases (9, 16, 20, 21). Associated congenital heart diseases (CHDs) have a reported incidence of around 5%. However, there could be an underestimation of the reported incidence both because most of the studies were retrospective and focused on genotype rather than on phenotype determination, and because usually there is a mild or late symptoms onset with subsequent missed or delayed diagnosis (9, 20, 21).
The aim of our study was to assess the prevalence and the phenotype of associated CHDs in patients with HSCR.
Materials and Methods
All consecutive HSCR patients admitted for further evaluation and/or surgical treatment to the G. Gaslini Institute between January 2010 and December 2013 were taken into account for this prospective observational study. Only those with a histological diagnosis of intestinal aganglionosis were included in the study and underwent cardiac screening (6). Exclusion criteria were uncertain HSCR diagnosis and/or refusal to sign the Informed Consent.
Enrolled patients and/or their parents were interviewed to collect personal and family history, length of aganglionosis, and any information regarding associated cardiac anomalies if already detected during previous phenotype screening.
Length-wise, four types of aganglionosis have been identified: short segment HSCR (S-HSCR) with aganglionosis extending up to the left descending colon, long segment HSCR (L-HSCR) with aganglionosis extending up to the right transverse/ascending colon, total colonic aganglionosis (TCA) with aganglionosis extending to the whole colon with or without small bowel involvement, and total intestinal aganglionosis (TIA) with aganglionosis extending over the jejunum with <20 cm of normoganglionic bowel (11).
For the purpose of our study, CHD was considered as “a gross structural abnormality of the heart or intrathoracic great vessels that is actually or potentially of functional significance,” as defined by Mitchell et al. in 1971. Moreover, CHD requiring a percutaneous or surgical intervention was described as major CHD (5, 7).
The cardiovascular screening included physical examination and an echocardiogram. Height, weight, and age were recorded in each patient as well as the non-invasive blood pressure measurement. Detailed echocardiograms were performed with a pulsed, continuous, and color-Doppler provided ultrasound system (iE33, Philips, Amsterdam, The Netherlands), using a 5 or 8 MHz transducer. Morphological variables were measured offline by two observers (Giulia Tuo and Maria Derchi) who used the so-called z-score for normalization of the cardiac structures dimensions to the body size (4, 15, 24).
Cardiac anatomy was routinely assessed by a segmental approach. Two-dimensional echocardiographic measurements included the end-sistolic diameter, respectively, of the aortic annulus, Valsalva sinuses, and ascending aorta. These dimensions were measured from the parasternal long-axis view with the “inner edge convention,” which uses the innermost bright edge reflection as a contour. The aortic root was considered dilated if the z-score was ≥2 and the degree of dilation was defined mild if the z-score was ≤3 (15). End-diastolic wall thickness and left ventricular internal dimensions were measured through the M-mode technique, which was performed also for calculating the shortening fraction. Diastolic ventricular function and valves function were assessed by both pulsed and continuous Doppler technique (2, 10, 12, 14).
For patients with a major CHD that required a trans-catheter or surgical treatment, we reported both clinical and echocardiographic details at the last follow-up examination.
Descriptive statistics were reported as percentages for categorical variables and as mean ± SD for continuous data. This study was approved by our institutional Ethics Committee and informed consent to view files, and echocardiographic reports was obtained by the parents.
Results
During the 5 years study period, 133 consecutive HSCR patients were admitted to the Department of Surgery of the Gaslini Institute and underwent cardiac screening. Median age at enrollment was 2.3 years (range 3 months to 25 years). Male to female ratio was 4:1. Twenty patients (15%) had ultralong HSCR (either TCA or TIA), 9 (7%) had L-HSCR, and 104 (78%) had S-HSCR. Eight HSCR patients had also Down’s syndrome (6%).
Eleven patients (8.3%) presented an associated CHD of whom six patients were already aware at the time of enrollment (Table 1). Nine out of 11 were affected by the S-HSCR, 2/11 by the TCA. 5/11 (45%) were also affected by Down’s syndrome, and 1/11 by Turner’s syndrome. CHDs were mostly represented by septal defects (atrial, ventricular, or atrioventricular septal defect) and patent ductus arteriosus (Table 1). The family history was always unremarkable for any CHD or arrhythmia. Six patients (4.5%) presented a major CHD. Four out of six were also carrying a chromosomal abnormality, and they all required a cardiac surgical procedure. At a mean follow-up of 9 years (±5.8 years), they were all asymptomatic and without any residual cardiac defects on echocardiogram.
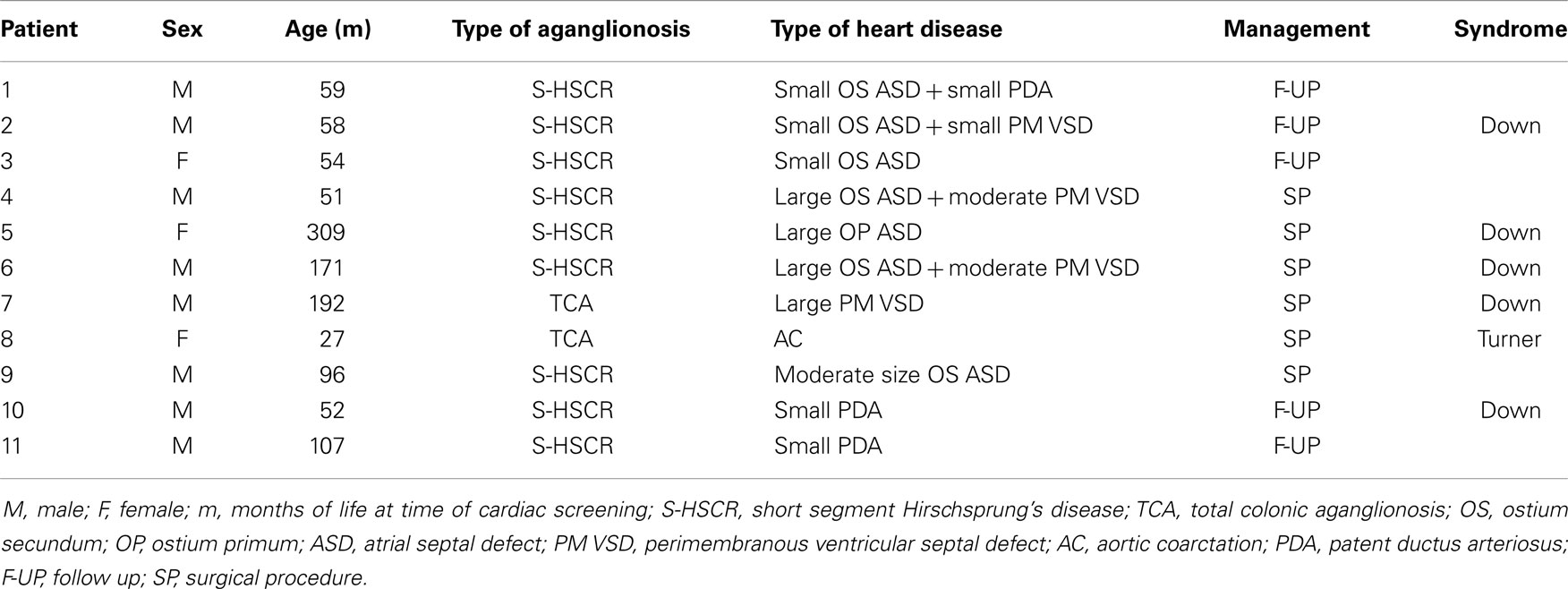
Table 1. Details of HSCR patients with associated congenital heart diseases.
In addition to the above-mentioned abnormalities, we observed mild dilatation of the aortic root in five patients. None of them had an associated chromosomal abnormality. They are all being followed up in the long term. None of the study population presented high blood pressure.
Discussion
Hirschsprung’s disease is the main genetic cause of functional intestinal obstruction with an incidence of 1/5000 live births. It is characterized by the absence of the enteric ganglia in a variable length of the intestine, and it is the consequence of multiple gene interactions, which affect the ability of the enteric neural crest cells to differentiate in the developing gut (1, 13, 17–19, 22, 23).
Although HSCR occurs as an isolated phenotype in at least 70% of cases, a number of associated congenital anomalies and syndromes have been retrospectively reported. In particular, cardiac defects occur with a mean incidence of 5% of cases of HSCR and, among them, septation defects (atrial, ventricular, or atrioventricular septal defect) and conotruncal developmental defects are the most often described (8, 9, 16, 20, 21, 25, 26).
Among our study population, we had an overall detection rate of associated CHDs of 8.3%, which is significantly higher than prevalence reported by previous authors as the result of retrospective assessments of surgical series or of systematic literature reviews (13, 20, 23). We may speculate that this discrepancy of prevalence is due by the absence or mild severity of symptoms related to the associated CHDs in most HSCR patients that may lead to a misdiagnosis and/or a delayed diagnosis. In fact, 5/11 (45%) patients were diagnosed with an associated CHD only at the time of our cardiac screening.
However, we could confirm literature data regarding the type of detected CHDs, as they were mostly represented by septal defects. This is in accordance to the embryologic role of neural crest-derived stem cells in the development of both ENS and cardiac outflow septation (9, 13, 18, 20, 21). Noteworthy, none of our patients presented conotruncal heart defects whose pathogenesis is similarly related to the abnormal neural crest cell proliferation and migration. On the other hand, five patients showed a mild dilatation of the aortic root. To the best of our knowledge, this echocardiographic finding has never been reported before and we think that it would be worthy of further investigations for understanding if there is some correlation between this aortic wall anomaly and the multiple gene mutations involved in HSCR.
Six out of 11 patients (5.4%) had a major CHD of whom 4 had also an associated chromosomal abnormality. The overall incidence of Down’s syndrome in our study population was 6% (8/133). Five out of eight patients (62%) with HSCR and Down’s syndrome presented also an associated CHD that required a surgical intervention in three cases. In 2009, the 30-years retrospective nationwide survey carried out in Japan showed an increased incidence of Down’s syndrome in patients affected by HSCR and interestingly also an increased incidence of associated cardiac anomalies in the last 10 years (27). We believe it is worth noting that Down’s syndrome patients affected by HSCR presented a higher incidence of associated CHD than that previously reported in the overall Down’s syndrome population (mean 40%) (3, 27).
If we do not consider HSCR patients with Down’s syndrome, CHDs were still present in 3.8% of the study population, which means that the prevalence of CHDs in isolated HSCR patients is at least three times that of the general population (5, 7).
On the ground of these considerations, we suggest to investigate all HSCR patients, regardless of sex, length of aganglionosis, and associated syndromes, in order to exclude a possible associated cardiac malformation. In particular, we suggest to routinely perform an ECG and an echocardiogram in all patients admit for further evaluation and/or treatment of HSCR.
Our institution is known as a referral center in Italy for HSCR and other intestinal dysganglionoses that could lead to an intrinsic subsequent risk of selection BIAS. This concern is supported by the twofolds higher than expected incidence of ultralong forms of the disease (17.9%). Nonetheless, the prevalence of Down’s syndrome (6%) as well as the male to female ratio (4:1) are coherent with literature data and confirm that our series is representative of the whole HSCR population.
To conclude, our study demonstrated a higher than previously reported prevalence of associated cardiac anomalies in HSCR patients. A better knowledge of clinical phenotype prompted the implementation of an updated diagnostic algorithm aimed at improving the outcome and the quality of life of our patients. In fact, early diagnosis of associated CHDs allows prompt establishment of adequate treatment and prevention of possible complications related to a delayed diagnosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research project was founded by the Italian Ministry of Health with Young Research Grant (Bando Giovani Ricercatori GR-2008-1135082) and Cinque per Mille and Ricerca Corrente funding. The funding organization had no role in study design, in data collection, management, and analysis, and in the decision to submit the manuscript for publication.
References
1. Dasgupta R, Langer JC. Hirschsprung disease. Curr Probl Surg (2004) 41(12):942–88. doi:10.1067/j.cpsurg.2004.09.004
2. Devereux RB, Lutas EM, Casale PN, Kligfield P, Eisenberg RR, Hammond IW, et al. Standardization of M-mode echocardiographic left ventricular anatomic measurements. J Am Coll Cardiol (1984) 4(6):1222–30. doi:10.1016/S0735-1097(84)80141-2
3. Freeman SB, Taft LF, Kenneth JD, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet (1998) 80(3):213–7. doi:10.1002/(SICI)1096-8628(19981116)80:3<213::AID-AJMG6>3.3.CO;2-#
4. Henry WL, Ware J, Gardin JM, Hepner SI, McKay J, Weiner M. Echocardiographic measurements in normal subjects. Growth related changes that occur between infancy and early adulthood. Circulation (1978) 57(2):278–85. doi:10.1161/01.CIR.57.2.278
5. Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol (2002) 39(12):1890–900. doi:10.1016/S0735-1097(02)01886-7
6. Martucciello G, Pini Prato A, Puri P, Holschneider AM, Meier-Ruge W, Jasonni V, et al. Controversies concerning diagnostic guidelines for anomalies of the enteric nervous system: a report from the fourth International Symposium on Hirschsprung’s disease and related neurocristopathies. J Pediatr Surg (2005) 40(10):1527–31. doi:10.1016/j.jpedsurg.2005.07.053
7. Mitchell SC, Korones SB, Berendes HW. Congenital heart disease in 56,109 births. Incidence and natural history. Circulation (1971) 43(3):323–32. doi:10.1161/01.CIR.43.3.323
8. Moore SW, Rode H, Millar AJ, Albertyn R, Cywes S. Familial aspects of Hirschsprung disease. Eur J Pediatr Surg (1991) 1(2):97–107. doi:10.1055/s-2008-1042468
9. Moore SW. The contribution of associated congenital anomalies in understanding Hirschsprung’s disease. Pediatr Surg Int (2006) 22(4):305–15. doi:10.1007/s00383-006-1655-2
10. Nidorf SM, Picard MH, Triulzi MO, Thomas JD, Newell J, King ME, et al. New perspectives in the assessment of cardiac chamber during devolopment and adulthood. J Am Coll Cardiol (1992) 19(5):983–8. doi:10.1016/0735-1097(92)90282-R
11. Nihoul-Feketè C, Ricour C, Martelli H, Lortat Jacob S, Pellerin D. Total colonic aganglionosis (with or without ileal involvement): a review of 27 cases. J Pediatr Surg (1986) 21(3):251–4. doi:10.1016/S0022-3468(86)80847-8
12. O’Leary PW, Durongpisitkul K, Cordes TM, Bailey KR, Hagler DJ, Tajik J, et al. Diastolic ventricular function in children: a Doppler echocardiographic study establishing normal values and predictors of increased ventricular end-diastolic pressure. Mayo Clin Proc (1998) 73(7):616–28. doi:10.4065/73.7.616
13. Panza E, Knowles CH, Graziano C, Thapar N, Burns AJ, Seri M, et al. Genetics of human enteric neuropathies. Prog Neurobiol (2012) 96(2):176–89. doi:10.1016/j.pneurobio.2012.01.001
14. Pearlman JD, Triulzi MO, King ME, Newell J, Weyman AE. Limits of normal left ventricular dimensions in growth and development: analysis of dimensions and variance in the two-dimensional echocardiograms of 268 normal healthy subjects. J Am Coll Cardiol (1988) 12(6):1432–41. doi:10.1016/S0735-1097(88)80006-8
15. Pettersen MD, Skeens ME, Humes RA. Regression equations for calculation of Z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: an echocardiographic study. J Am Soc Echocardiogr (2008) 21(8):922–34. doi:10.1016/j.echo.2008.02.006
16. Pini Prato A, Musso M, Ceccherini I, Mattioli G, Giunta C, Ghiggeri GM, et al. Hirschsprung disease and congenital anomalies of the kidney and urinary tract, a novel syndromic association. Medicine (Baltimore) (2009) 88(2):83–90. doi:10.1097/MD.0b013e31819cf5da
17. Kenny SE, Tam PKH, Garcia-Barcelo M, Phil M. Hirschprung’s disease. Semin Paediatr Surg (2010) 19(3):194–200. doi:10.1053/j.sempedsurg.2010.03.004
18. Bolande RP. The neurocristopathies; a unifying concept of disease arising in neural crest maldevelopment. Hum Pathol (1973) 5(4):409–29. doi:10.1016/S0046-8177(74)80021-3
19. Whitehouse F, Kernohan J. Myenteric plexuses in congenital megacolon; study of 11 cases. Arch Intern Med (1948) 82(1):75. doi:10.1001/archinte.1948.00220250085005
20. Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet (2008) 45(1):1–14. doi:10.1136/jmg.2007.053959
21. Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet (2001) 38(11):729–39. doi:10.1136/jmg.38.11.729
22. Taraviras S, Pachnis V. Development of the mammalian enteric nervous system. Curr Opin Genet Dev (1999) 9(3):321–7. doi:10.1016/S0959-437X(99)80048-3
23. Tam PK, Garcia-Barcelo M. Molecular genetics of Hirschsprung’s disease. Semin Pediatr Surg (2004) 13(4):236–48. doi:10.1053/j.sempedsurg.2004.10.011
24. Daubeney PE, Blackstone EH, Weintraub RG, Slavik Z, Scanlon J, Webber SA. Relationship of the dimension of cardiac structure to body surface size: an echocardiographic study in normal infants and children. Cardiol Young (1999) 9(4):402–10.
25. Ryan ET, Ecker JL, Christakis NA, Folkman J. Hirschprung’s disease: associated abnormalities and demography. J Pediatr Surg (1992) 27(1):76–81. doi:10.1016/0022-3468(92)90111-J
26. Scoll AM, Kirby ML. Signals controlling neural crest contributions to the heart. Interdiscip Rev Syst Biol Med (2009) 1(2):220–7. doi:10.1002/wsbm.8
Keywords: congenital heart disease, Hirschsprung’s disease, cardiac screening, neurocristopathy, echocardiography
Citation: Tuo G, Pini Prato A, Derchi M, Mosconi M, Mattioli G and Marasini M (2014) Hirschsprung’s disease and associated congenital heart defects: a prospective observational study from a single institution. Front. Pediatr. 2:99. doi: 10.3389/fped.2014.00099
Received: 02 July 2014; Paper pending published: 31 July 2014;
Accepted: 01 September 2014; Published online: 17 September 2014.
Edited by:
Jonathan Charles Jarvis, University of Liverpool, UKReviewed by:
Yves Durandy, Centre Chirurgical Marie Lannelongue, FranceJonathan Charles Jarvis, University of Liverpool, UK
Copyright: © 2014 Tuo, Pini Prato, Derchi, Mosconi, Mattioli and Marasini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Tuo, Cardiac Unit, Istituto Giannina Gaslini, Largo Gerolamo Gaslini 5, Genoa 16148, Italy e-mail: gtuo@libero.it